P2Y12 Inhibition beyond Thrombosis: Effects on Inflammation

 , , and
, , and
Abstract
1. Introduction
2. P2Y12 Receptors
2.1. Structure of P2Y12 Receptors
2.2. Expression of P2Y12 Receptor
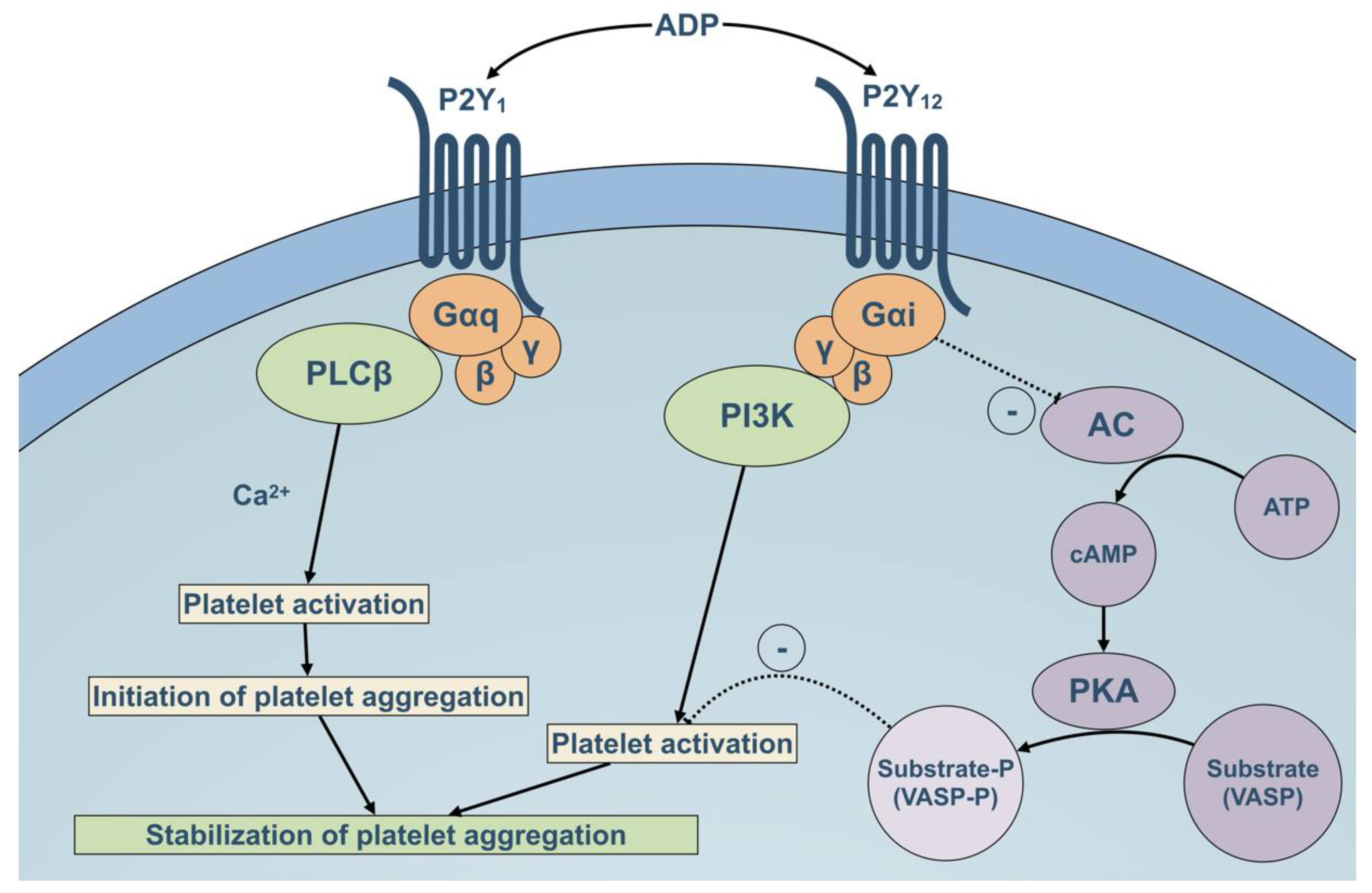
2.3. Role of P2Y12 Rreceptor in Platelet Activation Pathways
2.4. Role of Non-Platelet P2Y12 Receptors
3. P2Y12 Antagonists
3.1. Thienopyridines
3.2. Direct P2Y12 Inhibitors
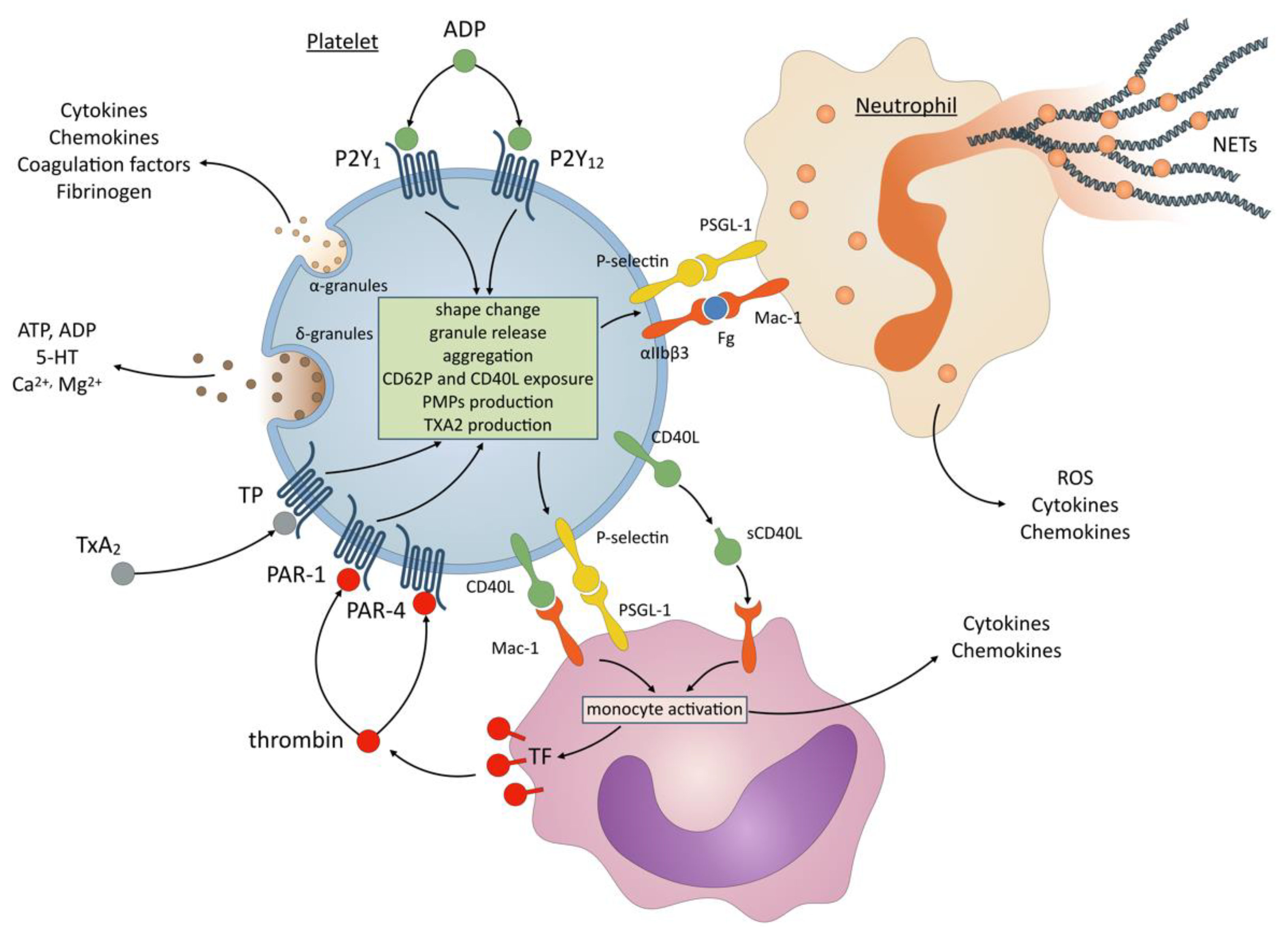
4. Effects of P2Y12 Inhibitors on Inflammation: Possible Molecular and Cellular Mechanisms
4.1. Thrombin Generation
4.2. Release of Inflammatory Mediators
4.3. Platelet-Leukocyte Interactions and Formation of Neutrophil Extracellular Traps (NETs)
4.4. Adenosine Mediated Effects
5. Current Evidence for P2Y12 Inhibition in Clinical Inflammatory Diseases and Syndromes
5.1. Sepsis and Sepsis-Induced Acute Lung Injury (ALI)
5.2. Asthma
5.3. Atherosclerosis
5.4. Cancer—Tumor Growth and Metastasis
5.4.1. In Vitro and Preclinical Studies on P2Y12 Inhibition in Cancer
5.4.2. Are anti P2Y12 Agents Protective or at Risk for Increased Cancer-Related Events?
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACS | Acute coronary syndrome |
| ADP | Adenosine diphosphate |
| ALI | Acute lung injury |
| APC | Antigen-presenting cells |
| ApoE | Apolipoprotein E |
| ARDS | Acute respiratory distress syndrome |
| ATP | Adenosine triphosphate |
| CAD | Coronary artery disease |
| cAMP | Cyclic adenosine monophosphate |
| CD40 | Cluster of differentiation 40 |
| COX | Cyclooxygenase |
| CREKA | Tumor-homing peptide Cys-Arg-Glu-Lys-Ala |
| CRP | C-reactive protein |
| CYP450 | Cytochrome P450 |
| DC | Dendritic cells |
| EL | Extracellular loop |
| ENT-1 | Equilibrative nucleoside transporter 1 |
| FDA | Food and Drug Administration |
| Gi | G-protein i |
| G-protein | Guanine nucleotide-binding protein |
| Gq | G-protein q |
| GPIIbIIIa | Glycoprotein αIIbβ3 |
| HMGB1 | High-mobility group box 1 |
| IL | Interleukin |
| IP3 | Inositol 1,4,5-trisphosphate |
| LPS | Lipopolysaccharide |
| MACE | Major adverse cardiovascular event |
| MCP-1 | Monocyte chemoattractant protein 1 |
| NETs | Neutrophil extracellular traps |
| PAR | Protease-activated receptors |
| PCI | Percutaneous coronary intervention |
| PDGF | Platelet-derived growth factor |
| PF4 | Platelet factor 4 |
| PI3K | Phosphoinositide-3-kinase |
| PKA | Protein kinase A |
| PLAs | Platelet leukocyte aggregates |
| RANTES | Regulated on activation, normal T-cell-expressed and -secreted |
| ROS | Reactive oxygen species |
| TCIPA | Tumor-cell-induced platelet activation |
| TF | Tissue factor |
| TGF | Transforming growth factor |
| TME | Tumor microenvironment |
| TNF | Tumor necrosis factor |
| TXA2 | Thromboxane A2 |
| VASP | Vasodilator-stimulated phosphoprotein |
| VEGF | Vascular endothelial growth factor |
| VSCMs | Vascular smooth muscle cells |
References
- Burnstock, G.; Ralevic, V. Purinergic Signaling and Blood Vessels in Health and Disease. Pharmacol. Rev. 2013, 66, 102–192. [Google Scholar] [PubMed]
- von Kügelgen, I. Pharmacology of P2Y receptors. Brain Res. Bull. 2019, 151, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, M. P2Y12 receptors: Structure and function. J. Thromb. Haemost. 2015, 13, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Gachet, C. P2Y(12) receptors in platelets and other hematopoietic and non-hematopoietic cells. Purinergic Signal. 2012, 8, 609–619. [Google Scholar] [CrossRef]
- Hechler, B.; Gachet, C. The P2 Receptors. Platelets in Thrombotic and Non-Thrombotic Disorders; Pathophysiology, Pharmacology and Therapeutics: An Update; Springer: Cham, Switzerland, 2017; pp. 187–202. [Google Scholar]
- Baqi, Y.; Müller, C.E. Antithrombotic P2Y12 receptor antagonists: Recent developments in drug discovery. Drug Discov. Today 2019, 24, 325–333. [Google Scholar] [CrossRef]
- Hechler, B.; Gachet, C. Purinergic Receptors in Thrombosis and Inflammation. Arterioscler. Thromb. Vasc Biol. 2015, 35, 2307–2315. [Google Scholar] [CrossRef]
- Thomas, M.R.; Storey, R.F. The role of platelets in inflammation. Thromb. Haemost. 2015, 114, 449–458. [Google Scholar]
- Palacios-Acedo, A.; Mège, D.; Crescence, L.; Dignat-George, F.; Dubois, C.; Panicot-Dubois, L. Platelets, Thrombo-Inflammation, and Cancer: Collaborating With the Enemy. Front. Immunol. 2019, 10, 1805. [Google Scholar] [CrossRef]
- Zhang, F.L.; Luo, L.; Gustafson, E.; Lachowicz, J.; Smith, M.; Qiao, X.; Liu, Y.H.; Chen, G.; Pramanik, B.; Laz, T.M.; et al. ADP Is the Cognate Ligand for the Orphan G Protein-coupled Receptor SP1999. J. Biol. Chem. 2000, 276, 8608–8615. [Google Scholar] [CrossRef]
- Hollopeter, G.; Jantzen, H.M.; Vincent, D.; Li, G.; England, L.; Ramakrishnan, V.; Yang, R.B.; Nurden, P.; Nurden, A.; Julius, D.; et al. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature 2001, 409, 202–207. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, J.; Gao, Z.G.; Zhang, D.; Zhu, L.; Han, G.; Moss, S.M.; Paoletta, S.; Kiselev, E.; Lu, W.; et al. Structure of the human P2Y12 receptor in complex with an antithrombotic drug. Nature 2014, 509, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, M. The platelet P2 receptor. In Platelets, 4th ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 259–277. [Google Scholar]
- von Kügelgen, I. Pharmacological profiles of cloned mammalian P2Y-receptor subtypes. Pharmacol. Therapeut. 2006, 110, 415–432. [Google Scholar] [CrossRef] [PubMed]
- Ohlmann, P.; Lecchi, A.; El-Tayeb, A.; Müller, C.E.; Cattaneo, M.; Gachet, C. The platelet P2Y(12) receptor under normal and pathological conditions. Assessment with the radiolabeled selective antagonist [(3)H]PSB-0413. Purinerg. Signal. 2012, 9, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.B.; Julius, D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Rauch, B.H.; Rosenkranz, A.C.; Ermler, S.; Böhm, A.; Driessen, J.; Fischer, J.W.; Sugidachi, A.; Jakubowski, J.A.; Schrör, K. Regulation of Functionally Active P2Y12 ADP Receptors by Thrombin in Human Smooth Muscle Cells and the Presence of P2Y12 in Carotid Artery Lesions. Arterioscler. Thromb. Vasc Biol. 2010, 30, 2434–2442. [Google Scholar] [CrossRef]
- Wihlborg, A.K.; Wang, L.; Braun, O.; Eyjolfsson, A.; Gustafsson, R.; Gudbjartsson, T.; Erlinge, D. ADP Receptor P2Y 12 Is Expressed in Vascular Smooth Muscle Cells and Stimulates Contraction in Human Blood Vessels. Arterioscler. Thromb. Vasc Biol. 2004, 24, 1810–1815. [Google Scholar] [CrossRef]
- Addi, A.; Cammarata, D.; Conley, P.B.; Boeynaems, J.M.; Robaye, B. Role of the P2Y 12 Receptor in the Modulation of Murine Dendritic Cell Function by ADP. J. Immunol. 2010, 185, 5900–5906. [Google Scholar] [CrossRef]
- Paruchuri, S.; Tashimo, H.; Feng, C.; Maekawa, A.; Xing, W.; Jiang, Y.; Kanaoka, Y.; Conley, P.; Boyce, J.A. Leukotriene E4-induced pulmonary inflammation is mediated by the P2Y12 receptor. J. Exp. Med. 2009, 206, 2543–2555. [Google Scholar] [CrossRef]
- Muniz, V.S.; Baptista-Dos-Reis, R.; Benjamim, C.F.; Mata-Santos, H.A.; Pyrrho, A.S.; Strauch, M.A.; Melo, P.A.; Vicentino, A.R.; Silva-Paiva, J.; Bandeira-Melo, C.; et al. Purinergic P2Y12 Receptor Activation in Eosinophils and the Schistosomal Host Response. PLoS ONE 2015, 10, e0139805. [Google Scholar] [CrossRef]
- Micklewright, J.; Layhadi, J.; Fountain, S. P2Y12receptor modulation of ADP-evoked intracellular Ca2+signalling in THP-1 human monocytic cells. Brit. J. Pharmacol. 2018, 175, 2483–2491. [Google Scholar] [CrossRef]
- Vemulapalli, H.; Albayati, S.; Patwa, V.C.; Tilley, D.G.; Tsygankov, A.Y.; Liverani, E. ADP exerts P2Y12 -dependent and P2Y12 -independent effects on primary human T cell responses to stimulation. J. Cell Commun. Signal. 2019, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jacobsen, S.W.; Bengtsson, A.; Erlinge, D. P2 receptor mRNA expression profiles in human lymphocytes, monocytes and CD34+ stem and progenitor cells. BMC Immunol. 2004, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Kronlage, M.; Song, J.; Sorokin, L.; Isfort, K.; Schwerdtle, T.; Leipziger, J.; Robaye, B.; Conley, P.; Kim, C.H.; Sargin, S.; et al. Autocrine Purinergic Receptor Signaling Is Essential for Macrophage Chemotaxis. Sci. Signal. 2010, 3, 55. [Google Scholar] [CrossRef]
- Su, X.; Floyd, D.H.; Hughes, A.; Xiang, J.; Schneider, J.G.; Uluckan, O.; Heller, E.; Deng, H.; Zou, W.; Craft, C.S.; et al. The ADP receptor P2RY12 regulates osteoclast function and pathologic bone remodeling. J. Clin. Investig. 2012, 122, 3579–3592. [Google Scholar] [CrossRef] [PubMed]
- Ballerini, P.; Dovizio, M.; Bruno, A.; Tacconelli, S.; Patrignani, P. P2Y12 Receptors in Tumorigenesis and Metastasis. Front. Pharmacol. 2018, 9, 66. [Google Scholar] [CrossRef] [PubMed]
- Reiner, M.F.; Akhmedov, A.; Stivala, S.; Keller, S.; Gaul, D.S.; Bonetti, N.R.; Savarese, G.; Glanzmann, M.; Zhu, C.; Ruf, W.; et al. Ticagrelor, but not clopidogrel, reduces arterial thrombosis via endothelial tissue factor suppression. Cardiovasc. Res. 2016, 113, 61–69. [Google Scholar] [CrossRef][Green Version]
- Haberstock-Debic, H.; Andre, P.; Mills, S.; Phillips, D.R.; Conley, P.B. A clopidogrel-insensitive inducible pool of P2Y12 receptors contributes to thrombus formation: Inhibition by elinogrel, a direct-acting, reversible P2Y12 antagonist. J. Pharmacol. Exp. Ther. 2011, 339, 54–61. [Google Scholar] [CrossRef]
- Cattaneo, M.; Canciani, M.; Lecchi, A.; Kinlough-Rathbone, R.; Packham, M.; Mannucci, P.; Mustard, J. Released adenosine diphosphate stabilizes thrombin-induced human platelet aggregates. Blood 1990, 75, 1081–1086. [Google Scholar] [CrossRef]
- Eckly, A.; Gendrault, J.L.; Hechler, B.; Cazenave, J.P.; Gachet, C. Differential Involvement of the P2Y1 and P2YT Receptors in the Morphological Changes of Platelet Aggregation. Thromb. Haemost. 2001, 85, 694–701. [Google Scholar] [CrossRef]
- Cattaneo, M. Bleeding manifestations of congenital and drug-induced defects of the platelet P2Y12 receptor for adenosine diphosphate. Thromb. Haemost. 2011, 105, 67–74. [Google Scholar] [CrossRef]
- McFadyen, J.D.; Schaff, M.; Peter, K. Current and future antiplatelet therapies: Emphasis on preserving haemostasis. Nat. Rev. Cardiol. 2018, 15, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Stefanini, L.; Roden, C.R.; Bergmeier, W. CalDAG-GEFI is at the nexus of calcium-dependent platelet activation. Blood 2009, 114, 2506–2514. [Google Scholar] [CrossRef] [PubMed]
- Ohlmann, P.; Laugwitz, K.; Nürnberg, B.; Spicher, K.; Schultz, G.; Cazenave, J.; Gachet, C. The human platelet ADP receptor activates G i2 proteins. Biochem. J. 1995, 312, 775–779. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.; Dangelmaier, C.; Jin, J.; Kim, Y.; Kunapuli, S. Role of Intracellular Signaling Events in ADP-induced Platelet Aggregation. Thromb. Haemost. 1999, 82, 1322–1326. [Google Scholar] [CrossRef]
- Aleil, B.; Ravanat, C.; Cazenave, J.; Rochoux, G.; Heitz, A.; Gachet, C. Flow cytometric analysis of intraplatelet VASP phosphorylation for the detection of clopidogrel resistance in patients with ischemic cardiovascular diseases. J. Thromb. Haemost. 2005, 3, 85–92. [Google Scholar] [CrossRef]
- Laine, M.; Panagides, V.; Frère, C.; Cuisset, T.; Gouarne, C.; Jouve, B.; Thuny, F.; Paganelli, F.; Alessi, M.C.; Mancini, J.; et al. Platelet reactivity inhibition following ticagrelor loading dose in patients undergoing percutaneous coronary intervention for acute coronary syndrome. J. Thromb. Haemost. 2019, 17, 2188–2195. [Google Scholar] [CrossRef]
- Gratacap, M.P.; Hérault, J.P.; Viala, C.; Ragab, A.; Savi, P.; Herbert, J.M.; Chap, H.; Plantavid, M.; Payrastre, B. FcγRIIA requires a Gi-dependent pathway for an efficient stimulation of phosphoinositide 3-kinase, calcium mobilization, and platelet aggregation. Blood 2000, 96, 3439–3446. [Google Scholar] [CrossRef]
- Trumel, C.; Payrastre, B.; Plantavid, M.; Hechler, B.; Viala, C.; Presek, P.; Martinson, E.A.; Cazenave, J.P.; Chap, H.; Gachet, C. A Key Role of Adenosine Diphosphate in the Irreversible Platelet Aggregation Induced by the PAR1-Activating Peptide Through the Late Activation of Phosphoinositide 3-Kinase. Blood 1999, 94, 4156–4165. [Google Scholar] [CrossRef]
- Jackson, S.; Yap, C.; Anderson, K. Phosphoinositide 3-kinases and the regulation of platelet function. Biochem. Soc. T 2004, 32, 387–392. [Google Scholar] [CrossRef]
- Gratacap, M.P.; Guillermet-Guibert, J.; Martin, V.; Chicanne, G.; Tronchère, H.; Gaits-Iacovoni, F.; Payrastre, B. Regulation and roles of PI3Kβ, a major actor in platelet signaling and functions. Adv. Enzym. Regul. 2010, 51, 106–116. [Google Scholar] [CrossRef]
- Dangelmaier, C.; Jin, J.; Smith, B.; Kunapuli, S. Potentiation of Thromboxane A2-induced Platelet Secretion by Gi Signaling through the Phosphoinositide-3 Kinase Pathway. Thromb. Haemost. 2001, 85, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Leon, C.; Alex, M.; Klocke, A.; Morgenstern, E.; Moosbauer, C.; Eckly, A.; Spannagl, M.; Gachet, C.; Engelmann, B. Platelet ADP receptors contribute to the initiation of intravascular coagulation. Blood 2004, 103, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Takano, K.; Asazuma, N.; Satoh, K.; Yatomi, Y.; Ozaki, Y. Collagen-induced generation of platelet-derived microparticles in whole blood is dependent on ADP released from red blood cells and calcium ions. Platelets 2004, 15, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Dorsam, R.; Tuluc, M.; Kunapuli, S. Role of protease-activated and ADP receptor subtypes in thrombin generation on human platelets. J. Thromb. Haemost. 2004, 2, 804–812. [Google Scholar] [CrossRef]
- van der Meijden, P.; Feijge, M.; Giesen, P.; Huijberts, M.; van Raak, L.; Heemskerk, J. Platelet P2Y12 receptors enhance signalling towards procoagulant activity and thrombin generation. Thromb. Haemost. 2005, 93, 1128–1136. [Google Scholar] [CrossRef]
- Liverani, E.; Rico, M.C.; Tsygankov, A.Y.; Kilpatrick, L.E.; Kunapuli, S.P. P2Y12 Receptor Modulates Sepsis-Induced Inflammation. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 961–971. [Google Scholar] [CrossRef]
- Lesyk, G.; Jurasz, P. Advances in Platelet Subpopulation Research. Front. Cardiovasc. Med. 2019, 6, 138. [Google Scholar] [CrossRef]
- Kotova, Y.; Ataullakhanov, F.; Panteleev, M. Formation of coated platelets is regulated by the dense granule secretion of adenosine 5′diphosphate acting via the P2Y12 receptor. J. Thromb. Haemost. 2008, 6, 1603–1605. [Google Scholar] [CrossRef]
- Kempton, C.L.; Hoffman, M.; Lenkowski, A.; Roberts, H.R.; Monroe, D.M. COAT Platelet Formation Is P2Y12-Dependent. Blood 2004, 104, 1569. [Google Scholar] [CrossRef]
- Gao, Y.; Yu, C.; Pi, S.; Mao, L.; Hu, B. The role of P2Y12 receptor in ischemic stroke of atherosclerotic origin. Cell. Mol. Life Sci. 2019, 76, 341–354. [Google Scholar] [CrossRef]
- Lee, C.; Hwang, I.; Park, C.S.; Lee, H.; Park, D.W.; Kang, S.J.; Lee, S.W.; Kim, Y.H.; Park, S.W.; Park, S.J. Comparison of Differential Expression of P2Y12 Receptor in Culprit Coronary Plaques in Patients With Acute Myocardial Infarction Versus Stable Angina Pectoris. Am. J. Cardiol. 2011, 108, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Satonaka, H.; Nagata, D.; Takahashi, M.; Kiyosue, A.; Myojo, M.; Fujita, D.; Ishimitsu, T.; Nagano, T.; Nagai, R.; Hirata, Y. Involvement of P2Y 12 receptor in vascular smooth muscle inflammatory changes via MCP-1 upregulation and monocyte adhesion. Am. J. Physiol Heart C 2015, 308, H853–H861. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Pi, S.; Baral, S.; Xia, Y.; He, Q.; Li, Y.; Jin, H.; Li, M.; Wang, M.; Mao, L.; et al. P2Y 12 Promotes Migration of Vascular Smooth Muscle Cells Through Cofilin Dephosphorylation During Atherogenesis. Arterioscler. Thromb. Vasc Biol. 2017, 37, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Cserép, C.; Pósfai, B.; Lénárt, N.; Fekete, R.; László, Z.I.; Lele, Z.; Orsolits, B.; Molnár, G.; Heindl, S.; Schwarcz, A.D.; et al. Microglia monitor and protect neuronal function via specialized somatic purinergic junctions. Science 2020, 367, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Patente, T.A.; Pinho, M.P.; Oliveira, A.A.; Evangelista, G.C.; Bergami-Santos, P.C.; Barbuto, J.A. Human Dendritic Cells: Their Heterogeneity and Clinical Application Potential in Cancer Immunotherapy. Front. Immunol. 2019, 9, 3176. [Google Scholar] [CrossRef]
- Liao, L.; Guo, Y.; Zhuang, X.; Li, W.; Zou, J.; Su, Q.; Zhao, J.; Liu, Y.; Liao, X.; Du, Z.; et al. Immunosuppressive Effect of Ticagrelor on Dendritic Cell Function: A New Therapeutic Target of Antiplatelet Agents in Cardiovascular Disease. J. Biomed. Nanotechnol. 2018, 14, 1665–1673. [Google Scholar] [CrossRef]
- Diehl, P.; Olivier, C.; Halscheid, C.; Helbing, T.; Bode, C.; Moser, M. Clopidogrel affects leukocyte dependent platelet aggregation by P2Y12 expressing leukocytes. Basic Res. Cardiol. 2009, 105, 379–387. [Google Scholar] [CrossRef]
- Suh, D.H.; Trinh, H.; Liu, J.N.; Pham, L.; Park, S.; Park, H.S.; Shin, Y. P2Y12 antagonist attenuates eosinophilic inflammation and airway hyperresponsiveness in a mouse model of asthma. J. Cell. Mol. Med. 2016, 20, 333–341. [Google Scholar] [CrossRef]
- Czajkowski, R.; Lei, L.; Sabal̵a, P.; Barańska, J. ADP-evoked phospholipase C stimulation and adenylyl cyclase inhibition in glioma C6 cells occur through two distinct nucleotide receptors, P2Y 1 and P2Y 12. FEBS Lett. 2002, 513, 179–183. [Google Scholar] [CrossRef]
- Jin, J.; Tomlinson, W.; Kirk, I.P.; Kim, Y.B.; Humphries, R.G.; Kunapuli, S.P. The C6-2B glioma cell P2Y AC receptor is pharmacologically and molecularly identical to the platelet P2Y 12 receptor. Brit. J. Pharmacol. 2001, 133, 521–528. [Google Scholar] [CrossRef]
- Sarangi, S.; Pandey, A.; Papa, A.L.; Sengupta, P.; Kopparam, J.; Dadwal, U.; Basu, S.; Sengupta, S. P2Y12 receptor inhibition augments cytotoxic effects of cisplatin in breast cancer. Med Oncol. (Northwood Lond. Engl.) 2013, 30, 567. [Google Scholar] [CrossRef] [PubMed]
- Fahmy, A.I.; Mekkawy, M.A.; Abou-Ali, A. Evaluation of adverse events involving bleeding associated with oral P2Y12 inhibitors use in the Food and Drug Administration adverse event reporting system. Int. J. Clin. Pharm. Ther. 2019, 57, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Becker, R.C.; Bassand, J.; Budaj, A.; Wojdyla, D.M.; James, S.K.; Cornel, J.H.; French, J.; Held, C.; Horrow, J.; Husted, S.; et al. Bleeding complications with the P2Y12 receptor antagonists clopidogrel and ticagrelor in the PLATelet inhibition and patient Outcomes (PLATO) trial. Eur. Heart J. 2011, 32, 2933–2944. [Google Scholar] [CrossRef] [PubMed]
- Godier, A.; Fontana, P.; Motte, S.; Steib, A.; Bonhomme, F.; Schlumberger, S.; Lecompte, T.; Rosencher, N.; Susen, S.; Vincentelli, A.; et al. Management of antiplatelet therapy in patients undergoing elective invasive procedures. Proposals from the French Working Group on perioperative haemostasis (GIHP) and the French Study Group on thrombosis and haemostasis (GFHT). In collaboration with the French Society for Anaesthesia and Intensive Care Medicine (SFAR). Anaesth. Crit. Care Pain Med. 2018, 37, 379–389. [Google Scholar]
- Cattaneo, M. P2Y12 antagonists. In Platelets, 4th ed.; Academic Press, Elsevier: Cambridge, MA, USA, 2019; pp. 937–956. [Google Scholar]
- Gross, L.; Aradi, D.; Sibbing, D. Pharmacology: Inhibitors of P2Y12. Platelets in Thrombotic and Non-Thrombotic Disorders; Springer: Cham, Switzerland, 2017; pp. 1253–1267. [Google Scholar]
- Wallentin, L. P2Y(12) inhibitors: Differences in properties and mechanisms of action and potential consequences for clinical use. Eur. Heart J. 2009, 30, 1964–1977. [Google Scholar] [CrossRef]
- Sharis, P.; Cannon, C.; Loscalzo, J. The Antiplatelet Effects of Ticlopidine and Clopidogrel. Ann. Intern. Med. 1998, 129, 394. [Google Scholar] [CrossRef]
- Gurbel, P.A.; Bliden, K.P.; Butler, K.; Tantry, U.S.; Gesheff, T.; Wei, C.; Teng, R.; Antonino, M.J.; Patil, S.B.; Karunakaran, A.; et al. Randomized Double-Blind Assessment of the ONSET and OFFSET of the Antiplatelet Effects of Ticagrelor Versus Clopidogrel in Patients With Stable Coronary Artery Disease. Circulation 2009, 120, 2577–2585. [Google Scholar] [CrossRef]
- Husted, S.E.; Storey, R.F.; Bliden, K.; Tantry, U.S.; Høimark, L.; Butler, K.; Wei, C.; Teng, R.; Gurbel, P.A. Pharmacokinetics and Pharmacodynamics of Ticagrelor in Patients with Stable Coronary Artery Disease. Clin. Pharm. 2012, 51, 397–409. [Google Scholar] [CrossRef]
- Husted, S.; Emanuelsson, H.; Heptinstall, S.; Sandset, P.; Wickens, M.; Peters, G. Pharmacodynamics, pharmacokinetics, and safety of the oral reversible P2Y12 antagonist AZD6140 with aspirin in patients with atherosclerosis: A double-blind comparison to clopidogrel with aspirin. Eur. Heart J. 2006, 27, 1038–1047. [Google Scholar] [CrossRef]
- Storey, R.F.; Husted, S.; Harrington, R.A.; Heptinstall, S.; Wilcox, R.G.; Peters, G.; Wickens, M.; Emanuelsson, H.; Gurbel, P.; Grande, P.; et al. Inhibition of platelet aggregation by AZD6140, a reversible oral P2Y12 receptor antagonist, compared with clopidogrel in patients with acute coronary syndromes. J. Am. Coll. Cardiol. 2007, 50, 1852–1856. [Google Scholar] [CrossRef]
- Dobesh, P.P.; Oestreich, J.H. Ticagrelor: Pharmacokinetics, Pharmacodynamics, Clinical Efficacy, and Safety. Pharm. J. Hum. Pharmacol. Drug Ther. 2014, 34, 1077–1090. [Google Scholar] [CrossRef] [PubMed]
- Franchi, F.; Rollini, F.; Muñiz-Lozano, A.; Cho, J.; Angiolillo, D.J. Cangrelor: A review on pharmacology and clinical trial development. Expert Rev. Cardiovasc. Ther. 2013, 11, 1279–1291. [Google Scholar] [CrossRef] [PubMed]
- Farid, N.A.; Kurihara, A.; Wrighton, S.A. Metabolism and Disposition of the Thienopyridine Antiplatelet Drugs Ticlopidine, Clopidogrel, and Prasugrel in Humans. J. Clin. Pharmacol. 2010, 50, 126–142. [Google Scholar] [CrossRef] [PubMed]
- Hulot, J.S.; Bura, A.; Villard, E.; Azizi, M.; Remones, V.; Goyenvalle, C.; Aiach, M.; Lechat, P.; Gaussem, P. Cytochrome P450 2C19 loss-of-function polymorphism is a major determinant of clopidogrel responsiveness in healthy subjects. Blood 2006, 108, 2244–2247. [Google Scholar] [CrossRef]
- Hagihara, K.; Kazui, M.; Kurihara, A.; Iwabuchi, H.; Ishikawa, M.; Kobayashi, H.; Tanaka, N.; Okazaki, O.; Farid, N.A.; Ikeda, T. Biotransformation of prasugrel, a novel thienopyridine antiplatelet agent, to the pharmacologically active metabolite. Drug Metab. Dispos. Biol. Fate Chem. 2010, 38, 898–904. [Google Scholar] [CrossRef]
- Wallentin, L.; Varenhorst, C.; James, S.; Erlinge, D.; Braun, O.; Jakubowski, J.; Sugidachi, A.; Winters, K.; Siegbahn, A. Prasugrel achieves greater and faster P2Y12receptor-mediated platelet inhibition than clopidogrel due to more efficient generation of its active metabolite in aspirin-treated patients with coronary artery disease. Eur. Heart J. 2007, 29, 21–30. [Google Scholar] [CrossRef]
- Sugidachi, A.; Ogawa, T.; Kurihara, A.; Hagihara, K.; Jakubowski, J.; Imoto; Niitsu, Y.; Asai, F. The greater in vivo antiplatelet effects of prasugrel as compared to clopidogrel reflect more efficient generation of its active metabolite with similar antiplatelet activity to that of clopidogrel’s active metabolite. J. Thromb. Haemost. 2007, 5, 1545–1551. [Google Scholar] [CrossRef]
- Hoffmann, K.; Lutz, D.; Straßburger, J.; Baqi, Y.; Müller, C.; von Kügelgen, I. Competitive mode and site of interaction of ticagrelor at the human platelet P2Y12 -receptor. J. Thromb. Haemost. 2014, 12, 1898–1905. [Google Scholar] [CrossRef]
- Giezen, V.J.; Nilsson, L.; Betsson, P.; Wissing, B.; Giordanetto, F.; Tomlinson, W.; Greasley, P. Ticagrelor binds to human P2Y 12 independently from ADP but antagonizes ADP-induced receptor signaling and platelet aggregation. J. Thromb. Haemost. 2009, 7, 1556–1565. [Google Scholar] [CrossRef]
- Teng, R.; Oliver, S.; Hayes, M.A.; Butler, K. Absorption, Distribution, Metabolism, and Excretion of Ticagrelor in Healthy Subjects. Drug Metab. Dispos. 2010, 38, 1514–1521. [Google Scholar] [CrossRef]
- Löffler, M.; Morote-Garcia, J.C.; Eltzschig, S.A.; Coe, I.R.; Eltzschig, H.K. Physiological Roles of Vascular Nucleoside Transporters. Arterioscler. Thromb. Vasc Biol. 2007, 27, 1004–1013. [Google Scholar] [CrossRef] [PubMed]
- Aungraheeta, R.; Conibear, A.; Butler, M.; Kelly, E.; Nylander, S.; Mumford, A.; Mundell, S.J. Inverse agonism at the P2Y12 receptor and ENT1 transporter blockade contribute to platelet inhibition by ticagrelor. Blood 2016, 128, 2717–2728. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, M.; Schulz, R.; Nylander, S. Adenosine-mediated effects of ticagrelor: Evidence and potential clinical relevance. J. Am. Coll. Cardiol. 2014, 63, 2503–2509. [Google Scholar] [CrossRef]
- Ow, K.; Parker, W.A.; Porter, M.M.; Hanson, J.; Judge, H.M.; Briffa, N.P.; Thomas, M.R.; Storey, R.F. Offset of ticagrelor prior to coronary artery bypass graft surgery for acute coronary syndromes: Effects on platelet function and cellular adenosine uptake. Platelets 2020, 1–7. [Google Scholar] [CrossRef]
- Ortega-Paz, L.; Brugaletta, S.; Ariotti, S.; Akkerhuis, M.K.; Karagiannis, A.; Windecker, S.; Valgimigli, M.; On behalf of the investigators. Adenosine and Ticagrelor Plasma Levels in Patients With and Without Ticagrelor-Related Dyspnea. Circulation 2018, 138, 646–648. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.; Maurel-Ribes, A.; Nauze, M.; N’Guyen, D.; Martinez, L.O.; Payrastre, B.; Sénard, J.M.; Galés, C.; Pons, V. Deciphering biased inverse agonism of cangrelor and ticagrelor at P2Y12 receptor. Cell. Mol. Life Sci. 2019, 76, 561–576. [Google Scholar] [CrossRef] [PubMed]
- Lancellotti, P.; Musumeci, L.; Jacques, N.; Servais, L.; Goffin, E.; Pirotte, B.; Oury, C. Antibacterial Activity of Ticagrelor in Conventional Antiplatelet Dosages Against Antibiotic-Resistant Gram-Positive Bacteria. JAMA Cardiol. 2019, 4, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Angiolillo, D.J.; Firstenberg, M.S.; Price, M.J.; Tummala, P.E.; Hutyra, M.; Welsby, I.J.; Voeltz, M.D.; Chandna, H.; Ramaiah, C.; Brtko, M.; et al. Bridging antiplatelet therapy with cangrelor in patients undergoing cardiac surgery: A randomized controlled trial. JAMA J. Am. Med Assoc. 2012, 307, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Steg, P.; Bhatt, D.L.; Hamm, C.W.; Stone, G.W.; Gibson, M.C.; Mahaffey, K.W.; Leonardi, S.; Liu, T.; Skerjanec, S.; Day, J.R.; et al. Effect of cangrelor on periprocedural outcomes in percutaneous coronary interventions: A pooled analysis of patient-level data. Lancet Lond Engl. 2013, 382, 1981–1992. [Google Scholar] [CrossRef]
- Ma, L.; Dorling, A. The roles of thrombin and protease-activated receptors in inflammation. Semin. Immunopathol. 2012, 34, 63–72. [Google Scholar] [CrossRef]
- Levi, M.; van der Poll, T.; Büller, H.R. Bidirectional Relation between Inflammation and Coagulation. Circulation 2004, 109, 2698–2704. [Google Scholar] [CrossRef] [PubMed]
- Naldini, A.; Carney, D.H.; Pucci, A.; Pasquali, A.; Carraro, F. Thrombin regulates the expression of proangiogenic cytokines via proteolytic activation of protease-activated receptor-1. Gen. Pharmacol. Vasc. Syst. 2000, 35, 255–259. [Google Scholar] [CrossRef]
- Naldini, A.; Carney, D.H.; Bocci, V.; Klimpel, K.D.; Asuncion, M.; Soares, L.E.; Klimpel, G.R. Thrombin Enhances T Cell Proliferative Responses and Cytokine Production. Cell. Immunol. 1993, 147, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.; Choi, Y.; DeGroot, E.; Samuels, I.; Creasey, A.; Aarden, L. Potential mechanisms for a proinflammatory vascular cytokine response to coagulation activation. J. Immunol. Baltim. Md. 1950 1998, 160, 5130–5135. [Google Scholar]
- Strande, J.L.; Phillips, S.A. Thrombin increases inflammatory cytokine and angiogenic growth factor secretion in human adipose cells in vitro. J. Inflamm. 2009, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Shankar, R.; de la Motte, C.; Poptic, E.; DiCorleto, P. Thrombin receptor-activating peptides differentially stimulate platelet-derived growth factor production, monocytic cell adhesion, and E-selectin expression in human umbilical vein endothelial cells. J. Biological. Chem. 1994, 269, 13936–13941. [Google Scholar]
- Iba, T.; Levy, J.H. Inflammation and thrombosis: Roles of neutrophils, platelets and endothelial cells and their interactions in thrombus formation during sepsis. J. Thromb. Haemost. 2018, 16, 231–241. [Google Scholar] [CrossRef]
- Norgard, N.B.; Hann, C.L.; Dale, G.L. Cangrelor Attenuates Coated-Platelet Formation. Clin. Appl. Thromb. Hemost. 2008, 15, 177–182. [Google Scholar] [CrossRef]
- Norgard, N.B.; Saya, S.; Hann, C.; Hennebry, T.; Schechter, E.; Dale, G. Clopidogrel attenuates coated-platelet production in patients undergoing elective coronary catheterization. J. Cardiovasc. Pharm. 2008, 52, 536–539. [Google Scholar] [CrossRef]
- Hérault, P.J.; Dol, F.; Gaich, C.; Bernat, A.; Herbert, M.J. Effect of Clopidogrel on Thrombin Generation in Platelet-Rich Plasma in the Rat. Thromb. Haemost. 1999, 81, 957–960. [Google Scholar] [CrossRef]
- Wang, X.; Deng, H.; Li, T.; Miao, S.; Xiao, Z.; Liu, M.; Liu, K.; Xiao, X. Clopidogrel reduces lipopolysaccharide-induced inflammation and neutrophil-platelet aggregates in an experimental endotoxemic model. J. Biochem. Mol. Toxicol. 2019, 33, 22279. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2563–2582. [Google Scholar] [CrossRef]
- Bester, J.; Pretorius, E. Effects of IL-1β, IL-6 and IL-8 on erythrocytes, platelets and clot viscoelasticity. Sci Rep. UK 2016, 6, 32188. [Google Scholar] [CrossRef] [PubMed]
- Lumadue, J.A.; Lanzkron, S.M.; Kennedy, S.D.; Kuhl, D.T.; Kickler, T.S. Cytokine Induction of Platelet Activation. Am. J. Clin. Pathol. 1996, 106, 795–798. [Google Scholar] [CrossRef]
- Assinger, A.; Schrottmaier, W.C.; Salzmann, M.; Rayes, J. Platelets in Sepsis: An Update on Experimental Models and Clinical Data. Front. Immunol. 2019, 10, 1687. [Google Scholar] [CrossRef] [PubMed]
- Slaba, I.; Kubes, P. Platelets in Thrombotic and Non-Thrombotic Disorders; Springer: Cham, Switzerland, 2017; pp. 489–512. [Google Scholar]
- Sanguigni, V.; Ferro, D.; Pignatelli, P.; Ben, M.; Nadia, T.; Saliola, M.; Sorge, R.; Violi, F. CD40 ligand enhances monocyte tissue factor expression and thrombin generation via oxidative stress in patients with hypercholesterolemia. J. Am. Coll. Cardiol. 2005, 45, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Liverani, E.; Kilpatrick, L.E.; Tsygankov, A.Y.; Kunapuli, S.P. The role of P2Y₁₂ receptor and activated platelets during inflammation. Curr. Drug Targets 2014, 15, 720–728. [Google Scholar] [CrossRef]
- Sexton, T.R.; Zhang, G.; Macaulay, T.E.; Callahan, L.A.; Charnigo, R.; Vsevolozhskaya, O.A.; Li, Z.; Smyth, S. Ticagrelor Reduces Thromboinflammatory Markers in Patients With Pneumonia. JACC. Basic Transl. Sci. 2018, 3, 435–449. [Google Scholar] [CrossRef]
- Hagiwara, S.; Iwasaka, H.; Hasegawa, A.; Oyama, M.; Imatomi, R.; Uchida, T.; Noguchi, T. Adenosine Diphosphate Receptor Antagonist Clopidogrel Sulfate Attenuates LPS-Induced Systemic Inflammation in a Rat Model. Shock (Augusta Ga.) 2011, 35, 289–292. [Google Scholar] [CrossRef]
- Thomas, M.R.; Outteridge, S.N.; Ajjan, R.A.; Phoenix, F.; Sangha, G.K.; Faulkner, R.E.; Ecob, R.; Judge, H.M.; Khan, H.; West, L.E.; et al. Platelet P2Y12 Inhibitors Reduce Systemic Inflammation and Its Prothrombotic Effects in an Experimental Human Model. Arterioscler. Thromb. Vasc Biol. 2015, 35, 2562–2570. [Google Scholar] [CrossRef]
- Thomas, M.R.; Storey, R.F. Effect of P2Y12 inhibitors on inflammation and immunity. Thromb. Haemost. 2015, 114, 490–497. [Google Scholar] [PubMed]
- Rossaint, J.; Margraf, A.; Zarbock, A. Role of Platelets in Leukocyte Recruitment and Resolution of Inflammation. Front. Immunol. 2018, 9, 2712. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Smyth, S.S. Platelets, 4th ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 295–310. [Google Scholar]
- Evangelista, V.; Totani, L.; Manfredi, A.A.; Maugeri, N. Platelets in Thrombotic and Non-Thrombotic Disorders; Springer: Berlin/Heidelberg, Germany, 2017; pp. 407–433. [Google Scholar]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Elaskalani, O.; Razak, N.; Metharom, P. Neutrophil extracellular traps induce aggregation of washed human platelets independently of extracellular DNA and histones. Cell Commun. Signal. 2018, 16, 24. [Google Scholar] [CrossRef]
- Etulain, J.; Martinod, K.; Wong, S.; Cifuni, S.M.; Schattner, M.; Wagner, D.D. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 2015, 126, 242–246. [Google Scholar] [CrossRef]
- Carestia, A.; Kaufman, T.; Schattner, M. Platelets: New Bricks in the Building of Neutrophil Extracellular Traps. Front. Immunol. 2016, 7, 271. [Google Scholar] [CrossRef]
- Zucoloto, A.Z.; Jenne, C.N. Platelet-Neutrophil Interplay: Insights Into Neutrophil Extracellular Trap (NET)-Driven Coagulation in Infection. Front. Cardiovasc. Med. 2019, 6, 85. [Google Scholar] [CrossRef]
- Kazzaz, N.M.; Sule, G.; Knight, J.S. Intercellular Interactions as Regulators of NETosis. Front. Immunol. 2016, 7, 453. [Google Scholar] [CrossRef]
- Rahman, M.; Gustafsson, D.; Wang, Y.; Thorlacius, H.; Braun, O. Ticagrelor reduces neutrophil recruitment and lung damage in abdominal sepsis. Platelets 2014, 25, 257–263. [Google Scholar] [CrossRef]
- Evangelista, V.; Manarini, S.; Dell’Elba, G.; Martelli, N.; Napoleone, E.; Santo, A.; Savi, P.; Lorenzet, R. Clopidogrel inhibits platelet-leukocyte adhesion and plateletdependent leukocyte activation. Thromb. Haemost. 2005, 94, 568–577. [Google Scholar]
- Judge, H.M.; Buckland, R.J.; Sugidachi, A.; Jakubowski, J.A.; Storey, R.F. The active metabolite of prasugrel effectively blocks the platelet P2Y12 receptor and inhibits procoagulant and pro-inflammatory platelet responses. Platelets 2008, 19, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Storey, R.; Judge, H.; Wilcox, R.; Heptinstall, S. Inhibition of ADP-induced P-selectin Expression and Platelet-Leukocyte Conjugate Formation by Clopidogrel and the P2Y12 Receptor Antagonist AR-C69931MX but not Aspirin. Thromb. Haemost. 2002, 88, 488–494. [Google Scholar] [PubMed]
- Klinkhardt, U. Clopidogrel but not aspirin reduces P-selectin expression and formation of platelet-leukocyte aggregates in patients with atherosclerotic vascular disease. Clin. Pharmacol. Ther. 2003, 73, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Théroux, P. Clopidogrel inhibits platelet-leukocyte interactions and thrombin receptor agonist peptide-induced platelet activation in patients with an acute coronary syndrome. J. Am. Coll. Cardiol. 2004, 43, 1982–1988. [Google Scholar] [CrossRef]
- Vigano, S.; Alatzoglou, D.; Irving, M.; Ménétrier-Caux, C.; Caux, C.; Romero, P.; Coukos, G. Targeting Adenosine in Cancer Immunotherapy to Enhance T-Cell Function. Front. Immunol. 2019, 10, 925. [Google Scholar] [CrossRef]
- Wolska, N.; Rozalski, M. Blood Platelet Adenosine Receptors as Potential Targets for Anti-Platelet Therapy. Int. J. Mol. Sci. 2019, 20, 5475. [Google Scholar] [CrossRef]
- Boncler, M.; Wzorek, J.; Wolska, N.; Polak, D.; Watala, C.; Rozalski, M. Adenosine receptor agonists deepen the inhibition of platelet aggregation by P2Y12 antagonists. Vasc. Pharmacol. 2018, 113, 47–56. [Google Scholar] [CrossRef]
- Winning, J.; Reichel, J.; Eisenhut, Y.; Hamacher, J.; Kohl, M.; Deigner, H.; Claus, R.A.; Bauer, M.; Lösche, W. Anti-platelet drugs and outcome in severe infection: Clinical impact and underlying mechanisms. Platelets 2009, 20, 50–57. [Google Scholar] [CrossRef]
- Storey, R.F.; James, S.K.; Siegbahn, A.; Varenhorst, C.; Held, C.; Ycas, J.; Husted, S.E.; Cannon, C.P.; Becker, R.C.; Steg, P.; et al. Lower mortality following pulmonary adverse events and sepsis with ticagrelor compared to clopidogrel in the PLATO study. Platelets 2013, 25, 517–525. [Google Scholar] [CrossRef]
- Tsai, M.J.; Ou, S.M.; Shih, C.J.; Chao, P.; Wang, L.F.; Shih, Y.N.; Li, S.Y.; Kuo, S.C.; Hsu, Y.T.; Chen, Y.T. Association of prior antiplatelet agents with mortality in sepsis patients: A nationwide population-based cohort study. Intensive Care Med. 2015, 41, 806–813. [Google Scholar] [CrossRef]
- Laidlaw, T.M.; Cahill, K.N.; Cardet, J.; Murphy, K.; Cui, J.; Dioneda, B.; Kothari, P.; Raby, B.A.; Israel, E.; Boyce, J. A trial of P2Y12 receptor inhibition with prasugrel identifies a potentially distinct endotype of patients with aspirin-exacerbated respiratory disease. J. Allergy Clin. Immun. 2018, 143, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Lussana, F.; Marco, D.F.; Terraneo, S.; Parati, M.; Razzari, C.; Scavone, M.; Femia, E.; Moro, A.; Centanni, S.; Cattaneo, M. Effect of prasugrel in patients with asthma: Results of PRINA, a randomized, double-blind, placebo-controlled, cross-over study. J. Thromb. Haemost. 2015, 13, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Raposeiras-Roubin, S.; Abu-Assi, E.; Muñoz-Pousa, I.; Cespón-Fernández, M.; Cobas-Paz, R.; Caneiro-Queija, B.; López-Rodríguez, E.; Pérez-Casares, L.; Jamhour-Chelh, K.; Castiñeira-Busto, M.; et al. Risk of cancer after an acute coronary syndrome according to the type of P2Y12 inhibitor. Thromb. Res. 2019, 174, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Leader, A.; Zelikson-Saporta, R.; Pereg, D.; Spectre, G.; Rozovski, U.; Raanani, P.; Hermoni, D.; Lishner, M. The Effect of Combined Aspirin and Clopidogrel Treatment on Cancer Incidence. Am. J. Med. 2017, 130, 826–832. [Google Scholar] [CrossRef]
- Hicks, B.M.; Murray, L.J.; Hughes, C.; Cardwell, C.R. Clopidogrel use and cancer-specific mortality: A population-based cohort study of colorectal, breast and prostate cancer patients. Pharmacoepidemiol. Drug Saf. 2015, 24, 830–840. [Google Scholar] [CrossRef]
- Elmariah, S.; Doros, G.; Benavente, O.R.; Bhatt, D.L.; Connolly, S.J.; Yusuf, S.; Steinhubl, S.R.; Liu, Y.; Hsieh, W.H.; Yeh, R.W.; et al. Impact of Clopidogrel Therapy on Mortality and Cancer in Patients With Cardiovascular and Cerebrovascular Disease: A Patient-Level Meta-Analysis. Circulation. Cardiovasc. Interv. 2018, 11, 005795. [Google Scholar] [CrossRef]
- Rodríguez-Miguel, A.; García-Rodríguez, L.A.; Gil, M.; Montoya, H.; Rodríguez-Martín, S.; de Abajo, F.J. Clopidogrel and Low-Dose Aspirin, Alone or Together, Reduce Risk of Colorectal Cancer. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2019, 17, 2024–2033. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Moldawer, L.L.; Opal, S.M.; Reinhart, K.; Turnbull, I.R.; Vincent, J.L. Sepsis and septic shock. Nat. Rev. Dis. Primers 2016, 2, 16045. [Google Scholar] [CrossRef]
- van der Poll, T.; van de Veerdonk, F.L.; Scicluna, B.P.; Netea, M.G. The immunopathology of sepsis and potential therapeutic targets. Nat. Rev. Immunol. 2017, 17, 407–420. [Google Scholar] [CrossRef]
- Vardon-Bounes, F.; Ruiz, S.; Gratacap, M.P.; Garcia, C.; Payrastre, B.; Minville, V. Platelets Are Critical Key Players in Sepsis. Int. J. Mol. Sci. 2019, 20, 3494. [Google Scholar] [CrossRef]
- Wang, Y.; Ouyang, Y.; Liu, B.; Ma, X.; Ding, R. Platelet activation and antiplatelet therapy in sepsis: A narrative review. Thromb. Res. 2018, 166, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute respiratory distress syndrome. Nat. Rev. Dis. Primers 2019, 5, 18. [Google Scholar] [CrossRef] [PubMed]
- Middleton, E.A.; Rondina, M.T.; Schwertz, H.; Zimmerman, G.A. Amicus or Adversary Revisited: Platelets in Acute Lung Injury and Acute Respiratory Distress Syndrome. Am. J. Resp. Cell Mol. 2018, 59, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Bozza, F.A.; ah, A.; Weyrich, A.S.; Zimmerman, G.A. Amicus or Adversary. Am. J. Resp. Cell Mol. 2009, 40, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Wallentin, L.; Becker, R.C.; Budaj, A.; Cannon, C.P.; Emanuelsson, H.; Held, C.; Horrow, J.; Husted, S.; James, S.; Katus, H.; et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N. Engl. J. Med. 2009, 361, 1045–1057. [Google Scholar] [CrossRef]
- Takeda, T.; Morita, H.; Saito, H.; Matsumoto, K.; Matsuda, A. Recent advances in understanding the roles of blood platelets in the pathogenesis of allergic inflammation and bronchial asthma. Allergol. Int. 2017, 67, 326–333. [Google Scholar] [CrossRef]
- Idzko, M.; Pitchford, S.; Page, C. Role of platelets in allergic airway inflammation. J. Allergy Clin. Immun. 2015, 135, 1416–1423. [Google Scholar] [CrossRef]
- Kowal, K.; Pampuch, A.; Kowal-Bielecka, O.; DuBuske, L.; Bodzenta-Łukaszyk, A. Platelet activation in allergic asthma patients during allergen challenge with Dermatophagoides pteronyssinus. Clin. Exp. Allergy 2006, 36, 426–432. [Google Scholar] [CrossRef]
- Gawaz, M.; Borst, O. 26 the Role of Platelets in Atherothrombosis. Platelets 2019, 459–467. [Google Scholar]
- Lievens, D.; von Hundelshausen, P. Platelets in atherosclerosis. Thromb. Haemost. 2011, 106, 827–838. [Google Scholar]
- Lebas, H.; Yahiaoui, K.; Martos, R.; Boulaftali, Y. Platelets Are at the Nexus of Vascular Diseases. Front. Cardiovasc. Med. 2019, 6, 132. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, Y.; Zhang, L.; Luo, X.; Li, J.; Chen, X.; Niu, H.; Wang, K.; Sun, Y.; Wang, X.; et al. Roles of Purinergic Receptor P2Y, G Protein–Coupled 12 in the Development of Atherosclerosis in Apolipoprotein E–Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Heim, C.; Gebhardt, J.; Ramsperger-Gleixner, M.; Jacobi, J.; Weyand, M.; Ensminger, S.M. Clopidogrel significantly lowers the development of atherosclerosis in ApoE-deficient mice in vivo. Heart Vessel. 2015, 31, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Afek, A.; Kogan, E.; Maysel-Auslender, S.; Mor, A.; Regev, E.; Rubinstein, A.; Keren, G.; George, J. Clopidogrel attenuates atheroma formation and induces a stable plaque phenotype in apolipoprotein E knockout mice. Microvasc. Res. 2009, 77, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Li, J.; Liang, X.; Zhang, S.; Liu, T.; Liu, J.; Arif, M.; Li, G. Ticagrelor suppresses oxidized low-density lipoprotein-induced endothelial cell apoptosis and alleviates atherosclerosis in ApoE-/- mice via downregulation of PCSK9. Mol. Med. Rep. 2018, 19, 1453–1462. [Google Scholar] [CrossRef] [PubMed]
- Ganbaatar, B.; Fukuda, D.; Salim, H.; Nishimoto, S.; Tanaka, K.; Higashikuni, Y.; Hirata, Y.; Yagi, S.; Soeki, T.; Sata, M. Ticagrelor, a P2Y12 antagonist, attenuates vascular dysfunction and inhibits atherogenesis in apolipoprotein-E-deficient mice. Atherosclerosis 2018, 275, 124–132. [Google Scholar] [CrossRef] [PubMed]
- West, L.E.; Steiner, T.; Judge, H.M.; Francis, S.E.; Storey, R.F. Vessel wall, not platelet, P2Y12 potentiates early atherogenesis. Cardiovasc. Res. 2014, 102, 429–435. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Haemmerle, M.; Stone, R.L.; Menter, D.G.; Afshar-Kharghan, V.; Sood, A.K. The Platelet Lifeline to Cancer: Challenges and Opportunities. Cancer Cell 2018, 33, 965–983. [Google Scholar] [CrossRef]
- Cho, M.; Noh, K.; Haemmerle, M.; Li, D.; Park, H.; Hu, Q.; Hisamatsu, T.; Mitamura, T.; Mak, S.; Kunapuli, S.; et al. Role of ADP receptors on platelets in the growth of ovarian cancer. Blood 2017, 130, 1235–1242. [Google Scholar] [CrossRef]
- Gareau, A.J.; Brien, C.; Gebremeskel, S.; Liwski, R.S.; Johnston, B.; Bezuhly, M. Ticagrelor inhibits platelet-tumor cell interactions and metastasis in human and murine breast cancer. Clin. Exp. Metastasis 2018, 35, 25–35. [Google Scholar] [CrossRef]
- Gebremeskel, S.; LeVatte, T.; Liwski, R.S.; Johnston, B.; Bezuhly, M. The reversible P2Y12 inhibitor ticagrelor inhibits metastasis and improves survival in mouse models of cancer. Int. J. Cancer 2015, 136, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, Y.; Li, D.; Zhang, L.; Wang, K.; Zuo, Y.; Gartner, K.T.; Liu, J. Platelet P2Y12 is involved in murine pulmonary metastasis. PLoS ONE 2013, 8, e80780. [Google Scholar] [CrossRef] [PubMed]
- Geranpayehvaghei, M.; Shi, Q.; Zhao, B.; Li, S.; Xu, J.; Taleb, M.; Qin, H.; Zhang, Y.; Khajeh, K.; Nie, G. Targeting Delivery of Platelets Inhibitor to Prevent Tumor Metastasis. Bioconjug. Chem. 2019, 30, 2349–2357. [Google Scholar] [CrossRef] [PubMed]
- Algra, A.M.; Rothwell, P.M. Effects of regular aspirin on long-term cancer incidence and metastasis: A systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol. 2012, 13, 518–527. [Google Scholar] [CrossRef]
- CAPRIE Steering Committee. A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). Lancet 1996, 348, 1329–1339. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Fox, K.A.; Hacke, W.; Berger, P.B.; Black, H.R.; Boden, W.E.; Cacoub, P.; Cohen, E.A.; Creager, M.A.; Easton, D.J.; et al. Clopidogrel and Aspirin versus Aspirin Alone for the Prevention of Atherothrombotic Events. N. Engl. J. Med. 2006, 354, 1706–1717. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Braunwald, E.; McCabe, C.H.; Montalescot, G.; Ruzyllo, W.; Gottlieb, S.; Neumann, F.J.; Ardissino, D.; Servi, S.; Murphy, S.A.; et al. Prasugrel versus Clopidogrel in Patients with Acute Coronary Syndromes. N. Engl. J. Med. 2007, 357, 2001–2015. [Google Scholar] [CrossRef]
- Roe, M.T.; Armstrong, P.W.; Fox, K.A.; White, H.D.; Prabhakaran, D.; Goodman, S.G.; Cornel, J.H.; Bhatt, D.L.; Clemmensen, P.; Martinez, F.; et al. Prasugrel versus Clopidogrel for Acute Coronary Syndromes without Revascularization. N. Engl J. Med. 2012, 367, 1297–1309. [Google Scholar] [CrossRef]
- Mauri, L.; Kereiakes, D.J.; Yeh, R.W.; Driscoll-Shempp, P.; Cutlip, D.E.; Steg, G.P.; Normand, S.L.T.; Braunwald, E.; Wiviott, S.D.; Cohen, D.J.; et al. Twelve or 30 months of dual antiplatelet therapy after drug-eluting stents. N. Engl. J. Med. 2014, 371, 2155–2166. [Google Scholar] [CrossRef]
- Bonaca, M.P.; Bhatt, D.L.; Cohen, M.; Steg, P.; Storey, R.F.; Jensen, E.C.; Magnani, G.; Bansilal, S.; Fish, P.M.; Im, K.; et al. Long-Term Use of Ticagrelor in Patients with Prior Myocardial Infarction. N. Engl. J. Med. 2015, 372, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Serebruany, V.L. Ticagrelor shift from PLATO to PEGASUS: Vanished mortality benefit, excess cancer deaths, massive discontinuations, and overshooting target events. Int. J. Cardiol. 2015, 201, 508–512. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. U.S. FDA Review Finds Long-Term Treatment with Blood- Thinning Medicine Plavix (Clopidogrel) Does not Change Risk of Death; FDA: Silver Spring, MD, USA, 2014.
- Serebruany, V.; Kim, M.; Thevathasan, C.; Marciniak, T. Assessing Cancer Signal during Oral Antiplatelet Therapy in the Food and Drug Administration Adverse Event Reporting System: Mission Impossible. TH Open Companion J. Thromb. Haemost. 2018, 2, 28–32. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kotronias, R.; Kwok, C.; Wong, C.; Kinnaird, T.; Zaman, A.; Mamas, M.A. Cancer Event Rate and Mortality with Thienopyridines: A Systematic Review and Meta-Analysis. Drug Saf. 2016, 40, 229–240. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug | Ticlopidine | Clopidogrel | Prasugrel | Ticagrelor | Cangrelor |
|---|---|---|---|---|---|
| Target | P2Y12 | P2Y12 | P2Y12 | P2Y12 ENT-1 | P2Y12 |
| P2Y12 receptor binding | Irreversible | Irreversible | Irreversible | Reversible | Reversible |
| Route of administration | Oral | Oral | Oral | Oral | Intravenous |
| Metabolism | Prodrug CYP450 | Prodrug Esterase CYP450 | Prodrug Intestinal Esterase CYP450 | Direct-Acting and CYP450 | Direct-Acting Dephosphorylation |
| Time to maximum IPA 1 | 3–4 days | 4–5 h | 2–4h | 2–4 h | 2 min |
| Steady-state IPA 2 | 20–30% | 33–64% | 43–73% | 82–95% | >80% |
| Offset of action 3 | 11–13 days | 5–7 days | 7–10 days | 3–5 days | 30–60 min |
| Study | Type of Study | Condition | Antiplatelet Drugs Evaluated | Effects of P2Y12 Antagonists |
|---|---|---|---|---|
| Winning et al. [135] | Observational 224 patients | CAP | ASA and thienopyridines | Lower use of ICU and shorter stay in hospital (thienopyridines plus ASA or thienopyridines alone |
| Storey et al. [136] | Observational post hoc analysis18,421 patients | ACS | Ticagrelor vs clopidogrel | Lower mortality risk following pulmonary events and sepsis |
| Tsai et al. [137] | Observational 683,421 patients | Sepsis | ASA and thienopyridines | Lower risk of mortality |
| XANTHIPPE Sexton et al. [113] | RCT 60 patients | Pneumonia (CAP, HAP) | Ticagrelor vs placebo | Reduced PLAs and IL-6 levels improved. Decreased supplemental oxygen requirements |
| Laidlaw et al. [138] | Crossover RCT 40 patients | AERD | Prasugrel vs placebo | No effect on clinical or inflammatory parameters |
| PRINA Lussana et al. [139] | Crossover RCT 26 patients | Chronic asthma | Prasugrel vs placebo | Decreased airway hyperresponsiveness |
| Raposeiras-Roubin et al. [140] | Observational 4229 patients | ACS | Ticagrelor, Clopidogrel and Prasugrel | Lower cancer risk ticagrelor vs clopidogrelNS clopidogrel vs prasugrel |
| Leader et al. [141] | Observational 3479 patients | ACS | ASA and clopidogrel | Lower cancer risk (clopidogrel plus ASA or clopidogrel alone) |
| Hicks et al. [142] | Observational 41,403 patients | Cancer (breast, colorectal, prostate) | Clopidogrel | No increase in cancer risk |
| Elmariah et al. [143] | Meta-analysis 48,000 patients | Cardiovascular and cerebrovascular disease | ASA and clopidogrel | No increase in cancer risk |
| Rodriguez-Miguel et al. [144] | Observational 75,491 patients | CRC | ASA and clopidogrel | Lower cancer risk (clopidogrel plus ASA or clopidogrel alone) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mansour, A.; Bachelot-Loza, C.; Nesseler, N.; Gaussem, P.; Gouin-Thibault, I. P2Y12 Inhibition beyond Thrombosis: Effects on Inflammation. Int. J. Mol. Sci. 2020, 21, 1391. https://doi.org/10.3390/ijms21041391
Mansour A, Bachelot-Loza C, Nesseler N, Gaussem P, Gouin-Thibault I. P2Y12 Inhibition beyond Thrombosis: Effects on Inflammation. International Journal of Molecular Sciences. 2020; 21(4):1391. https://doi.org/10.3390/ijms21041391
Chicago/Turabian StyleMansour, Alexandre, Christilla Bachelot-Loza, Nicolas Nesseler, Pascale Gaussem, and Isabelle Gouin-Thibault. 2020. "P2Y12 Inhibition beyond Thrombosis: Effects on Inflammation" International Journal of Molecular Sciences 21, no. 4: 1391. https://doi.org/10.3390/ijms21041391
APA StyleMansour, A., Bachelot-Loza, C., Nesseler, N., Gaussem, P., & Gouin-Thibault, I. (2020). P2Y12 Inhibition beyond Thrombosis: Effects on Inflammation. International Journal of Molecular Sciences, 21(4), 1391. https://doi.org/10.3390/ijms21041391