Hypoxia-Dependent Expression of TG2 Isoforms in Neuroblastoma Cells as Consequence of Different MYCN Amplification Status

 ,
,  , , ,
, , ,
Abstract
1. Introduction
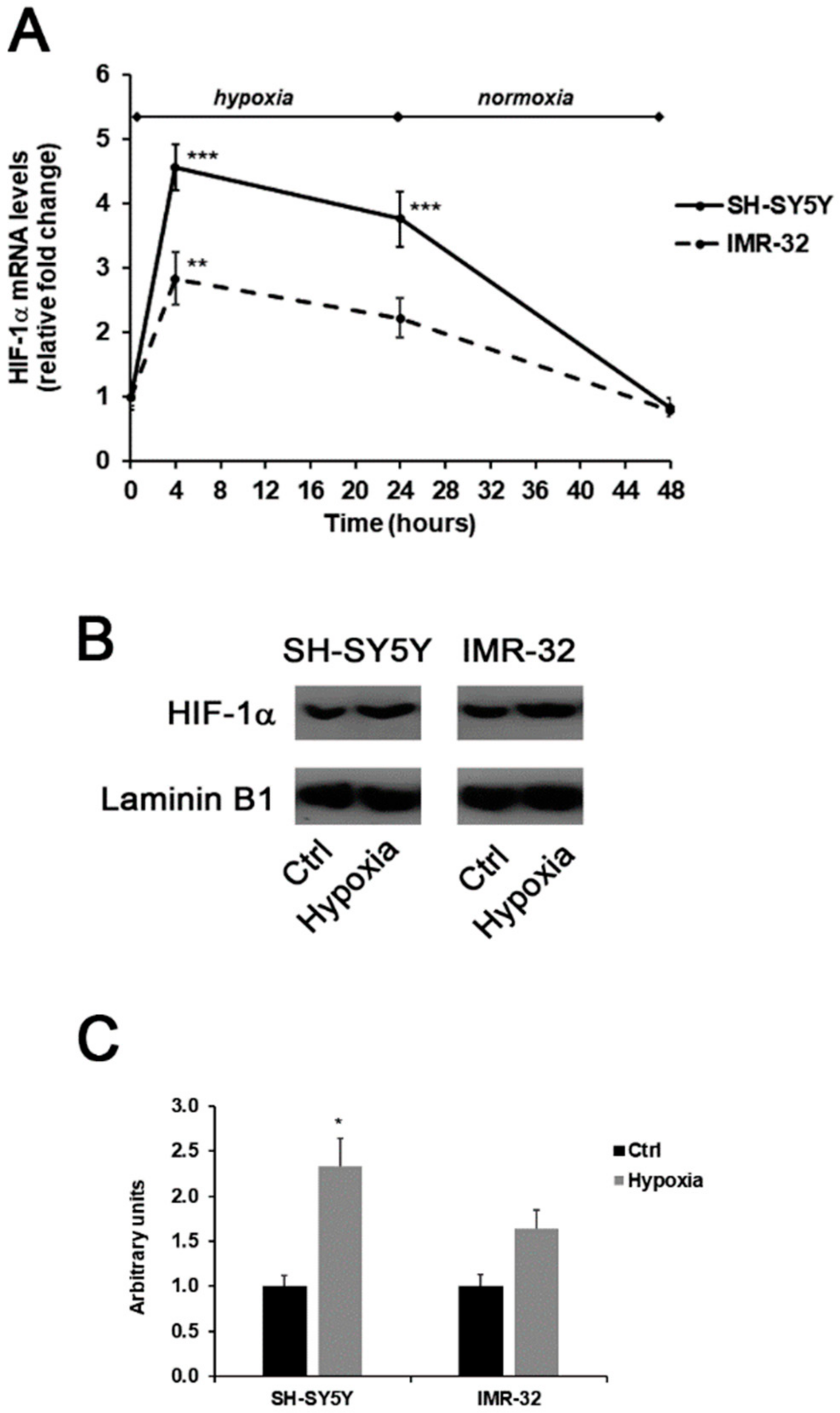
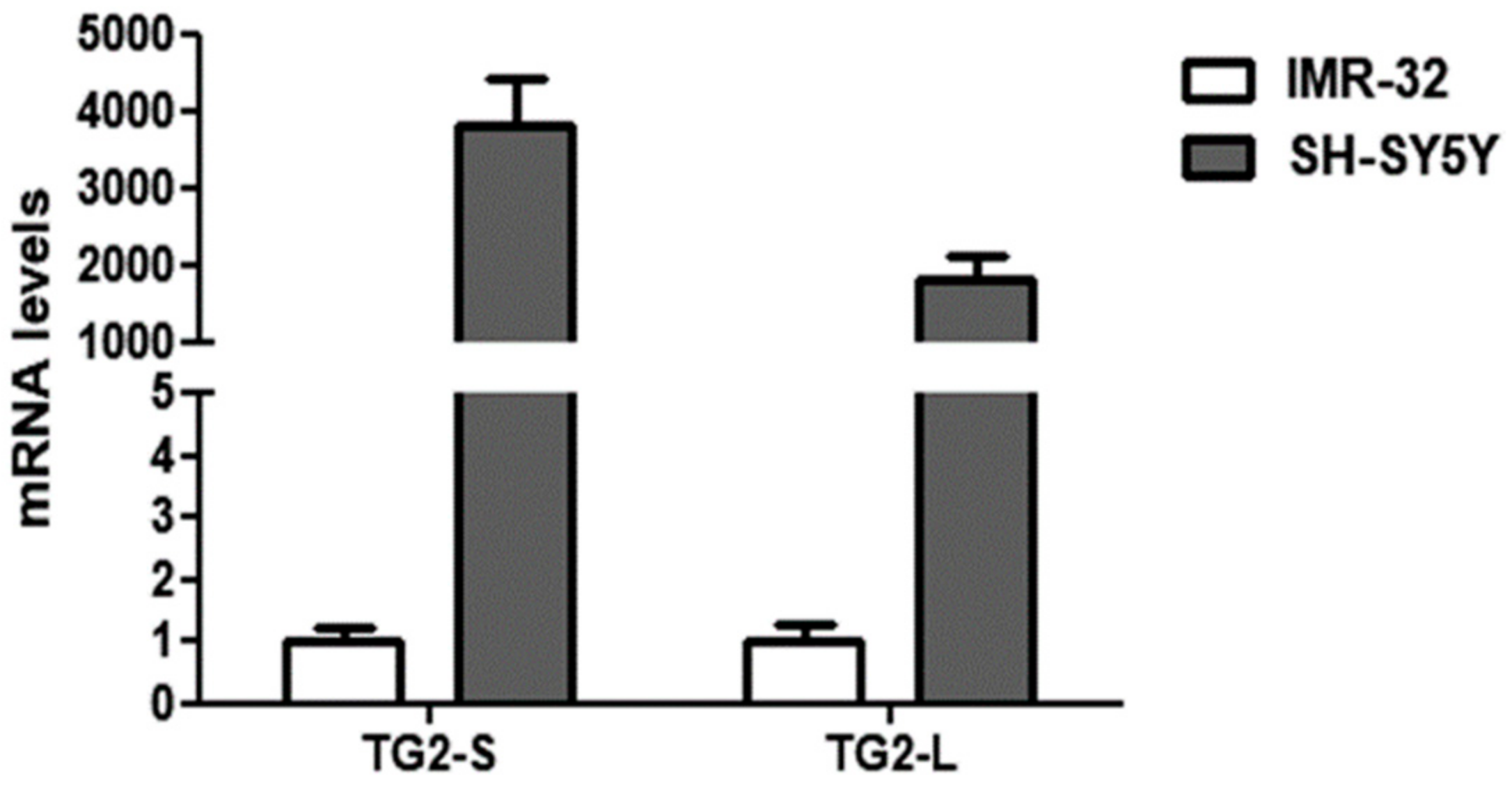
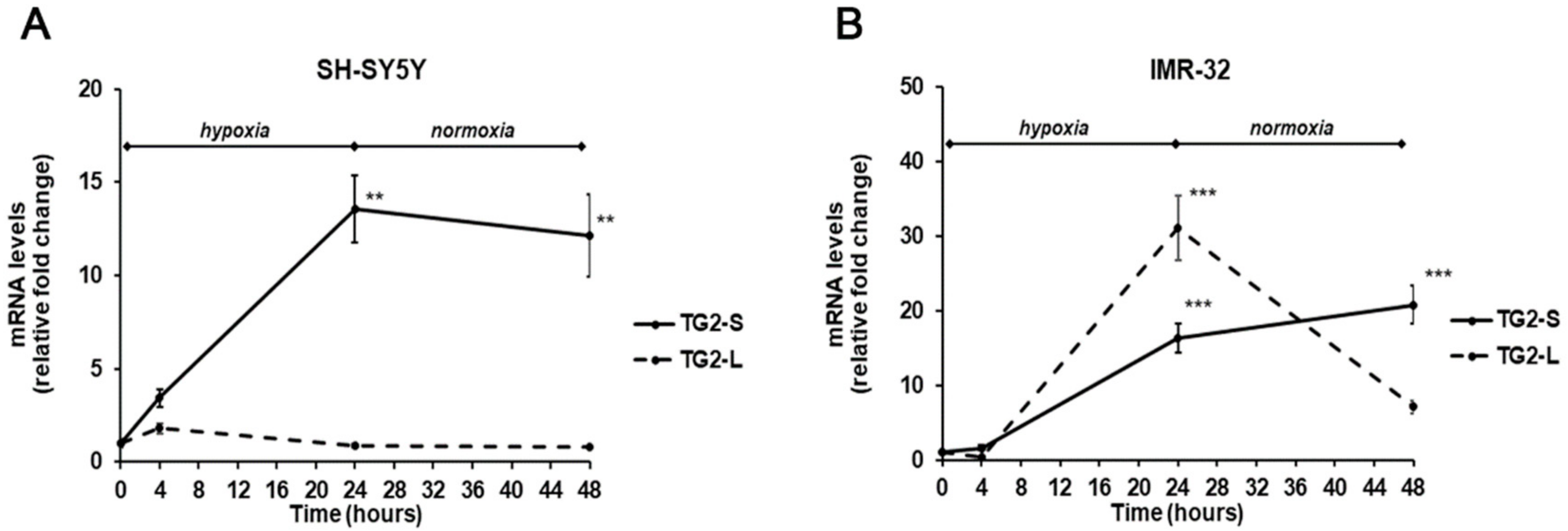
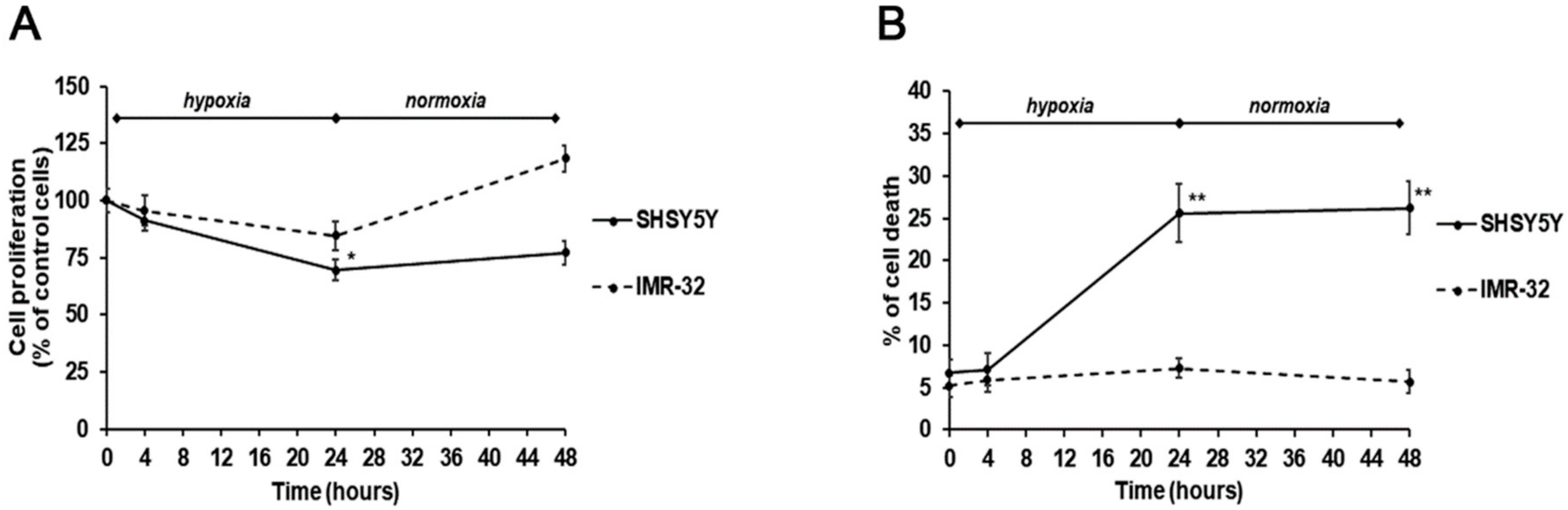
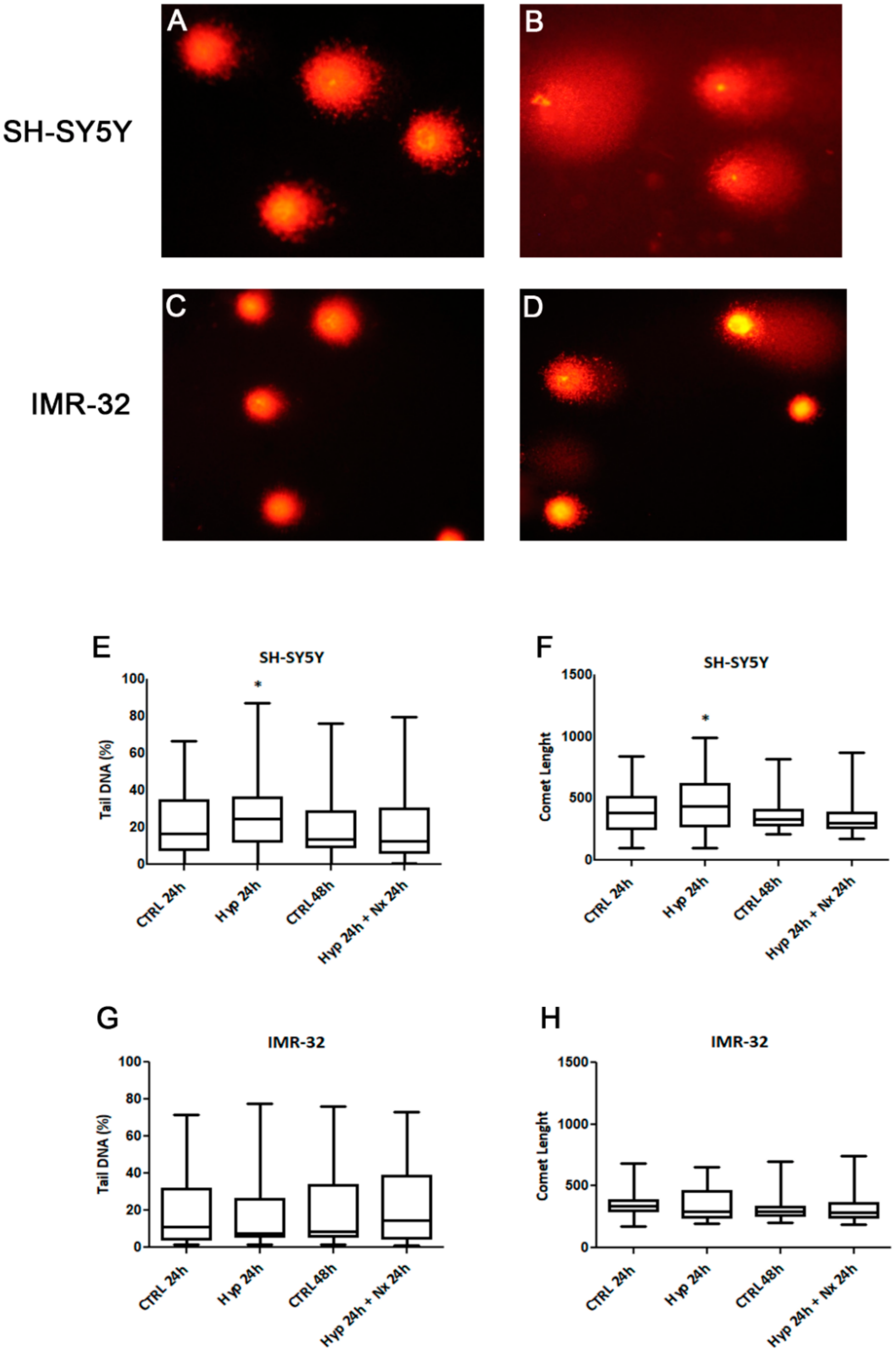
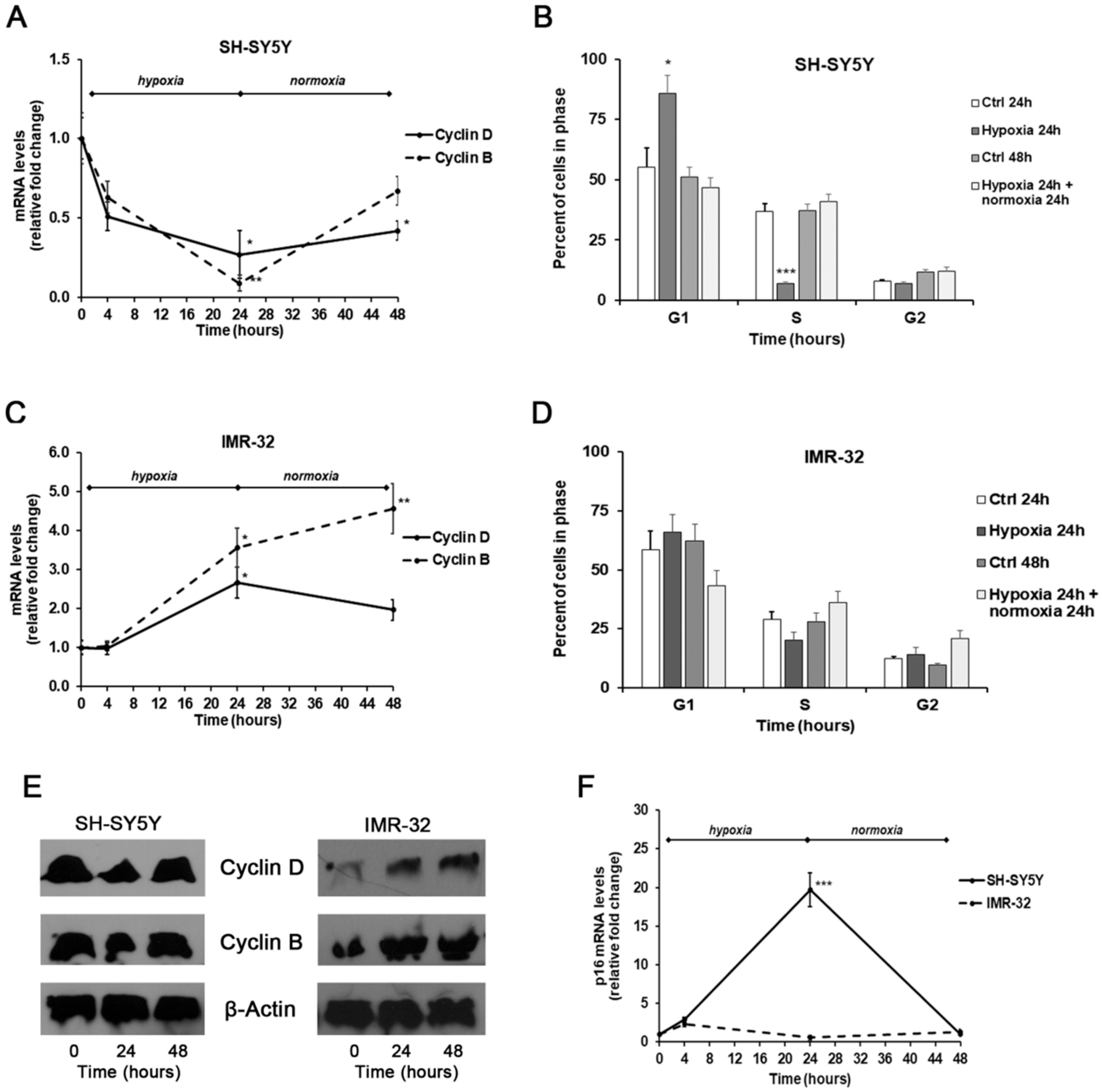
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Cell Culture and Treatment
3.3. Proliferation Assay and Cytotoxicity Study
3.4. Analysis of Gene Expression by Real-Time PCR
3.5. Western Blotting
3.6. Comet Assay
3.7. Cell Cycle Analysis by Flow Cytometry
3.8. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nurminskaya, M.V.; Belkin, A.M. Cellular functions of tissue transglutaminase. Int. Rev. Cell Mol. Biol. 2012, 294, 1–97. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, D.; Curro, M.; Ientile, R. Potential of transglutaminase 2 as a therapeutic target. Expert Opin. Ther. Targets 2010, 14, 989–1003. [Google Scholar] [CrossRef] [PubMed]
- Tabolacci, C.; De Martino, A.; Mischiati, C.; Feriotto, G.; Beninati, S. The Role of Tissue Transglutaminase in Cancer Cell Initiation, Survival and Progression. Med. Sci. 2019, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Fok, J.Y.; Mehta, K. Tissue transglutaminase induces the release of apoptosis inducing factor and results in apoptotic death of pancreatic cancer cells. Apoptosis 2007, 12, 1455–1463. [Google Scholar] [CrossRef]
- Aeschlimann, D.; Thomazy, V. Protein crosslinking in assembly and remodelling of extracellular matrices: The role of transglutaminases. Connect. Tissue Res. 2000, 41, 1–27. [Google Scholar] [CrossRef]
- Tee, A.E.; Marshall, G.M.; Liu, P.Y.; Xu, N.; Haber, M.; Norris, M.D.; Iismaa, S.E.; Liu, T. Opposing effects of two tissue transglutaminase protein isoforms in neuroblastoma cell differentiation. J. Biol. Chem. 2010, 285, 3561–3567. [Google Scholar] [CrossRef]
- Modak, S.; Cheung, N.K. Neuroblastoma: Therapeutic strategies for a clinical enigma. Cancer Treat. Rev. 2010, 36, 307–317. [Google Scholar] [CrossRef]
- Ruiz-Perez, M.V.; Henley, A.B.; Arsenian-Henriksson, M. The MYCN Protein in Health and Disease. Genes 2017, 8, 113. [Google Scholar] [CrossRef]
- Sasada, M.; Iyoda, T.; Asayama, T.; Suenaga, Y.; Sakai, S.; Kase, N.; Kodama, H.; Yokoi, S.; Isohama, Y.; Fukai, F. Inactivation of beta1 integrin induces proteasomal degradation of Myc oncoproteins. Oncotarget 2019, 10, 4960–4972. [Google Scholar] [CrossRef][Green Version]
- Beltran, H. The N-myc Oncogene: Maximizing its Targets, Regulation, and Therapeutic Potential. Mol. Cancer Res. MCR 2014, 12, 815–822. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef] [PubMed]
- Bragelmann, J.; Bohm, S.; Guthrie, M.R.; Mollaoglu, G.; Oliver, T.G.; Sos, M.L. Family matters: How MYC family oncogenes impact small cell lung cancer. Cell Cycle 2017, 16, 1489–1498. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Cubero, F.J.; Nevzorova, Y.A. c-MYC-Making Liver Sick: Role of c-MYC in Hepatic Cell Function, Homeostasis and Disease. Genes 2017, 8, 123. [Google Scholar] [CrossRef] [PubMed]
- Vega, M.E.; Schwarzbauer, J.E. Collaboration of fibronectin matrix with other extracellular signals in morphogenesis and differentiation. Curr. Opin. Cell Biol. 2016, 42, 1–6. [Google Scholar] [CrossRef]
- Wolfenson, H.; Lavelin, I.; Geiger, B. Dynamic regulation of the structure and functions of integrin adhesions. Dev. Cell 2013, 24, 447–458. [Google Scholar] [CrossRef]
- Bornstein, P.; Sage, E.H. Matricellular proteins: Extracellular modulators of cell function. Curr. Opin. Cell Biol. 2002, 14, 608–616. [Google Scholar] [CrossRef]
- Lutz, W.; Schwab, M. In vivo regulation of single copy and amplified N-myc in human neuroblastoma cells. Oncogene 1997, 15, 303–315. [Google Scholar] [CrossRef][Green Version]
- Tang, X.X.; Zhao, H.; Kung, B.; Kim, D.Y.; Hicks, S.L.; Cohn, S.L.; Cheung, N.K.; Seeger, R.C.; Evans, A.E.; Ikegaki, N. The MYCN enigma: Significance of MYCN expression in neuroblastoma. Cancer Res. 2006, 66, 2826–2833. [Google Scholar] [CrossRef]
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef]
- Kaczowka, P.; Wieczorek, A.; Czogala, M.; Ksiazek, T.; Szewczyk, K.; Balwierz, W. The role of N-Myc gene amplification in neuroblastoma childhood tumour—single-centre experience. Contemp. Oncol. 2018, 22, 223–228. [Google Scholar] [CrossRef]
- Montalto, A.S.; Curro, M.; Russo, T.; Visalli, G.; Impellizzeri, P.; Antonuccio, P.; Arena, S.; Borruto, F.A.; Scalfari, G.; Ientile, R.; et al. In vitro CO2-induced ROS production impairs cell cycle in SH-SY5Y neuroblastoma cells. Pediatric Surg. Int. 2013, 29, 51–59. [Google Scholar] [CrossRef]
- Curro, M.; Montalto, A.S.; Impellizzeri, P.; Montalto, E.; Risitano, R.; Russo, T.; Perrone, P.; Chirico, V.; Arrigo, T.; Salpietro, C.; et al. CO(2) pneumoperitoneum induces in vitro hypoxic response culminating in apoptosis of human neuroblastoma cells. J. Biol. Regul. Homeost. Agents 2014, 28, 497–506. [Google Scholar]
- Schwab, M.; Alitalo, K.; Klempnauer, K.H.; Varmus, H.E.; Bishop, J.M.; Gilbert, F.; Brodeur, G.; Goldstein, M.; Trent, J. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature 1983, 305, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Bhaskara, V.K.; Mohanam, I.; Rao, J.S.; Mohanam, S. Intermittent hypoxia regulates stem-like characteristics and differentiation of neuroblastoma cells. PLoS ONE 2012, 7, e30905. [Google Scholar] [CrossRef] [PubMed]
- Mabjeesh, N.J.; Amir, S. Hypoxia-inducible factor (HIF) in human tumorigenesis. Histol. Histopathol. 2007, 22, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Harrison, L. Tumor hypoxia: Causative factors, compensatory mechanisms, and cellular response. Oncologist 2004, 9 (Suppl. 5), 4–9. [Google Scholar] [CrossRef] [PubMed]
- Hockel, M.; Vaupel, P. Biological consequences of tumor hypoxia. Semin. Oncol. 2001, 28, 36–41. [Google Scholar] [CrossRef]
- Greijer, A.E.; van der Wall, E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J. Clin. Pathol. 2004, 57, 1009–1014. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008, 15, 678–685. [Google Scholar] [CrossRef]
- Jang, G.Y.; Jeon, J.H.; Cho, S.Y.; Shin, D.M.; Kim, C.W.; Jeong, E.M.; Bae, H.C.; Kim, T.W.; Lee, S.H.; Choi, Y.; et al. Transglutaminase 2 suppresses apoptosis by modulating caspase 3 and NF-kappaB activity in hypoxic tumor cells. Oncogene 2010, 29, 356–367. [Google Scholar] [CrossRef]
- Nezir, A.E.; Ulukan, B.; Telci, D. Transglutaminase 2: The Maestro of the Oncogenic Mediators in Renal Cell Carcinoma. Med. Sci. 2019, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Yao, S.; Luo, G.; Zhang, X. Identification of differentially expressed genes in human breast cancer cells induced by 4-hydroxyltamoxifen and elucidation of their pathophysiological relevance and mechanisms. Oncotarget 2018, 9, 2475–2501. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, N.; Ishii, H.; Mimori, K.; Tanaka, F.; Hitora, T.; Tei, M.; Sekimoto, M.; Doki, Y.; Mori, M. TGM2 is a novel marker for prognosis and therapeutic target in colorectal cancer. Ann. Surg. Oncol. 2010, 17, 967–972. [Google Scholar] [CrossRef]
- Park, K.S.; Kim, H.K.; Lee, J.H.; Choi, Y.B.; Park, S.Y.; Yang, S.H.; Kim, S.Y.; Hong, K.M. Transglutaminase 2 as a cisplatin resistance marker in non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2010, 136, 493–502. [Google Scholar] [CrossRef]
- Sodek, K.L.; Ringuette, M.J.; Brown, T.J. Compact spheroid formation by ovarian cancer cells is associated with contractile behavior and an invasive phenotype. Int. J. Cancer 2009, 124, 2060–2070. [Google Scholar] [CrossRef]
- Shan, H.; Zhou, X.; Chen, C. MicroRNA214 suppresses the viability, migration and invasion of human colorectal carcinoma cells via targeting transglutaminase 2. Mol. Med. Rep. 2019, 20, 1459–1467. [Google Scholar] [CrossRef]
- Kim, S.Y. New Insights into Development of Transglutaminase 2 Inhibitors as Pharmaceutical Lead Compounds. Med. Sci. 2018, 6, 87. [Google Scholar] [CrossRef]
- Lentini, A.; Forni, C.; Provenzano, B.; Beninati, S. Enhancement of transglutaminase activity and polyamine depletion in B16-F10 melanoma cells by flavonoids naringenin and hesperitin correlate to reduction of the in vivo metastatic potential. Amino Acids 2007, 32, 95–100. [Google Scholar] [CrossRef]
- Phatak, V.M.; Croft, S.M.; Setty, S.G.R.; Scarpellini, A.; Hughes, D.C.; Rees, R.; McArdle, S.; Verderio, E.A.M. Expression of transglutaminase-2 isoforms in normal human tissues and cancer cell lines: Dysregulation of alternative splicing in cancer. Amino Acids 2013, 44, 33–44. [Google Scholar] [CrossRef]
- Liu, T.; Tee, A.E.; Porro, A.; Smith, S.A.; Dwarte, T.; Liu, P.Y.; Iraci, N.; Sekyere, E.; Haber, M.; Norris, M.D.; et al. Activation of tissue transglutaminase transcription by histone deacetylase inhibition as a therapeutic approach for Myc oncogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 18682–18687. [Google Scholar] [CrossRef]
- Condello, S.; Caccamo, D.; Curro, M.; Ferlazzo, N.; Parisi, G.; Ientile, R. Transglutaminase 2 and NF-kappaB interplay during NGF-induced differentiation of neuroblastoma cells. Brain Res. 2008, 1207, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tucholski, J.; Lesort, M.; Johnson, G.V.W. Tissue transglutaminase is essential for neurite outgrowth in human neuroblastoma SH-SY5Y cells. Neuroscience 2001, 102, 481–491. [Google Scholar] [CrossRef]
- Song, Y.; Kirkpatrick, L.L.; Schilling, A.B.; Helseth, D.L.; Chabot, N.; Keillor, J.W.; Johnson, G.V.; Brady, S.T. Transglutaminase and polyamination of tubulin: Posttranslational modification for stabilizing axonal microtubules. Neuron 2013, 78, 109–123. [Google Scholar] [CrossRef]
- Begg, G.E.; Carrington, L.; Stokes, P.H.; Matthews, J.M.; Wouters, M.A.; Husain, A.; Lorand, L.; Iismaa, S.E.; Graham, R.M. Mechanism of allosteric regulation of transglutaminase 2 by GTP. Proc. Natl. Acad. Sci. USA 2006, 103, 19683–19688. [Google Scholar] [CrossRef]
- Holmquist, L.; Jogi, A.; Pahlman, S. Phenotypic persistence after reoxygenation of hypoxic neuroblastoma cells. Int. J. Cancer 2005, 116, 218–225. [Google Scholar] [CrossRef]
- Bindra, R.S.; Schaffer, P.J.; Meng, A.; Woo, J.; Maseide, K.; Roth, M.E.; Lizardi, P.; Hedley, D.W.; Bristow, R.G.; Glazer, P.M. Alterations in DNA repair gene expression under hypoxia: Elucidating the mechanisms of hypoxia-induced genetic instability. Ann. N. Y. Acad. Sci. 2005, 1059, 184–195. [Google Scholar] [CrossRef]
- Hui, A.S.; Bauer, A.L.; Striet, J.B.; Schnell, P.O.; Czyzyk-Krzeska, M.F. Calcium signaling stimulates translation of HIF-alpha during hypoxia. FASEB J. 2006, 20, 466–475. [Google Scholar] [CrossRef]
- Hu, X.; Zheng, W.; Zhu, Q.; Gu, L.; Du, Y.; Han, Z.; Zhang, X.; Carter, D.R.; Cheung, B.B.; Qiu, A.; et al. Increase in DNA Damage by MYCN Knockdown Through Regulating Nucleosome Organization and Chromatin State in Neuroblastoma. Front. Genet. 2019, 10, 684. [Google Scholar] [CrossRef]
- Bedessem, B.; Stephanou, A. A mathematical model of HiF-1alpha-mediated response to hypoxia on the G1/S transition. Math. Biosci. 2014, 248, 31–39. [Google Scholar] [CrossRef]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1alpha and HIF2alpha: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef]
- Pattyn, F.; Speleman, F.; De Paepe, A.; Vandesompele, J. RTPrimerDB: The Real-Time PCR primer and probe database. Nucleic Acids Res. 2003, 31, 122–123. [Google Scholar] [CrossRef]
- Caccamo, D.; Campisi, A.; Curro, M.; Aguennouz, M.; Volti, G.L.; Avola, R.; Ientile, R. Nuclear factor-kappa B activation is associated with glutamate-evoked tissue transglutaminase up-regulation in primary astrocyte cultures. J. Neurosci. Res. 2005, 82, 858–865. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| Cyclin B1 | 5′-ctgctgcctggtgaagag-3′ | 5′-cgcctgccatgttgatct-3′ |
| Cyclin D1 | 5′-tattgcgctgctaccgttga-3′ | 5′-ccaatagcagcaaacaatgtgaaa-3′ |
| HIF-1α | 5′-cgttccttcgatcagttgtc-3′ | 5′-tcagtggtggcagtggtagt-3′ |
| p16 | 5′-accagaggcagtaaccat-3′ | 5′-gtaggaccttcggtgact-3′ |
| TG2-L | 5′-ccttacggagtccaacctca-3′ | 5′-ccgtcttctgctcctcagtc-3′ |
| TG2-S | 5′-accgctgaggagtacgtctg-3′ | 5′-tcaacaaatgctccaggaa-3′ |
| β-Actin | 5′-ttgttacaggaagtcccttgcc-3′ | 5′-atgctatcacctcccctgtgtg-3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Currò, M.; Ferlazzo, N.; Giunta, M.L.; Montalto, A.S.; Russo, T.; Arena, S.; Impellizzeri, P.; Caccamo, D.; Romeo, C.; Ientile, R. Hypoxia-Dependent Expression of TG2 Isoforms in Neuroblastoma Cells as Consequence of Different MYCN Amplification Status. Int. J. Mol. Sci. 2020, 21, 1364. https://doi.org/10.3390/ijms21041364
Currò M, Ferlazzo N, Giunta ML, Montalto AS, Russo T, Arena S, Impellizzeri P, Caccamo D, Romeo C, Ientile R. Hypoxia-Dependent Expression of TG2 Isoforms in Neuroblastoma Cells as Consequence of Different MYCN Amplification Status. International Journal of Molecular Sciences. 2020; 21(4):1364. https://doi.org/10.3390/ijms21041364
Chicago/Turabian StyleCurrò, Monica, Nadia Ferlazzo, Maria Laura Giunta, Angela Simona Montalto, Tiziana Russo, Salvatore Arena, Pietro Impellizzeri, Daniela Caccamo, Carmelo Romeo, and Riccardo Ientile. 2020. "Hypoxia-Dependent Expression of TG2 Isoforms in Neuroblastoma Cells as Consequence of Different MYCN Amplification Status" International Journal of Molecular Sciences 21, no. 4: 1364. https://doi.org/10.3390/ijms21041364
APA StyleCurrò, M., Ferlazzo, N., Giunta, M. L., Montalto, A. S., Russo, T., Arena, S., Impellizzeri, P., Caccamo, D., Romeo, C., & Ientile, R. (2020). Hypoxia-Dependent Expression of TG2 Isoforms in Neuroblastoma Cells as Consequence of Different MYCN Amplification Status. International Journal of Molecular Sciences, 21(4), 1364. https://doi.org/10.3390/ijms21041364