M(VI) Oxidation State Stabilization in Iron, Cobalt and Nickel Heteroligand Metal Chelates Containing 3,7,11,15-Tetraazaporphine and Two Axial Oxo Ligands: Quantum-Chemical Simulation



Abstract
1. Introduction

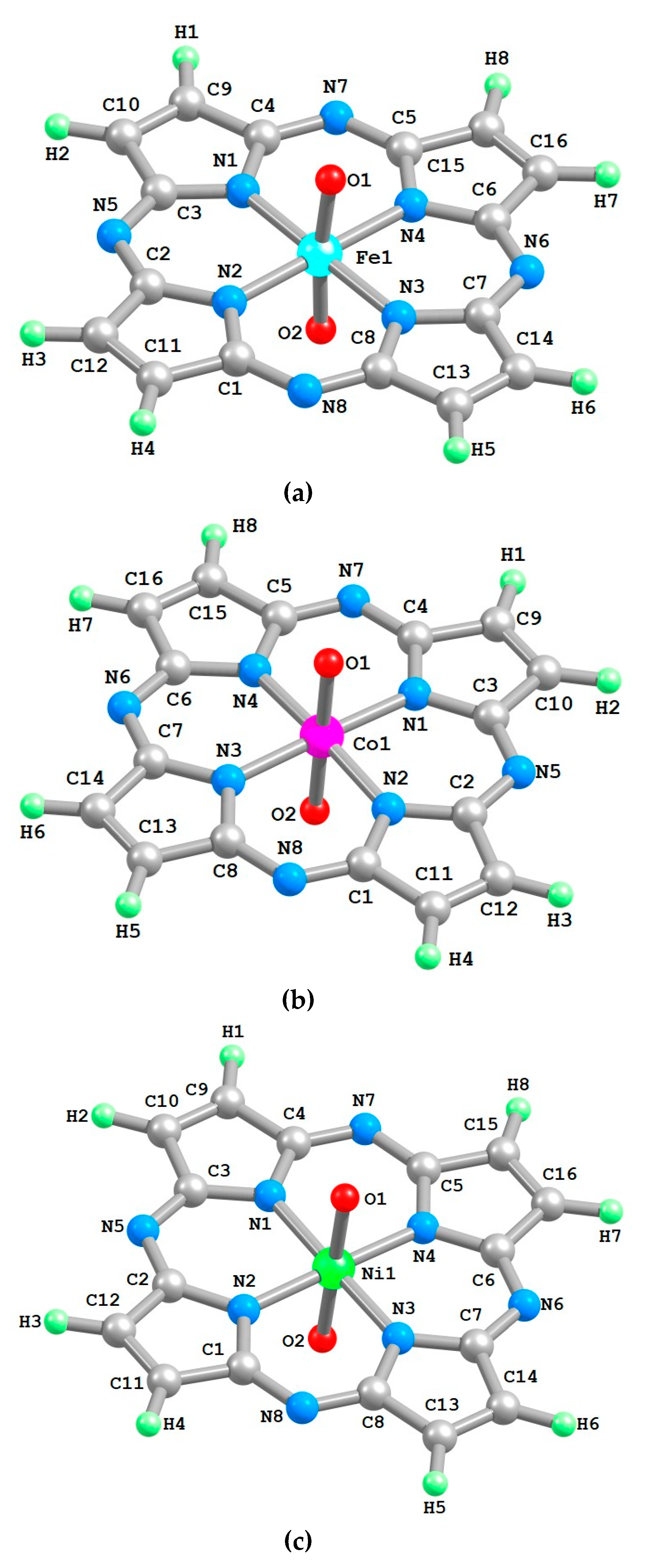
2. Results
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kiselev, Y.M.; Tretiyakov, Y.D. The problem of oxidation state stabilisation and some regularities of a Periodic system of the elements. Russ. Chem. Rev. 1999, 68, 365–377. [Google Scholar] [CrossRef]
- Kiselev, Y.M.; Kopelev, N.S.; Spitsyn, V.I.; Martynenko, L.I. Octavalent iron. Dokl. Akad. Nauk SSSR 1987, 292, 628–631. [Google Scholar]
- Perfil’ev, Y.D.; Kopelev, N.S.; Kiselev, Y.M.; Spitsyn, V.I. Mössbauer study of octavalent iron. Dokl. Akad. Nauk SSSR 1987, 296, 1406–1409. [Google Scholar]
- Kopelev, N.S.; Kiselev, Y.M.; Perfiliev, Y.D. Mössbauer spectroscopy of the oxocomplexes iron in higher oxidation states. J. Radioanal. Nucl. Chem. 1992, 157, 401–411. [Google Scholar] [CrossRef]
- Cotton, F.A.; Wilkinson, G.; Murillo, C.A.; Bochmann, M. Advanced Inorganic Chemistry, 6th ed.; Wiley: Hoboken, NJ, USA, 1999. [Google Scholar]
- Kasuda, K.; Tsutsui, M. Some new developments in the chemistry of metallophthalocyanines. Coord. Chem. Rev. 1980, 32, 67–95. [Google Scholar]
- Thomas, A.L. Phthalocyanines. Research & Applications; CRC Press: Boca Raton, FL, USA, 1990. [Google Scholar]
- Sliva, W.; Mianovska, B. Metalloporphyrin arrays. Transit. Met. Chem. 2000, 25, 491–504. [Google Scholar]
- Mamardashvili, G.M.; Mamardashvili, N.Z.; Koifman, O.I. Self-assembling systems based on porphirins. Russ. Chem. Rev. 2008, 77, 59–75. [Google Scholar] [CrossRef]
- Lomova, T.N. Axial Coordinated Metal Porphyrins in Science and Practice; URSS: Moscow, Russia, 2018. [Google Scholar]
- Mikhailov, O.V.; Chachkov, D.V. Novel oxidation state—Zinc(III) in chelate with 3,7,11,15-tetraazaporphine and one fluorine ligand: Quantum-chemical modeling. J. Porphyr. Phthalocya. 2019, 23, 685–689. [Google Scholar] [CrossRef]
- Chachkov, D.V.; Mikhailov, O.V. Combination of phthalocyanine and fluoride ligand for Zn(III) stabilization: Quantum-chemical consideration. Inorg. Chem. Commun. 2019, 108, 107526. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. About possibility of stabilization of unusual copper(IV) oxidation state in complexes with porphyrazine and two fluorine ligands: Quantum-chemical design. Inorg. Chem. Commun. 2019, 106, 219–224. [Google Scholar] [CrossRef]
- Schaefer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Schaefer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Hoe, W.M.; Cohen, A.; Handy, N.C. Assessment of a new local exchange functional OPTX. Chem. Phys. Lett. 2001, 341, 319–328. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Paulsen, H.; Duelund, L.; Winkler, H.; Toftlund, H.; Trautwein, A.X. Free Energy of Spin-Crossover Complexes Calculated with Density Functional Methods. Inorg. Chem. 2001, 40, 2201–2203. [Google Scholar] [CrossRef] [PubMed]
- Swart, M.; Groenhof, A.R.; Ehlers, A.W.; Lammertsma, K. Validation of Exchange—Correlation Functionals for Spin States of Iron Complexes. J. Phys. Chem. A 2004, 108, 5479–5483. [Google Scholar] [CrossRef]
- Swart, M.; Ehlers, A.W.; Lammertsma, K. Performance of the OPBE exchange-correlation functional. Mol. Phys. 2004, 102, 2467–2474. [Google Scholar] [CrossRef]
- Swart, M. Metal-ligand bonding in metallocenes: Differentiation between spin state, electrostatic and covalent bonding. Inorg. Chim. Acta 2007, 360, 179–189. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision A.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Ochterski, J.W. Thermochemistry in Gaussian; Gaussian, Inc.: Wallingford, CT, USA, 2000. [Google Scholar]
- Himmel, D.; Knapp, C.; Patzschke, M.; Riedel, S. How Far Can We Go? Quantum-Chemical Investigations of Oxidation State +IX. ChemPhysChem 2010, 11, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.S.; Truhlar, D.G. Oxidation state 10 exists. Angew. Chem. Int. Ed. 2016, 55, 9004–9006. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Structural Parameter | [FeL(O)2] | [CoL(O)2] | [NiL(O)2] |
|---|---|---|---|
| M–N bond lengths in chelate node MN4, pm | |||
| M1N1 | 193.8 | 192.2 | 191.6 |
| M1N2 | 193.8 | 193.2 | 191.6 |
| M1N3 | 196.3 | 194.2 | 191.6 |
| M1N4 | 196.3 | 193.2 | 191.6 |
| C–N bond lengths in 6-numbered chelate rings, pm | |||
| N1C3 | 136.3 | 136.4 | 136.5 |
| N1C4 | 136.4 | 136.4 | 136.5 |
| N2C1 | 136.4 | 136.3 | 136.5 |
| N2C2 | 136.3 | 136.2 | 136.5 |
| N3C7 | 136.2 | 136.7 | 136.5 |
| N3C8 | 136.1 | 136.7 | 136.5 |
| N4C5 | 136.1 | 136.2 | 136.5 |
| N4C6 | 136.2 | 136.3 | 136.5 |
| N5C2 | 132.7 | 132.4 | 132.1 |
| N5C3 | 132.7 | 132.4 | 132.1 |
| N6C6 | 132.7 | 132.4 | 132.1 |
| N6C7 | 132.7 | 132.3 | 132.1 |
| N7C4 | 132.7 | 132.4 | 132.1 |
| N7C5 | 132.7 | 132.4 | 132.1 |
| N8C1 | 132.7 | 132.4 | 132.1 |
| N8C8 | 132.7 | 132.3 | 132.1 |
| C–C bond lengths in 5-numbered chelate ring (C1N1C4C9C10C3), pm | |||
| C4C9 | 145.3 | 145.2 | 145.0 |
| C9C10 | 135.8 | 135.7 | 135.6 |
| C10C3 | 145.3 | 145.2 | 145.0 |
| M–O bond lengths, pm | |||
| M1O1 | 162.7 | 168.9 | 180.8 |
| M1O2 | 162.7 | 168.9 | 180.8 |
| Bond angles in chelate node MN4, deg | |||
| (N1M1N2) | 90.4 | 90.1 | 90.0 |
| (N2M1N3) | 90.0 | 89.9 | 90.0 |
| (N3M1N4) | 89.6 | 89.9 | 90.0 |
| (N4M1N1) | 90.0 | 90.1 | 90.0 |
| Bond angles sum (BAS), deg | 360.0 | 360.0 | 360.0 |
| Non-bond angles between N atoms in N4 grouping, deg | |||
| (N1N2N3) | 90.2 | 90.0 | 90.0 |
| (N2N3N4) | 89.8 | 89.8 | 90.0 |
| (N3N4N1) | 89.8 | 90.0 | 90.0 |
| (N4N1N2) | 90.2 | 90.2 | 90.0 |
| Non-bond angles sum (NBAS), deg | 360.0 | 360.0 | 360.0 |
| Bond angles in 6-numbered chelate ring (M1N1C4N7C5N4), deg | |||
| (M1N1C4) | 125.9 | 126.1 | 126.2 |
| (N1C4N7) | 128.4 | 128.3 | 128.5 |
| (C4N7C5) | 122.1 | 121.3 | 120.6 |
| (N7C5N4) | 127.9 | 128.1 | 128.5 |
| (C5N4M1) | 125.7 | 126.1 | 126.2 |
| (N4M1N1) | 90.0 | 90.1 | 90.0 |
| Bond angles sum (BAS61), deg | 720.0 | 720.0 | 720.0 |
| Bond angles in 6-numbered chelate ring (M1N2C2N5C3N1), deg | |||
| (M1N2C2) | 125.8 | 126.1 | 126.2 |
| (N2C2N5) | 128.1 | 128.1 | 128.5 |
| (C2N5C3) | 121.9 | 121.3 | 120.6 |
| (N5C3N1) | 128.1 | 128.3 | 128.5 |
| (C3N1M1) | 125.7 | 126.1 | 126.2 |
| (N1M1N2) | 90.4 | 90.1 | 90.0 |
| Bond angles sum (BAS62), deg | 720.0 | 720.0 | 720.0 |
| Bond angles in 6-numbered chelate ring (M1N3C8N8C1N2), deg | |||
| (M1N3C8) | 125.7 | 125.9 | 126.2 |
| (N3C8N8) | 127.9 | 128.2 | 128.5 |
| (C8N8C1) | 122.1 | 121.4 | 120.6 |
| (N8C1N2) | 128.4 | 128.5 | 128.5 |
| (C1N2M1) | 125.9 | 126.1 | 126.2 |
| (N2M1N3) | 90.0 | 89.9 | 90.0 |
| Bond angles sum (BAS63), deg | 720.0 | 720.0 | 720.0 |
| Bond angles in 6-numbered chelate ring (M1N4C6N6C7N3), deg | |||
| (M1N4C6) | 125.9 | 126.1 | 126.2 |
| (N4C6N6) | 128.2 | 128.5 | 128.5 |
| (C6N6C7) | 122.2 | 121.4 | 120.6 |
| (N6C7N3) | 128.2 | 128.2 | 128.5 |
| (C7N3M1) | 125.9 | 125.9 | 126.2 |
| (N3M1N4) | 89.6 | 89.9 | 90.0 |
| Bond angles sum (BAS64), deg | 720.0 | 720.0 | 720.0 |
| Bond angles in 5-numbered ring (C3N1C4C9C10), deg | |||
| (C3N1C4) | 108.3 | 107.8 | 107.4 |
| (N1C4C9) | 108.7 | 109.1 | 109.3 |
| (C4C9C10) | 107.1 | 107.0 | 107.0 |
| (C9C10C3) | 107.1 | 107.0 | 107.0 |
| (C10C3N1) | 108.8 | 109.1 | 109.1 |
| Bond angles sum (BAS51), deg | 540.0 | 540.0 | 540.0 |
| Bond angles between O, M and N atoms, deg | |||
| O1M1N1 | 92.4 | 94.9 | 89.2 |
| O1M1N2 | 92.5 | 90.0 | 90.8 |
| O1M1N3 | 87.6 | 85.1 | 89.2 |
| O1M1N4 | 87.5 | 90.0 | 90.8 |
| Bond angles between O, M and O atoms, deg | |||
| O1M1O2 | 173.1 | 170.1 | 180.0 |
| Complex | ΔH0f, 298, kJ/mole | S0f, 298, J/mole ∙K | ΔG0f, 298, kJ/mole |
|---|---|---|---|
| [FeL(O)2] | 438.2 | 758.7 | 663.4 |
| [CoL(O)2] | 616.3 | 759.8 | 842.1 |
| [NiL(O)2] | 780.9 | 784.3 | 999.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikhailov, O.V.; Chachkov, D.V. M(VI) Oxidation State Stabilization in Iron, Cobalt and Nickel Heteroligand Metal Chelates Containing 3,7,11,15-Tetraazaporphine and Two Axial Oxo Ligands: Quantum-Chemical Simulation. Int. J. Mol. Sci. 2020, 21, 1494. https://doi.org/10.3390/ijms21041494
Mikhailov OV, Chachkov DV. M(VI) Oxidation State Stabilization in Iron, Cobalt and Nickel Heteroligand Metal Chelates Containing 3,7,11,15-Tetraazaporphine and Two Axial Oxo Ligands: Quantum-Chemical Simulation. International Journal of Molecular Sciences. 2020; 21(4):1494. https://doi.org/10.3390/ijms21041494
Chicago/Turabian StyleMikhailov, Oleg V., and Denis V. Chachkov. 2020. "M(VI) Oxidation State Stabilization in Iron, Cobalt and Nickel Heteroligand Metal Chelates Containing 3,7,11,15-Tetraazaporphine and Two Axial Oxo Ligands: Quantum-Chemical Simulation" International Journal of Molecular Sciences 21, no. 4: 1494. https://doi.org/10.3390/ijms21041494
APA StyleMikhailov, O. V., & Chachkov, D. V. (2020). M(VI) Oxidation State Stabilization in Iron, Cobalt and Nickel Heteroligand Metal Chelates Containing 3,7,11,15-Tetraazaporphine and Two Axial Oxo Ligands: Quantum-Chemical Simulation. International Journal of Molecular Sciences, 21(4), 1494. https://doi.org/10.3390/ijms21041494