β2 Integrins—Multi-Functional Leukocyte Receptors in Health and Disease


Abstract
1. Introduction
2. Structure and Activation of β2 Integrins
2.1. The α Subunit
2.2. The β Subunit
2.3. Activation of β2 Integrins
2.4. Ligands of β2 Integrins
2.5. Expression Pattern of β2 Integrins
3. Cellular Functions of β2 Integrins
3.1. Cell Differentiation
3.2. Migration
3.3. Phagocytosis
3.4. β2 Integrin Signaling Events in APC
3.5. β2 Integrins in the Interaction of Immune Cells
3.5.1. Interaction of APC and T Cells
Composition of the Immunological Synapse
Role of LFA-1 in T Cell Activation
Regulation of LFA-1 activity on T Cells
Cytoskeletal Rearrangements in the Course of T Cell Stimulation
β2 integrin Activity and Cytoskeletal Rearrangements on the APC Side
3.5.2. Leukocyte/Target Cell Interaction
4. Pathophysiological Role of β2 Integrins in Human
5. Mouse Models to Study Functions of Distinct β2 Integrins
5.1. Functions of β2 Integrins in Infections
5.1.1. Viral Infections
5.1.2. Bacterial Infections
5.1.3. Fungal Infections
5.1.4. Metazoan Parasites
5.2. Functions of β2 Integrins in Autoimmunity
5.2.1. LFA-1
5.2.2. MAC-1
5.2.3. Other β2 Integrins
5.3. β2 Integrins and Tumor Development
5.3.1. Tumor Infiltration
5.3.2. Tumor Angiogenesis
5.3.3. Tumor-Specific Immune Responses
5.3.4. Interaction with Tumor Cells
5.3.5. Leukemia
5.3.6. β2 Integrin Expression by Tumor Cells
6. β2 Integrins as Therapeutic Targets
7. Concluding Remarks
Funding
Conflicts of Interest
Abbreviations
| ADAP | Fyn-binding protein |
| ADCC | Antibody-dependent cytotoxicity |
| ADMIDAS | Adjacent to metal-ion-dependent-adhesion-site |
| AP-1 | Activator protein-1 |
| APC | Antigen presenting cell |
| Arp2/3 | actin-related protein-2/3 |
| B cells | B lymphocytes |
| BCG | Bacillus Calmette–Guérin |
| BCL6 | B cell lymphoma 6 |
| BM-DC | Bone marrow-derived dendritic cells |
| BP | Bullous pemphigoid |
| Ca2 | Calcium |
| CALDAG–GEF I | Ca2+ and diacylglycerol regulated guanine nucleotide exchange factor I |
| Calf-1 | Calcium channel localization factor-1 |
| Cas-L | Crk-associated substrate lymphocyte-type |
| CD | Cluster of differentiation |
| CD40L | CD40 ligand |
| CDC42 | Cell division control protein 42 homolog |
| cSMAC | central supramolecular activation cluster |
| CIA | Collagen-induced arthritis |
| CLL | Chronic lymphatic leukemia |
| CMV | Cytomegalovirus |
| CTL | cytotoxic T lymphocytes |
| Cyr61 | Cysteine-rich angiogenic inducer 61 |
| CYTIP | Cytohesin-1 interacting protein |
| DC | Dendritic cells |
| DC-SIGN | Dendritic cell-specific ICAM-3-grabbing non-integrin |
| DENND1C | Differentially expressed in normal and neoplastic cells domain 1C |
| DOCK2 | Dedicator of cytokinesis 2 |
| EAE | Experimental autoimmune encephalomyelitis |
| EGF | Epidermal growth factor |
| EOC | Epithelial ovarian cancer |
| ERK | Extracellular signal-regulated kinase |
| ES | Embryonic stem cells |
| FcR | Fc receptor |
| FcγRIIA | Conventional type I transmembrane protein |
| FERMT3 | Fermitin family homolog 3 |
| Foxp3 | Forkhead-Box-Protein P3 |
| GEF | Guanine nucleotide exchange factor |
| h | Human |
| HSPC | Hematopoietic stem and progenitor cells |
| ICAM | Intercellular adhesion molecule |
| IFN-y | Interferon y |
| IL-6 | Interleukin-6 |
| IS | immunological synapse |
| ITGAM | Integrin subunit alpha M |
| JAB1 | Jun activating binding protein-1 |
| JAK | Janus kinase |
| JAM | Junctional adhesion molecule |
| LA-1 | Leukadherin-1 |
| LAD-I | Leukocyte adhesion deficiency type 1 |
| Lck | Lymphocyte cell-specific protein tyrosine kinase |
| LIMBS | Ligand-associated metal binding site |
| LLC | Lewis lung carcinoma |
| LPS | Lipopolysaccharide |
| MA-ARDS | Malaria-associated acute respiratory distress syndrome |
| MAC-1 | Macrophage antigen 1 |
| MAPK | Mitogen activated protein kinase |
| MHC | major histocompatibility complex |
| MIDAS | Metal ion-dependent-adhesion-site |
| MIP | Macrophage inflammatory protein |
| MOG | Myelin oligodendrocyte glycoprotein |
| m | Mouse |
| MPO | Myeloperoxidase |
| MS | Multiple sclerosis |
| Mst1 | Macrophage-Stimulating Protein |
| NFATc1 | nuclear factor of activated T cells, cytoplasmic 1 |
| NK cells | Natural killer cells |
| NOTCH1 | Neurogenic locus notch homolog protein 1 |
| NOX2 | NADPH Oxidase 2 |
| PI3K | Phosphoinositide 3-kinase |
| PIP5K1C | Phosphatidylinositol-4-phosphate 5-kinase type-1 gamma |
| PKC | Protein kinase C |
| PLC | Phospholipase C |
| PLD1 | Phospholipase D1 |
| PMN | Polymorphonuclear granulocytes |
| PRL | Phosphatase of regenerating liver 1 |
| PSI | Plexin-semaphorin-integrin |
| PSGL-1 | P-selectin glycoprotein ligand-1 |
| pSMAC | Peripheral supramolecular activation cluster |
| PTPRG | Protein tyrosine phosphatase receptor type g |
| RA | Rheumatoid arthritis |
| Rac1 | Ras-related C3 botulinum toxin substrate 1 |
| RACK1 | Receptor for activated C-kinase 1 |
| RAGE | Receptor for advanced glycation end products |
| RANKL | Receptor activator of nuclear factor kappa-Β ligand |
| Rap-1 | Ras-related protein 1 |
| Rap1B | Rap-1 binding protein |
| RAPL | Regulator of adhesion and polarization enriched in lymphocytes |
| RhoA | Ras homolog gene family, member A |
| RIAM | Rap-1-GTP interacting adaptor molecule |
| ROS | Reactive oxygen species |
| SHP-1 | Src homology region 2 domain-containing phosphatase 1 |
| SKAP55, | Src kinase-associated phosphoprotein 5 |
| SLE | Systemic lupus erythematosus |
| SNP | Single Nucleotide Polymorphism |
| SOCS-3 | Suppressor of cytokine signaling 3 |
| SPTAN1 | Spectrin alpha, non-erythrocytic 1 |
| STAT | Signal transducers and activators of transcription |
| STK4 | Serine/threonine protein kinase 4 |
| SYK | Spleen tyrosine kinase |
| T cells | T lymphocytes |
| TCR | T cell receptor |
| TEM | Transmission electron microscopy |
| TGLN2 | Taglin 2 |
| Thy-1 | Thymus cell antigen 1 |
| TLR | Toll-like Receptor |
| TME | Tumor microenvironment |
| TNF-α | Tumor necrosis factor alpha |
| Treg | Regulatory T cells |
| Tri12 | Trisomy 12 |
| VAV1 | Vav Guanine Nucleotide Exchange Factor 1 |
| VCAM-1 | Vascular cell adhesion protein 1 |
| VEGF | Vascular endothelial growth factor |
| WASp | Wiskott-Aldrich syndrome protein |
| WBC | White blood cells |
| WT | Wild type |
| γ/δ T cells | Gamma/delta T cells |
References
- Takada, Y.; Ye, X.; Simon, S. The integrins. Genome Biol. 2007, 8, 215. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.S.; Lu, N.; Denessiouk, K.; Heino, J.; Gullberg, D. Integrins during evolution: Evolutionary trees and model organisms. Biochim. Et Biophys. Acta 2009, 1788, 779–789. [Google Scholar] [CrossRef] [PubMed]
- LaFoya, B.; Munroe, J.A.; Miyamoto, A.; Detweiler, M.A.; Crow, J.J.; Gazdik, T.; Albig, A.R. Beyond the Matrix: The Many Non-ECM Ligands for Integrins. Int. J. Mol. Sci. 2018, 19, 449. [Google Scholar] [CrossRef]
- Conway, J.R.W.; Jacquemet, G. Cell matrix adhesion in cell migration. Essays Biochem. 2019, 63, 535–551. [Google Scholar] [CrossRef] [PubMed]
- Bros, M.; Haas, K.; Moll, L.; Grabbe, S. RhoA as a Key Regulator of Innate and Adaptive Immunity. Cells 2019, 8, 733. [Google Scholar] [CrossRef]
- Mitroulis, I.; Alexaki, V.I.; Kourtzelis, I.; Ziogas, A.; Hajishengallis, G.; Chavakis, T. Leukocyte integrins: Role in leukocyte recruitment and as therapeutic targets in inflammatory disease. Pharmacol. Ther. 2015, 147, 123–135. [Google Scholar] [CrossRef]
- Cabanillas, D.; Regairaz, L.; Deswarte, C.; Garcia, M.; Richard, M.E.; Casanova, J.L.; Bustamante, J.; Perez, L. Leukocyte Adhesion Deficiency Type 1 (LAD1) with Expressed but Nonfunctional CD11/CD18. J. Clin. Immunol 2016, 36, 627–630. [Google Scholar] [CrossRef]
- LaFlamme, S.E.; Mathew-Steiner, S.; Singh, N.; Colello-Borges, D.; Nieves, B. Integrin and microtubule crosstalk in the regulation of cellular processes. Cell. Mol. Life Sci. 2018, 75, 4177–4185. [Google Scholar] [CrossRef]
- Streuli, C.H.; Akhtar, N. Signal co-operation between integrins and other receptor systems. Biochem. J. 2009, 418, 491–506. [Google Scholar] [CrossRef]
- Humphries, M.J.; Symonds, E.J.; Mould, A.P. Mapping functional residues onto integrin crystal structures. Curr. Opin. Struct. Biol. 2003, 13, 236–243. [Google Scholar] [CrossRef]
- Chouhan, B.S.; Kapyla, J.; Denessiouk, K.; Denesyuk, A.; Heino, J.; Johnson, M.S. Early chordate origin of the vertebrate integrin alphaI domains. PLoS ONE 2014, 9, e112064. [Google Scholar] [CrossRef]
- Nunes, A.M.; Minetti, C.; Remeta, D.P.; Baum, J. Magnesium Activates Microsecond Dynamics to Regulate Integrin-Collagen Recognition. Structure 2018, 26, 1080–1090 e1085. [Google Scholar] [CrossRef]
- Sen, M.; Yuki, K.; Springer, T.A. An internal ligand-bound, metastable state of a leukocyte integrin, alphaXbeta2. J. Cell Biol. 2013, 203, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Shimaoka, M.; Salas, A.; Yang, W.; Weitz-Schmidt, G.; Springer, T.A. Small molecule integrin antagonists that bind to the beta2 subunit I-like domain and activate signals in one direction and block them in the other. Immunity 2003, 19, 391–402. [Google Scholar] [CrossRef]
- Xu, S.; Wang, J.; Wang, J.H.; Springer, T.A. Distinct recognition of complement iC3b by integrins alphaXbeta2 and alphaMbeta2. Proc. Natl. Acad. Sci. USA 2017, 114, 3403–3408. [Google Scholar] [CrossRef] [PubMed]
- Adorno-Cruz, V.; Liu, H. Regulation and functions of integrin alpha2 in cell adhesion and disease. Genes Dis. 2019, 6, 16–24. [Google Scholar] [CrossRef]
- Lim, J.; Hotchin, N.A. Signalling mechanisms of the leukocyte integrin alphaMbeta2: Current and future perspectives. Biol. Cell 2012, 104, 631–640. [Google Scholar] [CrossRef]
- Zhang, K.; Chen, J. The regulation of integrin function by divalent cations. Cell Adh. Migr. 2012, 6, 20–29. [Google Scholar] [CrossRef]
- Xiang, B.; Liu, Y.; Xie, L.; Zhao, Q.; Zhang, L.; Gan, X.; Yu, H. The osteoclasts attach to the bone surface where the extracellular calcium concentration decreases. Cell Biochem. Biophys. 2016, 74, 553–558. [Google Scholar] [CrossRef]
- Valdramidou, D.; Humphries, M.J.; Mould, A.P. Distinct roles of beta1 metal ion-dependent adhesion site (MIDAS), adjacent to MIDAS (ADMIDAS), and ligand-associated metal-binding site (LIMBS) cation-binding sites in ligand recognition by integrin alpha2beta1. J. Biol. Chem. 2008, 283, 32704–32714. [Google Scholar] [CrossRef]
- Rui, X.; Mehrbod, M.; Van Agthoven, J.F.; Anand, S.; Xiong, J.P.; Mofrad, M.R.; Arnaout, M.A. The alpha-subunit regulates stability of the metal ion at the ligand-associated metal ion-binding site in beta3 integrins. J. Biol. Chem. 2014, 289, 23256–23263. [Google Scholar] [CrossRef] [PubMed]
- Thome, S.; Begandt, D.; Pick, R.; Salvermoser, M.; Walzog, B. Intracellular beta2 integrin (CD11/CD18) interacting partners in neutrophil trafficking. Eur. J. Clin. Investig. 2018, 48 (Suppl. 2), e12966. [Google Scholar] [CrossRef]
- Muller, W.A. Getting leukocytes to the site of inflammation. Vet. Pathol 2013, 50, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Varga, G.; Balkow, S.; Wild, M.K.; Stadtbaeumer, A.; Krummen, M.; Rothoeft, T.; Higuchi, T.; Beissert, S.; Wethmar, K.; Scharffetter-Kochanek, K.; et al. Active MAC-1 (CD11b/CD18) on DCs inhibits full T-cell activation. Blood 2007, 109, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Yuki, K. Differential effects of volatile anesthetics on leukocyte integrin macrophage-1 antigen. J. Immunotoxicol. 2016, 13, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Block, H.; Stadtmann, A.; Riad, D.; Rossaint, J.; Sohlbach, C.; Germena, G.; Wu, D.; Simon, S.I.; Ley, K.; Zarbock, A. Gnb isoforms control a signaling pathway comprising Rac1, Plcbeta2, and Plcbeta3 leading to LFA-1 activation and neutrophil arrest in vivo. Blood 2016, 127, 314–324. [Google Scholar] [CrossRef]
- Jaskiewicz, A.; Pajak, B.; Orzechowski, A. The Many Faces of Rap1 GTPase. Int. J. Mol. Sci. 2018, 19, 2848. [Google Scholar] [CrossRef]
- Hashimoto, A.; Tanaka, M.; Takeda, S.; Ito, H.; Nagano, K. Cilostazol Induces PGI2 Production via Activation of the Downstream Epac-1/Rap1 Signaling Cascade to Increase Intracellular Calcium by PLCepsilon and to Activate p44/42 MAPK in Human Aortic Endothelial Cells. PLoS ONE 2015, 10, e0132835. [Google Scholar] [CrossRef]
- Katagiri, K.; Maeda, A.; Shimonaka, M.; Kinashi, T. RAPL, a Rap1-binding molecule that mediates Rap1-induced adhesion through spatial regulation of LFA-1. Nat. Immunol. 2003, 4, 741–748. [Google Scholar] [CrossRef]
- Shannon, M.J.; Pineau, J.; Griffie, J.; Aaron, J.; Peel, T.; Williamson, D.J.; Zamoyska, R.; Cope, A.P.; Cornish, G.H.; Owen, D.M. Differential nanoscale organisation of LFA-1 modulates T cell migration. J. Cell Sci. 2019. [Google Scholar] [CrossRef]
- Witte, A.; Meineke, B.; Sticht, J.; Philipsen, L.; Kuropka, B.; Muller, A.J.; Freund, C.; Schraven, B.; Kliche, S. D120 and K152 within the PH Domain of T Cell Adapter SKAP55 Regulate Plasma Membrane Targeting of SKAP55 and LFA-1 Affinity Modulation in Human T Lymphocytes. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef]
- Stadtmann, A.; Zarbock, A. The role of kindlin in neutrophil recruitment to inflammatory sites. Curr. Opin. Hematol. 2017, 24, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Wegener, K.L.; Partridge, A.W.; Han, J.; Pickford, A.R.; Liddington, R.C.; Ginsberg, M.H.; Campbell, I.D. Structural basis of integrin activation by talin. Cell 2007, 128, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Bardhan, K.; Weaver, J.D.; Sari, D.; Torres-Gomez, A.; Li, L.; Strauss, L.; Lafuente, E.M.; Boussiotis, V.A. The adaptor molecule RIAM integrates signaling events critical for integrin-mediated control of immune function and cancer progression. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Ley, K. Leukocyte arrest: Biomechanics and molecular mechanisms of beta2 integrin activation. Biorheology 2015, 52, 353–377. [Google Scholar] [CrossRef]
- Lefort, C.T.; Rossaint, J.; Moser, M.; Petrich, B.G.; Zarbock, A.; Monkley, S.J.; Critchley, D.R.; Ginsberg, M.H.; Fassler, R.; Ley, K. Distinct roles for talin-1 and kindlin-3 in LFA-1 extension and affinity regulation. Blood 2012, 119, 4275–4282. [Google Scholar] [CrossRef]
- Yago, T.; Zhang, N.; Zhao, L.; Abrams, C.S.; McEver, R.P. Selectins and chemokines use shared and distinct signals to activate beta2 integrins in neutrophils. Blood Adv. 2018, 2, 731–744. [Google Scholar] [CrossRef]
- Geiger, C.; Nagel, W.; Boehm, T.; van Kooyk, Y.; Figdor, C.G.; Kremmer, E.; Hogg, N.; Zeitlmann, L.; Dierks, H.; Weber, K.S.; et al. Cytohesin-1 regulates beta-2 integrin-mediated adhesion through both ARF-GEF function and interaction with LFA-1. Embo J. 2000, 19, 2525–2536. [Google Scholar] [CrossRef]
- Quast, T.; Tappertzhofen, B.; Schild, C.; Grell, J.; Czeloth, N.; Forster, R.; Alon, R.; Fraemohs, L.; Dreck, K.; Weber, C.; et al. Cytohesin-1 controls the activation of RhoA and modulates integrin-dependent adhesion and migration of dendritic cells. Blood 2009, 113, 5801–5810. [Google Scholar] [CrossRef]
- Rognoni, E.; Ruppert, R.; Fassler, R. The kindlin family: Functions, signaling properties and implications for human disease. J. Cell Sci. 2016, 129, 17–27. [Google Scholar] [CrossRef]
- Li, N.; Yang, H.; Wang, M.; Lu, S.; Zhang, Y.; Long, M. Ligand-specific binding forces of LFA-1 and Mac-1 in neutrophil adhesion and crawling. Mol. Biol. Cell 2018, 29, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Wingren, A.G.; Parra, E.; Varga, M.; Kalland, T.; Sjogren, H.O.; Hedlund, G.; Dohlsten, M. T Cell Activation Pathways: B7, LFA-3, and ICAM-1 Shape Unique T Cell Profiles. Crit. Rev. Immunol. 2017, 37, 463–481. [Google Scholar] [CrossRef] [PubMed]
- Atsaves, V.; Leventaki, V.; Rassidakis, G.Z.; Claret, F.X. AP-1 Transcription Factors as Regulators of Immune Responses in Cancer. Cancers (Basel) 2019, 11, 1037. [Google Scholar] [CrossRef]
- Pantarelli, C.; Welch, H.C.E. Rac-GTPases and Rac-GEFs in neutrophil adhesion, migration and recruitment. Eur. J. Clin. Investig. 2018, 48 (Suppl. 2), e12939. [Google Scholar] [CrossRef]
- Nurmi, S.M.; Autero, M.; Raunio, A.K.; Gahmberg, C.G.; Fagerholm, S.C. Phosphorylation of the LFA-1 integrin beta2-chain on Thr-758 leads to adhesion, Rac-1/Cdc42 activation, and stimulation of CD69 expression in human T cells. J. Biol. Chem. 2007, 282, 968–975. [Google Scholar] [CrossRef]
- Stadtmann, A.; Brinkhaus, L.; Mueller, H.; Rossaint, J.; Bolomini-Vittori, M.; Bergmeier, W.; Van Aken, H.; Wagner, D.D.; Laudanna, C.; Ley, K.; et al. Rap1a activation by CalDAG-GEFI and p38 MAPK is involved in E-selectin-dependent slow leukocyte rolling. Eur. J. Immunol. 2011, 41, 2074–2085. [Google Scholar] [CrossRef]
- Toffali, L.; Montresor, A.; Mirenda, M.; Scita, G.; Laudanna, C. SOS1, ARHGEF1, and DOCK2 rho-GEFs Mediate JAK-Dependent LFA-1 Activation by Chemokines. J. Immunol. 2017, 198, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Riteau, B.; Barber, D.F.; Long, E.O. Vav1 phosphorylation is induced by beta2 integrin engagement on natural killer cells upstream of actin cytoskeleton and lipid raft reorganization. J. Exp. Med. 2003, 198, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Burbach, B.J.; Srivastava, R.; Ingram, M.A.; Mitchell, J.S.; Shimizu, Y. The pleckstrin homology domain in the SKAP55 adapter protein defines the ability of the adapter protein ADAP to regulate integrin function and NF-kappaB activation. J. Immunol. 2011, 186, 6227–6237. [Google Scholar] [CrossRef]
- March, M.E.; Long, E.O. beta2 integrin induces TCRzeta-Syk-phospholipase C-gamma phosphorylation and paxillin-dependent granule polarization in human NK cells. J. Immunol. 2011, 186, 2998–3005. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Wang, P.; Petri, B.; Zhang, Y.; Tang, W.; Sun, L.; Kress, H.; Mann, T.; Shi, Y.; Kubes, P.; et al. Integrin-induced PIP5K1C kinase polarization regulates neutrophil polarization, directionality, and in vivo infiltration. Immunity 2010, 33, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Montresor, A.; Toffali, L.; Constantin, G.; Laudanna, C. Chemokines and the signaling modules regulating integrin affinity. Front. Immunol. 2012, 3, 127. [Google Scholar] [CrossRef] [PubMed]
- Urlaub, D.; Hofer, K.; Muller, M.L.; Watzl, C. LFA-1 Activation in NK Cells and Their Subsets: Influence of Receptors, Maturation, and Cytokine Stimulation. J. Immunol. 2017, 198, 1944–1951. [Google Scholar] [CrossRef] [PubMed]
- Kummer, D.; Ebnet, K. Junctional Adhesion Molecules (JAMs): The JAM-Integrin Connection. Cells 2018, 7, 25. [Google Scholar] [CrossRef]
- Podolnikova, N.P.; Podolnikov, A.V.; Haas, T.A.; Lishko, V.K.; Ugarova, T.P. Ligand recognition specificity of leukocyte integrin alphaMbeta2 (Mac-1, CD11b/CD18) and its functional consequences. Biochemistry 2015, 54, 1408–1420. [Google Scholar] [CrossRef]
- Lukacsi, S.; Nagy-Balo, Z.; Erdei, A.; Sandor, N.; Bajtay, Z. The role of CR3 (CD11b/CD18) and CR4 (CD11c/CD18) in complement-mediated phagocytosis and podosome formation by human phagocytes. Immunol. Lett. 2017, 189, 64–72. [Google Scholar] [CrossRef]
- Vorup-Jensen, T.; Jensen, R.K. Structural Immunology of Complement Receptors 3 and 4. Front. Immunol. 2018, 9, 2716. [Google Scholar] [CrossRef]
- Jacobson, A.C.; Weis, J.H. Comparative functional evolution of human and mouse CR1 and CR2. J. Immunol. 2008, 181, 2953–2959. [Google Scholar] [CrossRef]
- Humphries, J.D.; Byron, A.; Humphries, M.J. Integrin ligands at a glance. J. Cell Sci. 2006, 119, 3901–3903. [Google Scholar] [CrossRef]
- Yakubenko, V.P.; Yadav, S.P.; Ugarova, T.P. Integrin alphaDbeta2, an adhesion receptor up-regulated on macrophage foam cells, exhibits multiligand-binding properties. Blood 2006, 107, 1643–1650. [Google Scholar] [CrossRef]
- Yakubenko, V.P.; Cui, K.; Ardell, C.L.; Brown, K.E.; West, X.Z.; Gao, D.; Stefl, S.; Salomon, R.G.; Podrez, E.A.; Byzova, T.V. Oxidative modifications of extracellular matrix promote the second wave of inflammation via beta2 integrins. Blood 2018, 132, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Walling, B.L.; Kim, M. LFA-1 in T Cell Migration and Differentiation. Front. Immunol. 2018, 9, 952. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.; Hyun, Y.M.; Lambert-Emo, K.; Topham, D.J.; Kim, M. Visualization of integrin Mac-1 in vivo. J. Immunol. Methods 2015, 426, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Lishko, V.K.; Yakubenko, V.P.; Ugarova, T.P.; Podolnikova, N.P. Leukocyte integrin Mac-1 (CD11b/CD18, alphaMbeta2, CR3) acts as a functional receptor for platelet factor 4. J. Biol. Chem. 2018, 293, 6869–6882. [Google Scholar] [CrossRef]
- Singh-Jasuja, H.; Thiolat, A.; Ribon, M.; Boissier, M.C.; Bessis, N.; Rammensee, H.G.; Decker, P. The mouse dendritic cell marker CD11c is down-regulated upon cell activation through Toll-like receptor triggering. Immunobiology 2013, 218, 28–39. [Google Scholar] [CrossRef]
- Karnell, J.L.; Kumar, V.; Wang, J.; Wang, S.; Voynova, E.; Ettinger, R. Role of CD11c(+) T-bet(+) B cells in human health and disease. Cell. Immunol. 2017, 321, 40–45. [Google Scholar] [CrossRef]
- Aziz, M.H.; Cui, K.; Das, M.; Brown, K.E.; Ardell, C.L.; Febbraio, M.; Pluskota, E.; Han, J.; Wu, H.; Ballantyne, C.M.; et al. The Upregulation of Integrin alphaDbeta2 (CD11d/CD18) on Inflammatory Macrophages Promotes Macrophage Retention in Vascular Lesions and Development of Atherosclerosis. J. Immunol. 2017, 198, 4855–4867. [Google Scholar] [CrossRef]
- Yu, X.; Feng, B.; He, P.; Shan, L. From Chaos to Harmony: Responses and Signaling upon Microbial Pattern Recognition. Annu. Rev. Phytopathol. 2017, 55, 109–137. [Google Scholar] [CrossRef]
- Todd, J.L.; Palmer, S.M. Danger signals in regulating the immune response to solid organ transplantation. J. Clin. Investig. 2017, 127, 2464–2472. [Google Scholar] [CrossRef]
- Bose, T.O.; Colpitts, S.L.; Pham, Q.M.; Puddington, L.; Lefrancois, L. CD11a is essential for normal development of hematopoietic intermediates. J. Immunol. 2014, 193, 2863–2872. [Google Scholar] [CrossRef]
- Singh, K.; Gatzka, M.; Peters, T.; Borkner, L.; Hainzl, A.; Wang, H.; Sindrilaru, A.; Scharffetter-Kochanek, K. Reduced CD18 levels drive regulatory T cell conversion into Th17 cells in the CD18hypo PL/J mouse model of psoriasis. J. Immunol. 2013, 190, 2544–2553. [Google Scholar] [CrossRef] [PubMed]
- Marski, M.; Kandula, S.; Turner, J.R.; Abraham, C. CD18 is required for optimal development and function of CD4+CD25+ T regulatory cells. J. Immunol. 2005, 175, 7889–7897. [Google Scholar] [CrossRef]
- Meli, A.P.; Fontes, G.; Avery, D.T.; Leddon, S.A.; Tam, M.; Elliot, M.; Ballesteros-Tato, A.; Miller, J.; Stevenson, M.M.; Fowell, D.J.; et al. The Integrin LFA-1 Controls T Follicular Helper Cell Generation and Maintenance. Immunity 2016, 45, 831–846. [Google Scholar] [CrossRef] [PubMed]
- Jogdand, G.M.; Mohanty, S.; Devadas, S. Regulators of Tfh Cell Differentiation. Front. Immunol. 2016, 7, 520. [Google Scholar] [CrossRef] [PubMed]
- Vinuesa, C.G.; Linterman, M.A.; Yu, D.; MacLennan, I.C. Follicular Helper T Cells. Annu. Rev. Immunol. 2016, 34, 335–368. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Z.; Duan, N.; Zhu, G.; Schwarz, E.M.; Xie, C. Osteoblast-osteoclast interactions. Connect. Tissue Res. 2018, 59, 99–107. [Google Scholar] [CrossRef]
- Lampiasi, N.; Russo, R.; Zito, F. The Alternative Faces of Macrophage Generate Osteoclasts. Biomed. Res. Int. 2016, 2016, 9089610. [Google Scholar] [CrossRef]
- Park-Min, K.H.; Lee, E.Y.; Moskowitz, N.K.; Lim, E.; Lee, S.K.; Lorenzo, J.A.; Huang, C.; Melnick, A.M.; Purdue, P.E.; Goldring, S.R.; et al. Negative regulation of osteoclast precursor differentiation by CD11b and beta2 integrin-B-cell lymphoma 6 signaling. J. Bone Miner. Res. 2013, 28, 135–149. [Google Scholar] [CrossRef]
- Wu, H.; Rodgers, J.R.; Perrard, X.Y.; Perrard, J.L.; Prince, J.E.; Abe, Y.; Davis, B.K.; Dietsch, G.; Smith, C.W.; Ballantyne, C.M. Deficiency of CD11b or CD11d results in reduced staphylococcal enterotoxin-induced T cell response and T cell phenotypic changes. J. Immunol. 2004, 173, 297–306. [Google Scholar] [CrossRef]
- Meng, D.; Qin, Y.; Lu, N.; Fang, K.; Hu, Y.; Tian, Z.; Zhang, C. Kupffer Cells Promote the Differentiation of Adult Liver Hematopoietic Stem and Progenitor Cells into Lymphocytes via ICAM-1 and LFA-1 Interaction. Stem Cells Int. 2019, 2019, 4848279. [Google Scholar] [CrossRef]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Crozat, K.; Eidenschenk, C.; Jaeger, B.N.; Krebs, P.; Guia, S.; Beutler, B.; Vivier, E.; Ugolini, S. Impact of beta2 integrin deficiency on mouse natural killer cell development and function. Blood 2011, 117, 2874–2882. [Google Scholar] [CrossRef] [PubMed]
- Kourtzelis, I.; Mitroulis, I.; von Renesse, J.; Hajishengallis, G.; Chavakis, T. From leukocyte recruitment to resolution of inflammation: The cardinal role of integrins. J. Leukoc. Biol. 2017, 102, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Alon, R.; Feigelson, S.W. Chemokine-triggered leukocyte arrest: Force-regulated bi-directional integrin activation in quantal adhesive contacts. Curr. Opin. Cell Biol. 2012, 24, 670–676. [Google Scholar] [CrossRef]
- Heit, B.; Colarusso, P.; Kubes, P. Fundamentally different roles for LFA-1, Mac-1 and alpha4-integrin in neutrophil chemotaxis. J. Cell Sci. 2005, 118, 5205–5220. [Google Scholar] [CrossRef]
- Ding, Z.M.; Babensee, J.E.; Simon, S.I.; Lu, H.; Perrard, J.L.; Bullard, D.C.; Dai, X.Y.; Bromley, S.K.; Dustin, M.L.; Entman, M.L.; et al. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J. Immunol. 1999, 163, 5029–5038. [Google Scholar]
- Dunne, J.L.; Ballantyne, C.M.; Beaudet, A.L.; Ley, K. Control of leukocyte rolling velocity in TNF-alpha-induced inflammation by LFA-1 and Mac-1. Blood 2002, 99, 336–341. [Google Scholar] [CrossRef]
- Sumagin, R.; Prizant, H.; Lomakina, E.; Waugh, R.E.; Sarelius, I.H. LFA-1 and Mac-1 define characteristically different intralumenal crawling and emigration patterns for monocytes and neutrophils in situ. J. Immunol. 2010, 185, 7057–7066. [Google Scholar] [CrossRef]
- Trus, E.; Basta, S.; Gee, K. Who’s in charge here? Macrophage colony stimulating factor and granulocyte macrophage colony stimulating factor: Competing factors in macrophage polarization. Cytokine 2019, 127, 154939. [Google Scholar] [CrossRef]
- Cui, K.; Ardell, C.L.; Podolnikova, N.P.; Yakubenko, V.P. Distinct Migratory Properties of M1, M2, and Resident Macrophages Are Regulated by alphaDbeta2 and alphaMbeta2 Integrin-Mediated Adhesion. Front. Immunol. 2018, 9, 2650. [Google Scholar] [CrossRef]
- Wethmar, K.; Helmus, Y.; Luhn, K.; Jones, C.; Laskowska, A.; Varga, G.; Grabbe, S.; Lyck, R.; Engelhardt, B.; Bixel, M.G.; et al. Migration of immature mouse DC across resting endothelium is mediated by ICAM-2 but independent of beta2-integrins and murine DC-SIGN homologues. Eur. J. Immunol. 2006, 36, 2781–2794. [Google Scholar] [CrossRef] [PubMed]
- Heufler, C.; Ortner, D.; Hofer, S. Cybr, CYTIP or CASP: An attempt to pinpoint a molecule’s functions and names. Immunobiology 2008, 213, 729–732. [Google Scholar] [CrossRef] [PubMed]
- Balkow, S.; Heinz, S.; Schmidbauer, P.; Kolanus, W.; Holzmann, B.; Grabbe, S.; Laschinger, M. LFA-1 activity state on dendritic cells regulates contact duration with T cells and promotes T-cell priming. Blood 2010, 116, 1885–1894. [Google Scholar] [CrossRef] [PubMed]
- Theodoridis, A.A.; Eich, C.; Figdor, C.G.; Steinkasserer, A. Infection of dendritic cells with herpes simplex virus type 1 induces rapid degradation of CYTIP, thereby modulating adhesion and migration. Blood 2011, 118, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Grosche, L.; Drassner, C.; Muhl-Zurbes, P.; Kamm, L.; Le-Trilling, V.T.K.; Trilling, M.; Steinkasserer, A.; Heilingloh, C.S. Human Cytomegalovirus-Induced Degradation of CYTIP Modulates Dendritic Cell Adhesion and Migration. Front. Immunol. 2017, 8, 461. [Google Scholar] [CrossRef] [PubMed]
- Morrison, V.L.; James, M.J.; Grzes, K.; Cook, P.; Glass, D.G.; Savinko, T.; Lek, H.S.; Gawden-Bone, C.; Watts, C.; Millington, O.R.; et al. Loss of beta2-integrin-mediated cytoskeletal linkage reprogrammes dendritic cells to a mature migratory phenotype. Nat. Commun. 2014, 5, 5359. [Google Scholar] [CrossRef]
- Savinko, T.S.; Morrison, V.L.; Uotila, L.M.; Wolff, C.H.J.; Alenius, H.T.; Fagerholm, S.C. Functional Beta2-Integrins Restrict Skin Inflammation In Vivo. J. Investig. Dermatol. 2015, 135, 2249–2257. [Google Scholar] [CrossRef]
- Nishikimi, A.; Ishihara, S.; Ozawa, M.; Etoh, K.; Fukuda, M.; Kinashi, T.; Katagiri, K. Rab13 acts downstream of the kinase Mst1 to deliver the integrin LFA-1 to the cell surface for lymphocyte trafficking. Sci. Signal. 2014, 7, ra72. [Google Scholar] [CrossRef]
- Xu, X.; Jaeger, E.R.; Wang, X.; Lagler-Ferrez, E.; Batalov, S.; Mathis, N.L.; Wiltshire, T.; Walker, J.R.; Cooke, M.P.; Sauer, K.; et al. Mst1 directs Myosin IIa partitioning of low and higher affinity integrins during T cell migration. PLoS ONE 2014, 9, e105561. [Google Scholar] [CrossRef]
- Samuelsson, M.; Potrzebowska, K.; Lehtonen, J.; Beech, J.P.; Skorova, E.; Uronen-Hansson, H.; Svensson, L. RhoB controls the Rab11-mediated recycling and surface reappearance of LFA-1 in migrating T lymphocytes. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef]
- Takahashi, S.; Kubo, K.; Waguri, S.; Yabashi, A.; Shin, H.W.; Katoh, Y.; Nakayama, K. Rab11 regulates exocytosis of recycling vesicles at the plasma membrane. J. Cell Sci. 2012, 125, 4049–4057. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.T.; Freeley, M.; Skubis-Zegadlo, J.; Fazil, M.H.; Kelleher, D.; Fresser, F.; Baier, G.; Verma, N.K.; Long, A. Phosphorylation of Rab5a protein by protein kinase C is crucial for T-cell migration. J. Biol. Chem. 2014, 289, 19420–19434. [Google Scholar] [CrossRef] [PubMed]
- Teske, C.; Schweitzer, C.; Palamidessi, A.; Aust, D.E.; Scita, G.; Weitz, J.; Welsch, T. Modulation of RAB5A early endosome trafficking in response to KRas mediated macropinocytic fluxes in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2017, 493, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Rougerie, P.; Delon, J. Rho GTPases: Masters of T lymphocyte migration and activation. Immunol. Lett. 2012, 142, 1–13. [Google Scholar] [CrossRef]
- Jevnikar, Z.; Obermajer, N.; Pecar-Fonovic, U.; Karaoglanovic-Carmona, A.; Kos, J. Cathepsin X cleaves the beta2 cytoplasmic tail of LFA-1 inducing the intermediate affinity form of LFA-1 and alpha-actinin-1 binding. Eur. J. Immunol. 2009, 39, 3217–3227. [Google Scholar] [CrossRef]
- Rovira-Clave, X.; Angulo-Ibanez, M.; Reina, M.; Espel, E. The PDZ-binding domain of syndecan-2 inhibits LFA-1 high-affinity conformation. Cell. Signal. 2014, 26, 1489–1499. [Google Scholar] [CrossRef]
- Ueda, Y.; Kondo, N.; Ozawa, M.; Yasuda, K.; Tomiyama, T.; Kinashi, T. Sema3e/Plexin D1 Modulates Immunological Synapse and Migration of Thymocytes by Rap1 Inhibition. J. Immunol. 2016, 196, 3019–3031. [Google Scholar] [CrossRef]
- Mirenda, M.; Toffali, L.; Montresor, A.; Scardoni, G.; Sorio, C.; Laudanna, C. Protein tyrosine phosphatase receptor type gamma is a JAK phosphatase and negatively regulates leukocyte integrin activation. J. Immunol. 2015, 194, 2168–2179. [Google Scholar] [CrossRef]
- Chen, J.; Zhong, M.C.; Guo, H.; Davidson, D.; Mishel, S.; Lu, Y.; Rhee, I.; Perez-Quintero, L.A.; Zhang, S.; Cruz-Munoz, M.E.; et al. SLAMF7 is critical for phagocytosis of haematopoietic tumour cells via Mac-1 integrin. Nature 2017, 544, 493–497. [Google Scholar] [CrossRef]
- Hawley, K.L.; Olson, C.M., Jr.; Carreras-Gonzalez, A.; Navasa, N.; Anguita, J. Serum C3 Enhances Complement Receptor 3-Mediated Phagocytosis of Borrelia burgdorferi. Int. J. Biol. Sci. 2015, 11, 1269–1271. [Google Scholar] [CrossRef][Green Version]
- Zhou, M.; Todd, R.F., 3rd; van de Winkel, J.G.; Petty, H.R. Cocapping of the leukoadhesin molecules complement receptor type 3 and lymphocyte function-associated antigen-1 with Fc gamma receptor III on human neutrophils. Possible role of lectin-like interactions. J. Immunol. 1993, 150, 3030–3041. [Google Scholar] [PubMed]
- Saggu, G.; Okubo, K.; Chen, Y.; Vattepu, R.; Tsuboi, N.; Rosetti, F.; Cullere, X.; Washburn, N.; Tahir, S.; Rosado, A.M.; et al. Cis interaction between sialylated FcgammaRIIA and the alphaI-domain of Mac-1 limits antibody-mediated neutrophil recruitment. Nat. Commun. 2018, 9, 5058. [Google Scholar] [CrossRef] [PubMed]
- Jongstra-Bilen, J.; Harrison, R.; Grinstein, S. Fcgamma-receptors induce Mac-1 (CD11b/CD18) mobilization and accumulation in the phagocytic cup for optimal phagocytosis. J. Biol. Chem. 2003, 278, 45720–45729. [Google Scholar] [CrossRef] [PubMed]
- van Spriel, A.B.; Leusen, J.H.; van Egmond, M.; Dijkman, H.B.; Assmann, K.J.; Mayadas, T.N.; van de Winkel, J.G. Mac-1 (CD11b/CD18) is essential for Fc receptor-mediated neutrophil cytotoxicity and immunologic synapse formation. Blood 2001, 97, 2478–2486. [Google Scholar] [CrossRef]
- Wu, Z.; Zhang, Z.; Lei, Z.; Lei, P. CD14: Biology and role in the pathogenesis of disease. Cytokine Growth Factor Rev. 2019, 48, 24–31. [Google Scholar] [CrossRef]
- Zanoni, I.; Ostuni, R.; Marek, L.R.; Barresi, S.; Barbalat, R.; Barton, G.M.; Granucci, F.; Kagan, J.C. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell 2011, 147, 868–880. [Google Scholar] [CrossRef]
- Ling, G.S.; Bennett, J.; Woollard, K.J.; Szajna, M.; Fossati-Jimack, L.; Taylor, P.R.; Scott, D.; Franzoso, G.; Cook, H.T.; Botto, M. Integrin CD11b positively regulates TLR4-induced signalling pathways in dendritic cells but not in macrophages. Nat. Commun. 2014, 5, 3039. [Google Scholar] [CrossRef]
- Perera, P.Y.; Mayadas, T.N.; Takeuchi, O.; Akira, S.; Zaks-Zilberman, M.; Goyert, S.M.; Vogel, S.N. CD11b/CD18 acts in concert with CD14 and Toll-like receptor (TLR) 4 to elicit full lipopolysaccharide and taxol-inducible gene expression. J. Immunol. 2001, 166, 574–581. [Google Scholar] [CrossRef]
- Zhou, H.; Liao, J.; Aloor, J.; Nie, H.; Wilson, B.C.; Fessler, M.B.; Gao, H.M.; Hong, J.S. CD11b/CD18 (Mac-1) is a novel surface receptor for extracellular double-stranded RNA to mediate cellular inflammatory responses. J. Immunol. 2013, 190, 115–125. [Google Scholar] [CrossRef]
- Zhang, Q.; Lee, W.B.; Kang, J.S.; Kim, L.K.; Kim, Y.J. Integrin CD11b negatively regulates Mincle-induced signaling via the Lyn-SIRPalpha-SHP1 complex. Exp. Mol. Med. 2018, 50, e439. [Google Scholar] [CrossRef]
- Jiang, L.; Liu, G.; Ni, W.; Zhang, N.; Jie, J.; Xie, F.; Tai, G. The Combination of MBP and BCG-Induced Dendritic Cell Maturation through TLR2/TLR4 Promotes Th1 Activation In Vitro and Vivo. Mediat. Inflamm 2017, 2017, 1953680. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Qian, C.; Qian, L.; Ma, F.; Hou, J.; Chen, Y.; Wang, Q.; Cao, X. Integrin CD11b negatively regulates TLR9-triggered dendritic cell cross-priming by upregulating microRNA-146a. J. Immunol. 2012, 188, 5293–5302. [Google Scholar] [CrossRef] [PubMed]
- Yee, N.K.; Hamerman, J.A. beta(2) integrins inhibit TLR responses by regulating NF-kappaB pathway and p38 MAPK activation. Eur. J. Immunol. 2013, 43, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Boonyatecha, N.; Sangphech, N.; Wongchana, W.; Kueanjinda, P.; Palaga, T. Involvement of Notch signaling pathway in regulating IL-12 expression via c-Rel in activated macrophages. Mol. Immunol. 2012, 51, 255–262. [Google Scholar] [CrossRef]
- Morelli, A.E.; Larregina, A.T.; Shufesky, W.J.; Zahorchak, A.F.; Logar, A.J.; Papworth, G.D.; Wang, Z.; Watkins, S.C.; Falo, L.D., Jr.; Thomson, A.W. Internalization of circulating apoptotic cells by splenic marginal zone dendritic cells: Dependence on complement receptors and effect on cytokine production. Blood 2003, 101, 611–620. [Google Scholar] [CrossRef]
- Skoberne, M.; Somersan, S.; Almodovar, W.; Truong, T.; Petrova, K.; Henson, P.M.; Bhardwaj, N. The apoptotic-cell receptor CR3, but not alphavbeta5, is a regulator of human dendritic-cell immunostimulatory function. Blood 2006, 108, 947–955. [Google Scholar] [CrossRef]
- Ding, C.; Ma, Y.; Chen, X.; Liu, M.; Cai, Y.; Hu, X.; Xiang, D.; Nath, S.; Zhang, H.G.; Ye, H.; et al. Integrin CD11b negatively regulates BCR signalling to maintain autoreactive B cell tolerance. Nat. Commun. 2013, 4, 2813. [Google Scholar] [CrossRef]
- Zhang, M.; Han, Y.; Han, C.; Xu, S.; Bao, Y.; Chen, Z.; Gu, Y.; Xia, D.; Cao, X. The beta2 integrin CD11b attenuates polyinosinic:polycytidylic acid-induced hepatitis by negatively regulating natural killer cell functions. Hepatol. (Baltim. Md.) 2009, 50, 1606–1616. [Google Scholar] [CrossRef]
- Wooten, D.K.; Xie, X.; Bartos, D.; Busche, R.A.; Longmore, G.D.; Watowich, S.S. Cytokine signaling through Stat3 activates integrins, promotes adhesion, and induces growth arrest in the myeloid cell line 32D. J. Biol. Chem. 2000, 275, 26566–26575. [Google Scholar] [CrossRef]
- Roberts, A.L.; Furnrohr, B.G.; Vyse, T.J.; Rhodes, B. The complement receptor 3 (CD11b/CD18) agonist Leukadherin-1 suppresses human innate inflammatory signalling. Clin. Exp. Immunol. 2016, 185, 361–371. [Google Scholar] [CrossRef]
- Basu, R.; Huse, M. Mechanical Communication at the Immunological Synapse. Trends Cell Biol. 2017, 27, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Benvenuti, F. The Dendritic Cell Synapse: A Life Dedicated to T Cell Activation. Front. Immunol. 2016, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, B.; Mestre, D.; Martinez-Martin, N. The immunological synapse: A cause or consequence of T-cell receptor triggering? Immunology 2011, 133, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Comrie, W.A.; Burkhardt, J.K. Action and Traction: Cytoskeletal Control of Receptor Triggering at the Immunological Synapse. Front. Immunol. 2016, 7, 68. [Google Scholar] [CrossRef] [PubMed]
- Le Floc’h, A.; Huse, M. Molecular mechanisms and functional implications of polarized actin remodeling at the T cell immunological synapse. Cell. Mol. Life Sci. 2015, 72, 537–556. [Google Scholar] [CrossRef]
- Pettmann, J.; Santos, A.M.; Dushek, O.; Davis, S.J. Membrane Ultrastructure and T Cell Activation. Front. Immunol. 2018, 9, 2152. [Google Scholar] [CrossRef]
- Varga, G.; Nippe, N.; Balkow, S.; Peters, T.; Wild, M.K.; Seeliger, S.; Beissert, S.; Krummen, M.; Roth, J.; Sunderkotter, C.; et al. LFA-1 contributes to signal I of T-cell activation and to the production of T(h)1 cytokines. J. Investig. Dermatol. 2010, 130, 1005–1012. [Google Scholar] [CrossRef]
- Wang, Y.; Shibuya, K.; Yamashita, Y.; Shirakawa, J.; Shibata, K.; Kai, H.; Yokosuka, T.; Saito, T.; Honda, S.; Tahara-Hanaoka, S.; et al. LFA-1 decreases the antigen dose for T cell activation in vivo. Int. Immunol. 2008, 20, 1119–1127. [Google Scholar] [CrossRef]
- Palmer, E.; Drobek, A.; Stepanek, O. Opposing effects of actin signaling and LFA-1 on establishing the affinity threshold for inducing effector T-cell responses in mice. Eur. J. Immunol. 2016, 46, 1887–1901. [Google Scholar] [CrossRef]
- Hashimoto-Tane, A.; Sakuma, M.; Ike, H.; Yokosuka, T.; Kimura, Y.; Ohara, O.; Saito, T. Micro-adhesion rings surrounding TCR microclusters are essential for T cell activation. J. Exp. Med. 2016, 213, 1609–1625. [Google Scholar] [CrossRef]
- Azoulay-Alfaguter, I.; Strazza, M.; Peled, M.; Novak, H.K.; Muller, J.; Dustin, M.L.; Mor, A. The tyrosine phosphatase SHP-1 promotes T cell adhesion by activating the adaptor protein CrkII in the immunological synapse. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Kondo, N.; Ueda, Y.; Kita, T.; Ozawa, M.; Tomiyama, T.; Yasuda, K.; Lim, D.S.; Kinashi, T. NDR1-Dependent Regulation of Kindlin-3 Controls High-Affinity LFA-1 Binding and Immune Synapse Organization. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Garcon, F.; Okkenhaug, K. PI3Kdelta promotes CD4(+) T-cell interactions with antigen-presenting cells by increasing LFA-1 binding to ICAM-1. Immunol. Cell Biol. 2016, 94, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Feigelson, S.W.; Grabovsky, V.; Manevich-Mendelson, E.; Pasvolsky, R.; Shulman, Z.; Shinder, V.; Klein, E.; Etzioni, A.; Aker, M.; Alon, R. Kindlin-3 is required for the stabilization of TCR-stimulated LFA-1:ICAM-1 bonds critical for lymphocyte arrest and spreading on dendritic cells. Blood 2011, 117, 7042–7052. [Google Scholar] [CrossRef] [PubMed]
- Siokis, A.; Robert, P.A.; Demetriou, P.; Dustin, M.L.; Meyer-Hermann, M. F-Actin-Driven CD28-CD80 Localization in the Immune Synapse. Cell Rep. 2018, 24, 1151–1162. [Google Scholar] [CrossRef]
- Roy, N.H.; Burkhardt, J.K. The Actin Cytoskeleton: A Mechanical Intermediate for Signal Integration at the Immunological Synapse. Front. Cell Dev. Biol. 2018, 6, 116. [Google Scholar] [CrossRef]
- Murugesan, S.; Hong, J.; Yi, J.; Li, D.; Beach, J.R.; Shao, L.; Meinhardt, J.; Madison, G.; Wu, X.; Betzig, E.; et al. Formin-generated actomyosin arcs propel T cell receptor microcluster movement at the immune synapse. J. Cell Biol. 2016, 215, 383–399. [Google Scholar] [CrossRef]
- Santos, L.C.; Blair, D.A.; Kumari, S.; Cammer, M.; Iskratsch, T.; Herbin, O.; Alexandropoulos, K.; Dustin, M.L.; Sheetz, M.P. Actin polymerization-dependent activation of Cas-L promotes immunological synapse stability. Immunol. Cell Biol. 2016, 94, 981–993. [Google Scholar] [CrossRef]
- Borger, J.G.; Morrison, V.L.; Filby, A.; Garcia, C.; Uotila, L.M.; Simbari, F.; Fagerholm, S.C.; Zamoyska, R. Caveolin-1 Influences LFA-1 Redistribution upon TCR Stimulation in CD8 T Cells. J. Immunol. 2017, 199, 874–884. [Google Scholar] [CrossRef]
- Castro-Sanchez, P.; Ramirez-Munoz, R.; Martin-Cofreces, N.B.; Aguilar-Sopena, O.; Alegre-Gomez, S.; Hernandez-Perez, S.; Reyes, R.; Zeng, Q.; Cabanas, C.; Sanchez-Madrid, F.; et al. Phosphatase of Regenerating Liver-1 (PRL-1) Regulates Actin Dynamics During Immunological Synapse Assembly and T Cell Effector Function. Front. Immunol. 2018, 9, 2655. [Google Scholar] [CrossRef]
- Wabnitz, G.; Balta, E.; Samstag, Y. L-plastin regulates the stability of the immune synapse of naive and effector T-cells. Adv. Biol. Regul. 2017, 63, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Na, B.R.; Jun, C.D. TAGLN2-mediated actin stabilization at the immunological synapse: Implication for cytotoxic T cell control of target cells. Bmb Rep. 2015, 48, 369–370. [Google Scholar] [CrossRef] [PubMed]
- Meissner, J.M.; Sikorski, A.F.; Nawara, T.; Grzesiak, J.; Marycz, K.; Boguslawska, D.M.; Michalczyk, I.; Lecomte, M.C.; Machnicka, B. alphaII-spectrin in T cells is involved in the regulation of cell-cell contact leading to immunological synapse formation? PLoS ONE 2017, 12, e0189545. [Google Scholar] [CrossRef]
- Jankowska, K.I.; Williamson, E.K.; Roy, N.H.; Blumenthal, D.; Chandra, V.; Baumgart, T.; Burkhardt, J.K. Integrins Modulate T Cell Receptor Signaling by Constraining Actin Flow at the Immunological Synapse. Front. Immunol. 2018, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Markey, K.A.; Gartlan, K.H.; Kuns, R.D.; MacDonald, K.P.; Hill, G.R. Imaging the immunological synapse between dendritic cells and T cells. J. Immunol. Methods 2015, 423, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Malinova, D.; Fritzsche, M.; Nowosad, C.R.; Armer, H.; Munro, P.M.; Blundell, M.P.; Charras, G.; Tolar, P.; Bouma, G.; Thrasher, A.J. WASp-dependent actin cytoskeleton stability at the dendritic cell immunological synapse is required for extensive, functional T cell contacts. J. Leukoc. Biol. 2016, 99, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Wioland, H.; Guichard, B.; Senju, Y.; Myram, S.; Lappalainen, P.; Jegou, A.; Romet-Lemonne, G. ADF/Cofilin Accelerates Actin Dynamics by Severing Filaments and Promoting Their Depolymerization at Both Ends. Curr. Biol. Cb 2017, 27, 1956.e1957–1967.e1957. [Google Scholar] [CrossRef]
- Xu, Y.; Pektor, S.; Balkow, S.; Hemkemeyer, S.A.; Liu, Z.; Grobe, K.; Hanley, P.J.; Shen, L.; Bros, M.; Schmidt, T.; et al. Dendritic cell motility and T cell activation requires regulation of Rho-cofilin signaling by the Rho-GTPase activating protein myosin IXb. J. Immunol. 2014, 192, 3559–3568. [Google Scholar] [CrossRef]
- Comrie, W.A.; Li, S.; Boyle, S.; Burkhardt, J.K. The dendritic cell cytoskeleton promotes T cell adhesion and activation by constraining ICAM-1 mobility. J. Cell Biol. 2015, 208, 457–473. [Google Scholar] [CrossRef]
- Petit, A.E.; Demotte, N.; Scheid, B.; Wildmann, C.; Bigirimana, R.; Gordon-Alonso, M.; Carrasco, J.; Valitutti, S.; Godelaine, D.; van der Bruggen, P. A major secretory defect of tumour-infiltrating T lymphocytes due to galectin impairing LFA-1-mediated synapse completion. Nat. Commun. 2016, 7, 12242. [Google Scholar] [CrossRef]
- Ruvolo, P.P. Galectin 3 as a guardian of the tumor microenvironment. Biochim. Et Biophys. Acta 2016, 1863, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Jeon, B.N.; Kim, H.R.; Chung, Y.S.; Na, B.R.; Park, H.; Hong, C.; Fatima, Y.; Oh, H.; Kim, C.H.; Jun, C.D. Actin stabilizer TAGLN2 potentiates adoptive T cell therapy by boosting the inside-out costimulation via lymphocyte function-associated antigen-1. Oncoimmunology 2018, 7, e1500674. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.S.; Weyrich, A.S.; Zimmerman, G.A. Lessons from rare maladies: Leukocyte adhesion deficiency syndromes. Curr. Opin. Hematol. 2013, 20, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Levy-Mendelovich, S.; Rechavi, E.; Abuzaitoun, O.; Vernitsky, H.; Simon, A.J.; Lev, A.; Somech, R. Highlighting the problematic reliance on CD18 for diagnosing leukocyte adhesion deficiency type 1. Immunol Res. 2016, 64, 476–482. [Google Scholar] [CrossRef]
- Moutsopoulos, N.M.; Konkel, J.; Sarmadi, M.; Eskan, M.A.; Wild, T.; Dutzan, N.; Abusleme, L.; Zenobia, C.; Hosur, K.B.; Abe, T.; et al. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17-driven inflammatory bone loss. Sci. Transl. Med. 2014, 6, 229ra240. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Moutsopoulos, N.M. Role of bacteria in leukocyte adhesion deficiency-associated periodontitis. Microb. Pathog. 2016, 94, 21–26. [Google Scholar] [CrossRef]
- Wolach, B.; Gavrieli, R.; Wolach, O.; Stauber, T.; Abuzaitoun, O.; Kuperman, A.; Amir, Y.; Stepensky, P.; Somech, R.; Etzioni, A. Leucocyte adhesion deficiency-A multicentre national experience. Eur. J. Clin. Investig. 2019, 49, e13047. [Google Scholar] [CrossRef]
- Almarza Novoa, E.; Kasbekar, S.; Thrasher, A.J.; Kohn, D.B.; Sevilla, J.; Nguyen, T.; Schwartz, J.D.; Bueren, J.A. Leukocyte adhesion deficiency-I: A comprehensive review of all published cases. J. Allergy Clin. Immunol Pr. 2018, 6, 1418–1420 e1410. [Google Scholar] [CrossRef]
- Giger, U.; Boxer, L.A.; Simpson, P.J.; Lucchesi, B.R.; Todd, R.F., 3rd. Deficiency of leukocyte surface glycoproteins Mo1, LFA-1, and Leu M5 in a dog with recurrent bacterial infections: An animal model. Blood 1987, 69, 1622–1630. [Google Scholar]
- Hanna, S.; Etzioni, A. Leukocyte adhesion deficiencies. Ann. New York Acad. Sci. 2012, 1250, 50–55. [Google Scholar] [CrossRef]
- Sturla, L.; Puglielli, L.; Tonetti, M.; Berninsone, P.; Hirschberg, C.B.; De Flora, A.; Etzioni, A. Impairment of the Golgi GDP-L-fucose transport and unresponsiveness to fucose replacement therapy in LAD II patients. Pediatr. Res. 2001, 49, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Saultier, P.; Szepetowski, S.; Canault, M.; Falaise, C.; Poggi, M.; Suchon, P.; Barlogis, V.; Michel, G.; Loyau, S.; Jandrot-Perrus, M.; et al. Long-term management of leukocyte adhesion deficiency type III without hematopoietic stem cell transplantation. Haematologica 2018, 103, e264–e267. [Google Scholar] [CrossRef] [PubMed]
- Suratannon, N.; Yeetong, P.; Srichomthong, C.; Amarinthnukrowh, P.; Chatchatee, P.; Sosothikul, D.; van Hagen, P.M.; van der Burg, M.; Wentink, M.; Driessen, G.J.; et al. Adaptive immune defects in a patient with leukocyte adhesion deficiency type III with a novel mutation in FERMT3. Pediatr. Allergy Immunol. 2016, 27, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Stepensky, P.Y.; Wolach, B.; Gavrieli, R.; Rousso, S.; Ben Ami, T.; Goldman, V.; Rozovsky, K.; Hanna, S.; Etzioni, A.; Weintraub, M. Leukocyte adhesion deficiency type III: Clinical features and treatment with stem cell transplantation. J. Pediatr. Hematol. Oncol. 2015, 37, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.W.; Ballantyne, C.M.; Smith, C.W.; Montgomery, C.; Bradley, A.; O’Brien, W.E.; Beaudet, A.L. Gene targeting yields a CD18-mutant mouse for study of inflammation. J. Immunol. 1993, 151, 1571–1578. [Google Scholar] [PubMed]
- Shimizu, K.; Libby, P.; Shubiki, R.; Sakuma, M.; Wang, Y.; Asano, K.; Mitchell, R.N.; Simon, D.I. Leukocyte integrin Mac-1 promotes acute cardiac allograft rejection. Circulation 2008, 117, 1997–2008. [Google Scholar] [CrossRef][Green Version]
- Wang, H.; Peters, T.; Sindrilaru, A.; Scharffetter-Kochanek, K. Key role of macrophages in the pathogenesis of CD18 hypomorphic murine model of psoriasis. J. Investig. Dermatol. 2009, 129, 1100–1114. [Google Scholar] [CrossRef]
- Wang, H.; von Rohrscheidt, J.; Roehrbein, J.; Peters, T.; Sindrilaru, A.; Kess, D.; Preissner, K.T.; Scharffetter-Kochanek, K. Extracellular adherence protein of Staphylococcus aureus suppresses disease by inhibiting T-cell recruitment in a mouse model of psoriasis. J. Investig. Dermatol. 2010, 130, 743–754. [Google Scholar] [CrossRef]
- Scharffetter-Kochanek, K.; Lu, H.; Norman, K.; van Nood, N.; Munoz, F.; Grabbe, S.; McArthur, M.; Lorenzo, I.; Kaplan, S.; Ley, K.; et al. Spontaneous skin ulceration and defective T cell function in CD18 null mice. J. Exp. Med. 1998, 188, 119–131. [Google Scholar] [CrossRef]
- Leon-Rico, D.; Aldea, M.; Sanchez-Baltasar, R.; Mesa-Nunez, C.; Record, J.; Burns, S.O.; Santilli, G.; Thrasher, A.J.; Bueren, J.A.; Almarza, E. Lentiviral Vector-Mediated Correction of a Mouse Model of Leukocyte Adhesion Deficiency Type I. Hum. Gene 2016, 27, 668–678. [Google Scholar] [CrossRef]
- Sisco, M.; Chao, J.D.; Kim, I.; Mogford, J.E.; Mayadas, T.N.; Mustoe, T.A. Delayed wound healing in Mac-1-deficient mice is associated with normal monocyte recruitment. Wound Repair. Regen. 2007, 15, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Meakin, P.J.; Morrison, V.L.; Sneddon, C.C.; Savinko, T.; Uotila, L.; Jalicy, S.M.; Gabriel, J.L.; Kang, L.; Ashford, M.L.; Fagerholm, S.C. Mice Lacking beta2-Integrin Function Remain Glucose Tolerant in Spite of Insulin Resistance, Neutrophil Infiltration and Inflammation. PLoS ONE 2015, 10, e0138872. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef]
- Haasken, S.; Auger, J.L.; Binstadt, B.A. Absence of beta2 integrins impairs regulatory T cells and exacerbates CD4+ T cell-dependent autoimmune carditis. J. Immunol. 2011, 187, 2702–2710. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Miura, M.; Gronthos, S.; Allen, M.R.; Cao, C.; Uveges, T.E.; Bi, Y.; Ehirchiou, D.; Kortesidis, A.; Shi, S.; et al. Defective osteogenesis of the stromal stem cells predisposes CD18-null mice to osteoporosis. Proc. Natl. Acad. Sci. USA 2005, 102, 14022–14027. [Google Scholar] [CrossRef] [PubMed]
- Bose, T.O.; Pham, Q.M.; Jellison, E.R.; Mouries, J.; Ballantyne, C.M.; Lefrancois, L. CD11a regulates effector CD8 T cell differentiation and central memory development in response to infection with Listeria monocytogenes. Infect. Immun. 2013, 81, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Chackerian, A.A.; Parker, C.M.; Ballantyne, C.M.; Behar, S.M. The LFA-1 adhesion molecule is required for protective immunity during pulmonary Mycobacterium tuberculosis infection. J. Immunol. 2006, 176, 4914–4922. [Google Scholar] [CrossRef]
- Gorina, R.; Lyck, R.; Vestweber, D.; Engelhardt, B. beta2 integrin-mediated crawling on endothelial ICAM-1 and ICAM-2 is a prerequisite for transcellular neutrophil diapedesis across the inflamed blood-brain barrier. J. Immunol. 2014, 192, 324–337. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, X.; Liu, R.; Wang, L.; Qian, T.; Zheng, Y.; Deng, Y.; Huang, E.; Xu, F.; Wang, J.Y.; et al. B cells expressing CD11b effectively inhibit CD4+ T-cell responses and ameliorate experimental autoimmune hepatitis in mice. Hepatol. (Baltim. Md.) 2015, 62, 1563–1575. [Google Scholar] [CrossRef]
- Teschner, D.; Cholaszczynska, A.; Ries, F.; Beckert, H.; Theobald, M.; Grabbe, S.; Radsak, M.; Bros, M. CD11b Regulates Fungal Outgrowth but Not Neutrophil Recruitment in a Mouse Model of Invasive Pulmonary Aspergillosis. Front. Immunol. 2019, 10, 123. [Google Scholar] [CrossRef]
- Gao, X.P.; Liu, Q.; Broman, M.; Predescu, D.; Frey, R.S.; Malik, A.B. Inactivation of CD11b in a mouse transgenic model protects against sepsis-induced lung PMN infiltration and vascular injury. Physiol. Genom. 2005, 21, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Coxon, A.; Rieu, P.; Barkalow, F.J.; Askari, S.; Sharpe, A.H.; von Andrian, U.H.; Arnaout, M.A.; Mayadas, T.N. A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: A homeostatic mechanism in inflammation. Immunity 1996, 5, 653–666. [Google Scholar] [CrossRef]
- Faridi, M.H.; Khan, S.Q.; Zhao, W.; Lee, H.W.; Altintas, M.M.; Zhang, K.; Kumar, V.; Armstrong, A.R.; Carmona-Rivera, C.; Dorschner, J.M.; et al. CD11b activation suppresses TLR-dependent inflammation and autoimmunity in systemic lupus erythematosus. J. Clin. Investig. 2017, 127, 1271–1283. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, H.; Shi, C.; Erhardt, P.W.; Pavlovsky, A.; D, A.S.; Bledzka, K.; Ustinov, V.; Zhu, L.; Qin, J.; et al. Leukocyte integrin Mac-1 regulates thrombosis via interaction with platelet GPIbalpha. Nat. Commun. 2017, 8, 15559. [Google Scholar] [CrossRef] [PubMed]
- Underhill, D.M.; Ozinsky, A. Phagocytosis of microbes: Complexity in action. Annu. Rev. Immunol. 2002, 20, 825–852. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Liu, B.; Frost, J.L.; Hong, S.; Jin, M.; Ostaszewski, B.; Shankar, G.M.; Costantino, I.M.; Carroll, M.C.; Mayadas, T.N.; et al. Complement component C3 and complement receptor type 3 contribute to the phagocytosis and clearance of fibrillar Abeta by microglia. Glia 2012, 60, 993–1003. [Google Scholar] [CrossRef]
- Anderson, D.C.; Schmalsteig, F.C.; Finegold, M.J.; Hughes, B.J.; Rothlein, R.; Miller, L.J.; Kohl, S.; Tosi, M.F.; Jacobs, R.L.; Waldrop, T.C.; et al. The severe and moderate phenotypes of heritable Mac-1, LFA-1 deficiency: Their quantitative definition and relation to leukocyte dysfunction and clinical features. J. Infect. Dis. 1985, 152, 668–689. [Google Scholar] [CrossRef]
- Ley, K.; Hoffman, H.M.; Kubes, P.; Cassatella, M.A.; Zychlinsky, A.; Hedrick, C.C.; Catz, S.D. Neutrophils: New insights and open questions. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef]
- BoseDasgupta, S.; Pieters, J. Macrophage-microbe interaction: Lessons learned from the pathogen Mycobacterium tuberculosis. Semin. Immunopathol. 2018, 40, 577–591. [Google Scholar] [CrossRef]
- Gazendam, R.P.; van de Geer, A.; Roos, D.; van den Berg, T.K.; Kuijpers, T.W. How neutrophils kill fungi. Immunol. Rev. 2016, 273, 299–311. [Google Scholar] [CrossRef]
- Lu, H.; Ballantyne, C.; Smith, C.W. LFA-1 (CD11a/CD18) triggers hydrogen peroxide production by canine neutrophils. J. Leukoc. Biol. 2000, 68, 73–80. [Google Scholar] [PubMed]
- Allen, S.J.; Mott, K.R.; Chentoufi, A.A.; BenMohamed, L.; Wechsler, S.L.; Ballantyne, C.M.; Ghiasi, H. CD11c controls herpes simplex virus 1 responses to limit virus replication during primary infection. J. Virol. 2011, 85, 9945–9955. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Spear, P.G. Herpes simplex virus: Receptors and ligands for cell entry. Cell. Microbiol. 2004, 6, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Emoto, M.; Emoto, Y.; Brinkmann, V.; Miyamoto, M.; Yoshizawa, I.; Staber, M.; van Rooijen, N.; Hamann, A.; Kaufmann, S.H. Increased resistance of LFA-1-deficient mice to lipopolysaccharide-induced shock/liver injury in the presence of TNF-alpha and IL-12 is mediated by IL-10: A novel role for LFA-1 in the regulation of the proinflammatory and anti-inflammatory cytokine balance. J. Immunol. 2003, 171, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Basit, A.; Reutershan, J.; Morris, M.A.; Solga, M.; Rose, C.E., Jr.; Ley, K. ICAM-1 and LFA-1 play critical roles in LPS-induced neutrophil recruitment into the alveolar space. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L200–L207. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.R.; Han, X.; Soriano, S.G.; Yuki, K. The role of macrophage 1 antigen in polymicrobial sepsis. Shock (Augustaga.) 2014, 42, 532–539. [Google Scholar] [CrossRef]
- Echtenacher, B.; Mannel, D.N.; Hultner, L. Critical protective role of mast cells in a model of acute septic peritonitis. Nature 1996, 381, 75–77. [Google Scholar] [CrossRef]
- Rosenkranz, A.R.; Coxon, A.; Maurer, M.; Gurish, M.F.; Austen, K.F.; Friend, D.S.; Galli, S.J.; Mayadas, T.N. Impaired mast cell development and innate immunity in Mac-1 (CD11b/CD18, CR3)-deficient mice. J. Immunol. 1998, 161, 6463–6467. [Google Scholar]
- Hawkins, H.K.; Heffelfinger, S.C.; Anderson, D.C. Leukocyte adhesion deficiency: Clinical and postmortem observations. Pediatric Pathol. 1992, 12, 119–130. [Google Scholar] [CrossRef]
- Mizgerd, J.P.; Horwitz, B.H.; Quillen, H.C.; Scott, M.L.; Doerschuk, C.M. Effects of CD18 deficiency on the emigration of murine neutrophils during pneumonia. J. Immunol. 1999, 163, 995–999. [Google Scholar]
- Rijneveld, A.W.; de Vos, A.F.; Florquin, S.; Verbeek, J.S.; van der Poll, T. CD11b limits bacterial outgrowth and dissemination during murine pneumococcal pneumonia. J. Infect. Dis. 2005, 191, 1755–1760. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hu, C.; Mayadas-Norton, T.; Tanaka, K.; Chan, J.; Salgame, P. Mycobacterium tuberculosis infection in complement receptor 3-deficient mice. J. Immunol. 2000, 165, 2596–2602. [Google Scholar] [CrossRef] [PubMed]
- Mysak, J.; Podzimek, S.; Sommerova, P.; Lyuya-Mi, Y.; Bartova, J.; Janatova, T.; Prochazkova, J.; Duskova, J. Porphyromonas gingivalis: Major periodontopathic pathogen overview. J. Immunol. Res. 2014, 2014, 476068. [Google Scholar] [CrossRef]
- Wang, M.; Shakhatreh, M.A.; James, D.; Liang, S.; Nishiyama, S.; Yoshimura, F.; Demuth, D.R.; Hajishengallis, G. Fimbrial proteins of porphyromonas gingivalis mediate in vivo virulence and exploit TLR2 and complement receptor 3 to persist in macrophages. J. Immunol. 2007, 179, 2349–2358. [Google Scholar] [CrossRef] [PubMed]
- Zundler, S.; Neurath, M.F. Interleukin-12: Functional activities and implications for disease. Cytokine Growth Factor Rev. 2015, 26, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Shakhatreh, M.A.; Wang, M.; Liang, S. Complement receptor 3 blockade promotes IL-12-mediated clearance of Porphyromonas gingivalis and negates its virulence in vivo. J. Immunol. 2007, 179, 2359–2367. [Google Scholar] [CrossRef]
- Radoshevich, L.; Cossart, P. Listeria monocytogenes: Towards a complete picture of its physiology and pathogenesis. Nat. Rev. Microbiol. 2018, 16, 32–46. [Google Scholar] [CrossRef]
- Wu, H.; Prince, J.E.; Brayton, C.F.; Shah, C.; Zeve, D.; Gregory, S.H.; Smith, C.W.; Ballantyne, C.M. Host resistance of CD18 knockout mice against systemic infection with Listeria monocytogenes. Infect. Immun. 2003, 71, 5986–5993. [Google Scholar] [CrossRef]
- Miyamoto, M.; Emoto, M.; Emoto, Y.; Brinkmann, V.; Yoshizawa, I.; Seiler, P.; Aichele, P.; Kita, E.; Kaufmann, S.H. Neutrophilia in LFA-1-deficient mice confers resistance to listeriosis: Possible contribution of granulocyte-colony-stimulating factor and IL-17. J. Immunol. 2003, 170, 5228–5234. [Google Scholar] [CrossRef]
- Roberts, A.W. G-CSF: A key regulator of neutrophil production, but that’s not all! Growth Factors (Churswitzerland) 2005, 23, 33–41. [Google Scholar] [CrossRef]
- Emoto, M.; Miyamoto, M.; Emoto, Y.; Yoshizawa, I.; Brinkmann, V.; van Rooijen, N.; Kaufmann, S.H. Highly biased type 1 immune responses in mice deficient in LFA-1 in Listeria monocytogenes infection are caused by elevated IL-12 production by granulocytes. J. Immunol. 2003, 171, 3970–3976. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.H.; Cousens, L.P.; van Rooijen, N.; Dopp, E.A.; Carlos, T.M.; Wing, E.J. Complementary adhesion molecules promote neutrophil-Kupffer cell interaction and the elimination of bacteria taken up by the liver. J. Immunol. 2002, 168, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Guerau-de-Arellano, M.; Alroy, J.; Bullard, D.; Huber, B.T. Aggravated Lyme carditis in CD11a-/- and CD11c-/- mice. Infect. Immun. 2005, 73, 7637–7643. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, D.O.; Vieira-de-Abreu, A.; Arcanjo, A.F.; Bozza, P.T.; Zimmerman, G.A.; Castro-Faria-Neto, H.C. Integrin alphaDbeta2 (CD11d/CD18) Modulates Leukocyte Accumulation, Pathogen Clearance, and Pyroptosis in Experimental Salmonella Typhimurium Infection. Front. Immunol. 2018, 9, 1128. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; Shi, Y.; Ren, J. Research progresses of molecular mechanism of pyroptosis and its related diseases. Immunobiology 2019. [Google Scholar] [CrossRef]
- Gazendam, R.P.; van Hamme, J.L.; Tool, A.T.; Hoogenboezem, M.; van den Berg, J.M.; Prins, J.M.; Vitkov, L.; van de Veerdonk, F.L.; van den Berg, T.K.; Roos, D.; et al. Human Neutrophils Use Different Mechanisms to Kill Aspergillus fumigatus Conidia and Hyphae: Evidence from Phagocyte Defects. J. Immunol. 2016, 196, 1272–1283. [Google Scholar] [CrossRef]
- Rohm, M.; Grimm, M.J.; D’Auria, A.C.; Almyroudis, N.G.; Segal, B.H.; Urban, C.F. NADPH oxidase promotes neutrophil extracellular trap formation in pulmonary aspergillosis. Infect. Immun. 2014, 82, 1766–1777. [Google Scholar] [CrossRef]
- Urban, C.F.; Nett, J.E. Neutrophil extracellular traps in fungal infection. Semin. Cell Dev. Biol. 2019, 89, 47–57. [Google Scholar] [CrossRef]
- Clark, H.L.; Abbondante, S.; Minns, M.S.; Greenberg, E.N.; Sun, Y.; Pearlman, E. Protein Deiminase 4 and CR3 Regulate Aspergillus fumigatus and beta-Glucan-Induced Neutrophil Extracellular Trap Formation, but Hyphal Killing Is Dependent Only on CR3. Front. Immunol. 2018, 9, 1182. [Google Scholar] [CrossRef]
- Schmiedel, Y.; Zimmerli, S. Common invasive fungal diseases: An overview of invasive candidiasis, aspergillosis, cryptococcosis, and Pneumocystis pneumonia. Swiss Med. Wkly. 2016, 146, w14281. [Google Scholar] [CrossRef]
- O’Brien, X.M.; Reichner, J.S. Neutrophil Integrins and Matrix Ligands and NET Release. Front. Immunol. 2016, 7, 363. [Google Scholar] [CrossRef] [PubMed]
- Gazendam, R.P.; van Hamme, J.L.; Tool, A.T.; van Houdt, M.; Verkuijlen, P.J.; Herbst, M.; Liese, J.G.; van de Veerdonk, F.L.; Roos, D.; van den Berg, T.K.; et al. Two independent killing mechanisms of Candida albicans by human neutrophils: Evidence from innate immunity defects. Blood 2014, 124, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Soloviev, D.A.; Jawhara, S.; Fonzi, W.A. Regulation of innate immune response to Candida albicans infections by alphaMbeta2-Pra1p interaction. Infect. Immun. 2011, 79, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Ghorbani, M.; Farhoudi, R. Leishmaniasis in humans: Drug or vaccine therapy? Drug Des. Dev. Ther. 2018, 12, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Schonlau, F.; Scharffetter-Kochanek, K.; Grabbe, S.; Pietz, B.; Sorg, C.; Sunderkotter, C. In experimental leishmaniasis deficiency of CD18 results in parasite dissemination associated with altered macrophage functions and incomplete Th1 cell response. Eur. J. Immunol. 2000, 30, 2729–2740. [Google Scholar] [CrossRef]
- PrabhuDas, M.R.; Baldwin, C.L.; Bollyky, P.L.; Bowdish, D.M.E.; Drickamer, K.; Febbraio, M.; Herz, J.; Kobzik, L.; Krieger, M.; Loike, J.; et al. A Consensus Definitive Classification of Scavenger Receptors and Their Roles in Health and Disease. J. Immunol. 2017, 198, 3775–3789. [Google Scholar] [CrossRef]
- Olekhnovitch, R.; Bousso, P. Induction, Propagation, and Activity of Host Nitric Oxide: Lessons from Leishmania Infection. Trends Parasitol. 2015, 31, 653–664. [Google Scholar] [CrossRef]
- Ricardo-Carter, C.; Favila, M.; Polando, R.E.; Cotton, R.N.; Bogard Horner, K.; Condon, D.; Ballhorn, W.; Whitcomb, J.P.; Yadav, M.; Geister, R.L.; et al. Leishmania major inhibits IL-12 in macrophages by signalling through CR3 (CD11b/CD18) and down-regulation of ETS-mediated transcription. Parasite Immunol. 2013, 35, 409–420. [Google Scholar] [CrossRef]
- Carter, C.R.; Whitcomb, J.P.; Campbell, J.A.; Mukbel, R.M.; McDowell, M.A. Complement receptor 3 deficiency influences lesion progression during Leishmania major infection in BALB/c mice. Infect. Immun. 2009, 77, 5668–5675. [Google Scholar] [CrossRef]
- Okwor, I.; Uzonna, J.E. Pathways leading to interleukin-12 production and protective immunity in cutaneous leishmaniasis. Cell. Immunol. 2016, 309, 32–36. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Bunting, M.; Stafforini, D.M.; Harris, E.S.; McIntyre, T.M.; Prescott, S.M.; Frutuoso, V.S.; Amendoeira, F.C.; de Oliveira Nascimento, D.; Vieira-de-Abreu, A.; et al. Integrin alphaDbeta2 is dynamically expressed by inflamed macrophages and alters the natural history of lethal systemic infections. J. Immunol. 2008, 180, 590–600. [Google Scholar] [CrossRef]
- Perez-Mazliah, D.; Langhorne, J. CD4 T-cell subsets in malaria: TH1/TH2 revisited. Front. Immunol. 2014, 5, 671. [Google Scholar] [CrossRef] [PubMed]
- de Azevedo-Quintanilha, I.G.; Vieira-de-Abreu, A.; Ferreira, A.C.; Nascimento, D.O.; Siqueira, A.M.; Campbell, R.A.; Teixeira Ferreira, T.P.; Gutierrez, T.M.; Ribeiro, G.M. Integrin alphaDbeta2 (CD11d/CD18) mediates experimental malaria-associated acute respiratory distress syndrome (MA-ARDS). Malar. J. 2016, 15, 393. [Google Scholar] [CrossRef] [PubMed]
- Franz, J.; Tarantola, M.; Riethmuller, C. How tetraspanins shape endothelial and leukocyte nano-architecture during inflammation. Biochem. Soc. Trans. 2017, 45, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Patten, D.A.; Shetty, S. More Than Just a Removal Service: Scavenger Receptors in Leukocyte Trafficking. Front. Immunol. 2018, 9, 2904. [Google Scholar] [CrossRef] [PubMed]
- Ivetic, A.; Hoskins Green, H.L.; Hart, S.J. L-selectin: A Major Regulator of Leukocyte Adhesion, Migration and Signaling. Front. Immunol. 2019, 10, 1068. [Google Scholar] [CrossRef]
- Klann, J.E.; Kim, S.H.; Remedios, K.A.; He, Z.; Metz, P.J.; Lopez, J.; Tysl, T.; Olvera, J.G.; Ablack, J.N.; Cantor, J.M.; et al. Integrin Activation Controls Regulatory T Cell-Mediated Peripheral Tolerance. J. Immunol. 2018, 200, 4012–4023. [Google Scholar] [CrossRef]
- Lewis, K.L.; Reizis, B. Dendritic cells: Arbiters of immunity and immunological tolerance. Cold Spring Harb. Perspect. Biol. 2012, 4, a007401. [Google Scholar] [CrossRef]
- Ehirchiou, D.; Xiong, Y.; Xu, G.; Chen, W.; Shi, Y.; Zhang, L. CD11b facilitates the development of peripheral tolerance by suppressing Th17 differentiation. J. Exp. Med. 2007, 204, 1519–1524. [Google Scholar] [CrossRef]
- Arnaout, M.A. Biology and structure of leukocyte beta 2 integrins and their role in inflammation. F1000Res 2016, 5. [Google Scholar] [CrossRef]
- Subramanian, P.; Mitroulis, I.; Hajishengallis, G.; Chavakis, T. Regulation of tissue infiltration by neutrophils: Role of integrin alpha3beta1 and other factors. Curr. Opin. Hematol. 2016, 23, 36–43. [Google Scholar] [CrossRef] [PubMed]
- De Rose, D.U.; Giliani, S.; Notarangelo, L.D.; Lougaris, V.; Lanfranchi, A.; Moratto, D.; Martire, B.; Specchia, F.; Tommasini, A.; Plebani, A.; et al. Long term outcome of eight patients with type 1 Leukocyte Adhesion Deficiency (LAD-1): Not only infections, but high risk of autoimmune complications. Clin. Immunol. 2018, 191, 75–80. [Google Scholar] [CrossRef] [PubMed]
- D’Agata, I.D.; Paradis, K.; Chad, Z.; Bonny, Y.; Seidman, E. Leucocyte adhesion deficiency presenting as a chronic ileocolitis. Gut 1996, 39, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Uzel, G.; Kleiner, D.E.; Kuhns, D.B.; Holland, S.M. Dysfunctional LAD-1 neutrophils and colitis. Gastroenterology 2001, 121, 958–964. [Google Scholar] [CrossRef]
- Stevanin, M.; Busso, N.; Chobaz, V.; Pigni, M.; Ghassem-Zadeh, S.; Zhang, L.; Acha-Orbea, H.; Ehirchiou, D. CD11b regulates the Treg/Th17 balance in murine arthritis via IL-6. Eur. J. Immunol. 2017, 47, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Van Seventer, G.A.; Shimizu, Y.; Horgan, K.J.; Shaw, S. The LFA-1 ligand ICAM-1 provides an important costimulatory signal for T cell receptor-mediated activation of resting T cells. J. Immunol. 1990, 144, 4579–4586. [Google Scholar]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef] [PubMed]
- Gultner, S.; Kuhlmann, T.; Hesse, A.; Weber, J.P.; Riemer, C.; Baier, M.; Hutloff, A. Reduced Treg frequency in LFA-1-deficient mice allows enhanced T effector differentiation and pathology in EAE. Eur. J. Immunol. 2010, 40, 3403–3412. [Google Scholar] [CrossRef]
- Fu, J.H.; Zhou, C.C.; Mu, H.Q.; Nan, C.J.; Li, S.; Lu, D.Q. CD18 inhibits progression of kidney cancer by down-regulating Treg cell levels. Eur. Rev. Med. Pharm. Sci. 2019, 23, 2750–2755. [Google Scholar] [CrossRef]
- Koboziev, I.; Karlsson, F.; Ostanin, D.V.; Gray, L.; Davidson, M.; Zhang, S.; Grisham, M.B. Role of LFA-1 in the activation and trafficking of T cells: Implications in the induction of chronic colitis. Inflamm. Bowel Dis. 2012, 18, 2360–2370. [Google Scholar] [CrossRef]
- Dugger, K.J.; Zinn, K.R.; Weaver, C.; Bullard, D.C.; Barnum, S.R. Effector and suppressor roles for LFA-1 during the development of experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2009, 206, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Bravo, B.; Gallego, M.I.; Flores, A.I.; Bornstein, R.; Puente-Bedia, A.; Hernandez, J.; de la Torre, P.; Garcia-Zaragoza, E.; Perez-Tavarez, R.; Grande, J.; et al. Restrained Th17 response and myeloid cell infiltration into the central nervous system by human decidua-derived mesenchymal stem cells during experimental autoimmune encephalomyelitis. Stem Cell Res. 2016, 7, 43. [Google Scholar] [CrossRef] [PubMed]
- Gensterblum, E.; Renauer, P.; Coit, P.; Strickland, F.M.; Kilian, N.C.; Miller, S.; Ognenovski, M.; Wren, J.D.; Tsou, P.S.; Lewis, E.E.; et al. CD4+CD28+KIR+CD11a(hi) T cells correlate with disease activity and are characterized by a pro-inflammatory epigenetic and transcriptional profile in lupus patients. J. Autoimmun. 2018, 86, 19–28. [Google Scholar] [CrossRef]
- Wang, Y.; Shu, Y.; Xiao, Y.; Wang, Q.; Kanekura, T.; Li, Y.; Wang, J.; Zhao, M.; Lu, Q.; Xiao, R. Hypomethylation and overexpression of ITGAL (CD11a) in CD4(+) T cells in systemic sclerosis. Clin. Epigenetics 2014, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.X.; Zhang, F.; Yao, Q.M.; Yuan, T.; Xu, J.; Zhu, X.J. Expression of CD11a in lymphocyte subpopulation in immune thrombocytopenia. Int. J. Clin. Exp. Pathol. 2015, 8, 15642–15651. [Google Scholar] [PubMed]
- Guttman-Yassky, E.; Vugmeyster, Y.; Lowes, M.A.; Chamian, F.; Kikuchi, T.; Kagen, M.; Gilleaudeau, P.; Lee, E.; Hunte, B.; Howell, K.; et al. Blockade of CD11a by efalizumab in psoriasis patients induces a unique state of T-cell hyporesponsiveness. J. Investig. Dermatol. 2008, 128, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Schwab, N.; Ulzheimer, J.C.; Fox, R.J.; Schneider-Hohendorf, T.; Kieseier, B.C.; Monoranu, C.M.; Staugaitis, S.M.; Welch, W.; Jilek, S.; Du Pasquier, R.A.; et al. Fatal PML associated with efalizumab therapy: Insights into integrin alphaLbeta2 in JC virus control. Neurology 2012, 78, 458–467. [Google Scholar] [CrossRef]
- Sterry, W.; Bagot, M.; Ferrandiz, C.; Kragballe, K.; Papp, K.; Stingl, G. Immunosuppressive therapy in dermatology and PML. J. Dtsch. Derm. Ges. 2009, 7, 5. [Google Scholar] [CrossRef]
- Bullard, D.C.; Hu, X.; Schoeb, T.R.; Axtell, R.C.; Raman, C.; Barnum, S.R. Critical requirement of CD11b (Mac-1) on T cells and accessory cells for development of experimental autoimmune encephalomyelitis. J. Immunol. 2005, 175, 6327–6333. [Google Scholar] [CrossRef]
- Rosetti, F.; Tsuboi, N.; Chen, K.; Nishi, H.; Ernandez, T.; Sethi, S.; Croce, K.; Stavrakis, G.; Alcocer-Varela, J.; Gomez-Martin, D.; et al. Human lupus serum induces neutrophil-mediated organ damage in mice that is enabled by Mac-1 deficiency. J. Immunol. 2012, 189, 3714–3723. [Google Scholar] [CrossRef]
- McAdoo, S.P.; Pusey, C.D. Antiglomerular Basement Membrane Disease. Semin. Respir. Crit. Care Med. 2018, 39, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Rosenkranz, A.; Assmann, K.J.; Goodman, M.J.; Gutierrez-Ramos, J.C.; Carroll, M.C.; Cotran, R.S.; Mayadas, T.N. A role for Mac-1 (CDIIb/CD18) in immune complex-stimulated neutrophil function in vivo: Mac-1 deficiency abrogates sustained Fcgamma receptor-dependent neutrophil adhesion and complement-dependent proteinuria in acute glomerulonephritis. J. Exp. Med. 1997, 186, 1853–1863. [Google Scholar] [CrossRef] [PubMed]
- Bernard, P.; Antonicelli, F. Bullous Pemphigoid: A Review of its Diagnosis, Associations and Treatment. Am. J. Clin. Dermatol. 2017, 18, 513–528. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hwang, B.J.; Liu, Z.; Li, N.; Lough, K.; Williams, S.E.; Chen, J.; Burette, S.W.; Diaz, L.A.; Su, M.A.; et al. BP180 dysfunction triggers spontaneous skin inflammation in mice. Proc. Natl. Acad. Sci. USA 2018, 115, 6434–6439. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, M.; Li, N.; Diaz, L.A.; Mayadas, T.N. Differential roles for beta2 integrins in experimental autoimmune bullous pemphigoid. Blood 2006, 107, 1063–1069. [Google Scholar] [CrossRef]
- Lee, Y.H.; Bae, S.C. Association between the functional ITGAM rs1143679 G/A polymorphism and systemic lupus erythematosus/lupus nephritis or rheumatoid arthritis: An update meta-analysis. Rheumatol. Int. 2015, 35, 815–823. [Google Scholar] [CrossRef]
- Toller-Kawahisa, J.E.; Vigato-Ferreira, I.C.; Pancoto, J.A.; Mendes-Junior, C.T.; Martinez, E.Z.; Palomino, G.M.; Louzada-Junior, P.; Donadi, E.A.; Del Lama, J.E.; Marzocchi-Machado, C.M. The variant of CD11b, rs1143679 within ITGAM, is associated with systemic lupus erythematosus and clinical manifestations in Brazilian patients. Hum. Immunol. 2014, 75, 119–123. [Google Scholar] [CrossRef]
- Li, C.; Tong, F.; Ma, Y.; Qian, K.; Zhang, J.; Chen, X. Association of the CD11b rs1143679 polymorphism with systemic lupus erythematosus in the Han Chinese population. J. Int. Med. Res. 2018, 46, 1008–1014. [Google Scholar] [CrossRef]
- Nieuwenhuizen, N.E.; Kirstein, F.; Hoving, J.C.; Brombacher, F. House dust mite induced allergic airway disease is attenuated in CD11c(cre)IL-4Ralpha(-/l) degrees (x) mice. Sci. Rep. 2018, 8, 885. [Google Scholar] [CrossRef]
- Sadhu, C.; Ting, H.J.; Lipsky, B.; Hensley, K.; Garcia-Martinez, L.F.; Simon, S.I.; Staunton, D.E. CD11c/CD18: Novel ligands and a role in delayed-type hypersensitivity. J. Leukoc. Biol. 2007, 81, 1395–1403. [Google Scholar] [CrossRef]
- Miyashita, M.; Sasano, H.; Tamaki, K.; Hirakawa, H.; Takahashi, Y.; Nakagawa, S.; Watanabe, G.; Tada, H.; Suzuki, A.; Ohuchi, N.; et al. Prognostic significance of tumor-infiltrating CD8+ and FOXP3+ lymphocytes in residual tumors and alterations in these parameters after neoadjuvant chemotherapy in triple-negative breast cancer: A retrospective multicenter study. Breast Cancer Res. 2015, 17, 124. [Google Scholar] [CrossRef] [PubMed]
- Barbier, L.; Ferhat, M.; Salame, E.; Robin, A.; Herbelin, A.; Gombert, J.M.; Silvain, C.; Barbarin, A. Interleukin-1 Family Cytokines: Keystones in Liver Inflammatory Diseases. Front. Immunol. 2019, 10, 2014. [Google Scholar] [CrossRef] [PubMed]
- Glatigny, S.; Duhen, R.; Arbelaez, C.; Kumari, S.; Bettelli, E. Integrin alpha L controls the homing of regulatory T cells during CNS autoimmunity in the absence of integrin alpha 4. Sci. Rep. 2015, 5, 7834. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Wang, Y.; Shu, Y.; Lu, Q.; Xiao, R. Epigenetic mechanisms: An emerging role in pathogenesis and its therapeutic potential in systemic sclerosis. Int. J. Biochem. Cell Biol. 2015, 67, 92–100. [Google Scholar] [CrossRef]
- Sanchez-Blanco, C.; Clarke, F.; Cornish, G.H.; Depoil, D.; Thompson, S.J.; Dai, X.; Rawlings, D.J.; Dustin, M.L.; Zamoyska, R.; Cope, A.P.; et al. Protein tyrosine phosphatase PTPN22 regulates LFA-1 dependent Th1 responses. J. Autoimmun. 2018, 94, 45–55. [Google Scholar] [CrossRef]
- Singh, K.; Colmegna, I.; He, X.; Weyand, C.M.; Goronzy, J.J. Synoviocyte stimulation by the LFA-1-intercellular adhesion molecule-2-Ezrin-Akt pathway in rheumatoid arthritis. J. Immunol. 2008, 180, 1971–1978. [Google Scholar] [CrossRef]
- Watts, G.M.; Beurskens, F.J.; Martin-Padura, I.; Ballantyne, C.M.; Klickstein, L.B.; Brenner, M.B.; Lee, D.M. Manifestations of inflammatory arthritis are critically dependent on LFA-1. J. Immunol. 2005, 174, 3668–3675. [Google Scholar] [CrossRef]
- Esterhazy, D.; Loschko, J.; London, M.; Jove, V.; Oliveira, T.Y.; Mucida, D. Classical dendritic cells are required for dietary antigen-mediated induction of peripheral T(reg) cells and tolerance. Nat. Immunol. 2016, 17, 545–555. [Google Scholar] [CrossRef]
- Fagerholm, S.C.; MacPherson, M.; James, M.J.; Sevier-Guy, C.; Lau, C.S. The CD11b-integrin (ITGAM) and systemic lupus erythematosus. Lupus 2013, 22, 657–663. [Google Scholar] [CrossRef]
- Rosetti, F.; de la Cruz, A.; Crispin, J.C. Gene-function studies in systemic lupus erythematosus. Curr. Opin. Rheumatol. 2019, 31, 185–192. [Google Scholar] [CrossRef]
- Wang, L.; Li, Z.; Ciric, B.; Safavi, F.; Zhang, G.X.; Rostami, A. Selective depletion of CD11c(+) CD11b(+) dendritic cells partially abrogates tolerogenic effects of intravenous MOG in murine EAE. Eur. J. Immunol. 2016, 46, 2454–2466. [Google Scholar] [CrossRef] [PubMed]
- Besusso, D.; Saul, L.; Leech, M.D.; O’Connor, R.A.; MacDonald, A.S.; Anderton, S.M.; Mellanby, R.J. 1,25-Dihydroxyvitamin D3-Conditioned CD11c+ Dendritic Cells are Effective Initiators of CNS Autoimmune Disease. Front. Immunol. 2015, 6, 575. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.Y.; Soler, D.C.; Debanne, S.M.; Grozdev, I.; Rodriguez, M.E.; Feig, R.L.; Carman, T.L.; Gilkeson, R.C.; Orringer, C.E.; Kern, E.F.; et al. Psoriasis and cardiovascular risk factors: Increased serum myeloperoxidase and corresponding immunocellular overexpression by Cd11b(+) CD68(+) macrophages in skin lesions. Am. J. Transl. Res. 2013, 6, 16–27. [Google Scholar] [PubMed]
- van Pelt, J.P.; Kuijpers, S.H.; van de Kerkhof, P.C.; de Jong, E.M. The CD11b/CD18-integrin in the pathogenesis of psoriasis. J. Derm. Sci. 1998, 16, 135–143. [Google Scholar] [CrossRef]
- Sjogren, F.; Ljunghusen, O.; Baas, A.; Coble, B.I.; Stendahl, O. Expression and function of beta 2 integrin CD11B/CD18 on leukocytes from patients with psoriasis. Acta Derm. Venereol. 1999, 79, 105–110. [Google Scholar] [CrossRef]
- Ito, A.; Hong, C.; Oka, K.; Salazar, J.V.; Diehl, C.; Witztum, J.L.; Diaz, M.; Castrillo, A.; Bensinger, S.J.; Chan, L.; et al. Cholesterol Accumulation in CD11c(+) Immune Cells Is a Causal and Targetable Factor in Autoimmune Disease. Immunity 2016, 45, 1311–1326. [Google Scholar] [CrossRef]
- Thomas, A.P.; Dunn, T.N.; Oort, P.J.; Grino, M.; Adams, S.H. Inflammatory phenotyping identifies CD11d as a gene markedly induced in white adipose tissue in obese rodents and women. J. Nutr. 2011, 141, 1172–1180. [Google Scholar] [CrossRef]
- Lanitis, E.; Dangaj, D.; Irving, M.; Coukos, G. Mechanisms regulating T-cell infiltration and activity in solid tumors. Ann. Oncol. 2017, 28, xii18–xii32. [Google Scholar] [CrossRef]
- Prenen, H.; Mazzone, M. Tumor-associated macrophages: A short compendium. Cell. Mol. Life Sci. 2019, 76, 1447–1458. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Schupp, J.; Krebs, F.K.; Zimmer, N.; Trzeciak, E.; Schuppan, D.; Tuettenberg, A. Targeting myeloid cells in the tumor sustaining microenvironment. Cell. Immunol. 2019, 343, 103713. [Google Scholar] [CrossRef] [PubMed]
- Tcyganov, E.; Mastio, J.; Chen, E.; Gabrilovich, D.I. Plasticity of myeloid-derived suppressor cells in cancer. Curr. Opin. Immunol. 2018, 51, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Harjunpaa, H.; Llort Asens, M.; Guenther, C.; Fagerholm, S.C. Cell Adhesion Molecules and Their Roles and Regulation in the Immune and Tumor Microenvironment. Front. Immunol. 2019, 10, 1078. [Google Scholar] [CrossRef]
- Zhang, Q.Q.; Hu, X.W.; Liu, Y.L.; Ye, Z.J.; Gui, Y.H.; Zhou, D.L.; Qi, C.L.; He, X.D.; Wang, H.; Wang, L.J. CD11b deficiency suppresses intestinal tumor growth by reducing myeloid cell recruitment. Sci. Rep. 2015, 5, 15948. [Google Scholar] [CrossRef]
- Deronic, A.; Tahvili, S.; Leanderson, T.; Ivars, F. The anti-tumor effect of the quinoline-3-carboxamide tasquinimod: Blockade of recruitment of CD11b(+) Ly6C(hi) cells to tumor tissue reduces tumor growth. Bmc Cancer 2016, 16, 440. [Google Scholar] [CrossRef]
- Okita, Y.; Tanaka, H.; Ohira, M.; Muguruma, K.; Kubo, N.; Watanabe, M.; Fukushima, W.; Hirakawa, K. Role of tumor-infiltrating CD11b+ antigen-presenting cells in the progression of gastric cancer. J. Surg. Res. 2014, 186, 192–200. [Google Scholar] [CrossRef]
- Kim, K.J.; Lee, K.S.; Cho, H.J.; Kim, Y.H.; Yang, H.K.; Kim, W.H.; Kang, G.H. Prognostic implications of tumor-infiltrating FoxP3+ regulatory T cells and CD8+ cytotoxic T cells in microsatellite-unstable gastric cancers. Hum. Pathol. 2014, 45, 285–293. [Google Scholar] [CrossRef]
- Ahn, G.O.; Tseng, D.; Liao, C.H.; Dorie, M.J.; Czechowicz, A.; Brown, J.M. Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc. Natl. Acad. Sci. USA 2010, 107, 8363–8368. [Google Scholar] [CrossRef]
- Soloviev, D.A.; Hazen, S.L.; Szpak, D.; Bledzka, K.M.; Ballantyne, C.M.; Plow, E.F.; Pluskota, E. Dual role of the leukocyte integrin alphaMbeta2 in angiogenesis. J. Immunol. 2014, 193, 4712–4721. [Google Scholar] [CrossRef]
- Sorrentino, C.; Miele, L.; Porta, A.; Pinto, A.; Morello, S. Myeloid-derived suppressor cells contribute to A2B adenosine receptor-induced VEGF production and angiogenesis in a mouse melanoma model. Oncotarget 2015, 6, 27478–27489. [Google Scholar] [CrossRef] [PubMed]
- Schmaljohn, A.L.; Orlandi, C.; Lewis, G.K. Deciphering Fc-mediated Antiviral Antibody Functions in Animal Models. Front. Immunol. 2019, 10, 1602. [Google Scholar] [CrossRef] [PubMed]
- van Spriel, A.B.; van Ojik, H.H.; Bakker, A.; Jansen, M.J.; van de Winkel, J.G. Mac-1 (CD11b/CD18) is crucial for effective Fc receptor-mediated immunity to melanoma. Blood 2003, 101, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.T.; Mace, E.M.; Carisey, A.F.; Viswanath, D.I.; Christakou, A.E.; Wiklund, M.; Onfelt, B.; Orange, J.S. NK cells converge lytic granules to promote cytotoxicity and prevent bystander killing. J. Cell Biol. 2016, 215, 875–889. [Google Scholar] [CrossRef]
- Kumar, S. Natural killer cell cytotoxicity and its regulation by inhibitory receptors. Immunology 2018, 154, 383–393. [Google Scholar] [CrossRef]
- Dustin, M.L. The immunological synapse. Cancer Immunol. Res. 2014, 2, 1023–1033. [Google Scholar] [CrossRef]
- Wurzer, H.; Hoffmann, C.; Al Absi, A.; Thomas, C. Actin Cytoskeleton Straddling the Immunological Synapse between Cytotoxic Lymphocytes and Cancer Cells. Cells 2019, 8, 463. [Google Scholar] [CrossRef]
- Fu, C.; Tong, C.; Wang, M.; Gao, Y.; Zhang, Y.; Lu, S.; Liang, S.; Dong, C.; Long, M. Determining beta2-integrin and intercellular adhesion molecule 1 binding kinetics in tumor cell adhesion to leukocytes and endothelial cells by a gas-driven micropipette assay. J. Biol. Chem. 2011, 286, 34777–34787. [Google Scholar] [CrossRef]
- Liang, S.; Slattery, M.J.; Dong, C. Shear stress and shear rate differentially affect the multi-step process of leukocyte-facilitated melanoma adhesion. Exp. Cell Res. 2005, 310, 282–292. [Google Scholar] [CrossRef]
- Hartmann, T.N.; Grabovsky, V.; Wang, W.; Desch, P.; Rubenzer, G.; Wollner, S.; Binsky, I.; Vallon-Eberhard, A.; Sapoznikov, A.; Burger, M.; et al. Circulating B-cell chronic lymphocytic leukemia cells display impaired migration to lymph nodes and bone marrow. Cancer Res. 2009, 69, 3121–3130. [Google Scholar] [CrossRef]
- Goldin, L.R.; McMaster, M.L.; Rotunno, M.; Herman, S.E.; Jones, K.; Zhu, B.; Boland, J.; Burdett, L.; Hicks, B.; Ravichandran, S.; et al. Whole exome sequencing in families with CLL detects a variant in Integrin beta 2 associated with disease susceptibility. Blood 2016, 128, 2261–2263. [Google Scholar] [CrossRef] [PubMed]
- Riches, J.C.; O’Donovan, C.J.; Kingdon, S.J.; McClanahan, F.; Clear, A.J.; Neuberg, D.S.; Werner, L.; Croce, C.M.; Ramsay, A.G.; Rassenti, L.Z.; et al. Trisomy 12 chronic lymphocytic leukemia cells exhibit upregulation of integrin signaling that is modulated by NOTCH1 mutations. Blood 2014, 123, 4101–4110. [Google Scholar] [CrossRef] [PubMed]
- Hutterer, E.; Asslaber, D.; Caldana, C.; Krenn, P.W.; Zucchetto, A.; Gattei, V.; Greil, R.; Hartmann, T.N. CD18 (ITGB2) expression in chronic lymphocytic leukaemia is regulated by DNA methylation-dependent and -independent mechanisms. Br. J. Haematol. 2015, 169, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Till, K.J.; Harris, R.J.; Linford, A.; Spiller, D.G.; Zuzel, M.; Cawley, J.C. Cell motility in chronic lymphocytic leukemia: Defective Rap1 and alphaLbeta2 activation by chemokine. Cancer Res. 2008, 68, 8429–8436. [Google Scholar] [CrossRef] [PubMed]
- Pye, D.S.; Rubio, I.; Pusch, R.; Lin, K.; Pettitt, A.R.; Till, K.J. Chemokine unresponsiveness of chronic lymphocytic leukemia cells results from impaired endosomal recycling of Rap1 and is associated with a distinctive type of immunological anergy. J. Immunol. 2013, 191, 1496–1504. [Google Scholar] [CrossRef] [PubMed]
- Packham, G.; Krysov, S.; Allen, A.; Savelyeva, N.; Steele, A.J.; Forconi, F.; Stevenson, F.K. The outcome of B-cell receptor signaling in chronic lymphocytic leukemia: Proliferation or anergy. Haematologica 2014, 99, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Montresor, A.; Bolomini-Vittori, M.; Simon, S.I.; Rigo, A.; Vinante, F.; Laudanna, C. Comparative analysis of normal versus CLL B-lymphocytes reveals patient-specific variability in signaling mechanisms controlling LFA-1 activation by chemokines. Cancer Res. 2009, 69, 9281–9290. [Google Scholar] [CrossRef]
- Benedicto, A.; Marquez, J.; Herrero, A.; Olaso, E.; Kolaczkowska, E.; Arteta, B. Decreased expression of the beta2 integrin on tumor cells is associated with a reduction in liver metastasis of colorectal cancer in mice. Bmc Cancer 2017, 17, 827. [Google Scholar] [CrossRef]
- Senbanjo, L.T.; Chellaiah, M.A. CD44: A Multifunctional Cell Surface Adhesion Receptor Is a Regulator of Progression and Metastasis of Cancer Cells. Front. Cell Dev. Biol. 2017, 5, 18. [Google Scholar] [CrossRef]
- Wang, H.S.; Hung, Y.; Su, C.H.; Peng, S.T.; Guo, Y.J.; Lai, M.C.; Liu, C.Y.; Hsu, J.W. CD44 cross-linking induces integrin-mediated adhesion and transendothelial migration in breast cancer cell line by up-regulation of LFA-1 (alpha L beta2) and VLA-4 (alpha4beta1). Exp. Cell Res. 2005, 304, 116–126. [Google Scholar] [CrossRef]
- Yanguas, A.; Garasa, S.; Teijeira, A.; Auba, C.; Melero, I.; Rouzaut, A. ICAM-1-LFA-1 Dependent CD8+ T-Lymphocyte Aggregation in Tumor Tissue Prevents Recirculation to Draining Lymph Nodes. Front. Immunol. 2018, 9, 2084. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; March, M.E.; Lane, W.S.; Long, E.O. A signaling network stimulated by beta2 integrin promotes the polarization of lytic granules in cytotoxic cells. Sci. Signal. 2014, 7, ra96. [Google Scholar] [CrossRef] [PubMed]
- Anikeeva, N.; Somersalo, K.; Sims, T.N.; Thomas, V.K.; Dustin, M.L.; Sykulev, Y. Distinct role of lymphocyte function-associated antigen-1 in mediating effective cytolytic activity by cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. USA 2005, 102, 6437–6442. [Google Scholar] [CrossRef] [PubMed]
- Saed, G.M.; Fletcher, N.M.; Diamond, M.P.; Morris, R.T.; Gomez-Lopez, N.; Memaj, I. Novel expression of CD11b in epithelial ovarian cancer: Potential therapeutic target. Gynecol. Oncol. 2018, 148, 567–575. [Google Scholar] [CrossRef]
- Krueger, J.G.; Ochs, H.D.; Patel, P.; Gilkerson, E.; Guttman-Yassky, E.; Dummer, W. Effect of therapeutic integrin (CD11a) blockade with efalizumab on immune responses to model antigens in humans: Results of a randomized, single blind study. J. Investig. Dermatol. 2008, 128, 2615–2624. [Google Scholar] [CrossRef][Green Version]
- Jelcic, I.; Jelcic, I.; Faigle, W.; Sospedra, M.; Martin, R. Immunology of progressive multifocal leukoencephalopathy. J. Neurovirol. 2015, 21, 614–622. [Google Scholar] [CrossRef]
- Major, E.O. Progressive multifocal leukoencephalopathy in patients on immunomodulatory therapies. Annu. Rev. Med. 2010, 61, 35–47. [Google Scholar] [CrossRef]
- Schwab, N.; Schneider-Hohendorf, T.; Melzer, N.; Cutter, G.; Wiendl, H. Natalizumab-associated PML: Challenges with incidence, resulting risk, and risk stratification. Neurology 2017, 88, 1197–1205. [Google Scholar] [CrossRef]
- Ho, P.R.; Koendgen, H.; Campbell, N.; Haddock, B.; Richman, S.; Chang, I. Risk of natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: A retrospective analysis of data from four clinical studies. Lancet Neurol. 2017, 16, 925–933. [Google Scholar] [CrossRef]
- Akaishi, T.; Nakashima, I. Efficiency of antibody therapy in demyelinating diseases. Int. Immunol. 2017, 29, 327–335. [Google Scholar] [CrossRef]
- Nelson, S.M.; Nguyen, T.M.; McDonald, J.W.; MacDonald, J.K. Natalizumab for induction of remission in Crohn’s disease. Cochrane Database Syst. Rev. 2018, 8, CD006097. [Google Scholar] [CrossRef] [PubMed]
- Potin, D.; Launay, M.; Monatlik, F.; Malabre, P.; Fabreguettes, M.; Fouquet, A.; Maillet, M.; Nicolai, E.; Dorgeret, L.; Chevallier, F.; et al. Discovery and development of 5-[(5S,9R)-9-(4-cyanophenyl)-3-(3,5-dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-tria zaspiro [4.4]non-7-yl-methyl]-3-thiophenecarboxylic acid (BMS-587101)--a small molecule antagonist of leukocyte function associated antigen-1. J. Med. Chem. 2006, 49, 6946–6949. [Google Scholar] [CrossRef] [PubMed]
- Suchard, S.J.; Stetsko, D.K.; Davis, P.M.; Skala, S.; Potin, D.; Launay, M.; Dhar, T.G.; Barrish, J.C.; Susulic, V.; Shuster, D.J.; et al. An LFA-1 (alphaLbeta2) small-molecule antagonist reduces inflammation and joint destruction in murine models of arthritis. J. Immunol. 2010, 184, 3917–3926. [Google Scholar] [CrossRef] [PubMed]
- Celik, E.; Faridi, M.H.; Kumar, V.; Deep, S.; Moy, V.T.; Gupta, V. Agonist leukadherin-1 increases CD11b/CD18-dependent adhesion via membrane tethers. Biophys. J. 2013, 105, 2517–2527. [Google Scholar] [CrossRef] [PubMed]
- Jagarapu, J.; Kelchtermans, J.; Rong, M.; Chen, S.; Hehre, D.; Hummler, S.; Faridi, M.H.; Gupta, V.; Wu, S. Efficacy of Leukadherin-1 in the Prevention of Hyperoxia-Induced Lung Injury in Neonatal Rats. Am. J. Respir Cell Mol. Biol. 2015, 53, 793–801. [Google Scholar] [CrossRef]
- Khan, S.Q.; Guo, L.; Cimbaluk, D.J.; Elshabrawy, H.; Faridi, M.H.; Jolly, M.; George, J.F.; Agarwal, A.; Gupta, V. A Small Molecule beta2 Integrin Agonist Improves Chronic Kidney Allograft Survival by Reducing Leukocyte Recruitment and Accompanying Vasculopathy. Front. Med. (Lausanne) 2014, 1, 45. [Google Scholar] [CrossRef]
- Khan, S.Q.; Khan, I.; Gupta, V. CD11b Activity Modulates Pathogenesis of Lupus Nephritis. Front. Med. (Lausanne) 2018, 5, 52. [Google Scholar] [CrossRef]
- Kavanaugh, A.F.; Davis, L.S.; Jain, R.I.; Nichols, L.A.; Norris, S.H.; Lipsky, P.E. A phase I/II open label study of the safety and efficacy of an anti-ICAM-1 (intercellular adhesion molecule-1; CD54) monoclonal antibody in early rheumatoid arthritis. J. Rheumatol. 1996, 23, 1338–1344. [Google Scholar]
- Kavanaugh, A.F.; Davis, L.S.; Nichols, L.A.; Norris, S.H.; Rothlein, R.; Scharschmidt, L.A.; Lipsky, P.E. Treatment of refractory rheumatoid arthritis with a monoclonal antibody to intercellular adhesion molecule 1. Arthritis Rheum 1994, 37, 992–999. [Google Scholar] [CrossRef]






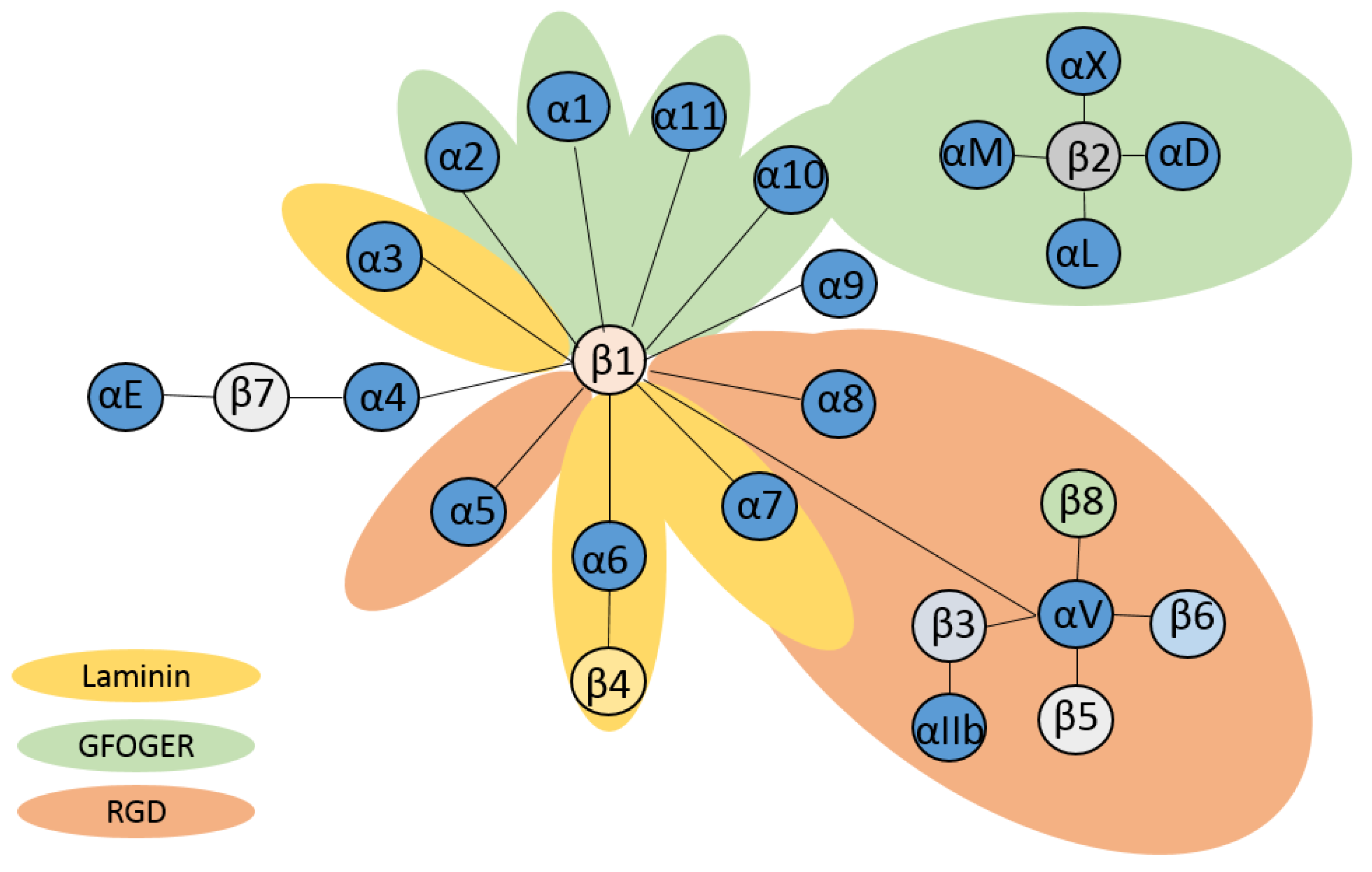{kind=link}
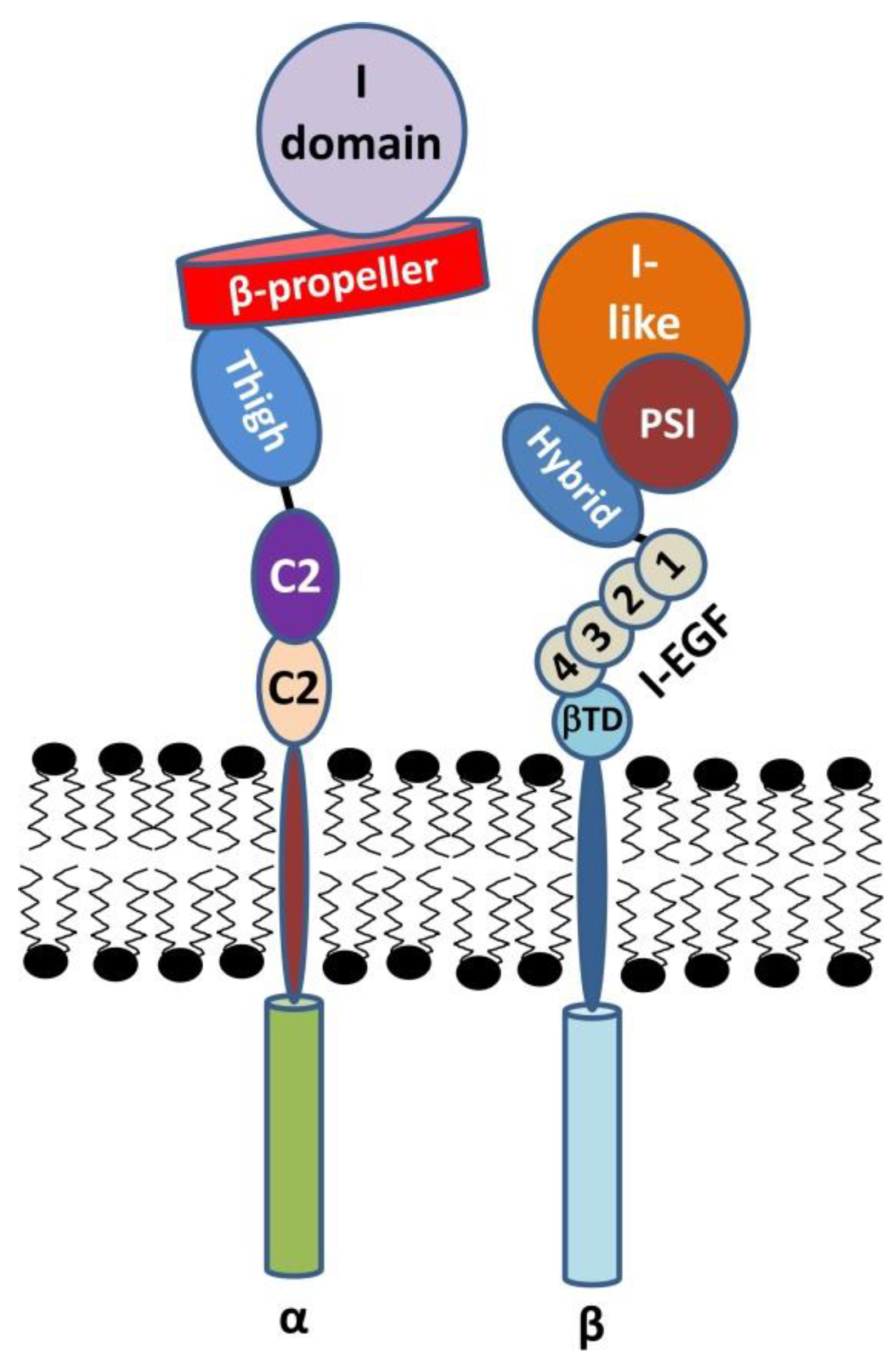{kind=link}
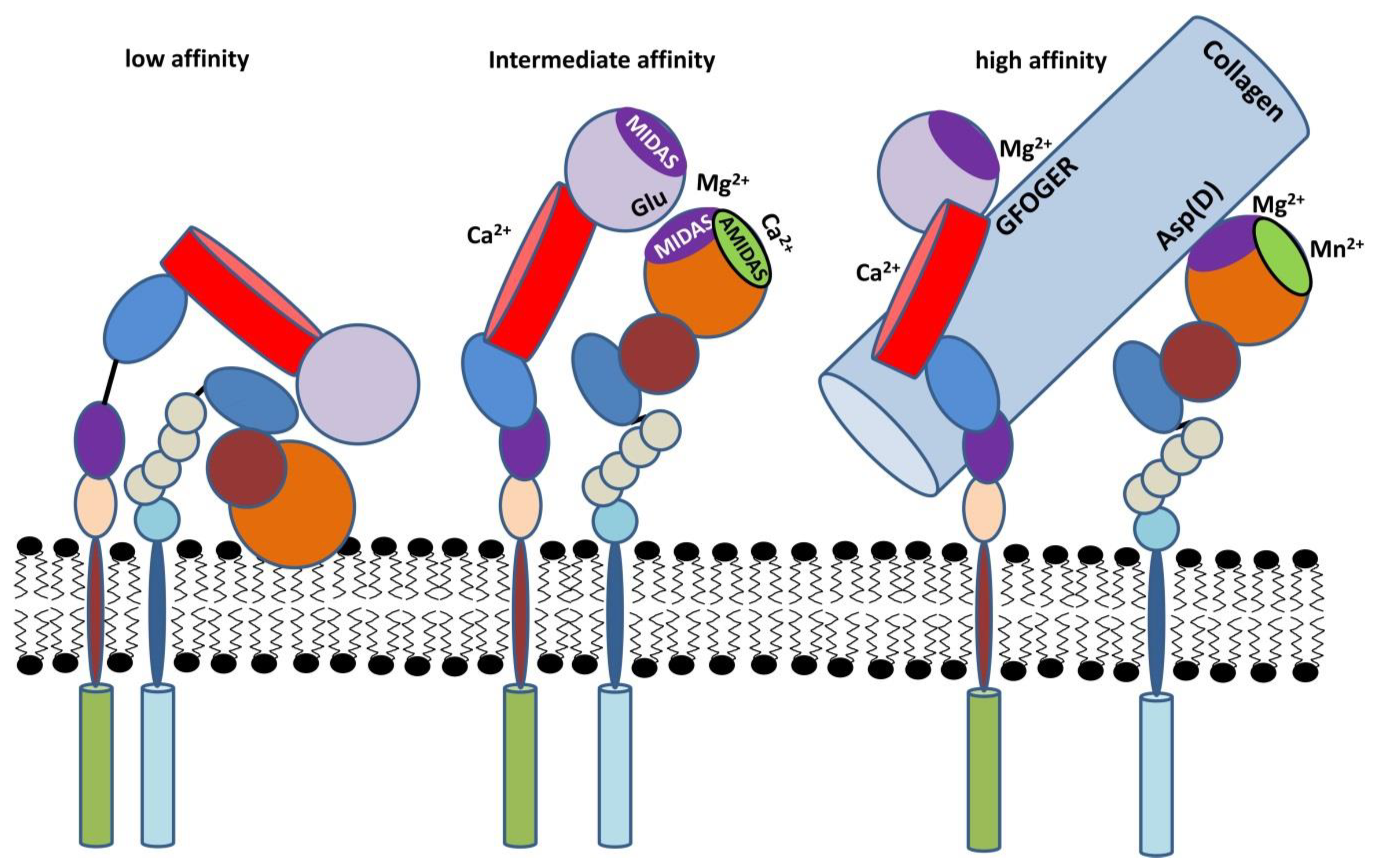{kind=link}
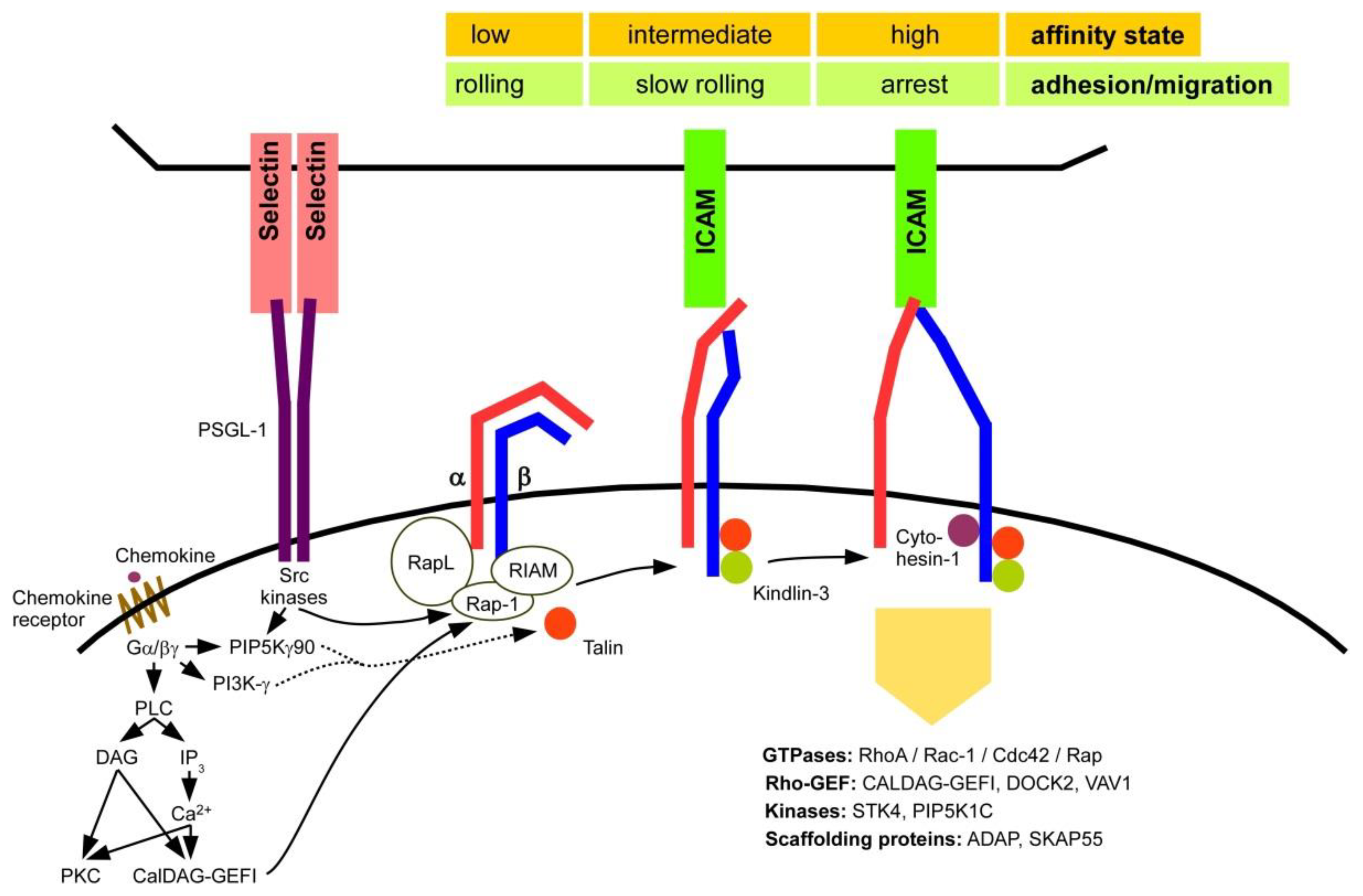{kind=link}
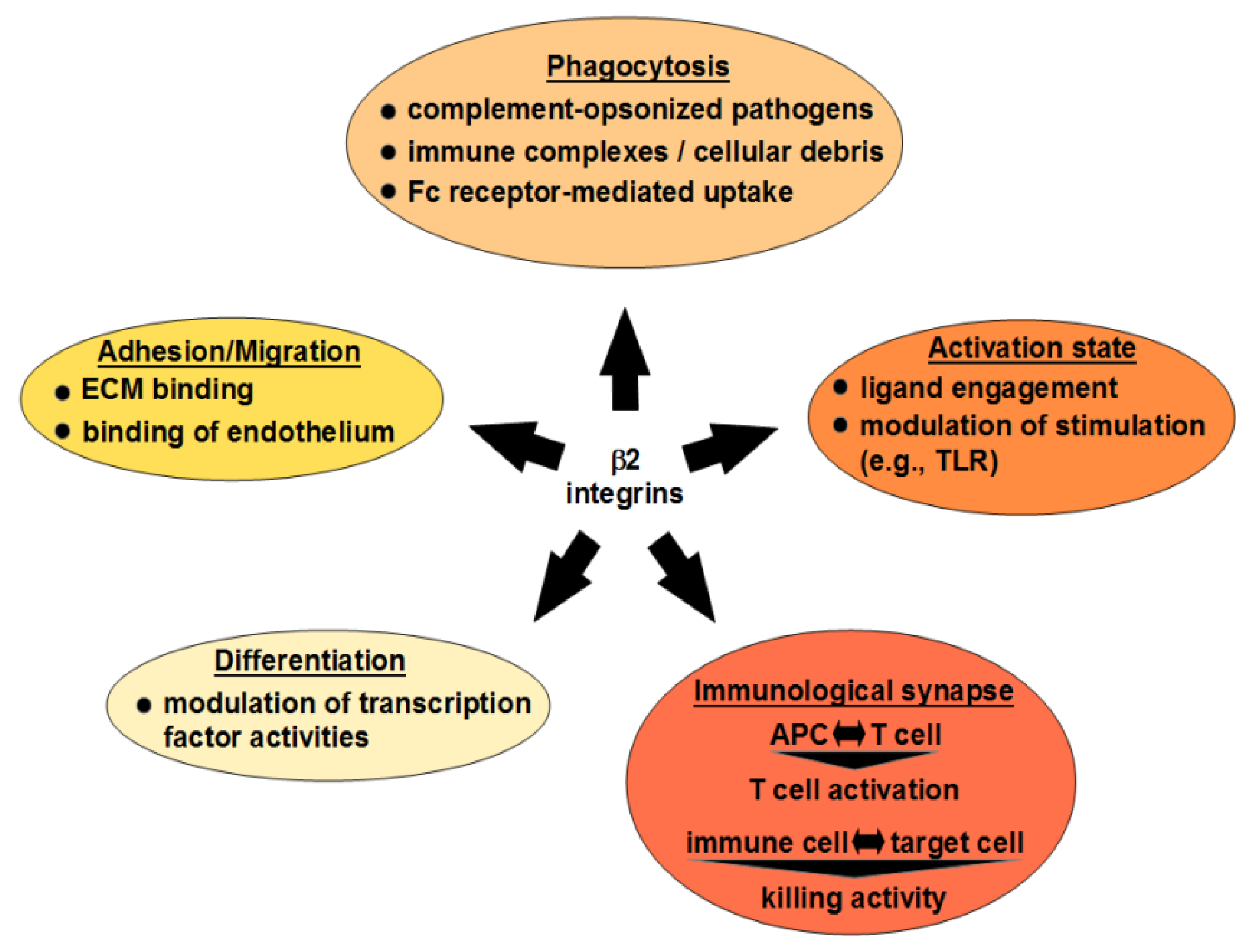{kind=link}
| Sub-Unit | Cell Type | Species 1 | Disease Model, Immune State | Observations | Reference |
|---|---|---|---|---|---|
| CD18 | - | h | LAD | LAD patients suffer from intestinal colitis, periodontitis, Type 1 Diabetes, autoimmune cytopenia | [166,252,253] |
| m | LAD | Chronic dermatitis and splenomegaly | [177,179] | ||
| CD11a | T cell | m | EAE | Pro- or anti-inflammatory functions, depending on the experimental setup | [258,276,283] |
| h | Systemic sclerosis | Expression correlates with severity of disease | [264,284] | ||
| h | Psoriasis | Blockade with Efalizumab induces T cell hyporesponsiveness and inhibits psoriasis pathogenesis | [266] | ||
| h | Autoimmune thrombocytopenia | High expression positively correlates with autoimmune thrombocytopenia pathogenesis | [189] | ||
| h | SLE | Expression correlates with SLE severity | [193] | ||
| h | RA | Expression correlates with RA pathogenesis | [285,286] | ||
| m | Expression is essential for CIA development | [287] | |||
| CD11b | DC | m | RA | Controls balance between Th17 and Treg via IL-6 | [255] |
| APC | m | Peripheral tolerance | Required for establishment of orally induced peripheral tolerance (suppresses IL-6/IL-17 induction) | [249,288] | |
| h | RA, SLE | Polymorphisms predispose for SLE and RA | [193,276,289,290] | ||
| m | SLE | Activation suppresses autoimmunity | [193] | ||
| DC | m | EAE | CD11b+ DC accumulate in CNS of MOG-immunized mice and present with tolerogenic phenotype (IL-10 and TGF-β secretion) | [291] | |
| T cells | m | CD11b+ TC are required for the EAE development | [269,292] | ||
| B cell | m | Experimental autoimmune hepatitis | CD11b+ B cells suppress T cell response by inhibiting TCR signaling | [189] | |
| - | h | Psoriasis | Frequency of CD11b+ cells in lesions correlates with MPO activity. CD11b expression in PMN/macrophages is elevated in pustular psoriasis. | [293,294,295] | |
| PMN | m | Bullous pemphigoid | CD11b is required for skin infiltration by PMN and inflammation development in anti-BP180 antibody-injected mice | [275] | |
| CD11c | DC | m | Autoantibody production | cholesterol accumulation in DC contributes to autoimmune processes | [296] |
| CD11d | - | h/m | Obesity | CD11d expression is elevated in white adipose tissue of obese humans/mice | [297] |
| Subunit | Cell Type | Species | Tumor Model | Observations | Reference |
|---|---|---|---|---|---|
| CD18 | S100A8+ myeloid cells | m | Lewis lung carcinoma (LLC) and MC38 colon adenocarcinoma | Tumors growing in CD18hypo, but not in CD11b-deficient mice were more sensitive to irradiation in comparison to WT mice | [309] |
| B cell | h | CLL | CD18 variant (E630K) enhances CLL susceptibility | [321,331] | |
| CD11a | NK, TC | h | - | Establishment of an intercellular synapse with cancer cell and targeted release of granules depend on LFA-1 | [331,332,333] |
| B cell | h | CLL | Impaired motility and accumulation in the blood due to defective Rap-1 GTPase signaling/diminished CD18 expression | [320,324] | |
| CD11aCD11b | PMN | m | Melanoma | ICAM-1 expressing melanoma cells bind PMN via LFA-1/MAC-1, and thus are carried across the vasculature forming metastasis | [319] |
| CD11b | PMN | m | B16F10 Melanoma, RM1 prostate cancer | CD11b−/− PMN fail to infiltrate tumor tissue and secrete VEGF needed for the neovascularization at lower extent | [310] |
| m | spontaneous intestinal adenoma | CD11b−/− myeloid cell infiltrate tumor mass at low extent, associated with diminished tumor growth and impaired Wnt/β-catenin activity in the tumor | [305] | ||
| m | Squamous cell carcinoma xenografts | Systemic application of CD11b blocking antibody increased anti-tumor response after radiation | [309] | ||
| h | Gastric cancer | Extent of CD11b+ cell infiltration correlated with tumor size, venous invasion, lymph node metastasis, general metastasis stage and FoxP3+ cell infiltration | [307] | ||
| h | Epithelial ovarian cancer (EOC) | de novo expression of MAC-1 on EOC cell lines | [334] | ||
| m | Melanoma | MAC-1 is essential for antibody-mediated antitumor responses | [313] | ||
| CD11c | APC | h | Gastric cancer | CD11c+ cell tumor infiltration correlated with tumor size. | [307] |
| CD11d | Macro-phage | m | Upregulation of CD11d/CD18 surface expression by myeloid cells is associated with their accumulation at the inflammation site and chronification of inflammation | [67] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bednarczyk, M.; Stege, H.; Grabbe, S.; Bros, M. β2 Integrins—Multi-Functional Leukocyte Receptors in Health and Disease. Int. J. Mol. Sci. 2020, 21, 1402. https://doi.org/10.3390/ijms21041402
Bednarczyk M, Stege H, Grabbe S, Bros M. β2 Integrins—Multi-Functional Leukocyte Receptors in Health and Disease. International Journal of Molecular Sciences. 2020; 21(4):1402. https://doi.org/10.3390/ijms21041402
Chicago/Turabian StyleBednarczyk, Monika, Henner Stege, Stephan Grabbe, and Matthias Bros. 2020. "β2 Integrins—Multi-Functional Leukocyte Receptors in Health and Disease" International Journal of Molecular Sciences 21, no. 4: 1402. https://doi.org/10.3390/ijms21041402
APA StyleBednarczyk, M., Stege, H., Grabbe, S., & Bros, M. (2020). β2 Integrins—Multi-Functional Leukocyte Receptors in Health and Disease. International Journal of Molecular Sciences, 21(4), 1402. https://doi.org/10.3390/ijms21041402