Abstract
Increased plasma homocysteine is a risk factor for several pathological disorders. The present review focused on the role of homocysteine (Hcy) in different population groups, especially in risk conditions (pregnancy, infancy, old age), and on its relevance as a marker or etiological factor of the diseases in these age groups, focusing on the nutritional treatment of elevated Hcy levels. In pregnancy, Hcy levels were investigated in relation to the increased risk of adverse pregnancy outcomes such as small size for gestational age at birth, preeclampsia, recurrent abortions, low birth weight, or intrauterine growth restriction. In pediatric populations, Hcy levels are important not only for cardiovascular disease, obesity, and renal disease, but the most interesting evidence concerns study of elevated levels of Hcy in autism spectrum disorder (ASD) and attention deficit hyperactivity disorder (ADHD). Finally, a focus on the principal pathologies of the elderly (cardiovascular and neurodegenerative disease, osteoporosis and physical function) is presented. The metabolism of Hcy is influenced by B vitamins, and Hcy-lowering vitamin treatments have been proposed. However, clinical trials have not reached a consensus about the effectiveness of vitamin supplementation on the reduction of Hcy levels and improvement of pathological condition, especially in elderly patients with overt pathologies, suggesting that other dietary and non-dietary factors are involved in high Hcy levels. The importance of novel experimental designs focusing on intra-individual variability as a complement to the typical case–control experimental designs and the study of interactions between different factors it should be emphasized.
1. Introduction
Homocysteine (Hcy) is a significant biomarker for overall health status and, although it is not clear whether the Hcy represents an indicator rather the etiology of disease, a direct relationship between its elevated fasting plasma levels and several pathological disorders, including bone health [1], neurodegenerative disease, [2], renal dysfunction [3], cognitive impairments [4], and congenital defect development [5], as well as its status of independent risk factor in coronary heart and cerebrovascular disease, is widely supported by scientific literature [6,7,8].
Hcy is a sulfur-containing amino acid not present in dietary protein or used for its endogenous synthesis, but represents an intermediate in methionine metabolism. Methionine is an essential sulfur-containing amino acid present in various protein foods such as meat, eggs, dairy products, and legumes. Figure 1 represents the pathways of Hcy metabolism, which include a system of transmethylation, remethylation, and transsulfuration paths. In most cells, by transmethylation route Hcy and methionine cycle metabolically, the methyl group on activated methionine (S-adenosyl-methionine or SAM) may be added to methyl acceptors (DNA, RNA, and protein) by methyltransferases, and the S-adenosyl-Hcy (SAH) is rapidly hydrolyzed to adenosine and Hcy, which could improve its concentration. Transmethylations, chemical reactions transferring a methyl group from one compound to another, are generally regulated by the intracellular concentration of involved compounds; thus, SAM and SAH concentrations determine a cell’s methylation balance. Once formed, Hcy can be recycled into methionine or converted into cysteine by remethylation and transsulfuration routes, respectively. Hcy is remethylated to methionine through two separate reactions catalyzed by three different enzymes. In all tissues, folic acid donates a methyl group across methylenetetrahydrofolate reductase (MTHFR) in a reaction catalyzed by methionine synthase, a vitamin-B12-dependent enzyme [9]. Otherwise, mainly in the human heart, liver, and kidneys, Hcy is remethylated using betaine, which donates a methyl group by betaine-Hcy S-methyltransferase (BHMT) by a route independent of the one-carbon metabolism. Betaine can be found in several dietary sources, including wheat germ or bran, spinach, beets, seafood, and legumes. Studies have confirmed betaine’s ability to reduce Hcy levels in the face of excess methionine intake, as well as the fact that low-dose betaine supplementation leads to immediate and long-term lowering of plasma Hcy in healthy men and women [10,11]. This remethylation process begins when there are low concentrations of Hcy and methionine [12]. Alternately, mainly in the liver, but also in the kidneys, small intestine, and pancreas [13], Hcy is enzymatically modified by cystathionine-β-synthase, a B6-dependent enzyme, to irreversibly form cysteine through the intermediate cystathionine. The transsulfuration route results in sulfur metabolites including GSH, a key cellular antioxidant, and hydrogen sulfide (H2S), acting like a gaseous signaling molecule. The transsulfuration path starts to function when the concentrations of Hcy and methionine increase, for example by post-prandial protein intake [14], or cysteine is needed.
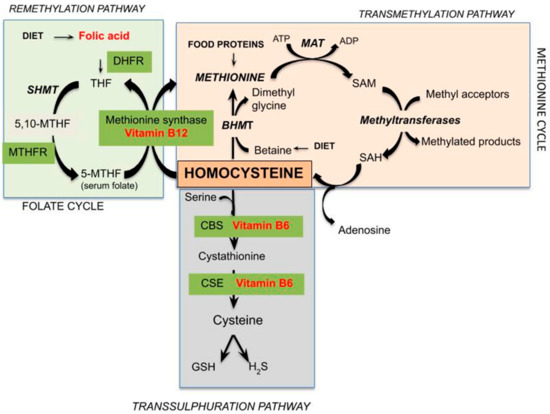
Figure 1.
Schematic representation of pathways of Hcy metabolism (DHFR = dihydrofolate reductase; THF = tetrahydrofolate; SHMT = serinehydroxymethyltransferase; MTHF = methylenetetrahydrofolate; MTHFR = 5,10-methylene-THF reductase; ATP = adenosine triphosphate; MAT = methionine adenosyltransferase; ADP = adenosine diphosphate; SAM = S-adenosylmethionine; SAH = S-adenosylHcy; BHMT = betaine-Hcy S-methyltransferase; CBS = cystathionine β-synthase; CSE = cystathionase; GSH = glutathione; H2S = hydrogen sulphide).
In humans, during protein synthesis, Hcy instead of methionine could be wrongly selected in the reaction catalyzed by methionyl-tRNA synthetase (MetRS), producing a reactive toxic metabolite of Hcy known as Hcy-thiolactone [15,16]. This compound is able to chemically modify proteins and to impair their normal function, generating N-homocysteinylated (N-Hcy) proteins. This increasingly prominent protein modification, called N-homocysteinylation, seems to exert cytotoxic, pro-inflammatory, prothrombotic, and pro-atherogenic properties, contributing to the cardiovascular and neurologic disorders of hyperhomocysteinemia (HHcy) [17,18]. Perła-Kaján et al. [19] demonstrated that Hcy-thiolactone-hydrolyzing enzymes, known as the paraoxonase (PON) multigene family, associated in the bloodstream with high-density lipoproteinis (PON1/HDL) detoxify homocysteine thiolactone. Therefore, genetic variation in PON1 could compromise this activity in humans. In serum or plasma, but not in urine, Hcy-thiolactone and Hcy can be interconverted by enzymatic and/or nonenzymatic reactions, indicating that interconversions do not contribute significantly to urinary Hcy-thiolactone or Hcy, while their presence is attributable to elimination or excretion from the blood. Hcy-thiolactone produced in the human body is efficiently cleared by the kidneys and excreted in the urine. The urinary Hcy-thiolactone levels are 100-fold higher than plasma levels. Approximate concentrations of Hcy-thiolactone (11–473.7 nmol/L; n = 19) were estimated in the urine of healthy volunteers with respect to plasma (<0.1–22.6 nmol/L; n = 20) [20]. The similar renal clearance of Hcy-thiolactone to that of creatinine indicates that Hcy-thiolactone is not reabsorbed in renal tubules; more than 95% is excreted in the urine. In contrast, 99% of Hcy is reabsorbed and 1% excreted in the urine [21]. Hcy-thiolactone can contribute significantly to Hcy pools, particularly in urine. As suggested by Jakubowsky [22], Hcy-thiolactone has adverse effects on physiological function, underscoring the importance of examining urinary and plasma Hcy-thiolactone in addition to Hcy in the context of human disease. Modification by Hcy-thiolactone seems to explain Hcy toxicity in autoimmune response, cellular toxicity, and atherosclerosis. This has been proposed as a mechanism in human subjects with HHcy caused by mutations in cystathionine β-synthase (CBS), the most common of the genetic causes of severe hyperhomocysteinemia, associated with increases up to 40-fold in fasting Hcy, and classic homocystinuria (congenital homocystinuria) or MTHFR genes [23] which has been associated with mild (13–24 µM) and moderate (25–60 µM) HHCy levels [9]. In neuronal degeneration and age-related neurodegenerative diseases, the Hcy neurotoxicity is due to troubled mechanisms of methylation and/or redox potentials triggering increased calcium influx [24], amyloid and tau protein accumulation [25,26], apoptosis, and neuronal death [27,28].
When Hcy metabolic pathways are deregulated, the intracellular Hcy enters the circulation, increasing plasma levels. At this stage, its fate is to bind to plasma proteins or to be eliminated via the renal route. Under normal conditions, Hcy is reabsorbed in the renal tubules and its urinary excretion is negligible, although there is no evidence that homocysteine is actively removed by the human kidney. The precise mechanism by which the glomerular filtration rate is related to plasma homocysteine concentration has not been definitively established, but there is a good degree of clinical evidence that HHcy does not cause renal insufficiency [29]. A comprehensive scheme representing Hcy transport across compartments was recently proposed by Hannibal and Blom [30]. At intracellular levels in its reducing condition, Hcy levels can be kept within a limited range of concentrations to enable adequate remethylation via methionine-synthase MS and to prevent accumulation of SAH through the concerted contributions of the methionine and folate cycles. The excess cellular Hcy (Hcy-SH) is exported into the bloodstream via an unknown cellular transporter(s). Several mechanisms have been proposed for the removal of Hcy to the extracellular compartment. It has been reported that an excess of Hcy in the extracellular compartment can be exported to the extracellular medium via transmembrane transport systems. Hcy shares carrier systems with cysteine in a preferable order of alanine–serine–cysteine (ASC) > aspartate and glutamate (XAG) = large branched-chain neutral amino acids (L) transporter systems in vascular endothelial cells and ASC > L > XAG in smooth muscle cells. The sodium-dependent ASC system plays a predominant role in Hcy transport in vascular cells. [31]. Moreover, three different amino acid transport systems, L, A, and y+L, have been identified for the transport of Hcy across the microvillous plasma membrane of the human placenta [32]. The specifity of Hcy transport is probably due to the ionic composition of the medium. In the more oxidizing environment found in plasma, homocysteine either reacts with protein Cys residues (R-SH) to homocysteinylate these targets (R-S-S-Hcy), or undergoes oxidization to form the disulfide homocystine (Hcy-Hcy) or mixed disulfides with cysteine (Hcy-Cys) and other low-molecular-weight thiols (Hcy-SR). Furthermore, the authors [30] hypothesized that homocystine and mixed disulfides derived from plasmatic Hcy oxidation can enter kidney and liver cells via another yet unidentified transporter, where the reducing environment of the cells can regenerate Hcy. Additionally, a proportion of the homocysteine is found in a protein-bound form, mainly bound to albumin, and enters the cells by endocytosis of these proteins. Sengupta et al. [33] proposed that homocysteine can be delivered to cells via its association with albumin, since albumin is known to bind to at least three endothelial cell membrane proteins, including transcytosed via plasmalemmal vesicles (gp 60), or endocytosed via receptor mediated endocytosis (gp30, gp18). Mudd et al. [34] described the Hcy and the major related disulfides present in normal human plasma. Figure 2 shows the several forms of Hcy present in human blood. The free sulfhydryl form, Hcy, is present in low amounts in blood (1%–2%) as a protein-bound mainly bound to albumin (about 70% to 85% in normal subjects) and soluble disulphides (30%–25%; e.g., homocystine or cysteine-SS-Hcy). The amount of total Hcy is the sum of these three components. Values obtained could vary by laboratory and collection method; some variable changes in Hcy have been observed post-prandially, so collection of fasting samples is recommended.
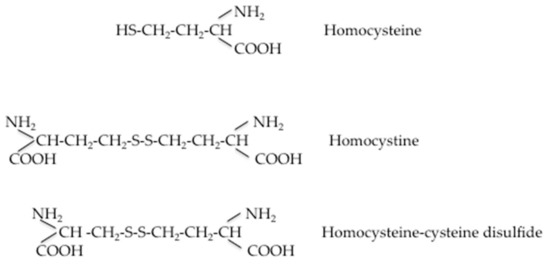
Figure 2.
Forms of Hcy present in human plasma.
To date, there is no general agreement on Hcy cut-off values. Since there a variety of factors affect Hcy levels, several cut-off points have been proposed [35], taking into account the imbalance of Hcy homeostasis in determining reference values. Jacobsen identified approximately 5 μmol/L and 25 μmol/L as ideal levels for an age range from 0 to 100 years [36]. Hcy reference range values within 4.7–14.6 μmol/L and 18.8–49.7 μmol/L have been reported for fasting and 6 h post-methionine load, respectively [37].
Even if correct functioning of the enzymes involved in the metabolic pathways of Hcy has an essential role in determining the plasma and urine levels of Hcy, several modifiable factors exert a strong effect on its concentration, thus characterizing a wide range of pathologies. These factors include adequate dietary intake of folate and vitamins B6 or B12, intake of proteins rich in methionine that help to regulate Hcy biochemical pathways, and unhealthy lifestyle choices such as alcohol abuse, high coffee intake, smoking habits, and use of drugs that may interfere with vitamin utilization and thus could increase the risk of HHcy-related pathologies [38]. What this means is that various diseases may have different etiologies in their respective relationships to plasma Hcy [39]. Considering its pivotal role, the nutritional management of elevated Hcy levels could provide powerful protection against its toxicity and unhealthy effects on human wellbeing. Several studies [40] have assessed the effects of folic acid, vitamin B6, and vitamin B12 supplements on elevated Hcy levels. Reductions in plasma Hcy concentrations in the range of about 25% to 30% and 10% to 15% were achieved in populations without or with folic acid fortification, respectively, upon dietary supplementation with folic acid and B12. A large prospective study reported a significant lowering effect on Hcy levels of daily supplementation combining folic acid, vitamin B6, and vitamin B12 [41]. Daily folic acid supplementation with 0.5 to 5.0 mg lowers plasma Hcy levels by about 25%. Daily supplementation of at least 0.4 mg vitamin B12 further reduces levels by about 7%, while vitamin B6 supplements can improve Hcy levels after methionine loading [42,43]. However, dose amount, bioavailability, and supplement type should be taken into account, as well as being adjusted for age, sex, and treatment assignment in analyzing the possible outcomes.
Additionally, various polymorphisms of the genes coding for MS, MTHFR, and CBS causing the low or absent activity rates of these proteins involved in Hcy clearance have an effect on circulating Hcy levels [44,45,46,47]. Kluijtmans et al. [47] reported at least five common functional polymorphisms that impair the activity of enzymes involved Hcy metabolism, including methylenetetrahydrofolate reductase [MTHFR] 677C>T and 1298A>C, methionine synthase 2756A>G, cystathionine β-synthase [CBS] 844ins68, and methionine synthase reductase 66A>G. These genetic variations affecting Hcy concentrations seem to be more significant in early-life than middle-aged subjects; it is possible that control of dietary factors would reduce homocysteine levels.
As already underlined, overproduction of Hcy could be harmful and is a possible cofactor in the etiology of multifactorial diseases, and may be a biomarker of chronic and acute inflammatory status and diseases, increasing the risk of other diseases and complication. Several studies and literature reviews have examined the role of Hcy in pathological conditions in specific population groups [48].
The aim of the present review article was to conduct a critical literature examination on the features of Hcy metabolism in different population groups at varying life stages, especially in risk conditions (pregnancy, infancy, old age), and on its relevance as a marker or etiological factor of diseases in these age groups, focusing on the nutritional treatment of elevated Hcy levels. Even if our review could not be exhaustive, we attempted to sum up what we consider the current state of the art of the most relevant and significant works about the role of Hcy in more vulnerable groups of people.
2. Pregnancy
The significance of HHcy levels in pregnancy has been investigated in relation to the increased risk of adverse pregnancy outcomes such as small size for gestational age at birth, preeclampsia, recurrent abortions, low birth weight, intrauterine growth restriction, and neural tube defects (NTDs) [49,50,51].
One systematic review [52] reported a correlation between HHcy levels and different complications in pregnancy; in particular, it was underlined that preeclampsia and placental abruption may occur more often in pregnant women with serum Hcy levels from 9.0 to 15.0 micromoles). In a previous study, Dodds et al. [53] demonstrated no significant correlation between Hcy levels in early pregnancy and risk of small size for gestational age at birth; meanwhile, a positive correlation between Hcy concentration at the top decile and pregnancy loss and preeclampsia was found (RR 2.1, 95% CI 1.2–3.6 and RR 2.7, 95% CI 1.4–5.0, respectively). The study did not observe a dose–response relationship according to quartile of Hcy concentration, due to the small sample size.
In one prospective study [54], Hcy levels and their effects on pregnancy and neonatal outcomes, including gestational diabetes, preeclampsia, gestational age at delivery, preterm birth, small size for gestational age, neonatal birth weight, and congenital abnormalities, were assessed in each trimester in 278 pregnant Korean women. Hcy levels were not significantly different among any of the trimesters (4.4–38.0 µmol/L), as observed by Bondevik et al. [55] in a group of Nepali women. No significant association was observed in Korean women between Hcy and any of the maternal or neonatal outcomes examined, including gestational age at delivery and birth weight of babies.
Mascarenhas et al. [56] suggested that the evaluation of Hcy levels during pregnancy should be done between 8 and 12 weeks to give a good indicator for pregnancy outcome. Increased serum Hcy in the first trimester of pregnancy is associated with history of pregnancy losses, hypertensive disorders of pregnancy, and preterm birth. This is also associated with hypertensive disorders of pregnancy, pregnancy loss, oligohydramnios, meconium stained amniotic fluid, and low birth weight in the current pregnancy.
Hcy levels during pregnancy are influenced both by the vitamin B nutritional status of women and by some specific physiological factors including increased glomerular filtration, rate hemodilution, and endocrinological changes occurring in this period, leading under normal conditions to a reduction of Hcy in the mid-pregnancy period [57]. Moreover Gaiday et al. [52] concluded that Hcy levels during pregnancy seem to also be linked to the geographical, cultural, and social characteristics of populations.
In the largest study (R Generation Study) about the associations of homocysteine, folate, and vitamin B12 levels and pregnancy outcomes, Bergen et al. [58] recruited 5805 women at gestational stage of 13.4 weeks (range: 11.4–16.5 weeks), and found a positive correlation between HHcy (highest quintile = 8.3 μmol/L) and birth weight (difference 110 g; p < 0.001), lower placental weight (difference 30 g; p < 0.001), and increased risk of small size for gestational age at birth (odds ratio (OR) 1.7; p = 0.006) in the lowest quintile (=5.8 μmol/L). Folate concentrations (lowest quintile = 9.2 nmol/L) were associated with lower placental weight (difference 26 g; p = 0.001) and birth weight (difference 125 g; p < 0.001), and increased risks of small size for gestational age at birth (OR 1.9; p = 0.002), prematurity (OR 2.2; p = 0.002), and preeclampsia (OR 2.1; p = 0.04) compared with the highest quintile (25.9 nmol/L). Interesting data have been recorded regarding folic acid supplementation: 18.8% of pregnant women (n = 1.091) did not use any folic acid supplements, 33.1% of women (n = 1.921) started folic acid supplementation in the pre-conceptional period, 24.5% (n = 1.422) started before 8 weeks of pregnancy, and data were missing for 23.7%.
As regards neural tube defects, low vitamin B12 and B9 status are risk factors for neural tube defects (NTDs) as they lead to an increase of Hcy levels [59,60,61]. The monitoring of Hcy levels might be important in understanding and following cases with NTDs.
Deb et al. [62] observed that mean Hcy levels were elevated in cases (15.71 ± 8.35 μmol/L) of NTD births compared to controls (12.87 ± 5.95 μmol/L) but were lower among the non-vegetarians (13.55 ± 6.64 μmol/L) than the vegetarians (14.78 ± 7.93 μmol/L). Due to low intake of vitamin B12, vegetarianism increased the risk of NTDs 1.6-fold (95% CI = 1.0–2.7), while folic acid supplementation demonstrated a protective effect for conceptions (OR = 0.59; 95% CI = 0.3–0.9). One meta-analysis [51] examinated 17 articles involving 3237 subjects and concluded that mothers with NTD offspring had significantly a higher mean log plasma Hcy level than mothers with normal offspring (log WMD: 0.06; 95% CI: 0.02–0.09, p = 0.001), but no positive association was found in studies conducted in countries with folic acid fortification.
Besides environmental and genetic factors, epigenetic mechanisms also play an important role in the etiology of multifactorial diseases such as NTDs. Iacobazzi et al. [63] described that when Hcy occurs at high levels, it competes with SAM for binding sites on DNA methyltransferase, leading to DNA hypomethylation with consequences for epigenetic programming. The reduction of DNA methylation during embryogenesis might be a genetic risk for NTDs. Zhao et al. [64] demonstrated a SAM/SAH reduction in mothers with NTD-affected offspring. Interesting evidence of the role of HHcy in determining neural tube defects (NTDs) recently emerged in a study by Zhang et al. [65] that suggested that higher levels of Hcy contribute to NTDs through upregulation of histone H3 N-homocysteinylation at Lys-79, leading to abnormal expression of selected NTC-related genes.
3. Children and Adolescents
Bailey et al. [66] defined cut-off levels for Hcy in healthy children and adolescents in the Canadian Laboratory Initiative for Pediatric Reference Intervals (CALIPER) program. The cut-off levels were as follows: 1–7 years: 2.7–7.6 μmol/L; 7–12 years: 3.4–8.4 μmol/L; 12–15 years: 4.7–10.4 μmol/L (males), 4.1–10.4 μmol/L (females); 15–19 years: 5.5–13.4 μmol/L (males), 4.9–11.9 μmol/L (females). As suggested by some authors [38,66,67], Hcy levels in children and adolescents are influenced by age, sex, ethnic differences in genetic polymorphisms of MTHFR, vitamin B12 deficiency, folate metabolism, and the laboratory test used to evaluate said levels. Table 1 collects pediatric reference intervals from several studies, showing the changes of Hcy and giving a picture of the variability found in healthy subjects. The majority of studies conducted on healthy children and adolescents have revealed that Hcy levels increase slightly as a function of age, ranging from 4.6 to 10.2 (μm/L) for 4–5 years and 16–19 years, respectively, in males, and from 3.8 to 8.33 (μm/L) for 9 years and 15-19 years, respectively, for females [68,69,70,71]. No trend with age was observed by Dávila-Rodriguez et al. [72], even though they found higher levels of Hcy in a group of 56 Mexican children (age: 2–9.99)―up to 11.94 ± 2.03 μm/L―compared to those of non–Hispanic Caucasican children reported in other studies and of other Latin-American pediatric groups [73,74]. Many studies have found gender differences in Hcy levels, with slightly higher Hcy levels in boys than in girls. This gender effect is enhanced during and after puberty (>15 years) [68,70,73]. The effect of puberty in increasing Hcy levels may be due to the different creatine turnover rates, with increased creatine production in boys related to their increase of muscle mass. Demand for creatine biosynthesis in men is usually greater than in women, and the link between creatine biosynthesis and Hcy metabolism is the enzyme guanidinoacetate methyltransferase [75]. Furthermore, increased Hcy levels were previously associated with increased androgen levels in boys [76], whereas estrogen levels were negatively correlated with Hcy levels in women [77]. Some authors have underlined that gender differences in Hcy concentrations may also be due to a greater efficacy in the remethylation and transmethylation phases in women, though the mechanism for this is still unknown. These phases represent important steps in Hcy metabolism, and the heightened efficacy of these steps in women may result in differences between males and females [78].

Table 1.
Total Hcy levels in healthy children and adolescents in relation to age and gender.
In addition to age and sex, folate and vitamin B status are the most important determinants of Hcy levels in children and adolescents. There are consistent reports of a strong relationship between HCy and serum folate in healthy children and adolescents, as reported by Gonzàlez-Gross et al. [79] (r = −0.328); meanwhile, the relationship with vitamin B12 biomarkers is weaker (r = −0.219 and −0.201 for cobalamin and holo-transcobalamin, respectively) (all p < 0.01) [79]. Other authors have obtained same results [68,69,75,80]. An interesting study [81,82] conducted on 26 hyperhomocysteinemic children (aged 6–15 years), 20 supplemented with 5mg oral of folic acid twice a week for 2 months and 6 children used as controls, showed that supplementation significantly decreased serum Hcy (p < 0.001) levels as well as systolic and diastolic pressure (p < 0.001 and p = 0.045, respectively) [78]. Moreover, significant improvements of total cholesterol levels from 183.8 (115–296 mg/dL) to 160.8 (109–265 mg/dL) (p < 0.05) were also observed [82].
The effect of lifestyle factors (e.g., diet and smoking) is poorly documented in children and adolescents. There is a positive association between Hcy level and sugar intake [83], as well as smoking habits [84]. A cross-sectional study on 483 subjects, 7–15 years of age and of both sexes [85], demonstrated that high serum Hcy levels were associated not only with age ≥12 years (prevalence ratio (PR) = 2.56; p < 0.01), being male (PR = 3.74; p < 0.01), high blood pressure (PR = 1.97; p < 0.01), low HDL-c levels (PR = 1.21; p = 0.03), high triglyceride levels (PR = 1.62; p = 0.03), and being overweight (PR = 2.32; p = 0.02), but also with poor intake of foods (PR = 1.46; p = 0.02) that protect against HHcy, such as dark green vegetables, whole and enriched grain products, legumes, and citrus fruit. Arouca et al. [86] found no positive correlation between high levels of Hcy and low adherence to the Mediterranean Diet or other “antioxidant diets” among adolescents in the Helena study.
Hcy concentration should also be expected to be affected by genetic background; however, data are scarce in children. In children (6–11 years) with familial hypercholesterolemia, an association between C677T mutation in the MTHFR gene and low serum folate and increased Hcy was found during cholestyramine treatment [87]. In a group of healthy young people (aged 3–18 years), the authors concluded that MTHFR genotype played a significant role in determining Hcy concentrations in subjects nutritionally stressed and aged over 10 years old [88].
The effect of MTHFR 677C>T polymorphism is enhanced by folate plasma levels. Caldeira-Araújo et al. [71] found that in a group of 9 year old children with high plasma folate levels, Hcy plasma concentrations did not differ significantly among the MTHFR 677 genotypes; the 17 year old adolescent subjects bearing the MTHFR 677TT genotype (with high folate levels) displayed significantly higher Hcy concentrations than those bearing the wild-type genotype. These results suggest that folate levels can modulate the expected correlations between genotype and Hcy levels.
3.1. HHcy and Cardiovascular Diseases
In recent years, research studies have investigated the role of HHcy levels in pediatric cardiovascular diseases, both to determine their relationship with the severity of diseases and to use Hcy as prognostic biomarker for increased risk of cardiovascular diseases. Recently, studies on cardiovascular disease in children and adolescents have examined postural tachycardia syndrome (PoTS) [89], congenital anomalies (congenital adrenal hyperplasia) [90], and heart failure [91]. PoTS is a common functional cardiovascular disease in children and adolescents, representing one of the main causes of syncope in pediatric populations, and it accounts for about 32.2% of all cases [92,93]. PoTS syncope is more common in adolescent females and often occurs after menarche [94]. The symptoms of PoTS include nausea, headache, palpitations, dizziness, chest discomfort, blurred vision, shortness of breath, and, in severe cases, syncope. The etiology of PoTS remains quite unclear; some authors suggest that this syndrome is mainly induced by an autonomic nerve dysfunction, and in particular by an increased baroreflex sensitivity (BRS), and BRS is correlated with PoTS symptoms [95,96]. Some studies on hypertensive patients have shown a relationship between HHcy levels and increased BRS [97], probably due to the regulation of oxidative stress mediated by Hcy. Based on these observations, Li et al. [89] demonstrated in a case–control study the role of Hcy in modulating PoTS symptoms in children, probably by interfering with BRS. The study was conducted on 35 children with PoTS (15 males and 20 females, age 6–13 years with a median of 11 years) and 30 controls (12 males and 18 female, age, 9–12 years with a median of 10 years). The results showed that Hcy plasma levels were significantly higher in the PoTS-affected subjects than the control group (9.78 (7.68, 15.31) μmol/L vs. 7.79 (7.46, 9.63) μmol/L, p < 0.05) as well as revealing a positive correlation with the PoTS symptom scores. Hcy can play an important role in modulating PoTS; nevertheless, further studies are needed to clarify how it could be used as an effective index to predict the short-term outcome of PoTS in children. At present, Hcy levels are suspected to play a role in etiology and control of these results in some cardiovascular diseases like PoTS, and some studies have shown that measurement of Hcy in children with acute heart failure (HF) and congenital adrenal hyperplasia (CHA) can be a very useful biomarker to predict the course of these diseases [90,91]. High levels of Hcy seem to determine adverse cardiac remodeling, both directly affecting myocardium or as a result of an independent vascular effect [98]. In a case–control study, El-Amousy et al. [91], investigating the prognostic value of Hcy levels and of highly sensitive troponin T, found children with HF with significantly higher Hcy plasma levels before HF treatment (11.150 ± 1.960 μmol/L), compared with after treatment (9.030 ± 1.616 μL·mol/L) and with the control group (6.69 ± 0.97 μL·mol/L). In patients with congestive heart failure, the authors suggested cut-off points of Hcy > 8.1 µmol/L and Hs-cTnT > 10 pg/mL levels as diagnostic predictor. The authors reported both plasma Hcy and serum hs-cTnT levels to be significantly (p < 0.001) increased in adverse outcomes (Hcy 13.23 ± 1.03 µmol/L; Hs-cTnT 110.50 ± 10.98 pg/mL) compared to favorable outcomes (10.96 ± 0.87 µmol/L and 69.78 ± 16.35 pg/mL, respectively). In addition, a significant increase of the mean levels of hs-cTnT and Hcy was associated with severity of HF (Class IV patients vs. Class III and Class II patients (p = 0.001). Results showed Hcy levels were correlated with clinical and echocardiographic data recording severity of HF, confirming previous findings on HF [99,100]. In this study, plasma Hcy and serum hs-cTnT levels were highlighted as good prognostic biomarkers in children with congenital heart diseases.
A case–control study conducted on 36 children with congenital adrenal hyperplasia (CAH), and 36 controls [90] evaluated serum levels of Hcy in relation to relation to carotid artery intima-media thickness (CA-IMT) and left ventricular (LV) function. Compared to the controls, high serum Hcy levels and positive correlation with CA-IMT and left ventricular (LV) function were present in CAH-affected children. Hcy levels were also significantly correlated with systolic (OR = 2.2; 95% CI: 1.10–1.18; p = 0.01) and diastolic blood pressures (OR = 2.9; 95% CI: 1.45–2.4; p = 0.01) and atherogenic index (OR = 2.6; 95% CI: 1.33–2.89; p = 0.01). The authors concluded that serum Hcy levels in children with CAH should be used as a biomarker for an increased risk in developing LV dysfunction and subclinical atherosclerosis.
As observed for autism spectrum disorder (ASD) studies, these studies had some limitations including relatively small number of patients and lack of evaluation of folate and B12 levels, or any possible mutation for genes encoding enzymes involved in the Hcy pathways. All these factors might lead to some bias and preclude an exhaustive evaluation of the role of Hcy in these diseases; thus, further studies are needed to overcome these shortcomings.
However, with regards to Hcy and cardiovascular disease in children and adolescents, some interesting conclusions emerged from the systematic review of Leal et al. [101], conducted on studies published between 1997 and 2011 on this topic. The authors underlined these fundamental conclusions: (a) HHcy is positively correlated with MTHFR 677T and heterozygous MTHFR 677T/1298C, with cardiovascular disease, and with low levels of folate and B12; (b) Hcy levels increase with age, being higher in adolescents than in children, and are higher in males than in females; (c) a possible association exisits between high Hcy levels in children and adolescents and a positive parental history for cardiovascular diseases; (d) there is a positive correlation between high Hcy and overweight and obesity in children and adolescents, with an increased risk for cardiovascular diseases (CVD). The correlation between HHcy levels and increased risk of CVD in children and adolescents is not yet well clarified. In their review, Leal et al. [101] found many studies concluding a positive parental history of CVD and HHcy in children and adolescents and studies showing HHcy levels in overweight or obese subjects [102]. The authors [101] suggested the need to investigate HHcy in conjunction with other parameters such as insulin levels (which have an inverse relationship with Hcy) and renal function to clarify the pathophysiological mechanism by which HHcy can increase the risk of CVD.
3.2. Obesity
In regards to the relationship between overweight and obesity and HHcy in children, a recent study [103] examined 138 children (46 normal weight, 40 overweight, and 52 obese) and observed that the median Hcy levels in overweight (16.7μmol/L, range: 11.2–2.5) and obese children (16.6 7 μmol/L, range: 13.3–22.4) were significantly higher than in normal weight children (7.3 μmol/L, range 5.5–10.5), p = 0.001. Debohkordi et al. [104], in a previous randomized double-blind controlled clinical trial on 60 obese and overweight children aged 5–12 years demonstrated that supplementation of 1 mg folic acid per day or 5 mg folic acid per day for 8 weeks had a positive effect on the reduction of HHcy and insulin resistance. Folic acid supplementation might represent a good strategy for decreasing high Hcy levels and related cardiovascular risk in obese and overweight children. However, further studies should be carried out with suggested lower levels of supplementation. In fact, the levels used in the randomized trial of Debohkordi et al. [104] were in excess of the upper levels of folic acid suggested by Dietary Reference Intakes [105,106] and are not safe for long-term supplementation. Overproduction of Hcy could be harmful and is a possible cofactor in the etiology of multifactorial diseases or a biomarker of chronic and acute inflammatory status and diseases, increasing the risk of other diseases and complication. The majority of studies on the relationship between Hcy levels and diseases in children and adolescents to date have been observational or case–control studies with a small number of subjects. At the present time, they provide data supporting a further in-depth investigation of the biochemical and metabolic mechanisms between metabolic Hcy and the etiology of some diseases.
3.3. HHcy and Renal Diseases
Hcys levels are dependent in children and adolescents with renal diseases. The kidney is a major organ that metabolizes Hcy, and most patients with chronic renal diseases show HHcy. HHcy in chronic renal disease is due to a decrease in intrarenal HCy clearance and/or a decrease in extrarenal Hcy clearance [107]. HHCy in renal diseases shows a similar pattern to atherosclerosis, i.e., production of lipid-laden macrophage, presence of cholesterol and cholesterol ester, intima thickening, elastic lamina disruption, and luminal platelet accumulation [108,109]. Nephrotic syndrome (NS), a common pediatric kidney disease, is associated with HHCy due to inhibition of the homocysteine remethylation process or disruption in cysteine clearance [110]. Most NS patients are resistant to steroid treatments and have evolved into focal segmental glomerulosclerosis (FSGS) due to the pathogenic effect of HHcy, which causes podocyte injury and glomerulosclerosis [111,112].
Merouani et al. [113] examined 29 children affected by different degrees of chronic renal failure (15 dialyzed, 14 not dialyzed) compared with 57 age-matched healthy children. Hcy concentrations were significantly higher (p < 0.0001) in patients than controls (17.3 and 6.8 μmol/L respectively) and 62.0% of patients and 5.2% of controls had HHcy (>95th percentile for controls: 14.0 μmol/L). Inversely, folate levels were significantly lower (p < 0.01) in children affected by chronic renal failure (9.9 nmol/L) than in controls (13.5 nmol/L). The authors [110] found a higher prevalence of HHcy in dialyzed children than in nondialyzed patients (87% vs. 35%), and higher levels of HHcy in dialyzed children with MTHFR mutations (28.5 μmol/L) compared to nondialyzed patients with the mutation (10.7 μmol/L p < 0.002). Child patients (n = 12, aged 11.0 ± 3.5) with acute glomerulonephritis had higher Hcy levels (from 9.4 ± 3.3 to 13.5 ± 2.8 μmol/L) compared to controls (n = 15, 8.4 ± 2.4 μmol/L) and lower vitamin B6 and RBC folate levels (p < 0.01) [104]. HHcy was also found in 26 renal transplanted children and adolescents (12.9 ± 4.8 μmol/L), with 73% of these patients showing Hcy levels above the 97th percentile of healthy children. Plasma HCy in these subjects was negatively correlated with plasma vitamin B12 (r = 0.40, p < 0.05) and creatine clearance (r = 0.53 p < 0.005), and plasma Hcy levels were higher in patients with a MTHFR 677TT/1298AA genotype [114,115]. The above-cited studies did not investigate the nutritional intake of folate, other B vitamins (B6 and B12), or of other micronutrients in children with renal diseases. As suggested by Viroonudomphol et al. [107], an adequate intake of folate, B6, and B12 with an increase of fruit, vegetables, wholegrain cereals, and legumes and/or an appropriate targeted supplementation could increase vitamin B status, reducing HHcy risk of end-stage renal diseases and reducing clinical complications and risk of cardiovascular events in renal transplant patients.
3.4. HHcy and Autism Spectrum Disorder (ASD)
Autism spectrum disorder (ASD) is a neurodevelopmental syndrome with a very complex pathogenesis, not yet completely understood even though gene–gene and gene–environment interactions that might play roles have been suggested [116]. Several studies have observed folate, Hcy, and glutathione metabolism anomalies in many children affected by ASD [117,118,119]. A great deal of research has demonstrated high levels of Hcy and low levels of folate, vitamin B6, and vitamin B12 in children affected by ASD [116,117,118,119,120,121]. In a case–control study, Altun et al. [121] focused on 105 children (3–12 years; 60 with confirmed ASD and 45 healthy controls) and observed significantly increased levels of Hcy in affected children compared to the control group (8.90 ± 0.19 nmol/L and 7.46 ± 0.21 nmol/L, respectively; p < 0.001). In contrast, the levels of vitamin B6, folate, and vitamin B12 were found to be significantly lower in the patients with ASD (25.17 ± 3.64 ng/mL, 121.16 ± 8.04 pg/mL, and 181.5 ± 41.61 pg/mL, respectively; 53.06 ± 7.95 ng/mL, 172.31 ± 17.19, pg/ml, and 382.06 ± 71.34 pg/mL, respectively; p < 0.001). Other studies confirmed higher levels of Hcy in ASD patients compared to healthy children in serum, plasma, and urine [118,122,123,124] with respect to control groups. In a clinical update, Fuentes-Albero et al. [120] reviewed current evidence aligning the severity of autism symptoms and HHcy. As for neurodegenerative diseases, one hypothesis of the significance of HHcy in the etiology of ASD is that Hcy is a powerful excitotoxin, and its metabolic products can cause damage in some proteins, generating “abnormal toxin proteins” and inducing an autoimmune response [125]. Some studies on the neurotoxicity of Hcy have demonstrated that Hcy can induce neuronal damage, causing activation of N-methyl-d-aspartate (NMDA) receptors and cell loss through excitotoxicity as well as apoptosis [124,125,126,127,128,129,130,131]. Additionally, Fulceri et al. [127] observed that autistic children often have gastrointestinal disorders and/or are picky eaters, and these factors could lead to an inadequate dietary intake, altered nutrient absorption, and micronutrient deficiency, particularly vitamin B deficiency, which causes increased plasma Hcy levels and worsening of autism. Along with HHcy, methylation impairment and decrease in glutathione redox status may be cofactors in autism pathology [128,129,130].
On the basis of the relationship between low folate levels, HHcy, and autism, Sun et al. [131] in an intervention study demonstrated that supplementation with 800 μg/day of folic acid in children (mean age 52.00 ± 13.13 months) improved autism symptoms related to receptive language, cognitive verbal/preverbal, sociability, affective expression, and communication, and that this treatment also improved the concentration of folic acid, reduced Hcy plasma levels, and normalized glutathione redox metabolism. These results, according to Józefczuck et al. [132], suggest serum Hcy levels as a useful biomarker for diagnosis of autism spectrum disorder in children as well as for the efficacy of treatment.
3.5. HHcy and Attention Deficit Hyperactivity Disorder (ADHD)
Interesting observations regarding levels of Hcy in relation to attention deficit hyperactivity disorder (ADHD) emerged from the study of Altun et al. [133] showing an opposite behaviour of Hcy levels with respect to ASD. ADHD is a neurobehavioral disorder characterized by symptoms of inattention and impulsivity, and is widespread among children. As ASD, ADHD is a multifactorial disorder and its etiology has not been clarified. Some studies have investigated the role of folate, B12, B6, and Hcy levels in many psychiatric diseases and depression [134,135,136,137] for the fundamental roles of these compounds in the carbon transfer metabolism, the necessary pathway to produce serotonin, catecholamines, and other monoamine neurotransmitters. These studies correlated high levels of Hcy and low levels of folate, B12, and B6 with the etiology and/or symptoms of diseases. Interesting results emerged from a study [133] conducted on 30 children and adolescents aged 6–15 years with ADHD and 30 controls (also aged 6–15). This study demonstrated lower folate levels in ADHD affected children with respect to the controls, as already observed in adults affected by ADHD [138], with a different behaviour of Hcy plasma levels observed in the majority of the studies. The authors explained that low Hcy levels could be an index of an excessive transformation to cystatin in the trans-sulfuration pathways for glutathione, taurine, and sulfate production, as already observed in Down’s syndrome patients [139]. Certain conditions or diseases that increase oxidative stress in the human body may increase hepatic glutathione synthesis, determining a decrease of plasma Hcy levels, which are critical for potential total body glutathione status. Low plasma levels of Hcy determine a reduction in cysteine and a very limited capacity of individuals to respond to oxidative stress [133]. With respect to the results of many studies indicating the correlation of low levels of vitamin B6, folate, and vitamin B12 with higher Hcy plasma levels, Altun et al. [133] observed lower levels of these compounds in ADHD patients compared to the control group. Thus, further studies are required to clarify the specific relationships between Hcy, folate, vitamin B12, and pyridoxine vitamin B6 levels for the assessment of ADHD.
4. Elderly
HHcy is common among elderly people and is often associated with cardiovascular diseases (CVD) (including coronary artery disease, stroke, and peripheral vascular disease), cognitive impairment, dementia, depression [140,141,142], and osteoporotic fractures [143]. In a large cohort of community-dwelling older men, HHcy appeared to be predictive of all-cause mortality, independent of frailty [136], an age-related clinical state characterized by a global impairment of physiological functions and involving multiple organ systems [144]. A meta-analysis of prospective studies indicated that an increase of 5 μmol/L Hcy increased by 27% the risk of all–cause mortality, by 32% the risk of cardiovascular mortality, and by 52% coronary heart disease mortality, and the risk was more elevated in elderly population [145]. In a long-term study [146] on middle-aged and elderly subjects, increased levels of Hcy (≥10.8 μmol/L) were significantly associated with all-cause mortality (p < 0.001) and CVD (p < 0.001) compared with decreased levels (Hcy < 10.8 μmol/L). As already described above, the plasma concentration of Hcy is the result of a close relationship between dietary habits, lifestyle, renal function, and predisposing genetic factors. The latter represent only a small fraction of the causes of HHcy. High levels of Hcy are frequently associated with a low dietary intake of folate and other B vitamins (B2, B6, and B12), and several studies have identified links between HHcy, some pathologies, and deficiencies in these micronutrients [147,148]. As an example, a study performed on 1350 elderly Taiwanese subjects aged 65–90 years with normal renal function showed that low plasma levels of folate and vitamin B6 or vitamin B12 were associated with a 3–5-fold increased risk of HHcy (>15 μmol/L) compared to patients with only one B vitamin insufficiency [147]. On the other hand, HHcy often occurs independently of vitamin status [149,150,151]. A recent review [152] reporting the impact of HHcy on aging and related diseases emphasized the importance of screening elderly subjects over 60 years for raised Hcy levels and of evaluating folate or vitamin B12 status for accurate nutritional requirements. The authors also underlined that supplementation with B vitamins, vitamin D, and antioxidants such as via a vitamin-rich diet “must be recommended to all people over 65 years of age”.
4.1. Cardiovascular Diseases
Some case–control studies have shown that 10% of all coronary artery disease is attributable to HHCy, and that an increase of HCy of 5 µmol/l raises the risk of ischemic heart disease by 84% [153]. Tang et al. [154] correlated plasma Hcy levels in patients (aged > 60 years old) with various ischemic cerebrovascular diseases (transient ischemic attack (TIA), large-artery atherosclerosis (LAA), and small-artery occlusion (SAO)). HHcy was closely related to occurrence of TIA; in fact, Hcy levels in the TIA group (17.05 ± 5.36 μmol/L) were significantly higher than those of the LAA (14.39 ± 5.22 μmol/L, p = 0.002) and SAO (13.54 ± 3.72 μmol/L, p = 0.000) groups, while no significant difference was found between the LAA and SAO groups. HHcy could play a key role in the occurrence and development of TIA through free radical production by Hcy oxidation. The high toxicity of these free radicals to vascular endothelial cells, could promote an increased synthesis of oxidized low-density lipoprotein (OxLDL). Thus, atherosclerosis progression could be promoted by induced inflammation and plaque instability. Cao et al. [155] sustained the involvement of HHcy in the pathological manifestation of atherosclerosis. In their study, the Hcy levels (ranged 5–37.1 μmol/L) of 1357 patients (aged 31–90 years) were significantly higher in the carotid/intracranial artery stenosis group than in the control (non-stenosis) group. The involvement of HHcy was probably via worsening the inflammation mechanism that led to the increase of unstable carotid plaques and the occurrence and development of acute cerebral infarction. Since inflammatory response plays an important role in the process of ischemic stroke, Hcy increases the risk of ischemic stroke. Recently, the results observed in a metabolomic study [156] conducted on 65 male patients with severe carotid artery stenosis (aged 60.2 ± 5.9 years) highlighted significantly higher levels of plasma Hcy but lower levels of cholesterol, high-density lipoprotein, and hemoglobin than 65 male control subjects (aged 63.3 ± 5.2) (p < 0.05).
As already mentioned, HHcy can be reduced by vitamin B group supplementation. Thus, if the relationship between Hcy and CVD is causal, supplementation could be expected to reduce the risk of CVD and stroke and this could be tested in randomized controlled trials. Some meta-analyses have been conducted to evaluate the effects of folic acid supplementation on stroke risk [157,158]. Lee et al. [157] identified 13 randomized controlled trials with a total of 39,005 adult participants and observed that folic acid supplementation did not reach statistical significance in reducing the risk of stroke (RR = 0.93; 95% CI, 0.85–1.03; p = 0.16). When folic acid supplementation was associated with B6 and B12, a potential mild benefit was observed (RR = 0.83; 95% CI, 0.71–0.97; p = 0.02), especially in men (trials with ratio men:women >2; RR = 0.84; 95% CI, 0.74–0.94; p = 0.003). Zeng et al. [158], examining 14 randomized trials with a total of 39,420 adult patients, stratifying trials according to the folate fortification, concluded that folic acid supplementation “might have a modest benefit on stroke prevention in regions without folate fortification”. In a study conducted on 390 healthy Chinese subjects aged 60–74 years, supplemented with vitamin C alone (control group) or vitamin C and B vitamins (treatment group) for 12 months, the authors reported that the Framingham risk score, a predictor of cardiovascular disease risk, was particularly reduced in elderly subjects with folate deficiency [159]. As pointed out by Ostrakhovitch and Tabibzadeh [152], although no consensus has yet been reached regarding the effectiveness of HHcy correction in preventing the development of vascular disease, Hcy-lowering therapy “might useful in lowering the risk of inflammation and vascular risk factors”. Several hypotheses have been advanced in the literature. Folate and vitamin B nutritional status could influence the response to supplementation. Loscalzo et al. [160] hypothesized that high levels of unmetabolized folate might also interfere with the effectiveness of B vitamin supplementation. Zhao et al. [161] underscored the importance of individual genetic background and nutritional status of patients. Their analysis of the data of 20,424 hypertensive adults enrolled in the China Stroke Primary Prevention Trial observed that in Chinese hypertensive patients, the effect of Hcy on the first stroke was significantly modified by the methylenetetrahydrofolate reductase C677T genotype and folic acid supplementation. In fact, folic acid intervention significantly reduced stroke risk in participants with the MTHFR CC/CT genotype and high Hcy levels (tertile 3; hazard ratio, 0.73; 0.55–0.97). It is also interesting to note that folic acid supplementation was significantly more effective in individuals with the MTHFR TT genotype without HHcy (<12.8 μmol/L), compared with individuals with HHcy in the same genotype. The authors speculated that differences in folate status in MTHFR enzyme activity such as long-standing injury between participants with CC/CT genotype and those with the TT genotype may have been responsible for the observed results, and speculated that higher doses of folic acid or longer supplementation periods would be needed.
4.2. Neurodegenerative Diseases
In addition to the known influence of Hcy on CVD, numerous diseases of the nervous system, such as cognitive decline, Alzheimer’s disease (AD), and Parkinson’s disease (PD) are correlated with HHcy. The risk of Alzheimer’s disease doubled with a plasma homocysteine level greater than 14 µmol/L, as observed by Seshadri et al. [162] in an 8 year follow-up study on 1092 subjects without dementia (667 women and 425 men; mean age, 76 years). Numerous mechanisms by which Hcy may act to promote these conditions have been proposed, such as excitotoxicity for neurons, increased generation of reactive oxygen species (ROS) production, inflammatory processes, and long-term levodopa treatment [35,48]. Hooshmand et al. [163], examining 2570 elderly individuals (mean age, 73.1 ± 10.4 years) over a 6 year period found that the methionine to Hcy status was associated with dementia development and structural brain changes, suggesting that a higher methionine to Hcy ratio may be significant in decreasing the rate of brain atrophy and decreasing the risk of dementia in older adults. Differences in vitamin B status can influence the grade of disease, although contrasting results are present in the literature. In the VITAGOG study [164], a two year supplementation with B vitamins was associated with a 30% reduction in Hcy and an improvement of cognitive decline. Douaud et al. [165], in a study performed on elderly subjects with increased dementia risk, found that B-vitamin supplementation slowed the atrophy of specific brain regions that are key components of Alzheimer’s disease and that are associated with cognitive decline. Similarly, other studies [166,167] have found that supplements containing folate, vitamin B6, and vitamin B12 in patients with HHcy could reduce Hcy levels and improve their cognitive function. Further studies have confirmed the efficacy of some vitamins in lowering Hcy, but not in the attenuation of pathological symptoms. In a meta-analysis of selected randomized controlled trials, including in total 679 patients with cognitive decline secondary to Alzheimer disease or dementia (age range: 74.6–79.1 years), upon intervention with folic acid along with vitamin B12 and/or B6, the authors observed a significant reduction in Hcy levels in the treated patients compared to the controls (pooled difference in means −3.626 µmol/L), but it was not associated with cognitive improvement [168]. In healthy men and women aged 55–65 years, after a multivitamin supplementation for 16 weeks, an improvement of blood biomarkers (reduced Hcy, increase of vitamin B6 and vitamin B12 levels) without effect on cognitive decline was observed [169]. Ma et al. [148] investigated the relationship between cognitive impairment and Hcy, folate, and vitamin B12 in a case–control study on elderly Chinese subjects (age ≥ 65 years: 112 with mild cognitive impairment (MCI), 89 Alzheimer’s disease (AD) patients, and 115 healthy control). Serum folate and vitamin B12 levels were significantly lower and the plasma Hcy level was higher in patients than in healthy controls, but no association existed between low vitamin B12 levels and AD or MCI (p > 0.05). The authors suggested that further studies using holotranscobalamin as a marker of vitamin B12 deficiency will be performed. Harrington [170] pointed out that “no single laboratory marker is suitable for the assessment of B12 status”, and holotranscobalamin can be a more reliable marker of B12 status than total vitamin B12 alone. The same author suggested using “algorithms or the combination of multiple markers to mitigate inherent limitations of each marker when used independently”. In a longitudinal cohort study on 38 subjects (men aged 81.3 ± 6 years) affected by AD and followed for an average period of 13 months, significant correlations between Hcy and plasma folate (rs = −0.58, p < 0.001) and vitamin B12 (rs = −0.42, p < 0.01), and between Hcy and dietary intake of folate (rs = −0.34, p < 0.04) were found at baseline. A decline of cognitive status associated with an increase of plasma level of Hcy (p=0.006) was observed at follow-up, but no association with vitamin B status was found [149]. Similarly, Bonetti et al. [150], in 318 elderly subjects (age ≥ 65 years) (44 normal cognition, 127 with cognitive impairment, 147 with dementia) found that HHcy (>15 μmol/L) was associated with a higher prevalence of cognitive and functional impairment and dementia (odds ratio (OR) = 1.98, 95% confidence interval (CI) = 1.13–3.48) independent of B group vitamin status. The authors suggested that HHcy cannot be explained only by vitamin status and that other dietary and non-dietary factors may contribute [150,151]. Lifestyle, coffee consumption, smoking, and alcohol use seemsto be associated with HHcy in Parkinson’s disease patients treated with levodopa [171]. Oulhaj et al. [172], analyzing the data of the VITACOG study, found that different omega-3 fatty acid status results in the trial participants influenced the effect of vitamin B treatment, and suggested that the interaction between the two nutrients on cognition and brain atrophy must be considered and that a complementary supplementation could delay the conversion from mild cognitive impairment to dementia.
4.3. Osteoporosis
Low bone mineral density (BMD) is a common geriatric problem, and it has been hypothesized that the metabolism of HCy is involved in osteoporosis, increasing the risk of osteoporotic fracture [1]. In the Framingham study [173] on 825 men and 1174 women aged from 59 to 91 years, with a follow up 12.3 years for men and 15.0 years for women, it was observed that HHcy (>20 μmol/L for men and >18 μmol/L for women) is associated with higher risk of bone fracture, almost 4 times as high for men and 1.9 times as high for women. Similarly, van Meurs [174] showed in two independent prospective studies that in 2406 subjects aged 55 years and older, the overall multivariable-adjusted relative risk of fracture was 1.4 (95% confidence interval, 1.2 to 1.6) for each increase of 1 S.D. in the natural-log-transformed Hcy level. An Hcy level in the highest age-specific quartile was associated with an increase in the risk of fracture twice as high as the risk in the lower three quartiles. Several mechanisms by which Hcy may act to promote the pathological status have been proposed [175,176,177,178,179,180]. In particular, as reviewed by Toohey et al. [181], HHcy can cause connective tissue pathology by conversion of Hcy thiolactone into mercaptopropionaldehyde via several routes, such as ascorbic acid depletion influencing collagen synthesis, interfering directly with collagen synthesis, causing abnormal cross-linking of collagen molecules, and triggering autoimmune response. Moreover, oxidative damage and the adverse effects of Hcy on endothelial-derived nitric oxide could increase risk of fracture [182,183,184]. MTHFR polymorphism C667 T could also be involved in the increase of osteoporotic fracture incidence [181], and a recently published meta-analysis showed an association between MTHFR C667 T polymorphism and reduced bone mineral density in the lumbar spine and femoral neck in Caucasians, post-menopausal women, and men, and with total BMD in women [185]. Ahn et al. [186] reported an association in 301 postmenopausal women between osteoporotic vertebral compression fractures and MTHFR and TS gene polymorphisms in the 3′-UTR region. Concerning the effect of supplementation, a systematic review with meta-analysis [187] concluded that vitamin B12 status may be associated with fracture risk, and evidence for an association between folate status and fracture risk is scarce. In the B-PROOF study [188,189], a randomized placebo-controlled trial of a 2 year intervention of vitamin B12 and folic acid supplementation in 2919 elderly subjects (aged ≥65 years) with moderate HHcy produced no observable effect of treatment on bone mineral density despite a significant reduction of Hcy (−4.2 ± 3.0 μmol/L, p < 0.001). Similarly, other studies have observed no association of Hcy-lowering treatment with B vitamins, bone turnover, and risk of fracture [187,188,189,190,191,192].
4.4. Physical Function
Several studies have highlighted the relationship between Hcy and muscle strength and physical function in the elderly, stressing that the decrease in muscle strength could also play a role in the risk of falls. In the MacArthur Studies of Successful Aging on 499 healthy and highly functional community-dwelling people (aged 70–79 years) [193], the authors observed that subjects with elevated plasma Hcy had an increased risk of functional decline over 3 years, with a 50% higher risk of being in the worst quartile of physical function (odds ratio = 1.5; 95% CI, 1.2–1.9) after multivariable adjustment. Similarly, in the Baltimore Longitudinal Study of Aging [194] on 1101 adults aged 50 years or older, in a follow-up of 4.7 years a significant inverse relationship between Hcy and grip strength was observed in women (β= −005, p = 0.031), while in men no significant results were observed, although an increase of 1 μmol/L in Hcy was associated with −0.10 kg decrease in grip strength. Oxidative damage of endothelial cells and deoxyribonucleic acid, telomere loss such as leukoaraiosis induced by HHCy, and bioavailability of nitric oxide [195] could be possible mechanisms involved in the relationship between Hcy and lowered physical function [193]. Physical performance is also related to poor vitamin B group nutritional status; moreover, Ao et al. [196] in an observational study performed on 65 women (aged 84 ± 7.1 years) observed low muscle and hand grip strength in subjects with low serum folate concentration and pointed to the importance of adequate folate nutrition to avoid impaired physical function. However, supplementation studies with these vitamins have reported no effect on physical performance, hand grip strength, and risk of falling, despite a significant reduction of Hcy [188,189]. Vidoni et al. [197], studying 774 adults aged 50 years or older, found that only Hcy and not vitamin B12 was associated with gait speed decline. The authors observed that this loss of association could have been due to the good state of nutrition in vitamin B12 of the studied sample, concluding that more studies are necessary in populations with greater variation in vitamin B12 concentrations, as well as a more in-depth study of the mechanisms. Additionally, Veeeranki [195] in his review on the potential implication of HHcy on muscle function and integrity highlighted the need for studies to provide deeper insights in this area.
5. Conclusions
Regarding the different pathological processes that can affect the human body at any stage of life, the main role of Hcy as an involved and predicting factor is clear. Despite extensive studies of Hcy’s role in several pathologies at different life stages, the exact mechanisms of action as well as the homeostasic regulation of this biomarker remain unresolved. Various common research approaches (cohort studies, case–control, randomized) and study designs do not take into account the relationships between nutrient intakes and the molecular and cellular responses of the organism that eventually result in health and disease; the requirements according to specific genetic profiles and the influences of age, sex, activity level, and metabolic status as well plasma nutrient levels might not reflect tissue storage and needs, or more generally the “optimal” biochemical status. Hcy levels during pregnancy even seem to be linked to the geographical, cultural, and social characteristics of populations, while for children and adolescents, regulated diet and modulated lifestyle decrease Hcy levels, and thus the determination of hyperhomocysteinemia in childhood and in adolescence could be an opportunity for a nutritional and lifestyle intervention for primary prevention of diseases. However, the results from clinical trials of Hcy-lowering vitamin treatments have not reached consensus about the effectiveness of vitamin supplementation on the reduction in Hcy levels and the improvement of pathological conditions, especially in elderly patients with overt pathologies. It is possible to speculate that these weaknesses explain why micronutrient supplementation studies continue to yield contradictory findings. The importance of novel experimental designs focusing on intra-individual variability as a complement to the typical case–control experimental designs and the study of interactions between different factors should be emphasized.
Author Contributions
All authors contributed equally to the work reported. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| Hcy | homocysteine |
| HHcy | hyperhomocysteinemia |
| MAT | methionine adenosyltransferase |
| SAM | S-adenosylmethionine |
| SAH | S-adenosylHcy |
| MTHFR | 5,10-methylene-THF reductase |
| BHMT | betaine-Hcy S-methyltransferase |
| GSH | reduced glutathione |
| MetRS | methionyl-tRNA synthetase |
| N-Hcy | N-homocysteinylated (N-Hcy) proteins |
| THF | tetrahydrofolate |
| DHFR | dihydrofolate reductase |
| SHMT | serine hydroxymethyltransferase |
| 5,10 MTHF | 5,10-methylenetetrahydrofolate |
| 5-MTHF | 5-methylenetetrahydrofolate |
| CBS | cystathionine β-synthase |
| CSE | cystathionase |
| PON | paraoxonase |
| HDL | high density lipoprotein |
| Hcy-SH | homocysteine |
| DMG | N,N-dimethylglycine |
| NTDPR | Neural Tube Defetc Prevalence Ratio |
| PoTS | Postural Tachycardia Syndrome |
| BRS | Baroreflex sensitivity |
| HF | Heart Failure |
| CHA | Congenital Adrenal Hyperplasia |
| Hs-cTnT | Highly sensitive cardiac troponin T |
| CHA | Congenital Adrenal Hyperplasia |
| CA-IMT | Carotid artery intima-media thickness |
| LV | Left ventricular |
| ASD | Autism spectrum disorder |
| CVD | Cardiovascular diseases |
| NS | Nephrotic Sindrome |
| FSGS | Focal Segmental Glomerulosclerosis |
| RBC | Red Blood Cell |
| NMDA | N-methyl-d-aspartate |
| ADHD | Attention Deficit Hyperactivity Disorder |
| TIA | Transient ischemic attack |
| LAA | Large-artery atherosclerosis |
| SAO | Small-artery occlusion |
| OxLDL | Oxidized low-density lipoprotein |
| ROS | Reactive Oxygen Species |
| MCI | Mild Cognitive Impairment |
| AD | Alzheimer’s disease |
| OR | Odd ratio |
| IC | Interval Confidence |
| BMD | Bone mineral Density |
References
- Behera, J.; Bala, J.; Nuru, M.; Tyagi, S.C.; Tyagi, N. Homocysteine as a Pathological Biomarker for Bone Disease. J. Cell. Physiol. 2017, 232, 2704–2709. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Tiwari, M.; Tiwari, R.K. Hyperhomocysteinemia: Impact on Neurodegenerative Diseases. Basic Clin. Pharmacol. Toxicol. 2015, 117, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Perna, A.F.; Ingrosso, D. Atherosclerosis determinants in renal disease: How much is homocysteine involved? Nephrol. Dial. Transplant. 2016, 31, 860–863. [Google Scholar] [CrossRef] [PubMed]
- Setién-Suero, E.; Suárez-Pinilla, M.; Suárez-Pinilla, P.; Crespo-Facorro, B.; Ayesa-Arriola, R. Homocysteine and cognition: A systematic review of 111 studies. Neurosci. Biobehav. Rev. 2016, 69, 280–298. [Google Scholar] [CrossRef] [PubMed]
- Nasri, K.; Ben Fradj, M.K.; Touati, A.; Aloui, M.; Ben Jemaa, N.; Masmoudi, A.; Elmay, M.V.; Omar, S.; Feki, M.; Kaabechi, N.; et al. Association of maternal homocysteine and vitamins status with the risk of neural tube defects in Tunisia: A case-control study. Birth Defects Res. A Clin. Mol. Teratol. 2015, 103, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, A.; Verdoia, M.; Cassetti, E.; Marino, P.; Suryapranata, H.; De Luca, G.; Novara Atherosclerosis Study Group (NAS). Relationship between homocysteine and coronary artery disease. Results from a large prospective cohort study. Thromb. Res. 2014, 134, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, P.; Alam, S.F. Role of homocysteine in the development of cardiovascular disease. Nutr. J. 2015, 14, 6. [Google Scholar] [CrossRef]
- Lehotský, J.; Tothová, B.; Kovalská, M.; Dobrota, D.; Beňová, A.; Kalenská, D.; Kaplán, P. Role of Homocysteine in the Ischemic Stroke and Development of Ischemic Tolerance. Front. Neurosci. 2016, 10, 538. [Google Scholar] [CrossRef]
- Esse, R.; Barroso, M.; Almeida, I.; Castro, R. The contribution of homocysteine metabolism disruption to endothelial dysfunction: State-of-the-art. Int. J. Mol. Sci. 2019, 20, 867. [Google Scholar] [CrossRef]
- Steenge, G.R.; Verhoef, P.; Katan, M.B. Betaine supplementation lowers plasma HCY in healthy men and women. J. Nutr. 2003, 133, 1291–1295. [Google Scholar] [CrossRef]
- Olthof, M.R.; van Vliet, T.; Boelsma, E.; Verhoef, P. Low dose betaine supplementation leads to immediate and long term lowering of plasma homocysteine in healthy men and women. J. Nutr. 2003, 133, 4135–4138. [Google Scholar] [CrossRef] [PubMed]
- McRae, M.P. Betaine supplementation decreases plasma homocysteine in healthy adult participants: A meta-analysis. J. Chiropr. Med. 2013, 12, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Zaric, L.B.; Obradovic, M.; Bajic, V.; Haidara, M.A.; Jovanovic, M.; Isenovic, E.R. Homocysteine and Hyperhomocysteinaemia. Curr. Med. Chem. 2019, 26, 2948–2961. [Google Scholar] [CrossRef] [PubMed]
- Verhoef, P.; van Vliet, T.; Olthof, M.R.; Katan, M.B. A high-protein diet increases postprandial but not fasting plasma total homocysteine concentrations: A dietary controlled, crossover trial in healthy volunteers. Am. J. Clin. Nutr. 2005, 82, 553–558. [Google Scholar] [CrossRef]
- Jakubowski, H. Quality control in tRNA charging—Editing of homocysteine. Acta Biochim. Pol. 2011, 58, 149–163. [Google Scholar] [CrossRef]
- Jakubowski, H. Homocysteine editing, thioester chemistry, coenzyme A, and the origin of coded peptide synthesis dagger. Life 2017, 7, 6. [Google Scholar] [CrossRef]
- Gurda, D.; Handschuh, L.; Kotkowiak, W.; Jakubowski, H. Homocysteine thiolactone and N-homocysteinylated protein induce pro-atherogenic changes in gene expression in human vascular endothelial cells. Amino Acids 2015, 47, 1319–1339. [Google Scholar] [CrossRef]
- Jakubowski, H. Calcium-dependent human serum homocysteine thiolactone hydrolase. A protective mechanism against protein N-homocysteinylation. J. Biol. Chem. 2000, 275, 3957–3962. [Google Scholar] [CrossRef]
- Perła-Kaján, J.; Borowczyk, K.; Głowacki, R.; Nygård, O.; Jakubowski, H. Paraoxonase 1 Q192R genotype and activity affect homocysteine thiolactone levels in humans. FASEB J. 2018, 32, 6019–6024. [Google Scholar] [CrossRef]
- Chwatko, G.; Jakubowski, H. Urinary Excretion of Homocysteine-Thiolactone in Humans. Clin. Chem. 2005, 51, 408–415. [Google Scholar] [CrossRef]
- Borowczyk, K.; Piechocka, J.; Głowacki, R.; Dhar, I.; Midtun, Ø.; Tell, G.S.; Ueland, P.M.; Jakubowski, H. Urinary excretion of homocysteine thiolactone and the risk of acute myocardial infarction in coronary artery disease patients: The WENBIT trial. J. Intern. Med. 2019, 285, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Homocysteine Modification in Protein Structure/Function and Human Disease. Physiol. Rev. 2019, 99, 555–604. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Pathophysiological Consequences of Homocysteine Excess. J. Nutr. 2006, 136, 1741–1749. [Google Scholar] [CrossRef] [PubMed]
- Robert, K.; Pages, C.; Ledru, A.; Delabar, J.; Caboche, J.; Janel, N. Regulation of extracellular signal-regulated kinase by homocysteine in hippocampus. Neuroscience 2005, 133, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, M.C.; Gurol, M.E.; Raju, S.; Diaz-Arrastia, R.; Locascio, J.J.; Tennis, M.; Hyman, B.T.; Growdon, J.H.; Greenberg, S.M.; Bottiglieri, T. Association of homocysteine with plasma amyloid beta protein in aging and neurodegenerative disease. Neurology 2005, 65, 1402–1408. [Google Scholar] [CrossRef] [PubMed]
- Leulliot, N.; Quevillon-Cheruel, S.; Sorel, I.; Li de La Sierra-Gallay, I.; Collinet, B.; Graille, M.; Blondeau, K.; Bettache, N.; Poupon, A.; Janin, J.; et al. Structure of protein phosphatase methyltransferase 1 (PPM1), a leucine carboxyl methyltransferase involved in the regulation of protein phosphatase 2A activity. J. Biol. Chem. 2004, 279, 8351–8358. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.I.; Ashline, D.; Dhitavat, S.; Ortiz, D.; Collins, S.C.; Shea, T.B.; Rogers, E. Folate deprivation induces neurodegeneration: Roles of oxidative stress and increased homocysteine. Neurobiol. Dis. 2003, 14, 32–42. [Google Scholar] [CrossRef]
- Kruman, I.I.; Culmsee, C.; Chan, S.L.; Kruman, Y.; Guo, Z.; Penix, L.; Mattson, M.P. Homocysteine elicits a DNA damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J. Neurosci. 2000, 20, 6920–6926. [Google Scholar] [CrossRef]
- Van Guldener, C. Why is homocysteine elevated in renal failure and what can be expected from homocysteine-lowering? Nephrol. Dial. Transplant. 2006, 21, 1161–1166. [Google Scholar] [CrossRef]
- Hannibal, L.; Blom, H.J. Homocysteine and disease: Causal associations or epiphenomenons. Mol. Aspects Med. 2017, 53, 36–42. [Google Scholar] [CrossRef]
- Jiang, X.; Yang, F.; Brailoiu, E.; Jakubowski, H.; Dun, N.J.; Schafer, A.I.; Yang, X.; Durante, W.; Wang, H. Differential regulation of homocysteine transport in vascular endothelial and smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1976–1983. [Google Scholar] [CrossRef]
- Tsitsiou, E.; Sibley, C.P.; D’Souza, S.W.; Catanescu, O.; Jacobsen, D.W.; Glazier, J.D. Homocysteine is transported by the microvillous plasma membrane of human placenta. J. Inherit. Metab. Dis. 2011, 34, 57–65. [Google Scholar] [CrossRef]
- Sengupta, S.; Chen, H.; Togawa, T.; DiBello, P.M.; Majors, A.K.; Büdy, B.; Ketterer, M.E.; Jacobsen, D.W. Albumin thiolate anion is an intermediate in the formation of albumin-S-S-homocysteine. J. Biol Chem. 2001, 276, 30111–30117. [Google Scholar] [CrossRef]
- Mudd, S.H.; Jenden, D.J.; Capdevila, A.; Roch, M.; Levy, H.L.; Wagner, C. Isolated hypermethioninemia: Measurements of S-adenosylmethionine and choline. Metabolism 2000, 49, 1542–1547. [Google Scholar] [CrossRef] [PubMed]
- Škovierová, H.; Vidomanová, E.; Mahmood, S.; Sopková, J.; Drgová, A.; Červeňová, T.; Halašová, E.; Lehotský, J. The Molecular and Cellular Effect of Homocysteine Metabolism Imbalance on Human Health. Int. J. Mol. Sci. 2016, 17, 1733. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, D.W. Total Plasma Homocysteine: The Mediator/Marker Controversy Continues. Clin. Chem. 2009, 55, 1742–1743. [Google Scholar] [CrossRef] [PubMed]
- Fokkema, M.R.; Weijer, J.M.; Dijck-Brouwer, D.A.; van Doormaal, J.J.; Muskiet, F.A. Influence of vitamin-optimized plasma homocysteine cutoff values on the prevalence of hyperhomocysteinemia in healthy adults. Clin Chem. 2001, 47, 1001–1007. [Google Scholar] [CrossRef]
- Refsum, H.; Smith, A.D.; Ueland, M.P.; Nexo, E.; Clarke, R.; McPartlin, J.; Johnston, C.; Engbaek, F.; Schneede, J.; McPartlin, C.; et al. Facts and recommendations about total homocysteine determinations: An expert opinion. Clin. Chem. 2004, 50, 3–32. [Google Scholar] [CrossRef]
- Selhub, J.; Jacques, P.F.; Dallal, G.; Choumenkovitch, S.; Rogers, G. The use of Blood Concentrations of Vitamins and their Respective Functional Indicators to Define Folate and Vitamin B12 Status. Food Nutr. Bull. 2008, 29 (Suppl. S1), S67–S73. [Google Scholar] [CrossRef]
- B-Vitamin Treatment Trialists’ Collaboration. Homocysteine-lowering trials for prevention of cardiovascular events: A review of the design and power of the large randomized trials. Am. Heart J. 2006, 151, 282–287. [Google Scholar] [CrossRef]
- Lonn, E.; Yusuf, S.; Arnold, M.J.; Sheridan, P.; Pogue, J.; Micks, M.; McQueen, M.J.; Probstfield, J.; Fodor, G.; Held, C.; et al. Heart Outcomes Prevention Evaluation (HOPE) 2 Investigators. Homocysteine lowering with folic acid and B vitamins in vascular disease. N. Engl. J. Med. 2006, 354, 1567–1577. [Google Scholar] [CrossRef] [PubMed]
- Homocysteine Lowering Trialists’ Collaboration. Dose-dependent effects of folic acid on blood concentrations of homocysteine: A meta-analysis of the randomized trials. Am. J. Clin. Nutr 2005, 82, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Lonn, E.; Held, C.; Arnold, J.M.; Probstfield, J.; McQueen, M.; Micks, M.; Pogue, J.; Sheridan, P.; Bosch, J.; Genest, J.; et al. Rationale, design and baseline characteristics of a large, simple, randomized trial of combined folic acid and vitamins B6 and B12 in high-risk patients: The Heart Outcomes Prevention Evaluation (HOPE)-2 trial. Can. J. Cardiol. 2006, 22, 47–53. [Google Scholar] [CrossRef][Green Version]
- Laraqui, A.; Allami, A.; Carrié, A.; Coiffard, A.S.; Benkouka, F.; Benjouad, A.; Bendriss, A.; Kadiri, N.; Bennouar, N.; Benomar, A.; et al. Influence of methionine synthase (A2756G) and methionine synthase reductase (A66G) polymorphisms on plasma homocysteine levels and relation to risk of coronary artery disease. Acta Cardiol. 2006, 61, 51–61. [Google Scholar] [CrossRef]
- Li, W.X.; Cheng, F.; Zhang, A.J.; Dai, S.X.; Li, G.H.; Lv, W.W.; Zhou, T.; Zhang, Q.; Zhang, H.; Zhang, T.; et al. Folate Deficiency and Gene Polymorphisms of MTHFR, MTR and MTRR Elevate the Hyperhomocysteinemia Risk. Clin. Lab. 2017, 63, 523–533. [Google Scholar] [CrossRef]
- Mendes, M.I.S.; Colaço, H.G.; Smith, D.E.C.; Ramos, R.J.J.F.; Pop, A.; van Dooren, S.J.M.; Tavares de Almeida, I.; Kluijtmans, L.A.J.; Janssen, M.C.H.; Rivera, I.; et al. Reduced response of cystathionine beta-synthase (CBS) to S-adenosylmethionine (SAM): Identification and functional analysis of CBS gene mutations in Homocystinuria patients. J. Inherit. Metab. Dis. 2014, 37, 245–254. [Google Scholar] [CrossRef]
- Kluijtmans, L.A.J.; Young, I.S.; Boreham, C.A.; Murray, L.; McMaster, D.; McNulty, H.; Strain, J.J.; McPartlin, J.; Scott, J.M.; Whitehead, A.S. Genetic and nutritional factors contributing to hyperhomocysteinemia in young adults. Blood 2003, 101, 2483–2488. [Google Scholar] [CrossRef]
- Tinelli, C.; Di Pino, A.; Ficulle, E.; Marcelli, S.; Feligioni, M. Hyperhomocysteinemia as a Risk Factor and Potential Nutraceutical Target for Certain Pathologies. Front. Nutr. 2019, 6, 49. [Google Scholar] [CrossRef]
- Eskes, T.K. Homocysteine and human reproduction. Clin. Exp. Obstet. Gynecol. 2000, 27, 157–167. [Google Scholar]
- Laskowska, M.; Oleszczuk, J. Homocysteine in pregnancies complicated by preeclampsia with and without IUGR: A comparison with normotensive pregnant women with isolated IUGR and healthy pregnant women. Open J. Obstet. Gynecol. 2011, 1, 191–196. [Google Scholar] [CrossRef]
- Yang, M.; Li, W.; Wan, Z.; Du, Y. Elevated Homocysteine Levels in Mothers With Neural Tube Defects: A Systematic Review and Meta-Analysis. J. Matern. Fetal Neonatal Med. 2017, 30, 2051–2057. [Google Scholar] [CrossRef] [PubMed]
- Gaiday, A.N.; Tussupkaliyev, A.B.T.; Bermagambetova, S.K.; Zhumagulova, S.S.; Sarsembayeva, L.K.; Dossimbetova, M.B.; ZhDaribay, Z. Effect of homocysteine on pregnancy: A systematic review. Chem. Biol. Interact. 2018, 293, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Doods, L.; Fell, D.B.; Armson, B.A.; Allen, A.C.; Nassar, B.A.; Perkins, S.; Joseph, K.S. Effect of homocysteine concentration in early pregnancy on gestational hypertensive disorders and other pregnancy outcomes. Clin Chem. 2008, 54, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Choi, R.; Choi, S.; Lim, Y.; Cho, Y.Y.; Kim, H.J.; Kim, S.W.; Chung, J.H.; Oh, S.Y.; Lee, S.Y. A Prospective Study on Serum Methylmalonic Acid and Homocysteine in Pregnant Women. Nutrients 2016, 8, 797. [Google Scholar] [CrossRef]
- Bondevik, G.T.; Scneede, J.; Refsum, H.; Lie, R.T.; Ulstein, M.; Kvåle, G. Homocysteine and methylmalonic acid levels in pregnant Nepali women. Should cobalamin supplementation be considered? Eur. J. Clin. Nutr. 2001, 55, 856–864. [Google Scholar] [CrossRef]
- Mascarenhas, M.; Habeebullah, S.; Sridhar, M.G. Revisiting the Role of First Trimester Homocysteine as an Index of Maternal and Fetal Outcome. J. Pregnancy 2014, 2014, 123024. [Google Scholar] [CrossRef]
- Murphy, M.M.; Fernadez-Ballart, J.D. Homocysteine in Pregnancy. Adv. Clin. Chem. 2011, 53, 105–107. [Google Scholar] [CrossRef]
- Bergen, N.; Jaddoe, V.; Timmermans, S.; Hofman, A.; Lindemans, J.; Russcher, H.; Raat, H.; Steegers-Theunissen, R.; Steegers, E. Homocysteine and folate concentrations in early pregnancy and the risk of adverse pregnancy outcomes: The Generation R Study. BJOG 2012, 119, 739–751. [Google Scholar] [CrossRef]
- Gupta, R.; Kumari, P.; Pandey, S.; Joshi, D.; Sharma, S.P.; Kumar, S.; Singh, R. Homocysteine and Vitamin B12: Other Causes of Neural Tube Defects in Eastern Uttar Pradesh and Western Bihar Population. Neurol. India 2018, 66, 1016–1019. [Google Scholar] [CrossRef]
- Peker, E.; Demir, N.; Tuncer, O.; Üstyol, L.; Balahoroğlu, R.; Kaba, S.; Karaman, K. The Levels of Vitamın B12, Folate and Homocysteine in Mothers and Their Babies With Neural Tube Defects. J. Matern. Fetal Neonatal Med. 2016, 29, 2944–2948. [Google Scholar] [CrossRef]
- Senousy, S.M.; Farag, M.K.; Gouda, A.S.; El Noury, M.A.; Dabbous, O.A.; Gaber, K.R. Association Between Biomarkers of Vitamin B12 Status and the Risk of Neural Tube Defects. J. Obstet. Gynaecol. Res. 2018, 44, 1902–1908. [Google Scholar] [CrossRef] [PubMed]
- Deb, R.; Arora, J.; Samtani, R.; Garg, G.; Saksena, D.; Sharma, N.; Kumar Kalla, A.; Nava Saraswathy, K. Folic acid, dietary habits, and homocysteine levels in relation to neural tube defects: A case-control study in North India. Birth Defects Res. 2018, 110, 1148–1152. [Google Scholar] [CrossRef] [PubMed]
- Iacobazzi, V.; Infantino, V.; Castegna, A.; Andria, G. Hyperhomocysteinemia: Related genetic diseases and congenital defects, abnormal DNA methylation and newborn screening issues. Mol. Genet. Metab. 2014, 113, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Mosley, B.S.; Cleves, M.A.; Melnyk, S.; James, S.J.; Hobbs, C.A. Neural tube defects and maternal biomarkers of folate, homocysteine, and glutathione metabolism. Birth Defects Res. Part A Clin. Mol. Teratol. 2006, 76, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bai, B.; Mei, X.; Wan, C.; Cao, H.; Li, D.; Wang, S.; Zhang, M.; Wang, Z.; Wu, J.; et al. Elevated H3K79 homocysteinylation causes abnormal gene expression during neural development and subsequent neural tube defects. Nat. Commun. 2018, 9, 3436. [Google Scholar] [CrossRef]
- Bailey, D.; Colantonio, D.; Kyriakopoulou, L.; Cohen, A.H.; Chan, M.K.; Armbruster, D.; Adeli, K. Marked Biological Variance in Endocrine and Biochemical Markers in Childhood: Establishment of Pediatric Reference Intervals Using Healthy Community Children from the CALIPER Cohort. Clin Chem. 2013, 59, 1393–1405. [Google Scholar] [CrossRef]
- Rogers, L.M.; Boy, E.; Miller, J.W.; Green, R.; Sabel, J.C.; Allen, L.H. High prevalence of cobalamin deficiency in Guatemalan schoolchildren: Associations with low plasma holotranscobalamin II and elevated serum methylmalonic acid and plasma homocysteine concentrations. Am. J. Clin. Nutr. 2003, 2, 433–440. [Google Scholar] [CrossRef][Green Version]
- de Laet, C.; Wautrecht, J.C.; Brasseur, D.; Dramaix, M.; Boeynaems, J.M.; Decuyper, J.; Kahn, A. Plasmahomocysteine concentration in a Belgian school-age population. Am. J. Clin. Nutr. 1999, 69, 968–972. [Google Scholar] [CrossRef]
- Bates, C.J.; Mansoor, M.A.; Gregory, J.; Pentieva, K.; Prentice, A. Correlates of plasma homocysteine, cysteine and cysteinyl-glycine in respondents in the British National Diet and Nutrition Survey of Young People Aged 4–18 Years, and a comparison with the Survey of People Aged 65 Years and Over. Br. J. Nutr. 2002, 87, 71–79. [Google Scholar] [CrossRef][Green Version]
- Must, A.; Jacques, P.F.; Rogers, G.; Rosenberg, I.H.; Selhub, J. Serum total homocysteine concentrations in children and adolescents: Results from the third National Health and Nutrition Examination Survey (NHANES III). J. Nutr. 2003, 133, 2643–2649. [Google Scholar] [CrossRef]
- Caldeira-Araújo, H.; Ramos, R.; Florindo, C.; Rivera, I.; Castro, R.; Tavares de Almeida, I. Homocysteine Metabolism in Children and Adolescents: Influence of Age on Plasma Biomarkers and Correspondent Genotype Interactions. Nutrients 2019, 11, 646. [Google Scholar] [CrossRef]
- Dávila-Rodríguez, M.I.; Novelo-Huerta, H.I.; Márquez-Solís, R.; Cortés-Gutiérrez, E.; Pérez-Cortés, P.; Cerda-Flores, R.M. Nutritional indicators in children with acute lymphoblastic leukemia. Rev. Med. Inst. Mex. Seguro Soc. 2010, 48, 639–644. [Google Scholar] [PubMed]
- Ribas de Farias Costa, P.; Kinra, S.; D’Almeida, V.; Oliveira Assis, A.M. Serum homocysteine and cysteine levels and changes in the lipid profile of children and adolescents over a 12-month follow-up period. Clin. Nutr. ESPEN 2017, 21, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Osganian, S.K.; Stampfer, M.J.; Spiegelman, D.; Rimm, E.; Cutler, J.A.; Feldman, H.A.; Montgomery, D.H.; Webber, L.S.; Lytle, L.A.; Bausserman, L.; et al. Distribution of and Factors Associated With Serum Homocysteine Levels in Children: Child and Adolescent Trial for Cardiovascular Health. JAMA 1999, 281, 1189–1196. [Google Scholar] [CrossRef]
- Rauh, M.; Verwied, S.; Knerr, I.; Dörr, H.G.; Sönnichsen, A.; Koletzko, B. Homocysteine concentrations in a German cohort of 500 individuals: Reference ranges and determinants of plasma levels in healthy children and their parents. Amino Acids 2001, 20, 409–418. [Google Scholar] [CrossRef]
- Giltay, E.J.; Gooren, L.J.G.; Emeis, J.J.; Kooistra, T.; Stehouwer, C.D. Oral, but not transdermal, administration of estrogens lowers tissue-type plasminogen activator levels in humans without affecting endothelial synthesis. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1396–1403. [Google Scholar] [CrossRef] [PubMed]
- Lien, E.A.; Nedrebø, B.G.; Varhaug, J.E.; Nygård, O.; Aakvaag, A.; Ueland, P.M. Plasma Total Homocysteine Levels during Short-Term Iatrogenic Hypothyroidism. J. Clin. Endocrinol. Metab. 2000, 85, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Fukagawa, N.K.; Martin, J.M.; Wurthmann, A.; Prue, A.H.; Ebenstein, D.; O’Rourke, B. Sex-related differences in methionine metabolism and plasma homocysteine concentrations. Am. J. Clin. Nutr. 2000, 72, 22–29. [Google Scholar] [CrossRef] [PubMed][Green Version]
- González-Gross, M.; Benser, J.; Breidenassel, C.; Albers, U.; Huybrechts, I.; Valtueña, J.; Spinneker, A.; Segoviano, M.; Widhalm, K.; Molnar, D.; et al. HELENA Study group. Gender and age influence blood folate, vitamin B12, vitamin B6, and homocysteine levels in European adolescents: The Helena Study. Nutr. Res. 2012, 32, 817–826. [Google Scholar] [CrossRef]
- Bjørke Monsen, A.L.; Ueland, P.M. Homocysteine and methylmalonic acid in diagnosis and risk assessment from infancy to adolescence. Am. J. Clin. Nutr. 2003, 78, 7–21. [Google Scholar] [CrossRef]
- Papandreou, D.; Rousso, I.; Malindretos, P.; Makedou, A.; Arvanitidou, M. Effects of oral folate supplementation on serum total homocysteine and cholesterol levels in hyperhomocysteinemic children. Nutr. Clin. Pract. 2010, 25, 390–393. [Google Scholar] [CrossRef] [PubMed]
- Papandreou, D.; Malindretos, P.; Arvanitidou, M.; Makedou, A.; Rousso, I. Homocysteine lowering with folic acid supplements in children: Effects on blood pressure. Int. J. Food Sci. Nutr. 2010, 61, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Tonstad, S.; Refsum, H.; Sivertsen, M.; Christophersen, B.; Ose, L.; Ueland, P.M. Relation of total homocysteine and lipid levels in children to premature cardiovascular death in male relatives. Pediatr. Res. 1996, 40, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Crawley, H.F.; While, D. The diet and body weight of British teenage smokers at 16–17 y. Eur. J. Clin. Nutr. 1995, 49, 904–914. [Google Scholar]
- Ribas de Farias Costa, P.; Kinra, S.; D’Almeida, V.; Assis, A.M.O. Serum homocysteine and cysteine levels and associated factors in children and adolescents. Nutr. Clín. Diet. Hosp. 2017, 37, 106–116. [Google Scholar]
- Arouca, A.B.; Santaliestra-Pasías, A.M.; Moreno, L.A.; Marcos, A.; Widhalm, K.; Molnár, D.; Manios, Y.; Gottrand, F.; Kafatos, A.; Kersting, M.; et al. Diet as a moderator in the association of sedentary behaviors with inflammatory biomarkers among adolescents in the HELENA study. Eur. J. Nutr. 2019, 58, 2051–2065. [Google Scholar] [CrossRef]
- Tonstad, S.; Refsum, H.; Ose, L.; Ueland, P.M. The C677T mutation in the methylenetetrahydrofolate reductase gene predisposes to hyperhomocysteinemia in children with familial hypercholesterolemia treated with cholestyramine. J. Pediatr. 1998, 132, 365–368. [Google Scholar] [CrossRef]
- Delvin, E.E.; Rozen, R.; Merouani, A.; Genest, J., Jr.; Lambert, M. Influence of methylenetetrahydrofolate reductase genotype, age, vitamin B-12, and folate status on plasma homocysteine in children. Am. J. Clin. Nutr. 2000, 72, 1469–1473. [Google Scholar] [CrossRef][Green Version]
- Li, Y.; He, B.; Li, H.; Zhang, Q.; Tang, C.; Du, J.; Jin, H. Plasma Homocysteine Level in Children With Postural Tachycardia Syndrome. Front. Pediatr. 2018, 6, 375. [Google Scholar] [CrossRef]
- Metwalley, K.A.; Farghaly, H.S.; Abdelhamid, A. Homocysteine Level in Children with Classic Congenital Adrenal Hyperplasia: Relationship to Carotid Intimal Wall Thickness and Left Ventricular Function. Horm. Res. Paediatr. 2018, 90, 228–235. [Google Scholar] [CrossRef]
- El-Amrousy, D.; Hassan, S.; Hodeib, H. Prognostic value of homocysteine and highly sensitive cardiac troponin T in children with acute heart failure. J. Saudi Heart Assoc. 2017, 30, 198–204. [Google Scholar] [CrossRef]
- Johnson, J.N.; Mack, K.J.; Kuntz, N.L.; Brands, C.K.; Porter, C.J.; Fischer, P.R. Postural orthostatic tachycardia syndrome: A clinical review. Pediatr. Neurol. 2010, 42, 77–85. [Google Scholar] [CrossRef]
- Shen, W.K.; Sheldon, R.S.; Benditt, D.G.; Cohen, M.I.; Forman, D.E.; Goldberger, Z.D.; Grubb, B.P.; Hamdan M, H.; Krahn, A.D.; Link, M.S.; et al. 2017 ACC/AHA/HRS guideline for the evaluation and management of patients with syncope: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J. Am. Coll. Cardiol. 2017, 70, e39–e110. [Google Scholar] [CrossRef]
- Kanjwal, Y.; Kosinski, D.; Grubb, B.P. The postural orthostatic tachycardia syndrome: Definitions, diagnosis, and management. Pacing Clin. Electrophysiol. 2003, 26, 1747–1757. [Google Scholar] [CrossRef]
- Li, H.; Liao, Y.; Wang, Y.; Liu, P.; Sun, C.; Chen, Y.; Tang, C.; Jin, H.; Du, J. Baroreflex sensitivity predicts short-term outcome of postural tachycardia syndrome in children. PLoS ONE 2016, 11, e0167525. [Google Scholar] [CrossRef]
- Muenter Swift, N.; Charkoudian, N.; Dotson, R.M.; Suarez, G.A.; Low, P.A. Baroreflex control of muscle sympathetic nerve activity in postural orthostatic tachycardia syndrome. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, 1226–1233. [Google Scholar] [CrossRef]
- Tayama, J.; Munakata, M.; Yoshinaga, K.; Toyota, T. Higher plasma homocysteine concentration is associated with more advanced systemic arterial stiffness and greater blood pressure response to stress in hypertensive patients. Hypertens. Res. 2006, 29, 403–409. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kennedy, R.H.; Owings, R.; Shekhawat, N.; Joseph, J. Acute negative inotropic effects of homocysteine are mediated via the endothelium. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, 812–817. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Herrmann, M.; Kindermann, I.; Muller, S.; Herrrmann, W. Relationship of plasma homocysteine with the severity of chronic heart failure. Clin Chem. 2005, 10, 1512–1515. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gibelin, P.; Serre, S.; Candito, M.; Houcher, B.; Berthier, F.; Baudouy, M. Prognostic value of homocysteinemia in patients with congestive heart failure. Clin. Chem. Lab. Med. 2006, 44, 813–816. [Google Scholar] [CrossRef]
- Leal, A.A.; Palmeira, A.C.; Castro, G.M.; Simões, M.O.; Ramos, A.T.; Medeiros, C.C. Homocysteine: Cardiovascular risk factor in children and adolescents? Rev. Assoc. Med. Bras. 2013, 59, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Brasileiro, R.S.; Escrivao, M.A.; Taddei, J.A.; D’Almeida, V.; Ancona-Lopez, F.; Carvalhaes, J.T. Plasma total homocysteine in Brazilian overweight and non-overweight adolescents: A case-control study. Nutr. Hosp. 2005, 20, 313–319. [Google Scholar] [PubMed]
- Kumar, K.J.; Saldanha, K.; Sushma, K.; Murthy, D.S.; Vishwanath, P.A. Prospective Study of Homocysteine and its relation to Body Mass Index and Lipid Profile in School Children. Indian Pediatr. 2017, 54, 935–937. [Google Scholar] [CrossRef] [PubMed]
- Dehkordi, E.H.; Sedehi, M.; Shahraki, Z.G.; Najafi, R. Effect of folic acid on homocysteine and insulin resistance of overweight and obese children and adolescents. Adv. Biomed. Res. 2016, 5, 88. [Google Scholar] [CrossRef]
- Institute of Medicine (US) Standing Committee on the Scientific Evaluation of Dietary Reference Intakes and its Panel on Folate, Other B Vitamins, and Choline. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline; The National Academies Press: Washington, DC, USA, 1998. [Google Scholar]
- EFSA Panel on Dietetic Products, Nutrition and Allergies (NDA). Scientific Opinion on Dietary Reference Values for folate. EFSA J. 2014, 12, 3893. [Google Scholar] [CrossRef]
- Viroonudompho, D.; Kanjanachumpol, S.; Sirisate, S. Homocysteine, Vitamin B12 and Folic Acid in Children with Acute Glomerulonephritis. World J. Eng. Technol. 2018, 6, 661–670. [Google Scholar] [CrossRef][Green Version]
- Diamond, J.R. Analogous Pathobiologic Mechanism in Glomerulosclerosis and Atherosclerosis. Kidney Int. 1991, 31, 29–34. [Google Scholar]
- Chen, P.; Poddar, R.; Tipa, E.V.; Dibello, P.M.; Moraveca, C.D.; Robinson, K.; Green, R.; Kruger, W.D.; Garrow, T.A.; Jacobsen, D.W. Homocysteine Metabolism in Cardiovascular Cells and Tissues: Implication for Hyperhomocysteinemia and Cardiovascular Disease. Adv. Enzym. Regul. 1999, 39, 93–109. [Google Scholar] [CrossRef]
- Orimadegun, B.E.; Orimadegun, A.E.; Ademola, A.D.; Agbedana, E.O. Plasma homocysteine and B vitamins levels in Nigerian children with nephrotic syndrome. Pan Afr. Med. J. 2014, 18, 107. [Google Scholar] [CrossRef]
- Zhang, C.; Boini, K.M.; Xia, M.; Abais, J.M.; Li, X.; Liu, Q.; Li, P.L. Activation of nalp3 inflammasomes turns on podocyte injury and glomerular sclerosis in hyperhomocysteinemia. Hypertension 2012, 60, 154–162. [Google Scholar] [CrossRef]
- Dowie, M.L.; Gallobois, C.; Parekh, R.S.; Noone, D.G. Nephrotic syndrome in infants and children: Pathophysiology and management. Paediatr. Int. Child Health 2017, 37, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Merouani, A.; Lambert, M.; Delvin, E.E.; Genest, J., Jr.; Robitaille, P.; Rozen, R. Plasma homocysteine concentration in children with chronic renal failure. Pediatr. Nephrol. 2001, 16, 805–811. [Google Scholar] [CrossRef]
- Aldaámiz-Echevarría, L.; Sanjurio, P.; Vallo, A.; Pérez-Nanclares, G.; Gimeno, P.; Rueda, M.; Ruiz, I.; Urreizti, R.; Rodriguez-Soriano, J. Hyperhomocysteinemia in children with renal transplants. Pediatr. Nephrol. 2002, 17, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Tordjman, S.; Somogyi, E.; Coulon, N.; Kermarrec, S.; Cohen, D.; Bronsard, G.; Bonnot, O.; Weismann-Arcache, C.; Botbol, M.; Lauth, B.; et al. Gene X Environment interactions in autism spectrum disorders: Role of epigenetic mechanisms. Front. Psychiatry 2014, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Main, P.A.; Angley, M.T.; O’Doherty, C.E.; Thomas, P.; Fenech, M. The potential role of the antioxidant and detoxification properties of glutathione in autism spectrum disorders: A systematic review and meta-analysis. Nutr. Metab. 2012, 9, 35. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.; Sequeira, J.M.; Quadros, E.V. The metabolic basis for developmental disorders due to defective folate transport. Biochimie 2016, 126, 31–42. [Google Scholar] [CrossRef]
- Ali, A.; Waly, M.I.; Al-Farsi, Y.M.; Essa, M.M.; Al-Sharbati, M.M.; Deth, R.C. Hyperhomocysteinemia among Omani autistic children: A case-control study. Acta Biochim. Pol. 2011, 58, 547–551. [Google Scholar] [CrossRef]
- Al-Farsi, Y.M.; Waly, M.I.; Deth, R.C.; Al-Sharbati, M.M.; Al-Shafaee, M.; Al-Farsi, O.; Al-Khaduri, M.M. Low folate and vitamin B12 nourishment is common in Omani children with newly diagnosed autism. Nutrition 2013, 29, 537–541. [Google Scholar] [CrossRef]
- Fuentes-Albero, M.; Cauli, O. Homocysteine Levels in Autism Spectrum Disorder: A Clinical Update. Endocr. Metab. Immune Disord. Drug Targets 2018, 18, 289–296. [Google Scholar] [CrossRef]
- Altun, H.; Kurutaş, E.B.; Şahin, N.; Güngör, O.; Findikli, E. The Levels of Vitamin D, Vitamin D Receptor, Homocysteine and Complex B Vitamin in Children with Autism Spectrum Disorders. Clin. Psychopharmacol. Neurosci. 2018, 16, 383–390. [Google Scholar] [CrossRef]
- Paşca, S.P.; Nemeş, B.; Vlase, L.; Gagyi, C.E.; Dronca, E.; Miu, A.C.; Dronca, M. High levels of homocysteine and low serum paraoxonase 1 arylesterase activity in children with autism. Life Sci. 2006, 78, 2244–2248. [Google Scholar] [CrossRef]
- Kałużna-Czaplińska, J.; Michalska, M.; Rynkowski, J. Homocysteine level in urine of autistic and healthy children. Acta Biochim. Pol. 2011, 58, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Kałużna-Czaplińska, J.; Żurawicz, E.; Michalska, M.; Rynkowski, J. A focus on homocysteine in autism. Acta Biochim. Pol. 2013, 60, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Shea, T.B. Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci. 2003, 26, 137–146. [Google Scholar] [CrossRef]
- Ho, P.I.; Ortiz, D.; Rogers, E.; Shea, T.B. Multiple aspects of homocysteine neurotoxicity: Glutamate excitotoxicity, kinase hyperactivation and DNA damage. J. Neurosci. Res. 2002, 70, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Fulceri, F.; Morelli, M.; Santocchi, E.; Cena, H.; Del Bianco, T.; Narzisi, A.; Calderoni, S.; Muratori, F. Gastrointestinal symptoms and behavioral problems in preschoolers with Autism Spectrum Disorder. Dig. Liver Dis. 2016, 48, 248–254. [Google Scholar] [CrossRef]
- Chauhan, A.; Audhya, T.; Chauhan, V. Brain region-specific glutathione redox imbalance in autism. Neurochem. Res. 2012, 37, 1681–1689. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Frye, R.E. Evidence linking oxidative stress, mitochondrial dysfunction, and inflammation in the brain of individuals with autism. Front. Physiol. 2014, 5, 150. [Google Scholar] [CrossRef]
- Frye, R.E.; Melnyk, S.; Fuchs, G.; Reid, T.; Jernigan, S.; Pavliv, O.; Hubanks, A.; Gaylor, D.W.; Walters, L.; James, S.J. Effectiveness of methylcobalamin and folinic acid treatment on adaptive behavior in children with autistic disorder is related to glutathione redox status. Autism Res. Treat. 2013, 2013, 609705. [Google Scholar] [CrossRef]
- Sun, C.; Zou, M.; Zhao, D.; Xia, W.; Wu, L. Efficacy of Folic Acid Supplementation in Autistic Children Participating in Structured Teaching: An Open-Label Trial. Nutrients 2016, 8, 337. [Google Scholar] [CrossRef]
- Józefczuk, J.; Kasprozycka, W.; Czarnecki, R.; Graczyk, A.; Józefczuk, P.; Magda, K.; Lampart, U. Homocysteine as a Diagnostic and Etiopathogenic Factor in Children with Autism Spectrum Disorder. J. Med. Food 2017, 20, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Altun, H.; Şahin, N.; Belge Kurutaş, E.; Güngör, O. Homocysteine, Pyridoxine, Folate and Vitamin B12 Levels in Children with Attention Deficit Hyperactivity Disorder. Psychiatr. Danub. 2018, 30, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Bottiglieri, T. Folate, vitamin B12, and neuropsychiatric disorders. Nutr. Rev. 1996, 54, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Ezzaher, A.; Mouhamed, D.H.; Mechri, A.; Omezzine, A.; Neffati, F.; Douki, W.; Bouslama, A.; Gaha, L.; Najjar, M.F. Hyperhomocysteinemia in Tunisian bipolarI patients. Psychiatry Clin. Neurosci. 2011, 65, 664–671. [Google Scholar] [CrossRef]
- Kim, H.; Lee, K.J. Serum homocysteine levels are correlated with behavioral and psychological symptoms of Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2014, 10, 1887–1896. [Google Scholar] [CrossRef]
- Stanger, O.; Fowler, B.; Piertzik, K.; Huemer, M.; Haschke-Becher, E.; Semmler, A.; Lorenzl, S.; Linnebank, M. Homocysteine, folate and vitamin B12 in neuropsychiatric diseases: Review and treatment recommendations. Expert Rev. Neurother. 2009, 9, 1393–1412. [Google Scholar] [CrossRef]
- Karababa, İ.F.; Savas, S.N.; Selek, S.; Cicek, E.; Cicek, E.I.; Asoglu, M.; Bayazit, H.; Kandemir, H.; Kati, M.; Ulas, T. Homocysteine levels and oxidative stress parameters in patients with adult ADHD. J. Atten. Disord. 2017, 21, 487–493. [Google Scholar] [CrossRef]
- Kumar, S.; Gane, B.; Rao, R.; Bhat, V. Folate and Homocysteine metabolism in Indian children with Down syndrome. Curr. Pediatr. 2014, 18, 11–14. [Google Scholar]
- Ravaglia, G.; Forti, P.; Maioli, F.; Vettori, C.; Grossi, G.; Bargossi, A.M.; Caldarera, M.; Franceschi, C.; Facchini, A.; Mariani, E.; et al. Elevated plasma homocysteine levels in centenarians are not associated with cognitive impairment. Mech. Ageing Dev. 2000, 121, 251–256. [Google Scholar] [CrossRef]
- Wong, Y.Y.E.; Almeida, O.P.; McCaul, K.A.; Yeap, B.B.; Hankey, G.J.; Flicker, L. Homocysteine, frailty, and all-cause mortality in older men: The health in Men study. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2013, 68, 590–598. [Google Scholar] [CrossRef]
- Kuo, H.K.; Sorond, F.A.; Chen, J.H.; Hashmi, A.; Milberg, W.P.; Lipsitz, L.A. The role of homocysteine in multisystem age-related problems: A systematic review. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2005, 60, 1190–1201. [Google Scholar] [CrossRef] [PubMed]
- Dhonukshe-Rutten, R.A.; Pluijm, S.M.; de Groot, L.C.; Lips, P.; Smit, J.H.; van Staveren, W.A. Homocysteine and Vitamin B12 Status Relate to Bone Turnover Markers, Broadband Ultrasound Attenuation, and Fractures in Healthy Elderly People. J. Bone Miner. Res. 2005, 20, 921–929. [Google Scholar] [CrossRef]
- Clegg, A.; Young, J.; Iliffe, S.; Rikkert, M.O.; Rockwood, K. Frailty in elderly people. Lancet 2013, 381, 752–762. [Google Scholar] [CrossRef]
- Peng, H.Y.; Man, C.F.; Xu, J.; Fan, Y. Elevated homocysteine levels and risk of cardiovascular and all-cause mortality: A meta-analysis of prospective studies. J. Zhejiang Univ. Sci. B 2015, 16, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Petersen, J.F.; Larsen, B.S.; Sabbah, M.; Nielsen, O.W.; Kumarathurai, P.; Sajadieh, A. Long-term prognostic significance of homocysteine in middle-aged and elderly. Biomarkers 2016, 21, 490–496. [Google Scholar] [CrossRef]
- Chen, K.J.; Pan, W.H.; Yang, F.L.; Wei, I.L.; Shaw, N.S.; Lin, B.F. Association of B vitamins status and homocysteine levels in elderly Taiwanese. Asia Pac. J. Clin. Nutr. 2005, 14, 250–255. [Google Scholar] [PubMed]
- Ma, F.; Wu, T.; Zhao, J.; Ji, L.; Song, A.; Zhang, M.; Huang, G. Plasma Homocysteine and Serum Folate and Vitamin B12 Levels in Mild Cognitive Impairment and Alzheimer’s Disease: A Case-Control Study. Nutrients 2017, 9, 725. [Google Scholar] [CrossRef]
- Selhub, J.; Jacques, P.F.; Wilson, P.W.F.; Rush, D.; Rosenberg, I.H. Vitamin status and intake as primary determinants of homocysteinemia in an elderly population. JAMA 1993, 270, 2693–2698. [Google Scholar] [CrossRef] [PubMed]
- Bonetti, F.; Brombo, G.; Magon, S.; Zuliani, G. Cognitive Status According to Homocysteine and B-Group Vitamins in Elderly Adults. J. Am. Geriatr. Soc. 2015, 63, 1158–1163. [Google Scholar] [CrossRef] [PubMed]
- Farina, N.; Jernerén, F.; Turner, C.; Hart, K.; Tabet, N. Homocysteine concentrations in the cognitive progression of Alzheimer’s disease. Nutrients 2017, 9, 725. [Google Scholar] [CrossRef]
- Ostrakhovitch, E.; Tabibzadeh, S. Homocysteine and age-associated disorders. Ageing Res. Rev. 2019, 49, 144–164. [Google Scholar] [CrossRef]
- Homocysteine Studies Collaboration. Homocysteine and risk of ischemicheart disease and stroke: A meta-analysis. JAMA 2002, 288, 2015–2022. [Google Scholar] [CrossRef]
- Tang, Y.; Geng, D. Associations of plasma LP(a), Hcy and D-D levels with the subtype of ischemic cerebrovascular disease. Medicine 2019, 98, e14910. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Guo, Y.; Zhu, Z. Study of the Inflammatory Mechanisms in Hyperhomocysteinemia on Large-Artery Atherosclerosis Based on Hypersensitive C-Reactive Protein—A Study from Southern China. J. Stroke Cerebrovasc. Dis. 2019, 28, 1816–1823. [Google Scholar] [CrossRef]
- Lee, T.H.; Cheng, M.L.; Shiao, M.S.; Lin, C.N. Metabolomics study in severe extracranial carotid artery stenosis. BMC Neurol. 2019, 19, 138. [Google Scholar] [CrossRef]
- Lee, M.; Hong, K.S.; Chang, S.C.; Saver, J.L. Efficacy of homocysteine-lowering therapy with folic Acid in stroke prevention: A meta-analysis. Stroke 2010, 41, 1205–1212. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Xu, C.H.; Xu, Y.N.; Wang, Y.L.; Wang, M. The effect of folate fortification on folic acid-based homocysteine-lowering intervention and stroke risk: A meta-analysis. Public Health Nutr. 2015, 18, 1514–1521. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, H.; Zhou, Y.; Jin, L.; Liu, J. Low-dose B vitamins supplementation ameliorates cardiovascular risk: A double-blind randomized controlled trial in healthy Chinese elderly. Eur. J. Nutr. 2015, 54, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Loscalzo, J. Homocysteine trials-clear outcomes for complex reasons. N. Engl. J. Med. 2006, 354, 1629–1632. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, X.; He, M.; Qin, X.; Tang, G.; Huo, Y.; Li, J.; Fu, J.; Huang, X.; Cheng, X.; et al. Homocysteine and Stroke Risk: Modifying Effect of Methylenetetrahydrofolate Reductase C677T Polymorphism and Folic Acid Intervention. Stroke 2017, 48, 1183–1190. [Google Scholar] [CrossRef]
- Seshadri, S.; Beiser, A.; Selhub, J.; Jacques, P.F.; Rosenberg, I.H.; D’Agostino, R.B.; Wilson, P.W.; Wolf, P.A. Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N. Engl. J. Med. 2002, 346, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Hooshmand, B.; Refsum, H.; Smith, A.D.; Kalpouzos, G.; Mangialasche, F.; von Arnim, C.A.F.; Kåreholt, I.; Kivipelto, M.; Fratiglioni, L. Association of Methionine to Homocysteine Status With Brain Magnetic Resonance Imaging Measures and Risk of Dementia. JAMA Psychiatry 2019, 76, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- de Jager, C.A.; Oulhaj, A.; Jacoby, R.; Refsum, H.; Smith, A.D. Cognitive and clinical outcomes of homocysteine-lowering B-vitamin treatment in mild cognitive impairment: A randomized controlled trial. Int. J. Geriatr. Psychiatry 2012, 27, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Douaud, G.; Refsum, H.; de Jager, C.A.; Jacoby, R.; Nichols, T.E.; Smith, S.M.; Smith, A.D. Preventing Alzheimer’s disease-related gray matter atrophy by B-vitamin treatment. Proc. Natl. Acad. Sci. USA 2013, 110, 9523–9528. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Kong, H.; Pang, W.; Yang, H.; Lu, H.; Huang, C.; Jiang, Y. B vitamin supplementation improves cognitive function in the middle aged and elderly with hyperhomocysteinemia. Nutr. Neurosci. 2016, 19, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Kim, S.Y.; Sok, S.R. Effects of Multivitamin Supplements on Cognitive Function, Serum Homocysteine Level, and Depression of Korean Older Adults With Mild Cognitive Impairment in Care Facilities. J. Nurs. Scholarsh. 2016, 48, 223–231. [Google Scholar] [CrossRef]
- Zhang, D.M.; Ye, J.X.; Mu, J.S.; Cui, X.P. Efficacy of Vitamin B Supplementation on Cognition in Elderly Patients With Cognitive-Related Diseases. J. Geriatr. Psychiatry Neurol. 2017, 30, 50–59. [Google Scholar] [CrossRef]
- Harris, E.; Macpherson, H.; Pipingas, A. Improved blood biomarkers but no cognitive effects from 16 weeks of multivitamin supplementation in healthy older adults. Nutrients 2015, 7, 3796–3812. [Google Scholar] [CrossRef]
- Harrington, D.J. Laboratory assessment of vitamin B12 status. J. Clin. Pathol. 2017, 70, 168–173. [Google Scholar] [CrossRef]
- Siniscalchi, A.; Gallelli, L.; Mercuri, N.B.; Ibbadu, G.F.; De Sarro, G. Role of lifestyle factors on plasma homocysteine levels in Parkison’s disease patients treated with levodopa. Nutr. Neurosci. 2006, 9, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Oulhaj, A.; Jernerén, F.; Refsum, H.; Smith, A.D.; de Jager, C.A. Omega-3 Fatty Acid Status Enhances the Prevention of Cognitive Decline by B Vitamins in Mild Cognitive Impairment. J. Alzheimer’s Dis. 2016, 50, 547–557. [Google Scholar] [CrossRef] [PubMed]
- McLean, R.R.; Jacques, P.F.; Selhub, J.; Tucker, K.L.; Samelson, E.J.; Broe, K.E.; Hannan, M.T.; Cupples, L.A.; Kiel, D.P. Homocysteine as a predictive factor for hip fracture in older persons. N. Engl. J. Med. 2004, 350, 2042–2049. [Google Scholar] [CrossRef]
- van Meurs, J.B.; Dhonukshe-Rutten, R.A.; Pluijm, S.M.; van der Klift, M.; de Jonge, R.; Lindemans, J.; de Groot, L.C.; Hofman, A.; Witteman, J.C.; van Leeuwen, J.P.; et al. Homocysteine levels and the risk of osteoporotic fracture. N. Engl. J. Med. 2004, 350, 2033–2041. [Google Scholar] [CrossRef] [PubMed]
- Akalin, A.; Alatas, O.; Colak, O. Relation of plasma homocysteine levels to atherosclerotic vascular disease and inflammation markers in type 2 diabetic patients. Eur. J. Endocrinol. 2008, 158, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Undas, A.; Brozek, J.; Szczeklik, A. Homocysteine and thrombosis: From basic science to clinical evidence. Thromb. Haemost. 2005, 94, 907–915.64. [Google Scholar] [CrossRef]
- Austin, R.C.; Lentz, S.R.; Werstuck, G.H. Role of hyperhomocysteinemia in endothelial dysfunction and atherothrombotic disease. Cell Death Differ. 2004, 11 (Suppl. S1), S56–S64. [Google Scholar] [CrossRef]
- Zhang, C.; Cai, Y.; Adachi, M.T.; Oshiro, S.; Aso, T.; Kaufman, R.J.; Kitajima, S. Homocysteine induces programmed cell death in human vascular endothelial cells through activation of the unfolded protein response. J. Biol. Chem. 2001, 276, 35867–35874. [Google Scholar] [CrossRef]
- Skurk, C.; Walsh, K. Death receptor induced apoptosis: A new mechanism of homocysteine-mediated endothelial cell cytotoxicity. Hypertension 2004, 43, 1168–1170. [Google Scholar] [CrossRef]
- Sauls, D.L.; Lockhart, E.; Warren, M.E.; Lenkowski, A.; Wilhelm, S.E.; Hoffman, M. Modification of fibrinogen by homocysteine thiolactone increases resistance to fibrinolysis: A potential mechanism of the thrombotic tendency in hyperhomocysteinemia. Biochemistry 2006, 45, 2480–2487. [Google Scholar] [CrossRef]
- Toohey, J.L. Hcy Toxicity in Connective Tissue: Theories, Old and New. Connect. Tissue Res. 2008, 49, 57–61. [Google Scholar] [CrossRef]
- Parhami, F. Possible role of oxidized lipids in osteoporosis: Could hyperlipidemia be a risk factor? Prostaglandins Leukot. Essent. Fat. Acids 2003, 68, 373–378. [Google Scholar] [CrossRef]
- Armour, K.E.; Armour, K.J.; Gallagher, M.E.; Godecke, A.; Helfrich, M.H.; Reid, D.M.; Ralston, S.H. Defective bone formation and anabolic response to exogenous estrogen in mice with targeted disruption of endothelial nitric oxide synthase. Endocrinology 2001, 142, 760–766. [Google Scholar] [CrossRef]
- Saito, M.; Marumo, K. The Effects of Homocysteine on the Skeleton. Curr. Osteoporos. Rep. 2018, 16, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Z.; Wang, W.; Liu, Y.L.; He, X.F. Association between the methylenetetrahydrofolate reductase c.677C>T polymorphism and bone mineral density: An updated meta-analysis. Mol. Genet. Genom. 2016, 291, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Ahn, T.K.; Kim, J.O.; Kim, H.W.; Park, H.S.; Shim, J.H.; Ropper, A.E.; Han, I.B.; Kim, N.K. 3′-UTR Polymorphisms of MTHFR and TS Associated with Osteoporotic Vertebral Compression Fracture Susceptibility in Postmenopausal Women. Int. J. Mol. Sci. 2018, 19, 824. [Google Scholar] [CrossRef] [PubMed]
- van Wijngaarden, J.P.; Doets, E.L.; Szczecińska, A.; Souverein, O.W.; Duffy, M.E.; Dullemeijer, C.; Cavelaars, A.E.; Pietruszka, B.; Van’t Veer, P.; Brzozowska, A.; et al. Vitamin B12, folate, homocysteine, and bone health in adults and elderly people: A systematic review with meta-analyses. J. Nutr. Metab. 2013, 2013, 486186. [Google Scholar] [CrossRef] [PubMed]
- Enneman, A.W.; Swart, K.M.; van Wijngaarden, J.P.; van Dijk, S.C.; Ham, A.C.; Brouwer-Brolsma, E.M.; van der Zwaluw, N.L.; Dhonukshe-Rutten, R.A.; van der Cammen, T.J.; de Groot, L.C.; et al. Effect of Vitamin B12 and Folic Acid Supplementation on Bone Mineral Density and Quantitative Ultrasound Parameters in Older People with an Elevated Plasma Homocysteine Level: B-PROOF, a Randomized Controlled Trial. Calcif. Tissue Int. 2015, 96, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Swart, K.M.; Ham, A.C.; van Wijngaarden, J.P.; Enneman, A.W.; van Dijk, S.C.; Sohl, E.; Brouwer-Brolsma, E.M.; van der Zwaluw, N.L.; Zillikens, M.C.; Dhonukshe-Rutten, R.A.; et al. A Randomized Controlled Trial to Examine the Effect of 2-Year Vitamin B12 and Folic Acid Supplementation on Physical Performance, Strength, and Falling: Additional Findings from the B-PROOF Study. Calcif. Tissue Int. 2016, 98, 8–27. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Keser, I.; Ilich, J.Z.; Vrkić, N.; Giljević, Z.; Colić Barić, I. Folic acid and vitamin B12 supplementation lowers plasma homocysteine but has no effect on serum bone turnover markers in elderly women: A randomized, double-blind, placebo-controlled trial. Nutr. Res. 2013, 33, 211–219. [Google Scholar] [CrossRef]
- Stone, K.L.; Lui, L.Y.; Christen, W.G.; Troen, A.M.; Bauer, D.C.; Kado, D.; Schambach, C.; Cummings, S.R.; Manson, J.E. Effect of Combination Folic Acid, Vitamin B6, and Vitamin B12 Supplementation on Fracture Risk in Women: A Randomized, Controlled Trial. J. Bone Miner. Res. 2017, 32, 2331–2338. [Google Scholar] [CrossRef]
- Lopez, M.G.; Baron, J.A.; Omsland, T.K.; Søgaard, A.J.; Meyer, H.E. Homocysteine-Lowering Treatment and the Risk of Fracture: Secondary Analysis of a Randomized Controlled Trial and an Updated Meta-Analysis. JBMR Plus 2018, 2, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Kado, D.M.; Bucur, A.; Selhub, J.; Rowe, J.W.; Seeman, T. Homocysteine levels and decline in physical function: MacArthur Studies of Successful Aging. Am. J. Med. 2002, 113, 537–542. [Google Scholar] [CrossRef]
- Vidoni, M.L.; Pettee Gabriel, K.; Luo, S.T.; Simonsick, E.M.; Day, R.S. Relationship between Homocysteine and Muscle Strength Decline: The Baltimore Longitudinal Study of Aging. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2018, 73, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Veeranki, S.; Tyagi, S.C. Defective homocysteine metabolism: Potential implications for skeletal muscle malfunction. Int. J. Mol. Sci. 2013, 14, 15074–15091. [Google Scholar] [CrossRef]
- Ao, M.; Inuiya, N.; Ohta, J.; Kurose, S.; Takaoka, H.; Abe, Y.; Niki, N.; Inoue, S.; Tanaka, S.; Miyawaki, T.; et al. Relationship between Homocysteine, Folate, Vitamin B12 and Physical Performance in the Institutionalized Elderly. J. Nutr. Sci. Vitaminol. 2019, 65, 1–7. [Google Scholar] [CrossRef]
- Vidoni, M.L.; Pettee Gabriel, K.; Luo, S.T.; Simonsick, E.M.; Day, R.S. Vitamin B12 and Homocysteine Associations with Gait Speed in Older Adults: The Baltimore Longitudinal Study of Aging. J. Nutr. Health Aging 2017, 21, 1321–1328. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).