Endothelial Semaphorin 3F Maintains Endothelial Barrier Function and Inhibits Monocyte Migration

 , , and
, , and
Abstract
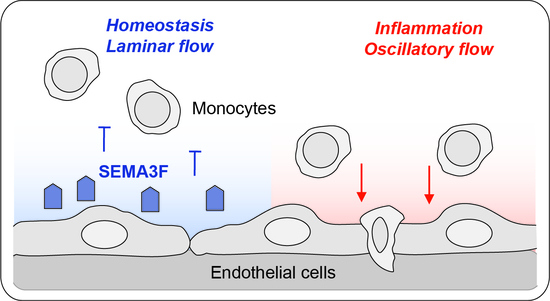
1. Introduction
2. Results
2.1. Semaphorin 3F Is the Highest Expressed Class III Semaphorin in Endothelial Cells
2.2. Expression of SEMA3F Is Regulated by Inflammatory Cytokines and Shear Stress
2.3. Loss of SEMA3F Impairs Endothelial Barrier Function
2.4. SEMA3F Itself Works as an Inhibitor of Migration for Activated Monocytes
3. Discussion
4. Materials and Methods
4.1. Access of Gene Expression Omnibus Data
4.2. Cell Culture
4.3. Laminar or Oscillatory Shear Stress Culture Conditions
4.4. Lentiviral Vector for KLF2 Overexpression or SEMA3F or KLF2 Knockdown
4.5. Quantitative Polymerase Chain Reaction
4.6. Western Blot Analysis
4.7. Immunofluorescence Staining
4.8. Electric Cell–Substrate Impedance Sensing
4.9. Permeability of 3D Endothelial Culture
4.10. Transwell Migration
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| EC | Endothelial cells |
| SEMA | Semaphorin |
| TNF | Tumor necrosis factor |
| HUVEC | Human umbilical vein endothelial cell |
| ECM | Extracellular matrix |
| KLF | Kruppel like factor |
References
- Cines, D.B.; Pollak, E.S.; Buck, C.A.; Loscalzo, J.; Zimmerman, G.A.; McEver, R.P.; Pober, J.S.; Wick, T.M.; Konkle, B.A.; Schwartz, B.S.; et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood 1998, 91, 3527–3561. [Google Scholar]
- Carmeliet, P. Blood vessels and nerves: Common signals, pathways and diseases. Nat. Rev. Genet. 2003, 4, 710–720. [Google Scholar] [CrossRef]
- Carmeliet, P.; Tessier-Lavigne, M. Common mechanisms of nerve and blood vessel wiring. Nature 2005, 436, 193–200. [Google Scholar] [CrossRef]
- Melani, M.; Weinstein, B.M. Common factors regulating patterning of the nervous and vascular systems. Annu. Rev. Cell. Dev. Biol. 2010, 26, 639–665. [Google Scholar] [CrossRef]
- Treps, L.; Le Guelte, A.; Gavard, J. Emerging roles of Semaphorins in the regulation of epithelial and endothelial junctions. Tissue Barriers 2013, 1, e23272. [Google Scholar] [CrossRef]
- Zhang, H.; Vreeken, D.; Bruikman, C.S.; van Zonneveld, A.J.; van Gils, J.M. Understanding netrins and semaphorins in mature endothelial cell biology. Pharmacol. Res. 2018, 137, 1–10. [Google Scholar] [CrossRef]
- Cora, D.; Astanina, E.; Giraudo, E.; Bussolino, F. Semaphorins in cardiovascular medicine. Trends Mol. Med. 2014, 20, 589–598. [Google Scholar] [CrossRef]
- Worzfeld, T.; Offermanns, S. Semaphorins and plexins as therapeutic targets. Nat. Rev. Drug Discov. 2014, 13, 603–621. [Google Scholar] [CrossRef]
- Janssen, B.J.; Malinauskas, T.; Weir, G.A.; Cader, M.Z.; Siebold, C.; Jones, E.Y. Neuropilins lock secreted semaphorins onto plexins in a ternary signaling complex. Nat. Struct. Mol. Biol. 2012, 19, 1293–1299. [Google Scholar] [CrossRef]
- Zanata, S.M.; Hovatta, I.; Rohm, B.; Puschel, A.W. Antagonistic effects of Rnd1 and RhoD GTPases regulate receptor activity in Semaphorin 3A-induced cytoskeletal collapse. J. Neurosci. 2002, 22, 471–477. [Google Scholar] [CrossRef]
- Wang, H.; Hota, P.K.; Tong, Y.; Li, B.; Shen, L.; Nedyalkova, L.; Borthakur, S.; Kim, S.; Tempel, W.; Buck, M.; et al. Structural basis of Rnd1 binding to plexin Rho GTPase binding domains (RBDs). J. Biol. Chem. 2011, 286, 26093–26106. [Google Scholar] [CrossRef] [PubMed]
- Millan, J.; Cain, R.J.; Reglero-Real, N.; Bigarella, C.; Marcos-Ramiro, B.; Fernandez-Martin, L.; Correas, I.; Ridley, A.J. Adherens junctions connect stress fibres between adjacent endothelial cells. BMC Biol. 2010, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Tiruppathi, C.; Malik, A.B.; Del Vecchio, P.J.; Keese, C.R.; Giaever, I. Electrical method for detection of endothelial cell shape change in real time: Assessment of endothelial barrier function. Proc. Natl. Acad. Sci. USA 1992, 89, 7919–7923. [Google Scholar] [CrossRef]
- Junaid, A.; Tang, H.; van Reeuwijk, A.; Abouleila, Y.; Wuelfroth, P.; van Duinen, V.; Stam, W.; van Zonneveld, A.J.; Hankemeier, T.; Mashaghi, A. Ebola Hemorrhagic Shock Syndrome-on-a-Chip. iScience 2019, 23, 100765. [Google Scholar] [CrossRef]
- Van Gils, J.M.; Ramkhelawon, B.; Fernandes, L.; Stewart, M.C.; Guo, L.; Seibert, T.; Menezes, G.B.; Cara, D.C.; Chow, C.; Kinane, T.B.; et al. Endothelial expression of guidance cues in vessel wall homeostasis dysregulation under proatherosclerotic conditions. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 911–919. [Google Scholar] [CrossRef]
- Nobes, C.D.; Lauritzen, I.; Mattei, M.G.; Paris, S.; Hall, A.; Chardin, P. A new member of the Rho family, Rnd1, promotes disassembly of actin filament structures and loss of cell adhesion. J. Cell Biol. 1998, 141, 187–197. [Google Scholar] [CrossRef]
- Bielenberg, D.R.; Shimizu, A.; Klagsbrun, M. Semaphorin-induced cytoskeletal collapse and repulsion of endothelial cells. Methods Enzymol. 2008, 443, 299–314. [Google Scholar] [CrossRef]
- Le Guelte, A.; Galan-Moya, E.M.; Dwyer, J.; Treps, L.; Kettler, G.; Hebda, J.K.; Dubois, S.; Auffray, C.; Chneiweiss, H.; Bidere, N.; et al. Semaphorin 3A elevates endothelial cell permeability through PP2A inactivation. J. Cell Sci. 2012, 125, 4137–4146. [Google Scholar] [CrossRef]
- Hou, S.T.; Nilchi, L.; Li, X.; Gangaraju, S.; Jiang, S.X.; Aylsworth, A.; Monette, R.; Slinn, J. Semaphorin3A elevates vascular permeability and contributes to cerebral ischemia-induced brain damage. Sci. Rep. 2015, 5, 7890. [Google Scholar] [CrossRef]
- Cerani, A.; Tetreault, N.; Menard, C.; Lapalme, E.; Patel, C.; Sitaras, N.; Beaudoin, F.; Leboeuf, D.; De Guire, V.; Binet, F.; et al. Neuron-derived semaphorin 3A is an early inducer of vascular permeability in diabetic retinopathy via neuropilin-1. Cell Metab. 2013, 18, 505–518. [Google Scholar] [CrossRef]
- Acevedo, L.M.; Barillas, S.; Weis, S.M.; Gothert, J.R.; Cheresh, D.A. Semaphorin 3A suppresses VEGF-mediated angiogenesis yet acts as a vascular permeability factor. Blood 2008, 111, 2674–2680. [Google Scholar] [CrossRef]
- Reichert, S.; Scheid, S.; Roth, T.; Herkel, M.; Petrova, D.; Linden, A.; Weberbauer, M.; Esser, J.; Diehl, P.; Grundmann, S.; et al. Semaphorin 3F Promotes Transendothelial Migration of Leukocytes in the Inflammatory Response After Survived Cardiac Arrest. Inflammation 2019, 42, 1252–1264. [Google Scholar] [CrossRef]
- Shimizu, A.; Nakayama, H.; Wang, P.; Konig, C.; Akino, T.; Sandlund, J.; Coma, S.; Italiano, J.E., Jr.; Mammoto, A.; Bielenberg, D.R.; et al. Netrin-1 promotes glioblastoma cell invasiveness and angiogenesis by multiple pathways including activation of RhoA, cathepsin B, and cAMP-response element-binding protein. J. Biol. Chem. 2013, 288, 2210–2222. [Google Scholar] [CrossRef]
- Shimizu, A.; Mammoto, A.; Italiano, J.E., Jr.; Pravda, E.; Dudley, A.C.; Ingber, D.E.; Klagsbrun, M. ABL2/ARG tyrosine kinase mediates SEMA3F-induced RhoA inactivation and cytoskeleton collapse in human glioma cells. J. Biol. Chem. 2008, 283, 27230–27238. [Google Scholar] [CrossRef]
- Tomita, H.; Hagaman, J.; Friedman, M.H.; Maeda, N. Relationship between hemodynamics and atherosclerosis in aortic arches of apolipoprotein E-null mice on 129S6/SvEvTac and C57BL/6J genetic backgrounds. Atherosclerosis 2012, 220, 78–85. [Google Scholar] [CrossRef]
- Chiu, J.J.; Chien, S. Effects of disturbed flow on vascular endothelium: Pathophysiological basis and clinical perspectives. Physiol. Rev. 2011, 91, 327–387. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Effects of shear stress on endothelial cells: Go with the flow. Acta Physiol. 2017, 219, 382–408. [Google Scholar] [CrossRef]
- Qiao, C.; Meng, F.; Jang, I.; Jo, H.; Chen, Y.E.; Zhang, J. Deep transcriptomic profiling reveals the similarity between endothelial cells cultured under static and oscillatory shear stress conditions. Physiol. Genom. 2016, 48, 660–666. [Google Scholar] [CrossRef]
- Dekker, R.J.; Boon, R.A.; Rondaij, M.G.; Kragt, A.; Volger, O.L.; Elderkamp, Y.W.; Meijers, J.C.; Voorberg, J.; Pannekoek, H.; Horrevoets, A.J. KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood 2006, 107, 4354–4363. [Google Scholar] [CrossRef]
- Clarhaut, J.; Gemmill, R.M.; Potiron, V.A.; Ait-Si-Ali, S.; Imbert, J.; Drabkin, H.A.; Roche, J. ZEB-1, a repressor of the semaphorin 3F tumor suppressor gene in lung cancer cells. Neoplasia 2009, 11, 157–166. [Google Scholar] [CrossRef]
- Coma, S.; Amin, D.N.; Shimizu, A.; Lasorella, A.; Iavarone, A.; Klagsbrun, M. Id2 promotes tumor cell migration and invasion through transcriptional repression of semaphorin 3F. Cancer Res. 2010, 70, 3823–3832. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, L.; Pappan, L.; Galliher-Beckley, A.; Shi, J. IL-1beta promotes stemness and invasiveness of colon cancer cells through Zeb1 activation. Mol. Cancer 2012, 11, 87. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Lin, Y.; Bacanamwo, M.; Chang, L.; Chai, R.; Massud, I.; Zhang, J.; Garcia-Barrio, M.T.; Thompson, W.E.; Chen, Y.E. Interleukin-1 beta-induced Id2 gene expression is mediated by Egr-1 in vascular smooth muscle cells. Cardiovasc. Res. 2007, 76, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.; Egbrink, M.G.o. The endothelial glycocalyx: Composition, functions, and visualization. Pflug. Arch. 2007, 454, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Rabelink, T.J.; van den Berg, B.M.; Garsen, M.; Wang, G.; Elkin, M.; van der Vlag, J. Heparanase: Roles in cell survival, extracellular matrix remodelling and the development of kidney disease. Nat. Rev. Nephrol. 2017, 13, 201–212. [Google Scholar] [CrossRef] [PubMed]
- De Wit, J.; Verhaagen, J. Proteoglycans as modulators of axon guidance cue function. Adv. Exp. Med. Biol. 2007, 600, 73–89. [Google Scholar] [CrossRef]
- Kantor, D.B.; Chivatakarn, O.; Peer, K.L.; Oster, S.F.; Inatani, M.; Hansen, M.J.; Flanagan, J.G.; Yamaguchi, Y.; Sretavan, D.W.; Giger, R.J.; et al. Semaphorin 5A is a bifunctional axon guidance cue regulated by heparan and chondroitin sulfate proteoglycans. Neuron 2004, 44, 961–975. [Google Scholar] [CrossRef]
- De Wit, J.; De Winter, F.; Klooster, J.; Verhaagen, J. Semaphorin 3A displays a punctate distribution on the surface of neuronal cells and interacts with proteoglycans in the extracellular matrix. Mol. Cell. Neurosci. 2005, 29, 40–55. [Google Scholar] [CrossRef]
- Alamri, A.; Rahman, R.; Zhang, M.; Alamri, A.; Gounni, A.S.; Kung, S.K.P. Semaphorin-3E Produced by Immature Dendritic Cells Regulates Activated Natural Killer Cells Migration. Front. Immunol. 2018, 9, 1005. [Google Scholar] [CrossRef]
- Teng, Y.; Yin, Z.; Li, J.; Li, K.; Li, X.; Zhang, Y. Adenovirus-mediated delivery of Sema3A alleviates rheumatoid arthritis in a serum-transfer induced mouse model. Oncotarget 2017, 8, 66270–66280. [Google Scholar] [CrossRef]
- Movassagh, H.; Shan, L.; Duke-Cohan, J.S.; Halayko, A.J.; Uzonna, J.E.; Gounni, A.S. Semaphorin 3E Alleviates Hallmarks of House Dust Mite-Induced Allergic Airway Disease. Am. J. Pathol. 2017, 187, 1566–1576. [Google Scholar] [CrossRef] [PubMed]
- Rienks, M.; Carai, P.; Bitsch, N.; Schellings, M.; Vanhaverbeke, M.; Verjans, J.; Cuijpers, I.; Heymans, S.; Papageorgiou, A. Sema3A promotes the resolution of cardiac inflammation after myocardial infarction. Basic Res. Cardiol. 2017, 112, 42. [Google Scholar] [CrossRef] [PubMed]
- Movassagh, H.; Saati, A.; Nandagopal, S.; Mohammed, A.; Tatari, N.; Shan, L.; Duke-Cohan, J.S.; Fowke, K.R.; Lin, F.; Gounni, A.S. Chemorepellent Semaphorin 3E Negatively Regulates Neutrophil Migration In Vitro and In Vivo. J. Immunol. 2017, 198, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Wanschel, A.; Seibert, T.; Hewing, B.; Ramkhelawon, B.; Ray, T.D.; van Gils, J.M.; Rayner, K.J.; Feig, J.E.; O’Brien, E.R.; Fisher, E.A.; et al. Neuroimmune guidance cue Semaphorin 3E is expressed in atherosclerotic plaques and regulates macrophage retention. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Curreli, S.; Wong, B.S.; Latinovic, O.; Konstantopoulos, K.; Stamatos, N.M. Class 3 semaphorins induce F-actin reorganization in human dendritic cells: Role in cell migration. J. Leukoc. Biol. 2016, 100, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Duncan, G.S.; Andrew, D.P.; Takimoto, H.; Kaufman, S.A.; Yoshida, H.; Spellberg, J.; de la Pompa, J.L.; Elia, A.; Wakeham, A.; Karan-Tamir, B.; et al. Genetic evidence for functional redundancy of Platelet/Endothelial cell adhesion molecule-1 (PECAM-1): CD31-deficient mice reveal PECAM-1-dependent and PECAM-1-independent functions. J. Immunol. 1999, 162, 3022–3030. [Google Scholar]
- Liao, F.; Huynh, H.K.; Eiroa, A.; Greene, T.; Polizzi, E.; Muller, W.A. Migration of monocytes across endothelium and passage through extracellular matrix involve separate molecular domains of PECAM-1. J. Exp. Med. 1995, 182, 1337–1343. [Google Scholar] [CrossRef]
- Ferrero, E.; Ferrero, M.E.; Pardi, R.; Zocchi, M.R. The platelet endothelial cell adhesion molecule-1 (PECAM1) contributes to endothelial barrier function. FEBS Lett. 1995, 374, 323–326. [Google Scholar] [CrossRef]
- Dangerfield, J.; Larbi, K.Y.; Huang, M.T.; Dewar, A.; Nourshargh, S. PECAM-1 (CD31) homophilic interaction up-regulates alpha6beta1 on transmigrated neutrophils in vivo and plays a functional role in the ability of alpha6 integrins to mediate leukocyte migration through the perivascular basement membrane. J. Exp. Med. 2002, 196, 1201–1211. [Google Scholar] [CrossRef]
- Hruz, T.; Laule, O.; Szabo, G.; Wessendorp, F.; Bleuler, S.; Oertle, L.; Widmayer, P.; Gruissem, W.; Zimmermann, P. Genevestigator v3: A reference expression database for the meta-analysis of transcriptomes. Adv. Bioinform. 2008, 2008, 420747. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009; ISBN 978-3-319-24277-4. [Google Scholar]
- Hong, J.; Kandasamy, K.; Marimuthu, M.; Choi, C.S.; Kim, S. Electrical cell-substrate impedance sensing as a non-invasive tool for cancer cell study. Analyst 2011, 136, 237–245. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Sequence | Reverse Sequence |
|---|---|---|
| GAPDH | CCTGCACCACCAACTGCTTA | GGCCATCCACAGTCTTCTGAG |
| SEMA3A | CTTGCATTCATCTCTTCTGGTGT | GTGCCAAGGCTGAAATTATCCT |
| SEMA3B | ACATTGGTACTGAGTGCATGAAC | GCCATCCTCTATCCTTCCTGG |
| SEMA3C | GTATGTCTGTGGGAGTGGCG | ACGTTGGGGTTGAAAGAGCA |
| SEMA3D | TCAGAGCACTACTGGCTCAAT | ATCGGAGGTACTGCCTTCTTG |
| SEMA3E | TGTCCTTTTGACCCCAGCTC | CGTCATGCTCAGTGCGGATA |
| SEMA3F | CCACAGCGCATCGAGGAAT | CATGGGGTTGTAGGCACCTG |
| SEMA3G | TGGCTCGAACCATGTCACTG | CATTTGTTCACCAGCACCCG |
| KLF2 | CTACACCAAGAGTTCGCATCTG | CCGTGTGCTTTCGGTAGTG |
| NOS3 | TGATGGCGAAGCGAGTGAAG | ACTCATCCATACACAGGACCC |
| NRP2 | GCTGGCTATATCACCTCTCCC | TCTCGATTTCAAAGTGAGGGTTG |
| PLXNA1 | CGTGCTGTTCACTGTGTTCG | ACTGGATGCGCTCCTTAATCT |
| PLXNA3 | CGGACATGTTCAGTCTCGTGTA | CGCTGACGAAGCCGTAGAT |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Vreeken, D.; Junaid, A.; Wang, G.; Sol, W.M.P.J.; de Bruin, R.G.; van Zonneveld, A.J.; van Gils, J.M. Endothelial Semaphorin 3F Maintains Endothelial Barrier Function and Inhibits Monocyte Migration. Int. J. Mol. Sci. 2020, 21, 1471. https://doi.org/10.3390/ijms21041471
Zhang H, Vreeken D, Junaid A, Wang G, Sol WMPJ, de Bruin RG, van Zonneveld AJ, van Gils JM. Endothelial Semaphorin 3F Maintains Endothelial Barrier Function and Inhibits Monocyte Migration. International Journal of Molecular Sciences. 2020; 21(4):1471. https://doi.org/10.3390/ijms21041471
Chicago/Turabian StyleZhang, Huayu, Dianne Vreeken, Abidemi Junaid, Gangqi Wang, Wendy M. P. J. Sol, Ruben G. de Bruin, Anton Jan van Zonneveld, and Janine M. van Gils. 2020. "Endothelial Semaphorin 3F Maintains Endothelial Barrier Function and Inhibits Monocyte Migration" International Journal of Molecular Sciences 21, no. 4: 1471. https://doi.org/10.3390/ijms21041471
APA StyleZhang, H., Vreeken, D., Junaid, A., Wang, G., Sol, W. M. P. J., de Bruin, R. G., van Zonneveld, A. J., & van Gils, J. M. (2020). Endothelial Semaphorin 3F Maintains Endothelial Barrier Function and Inhibits Monocyte Migration. International Journal of Molecular Sciences, 21(4), 1471. https://doi.org/10.3390/ijms21041471