RNA Splicing Defects in Hypertrophic Cardiomyopathy: Implications for Diagnosis and Therapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
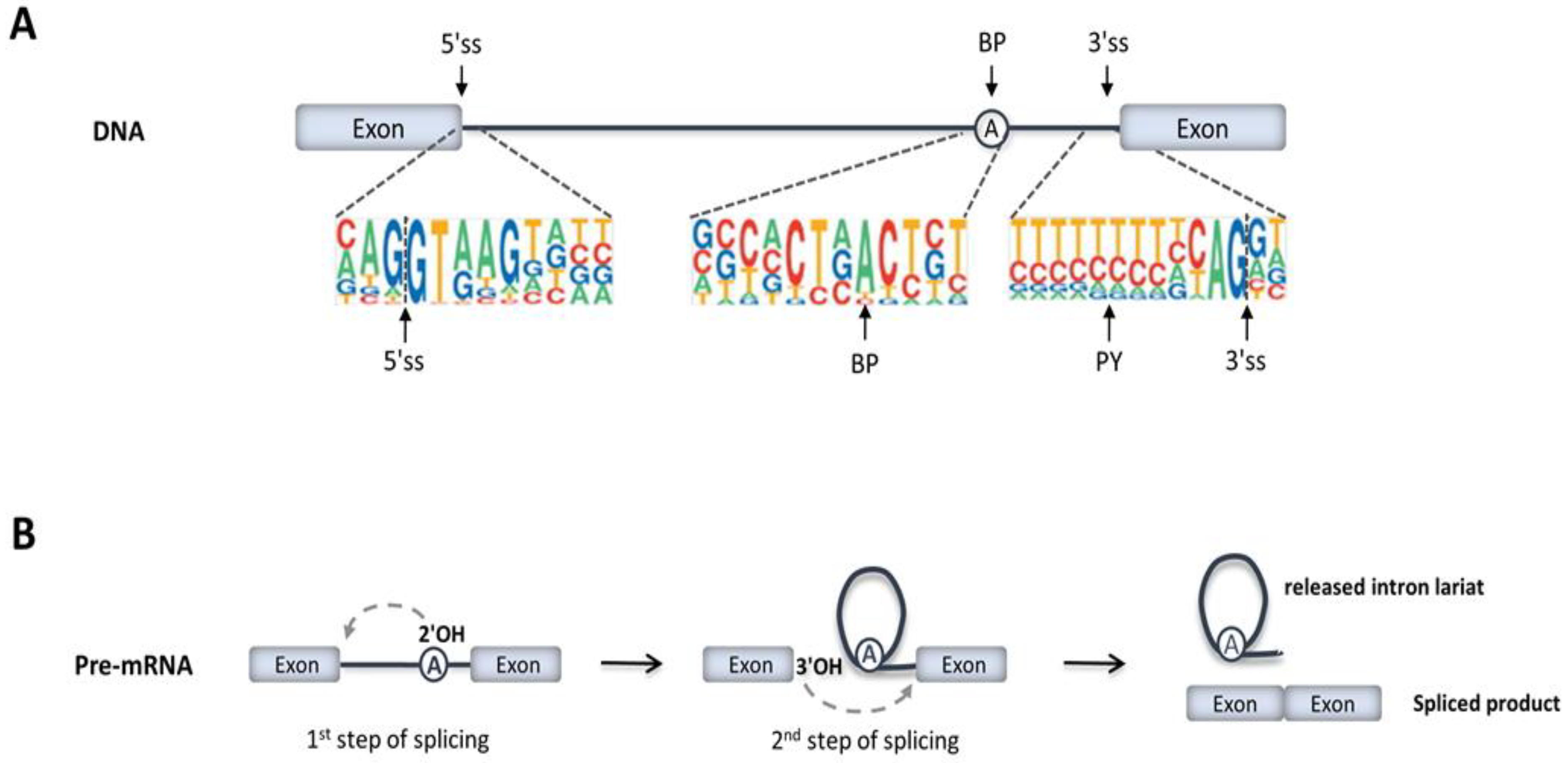
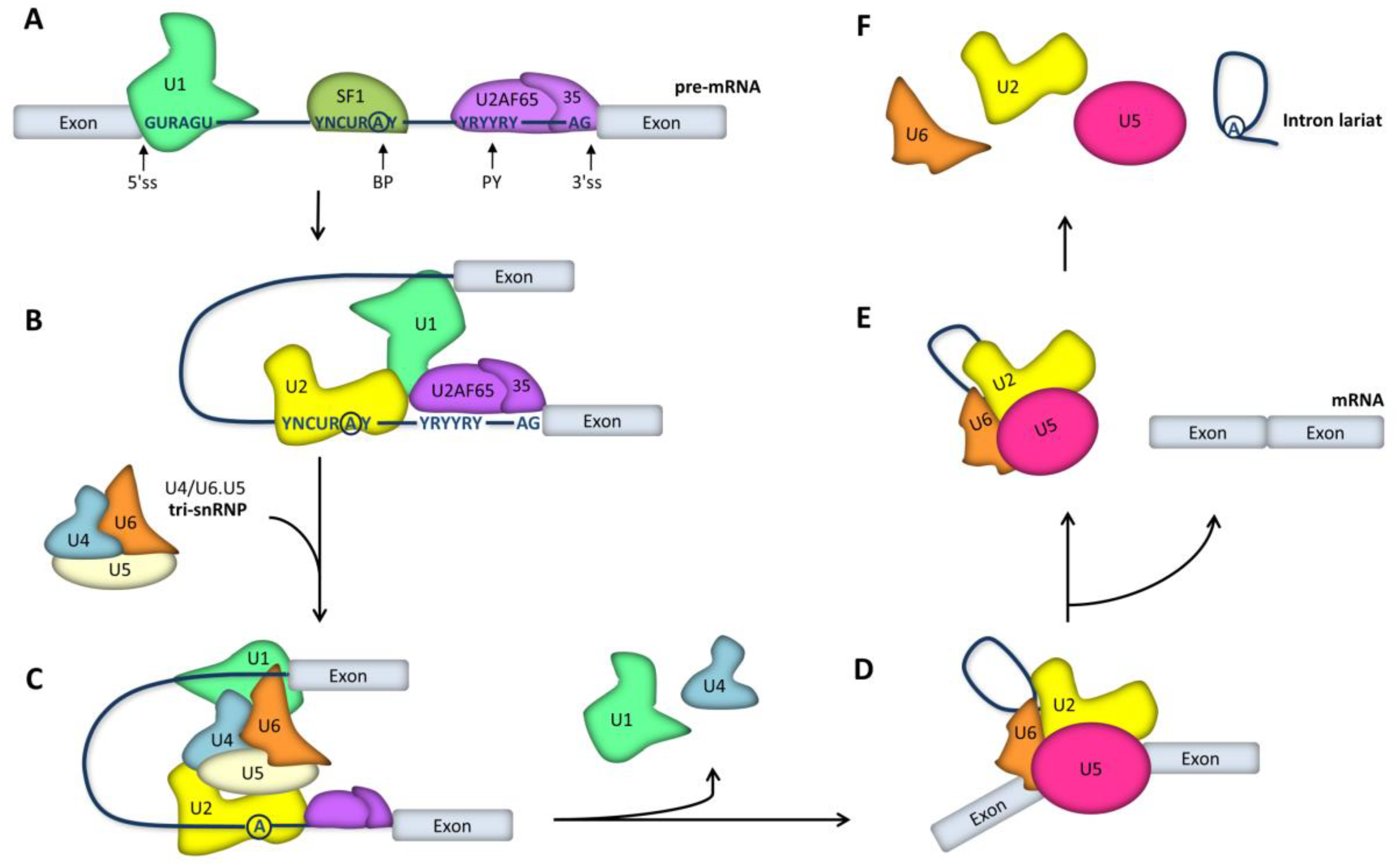
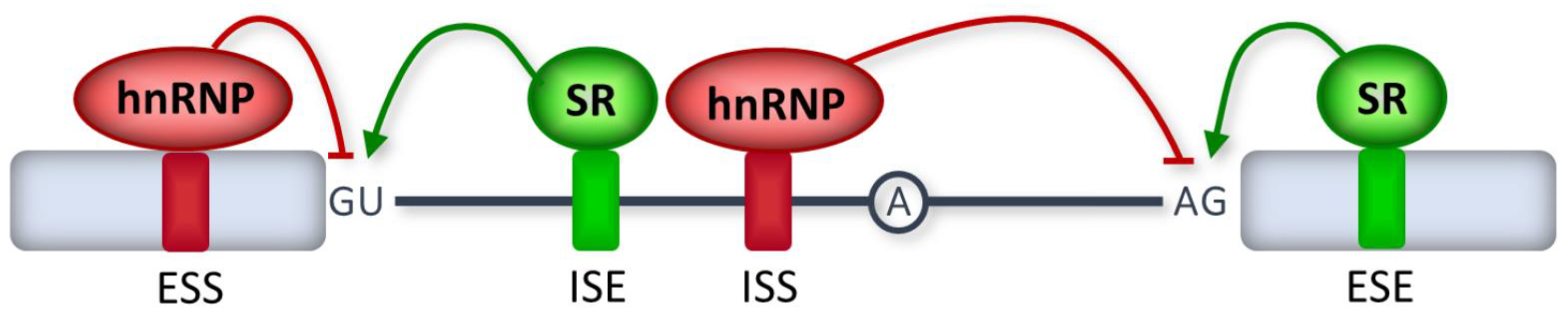
2. RNA Splicing Mechanisms
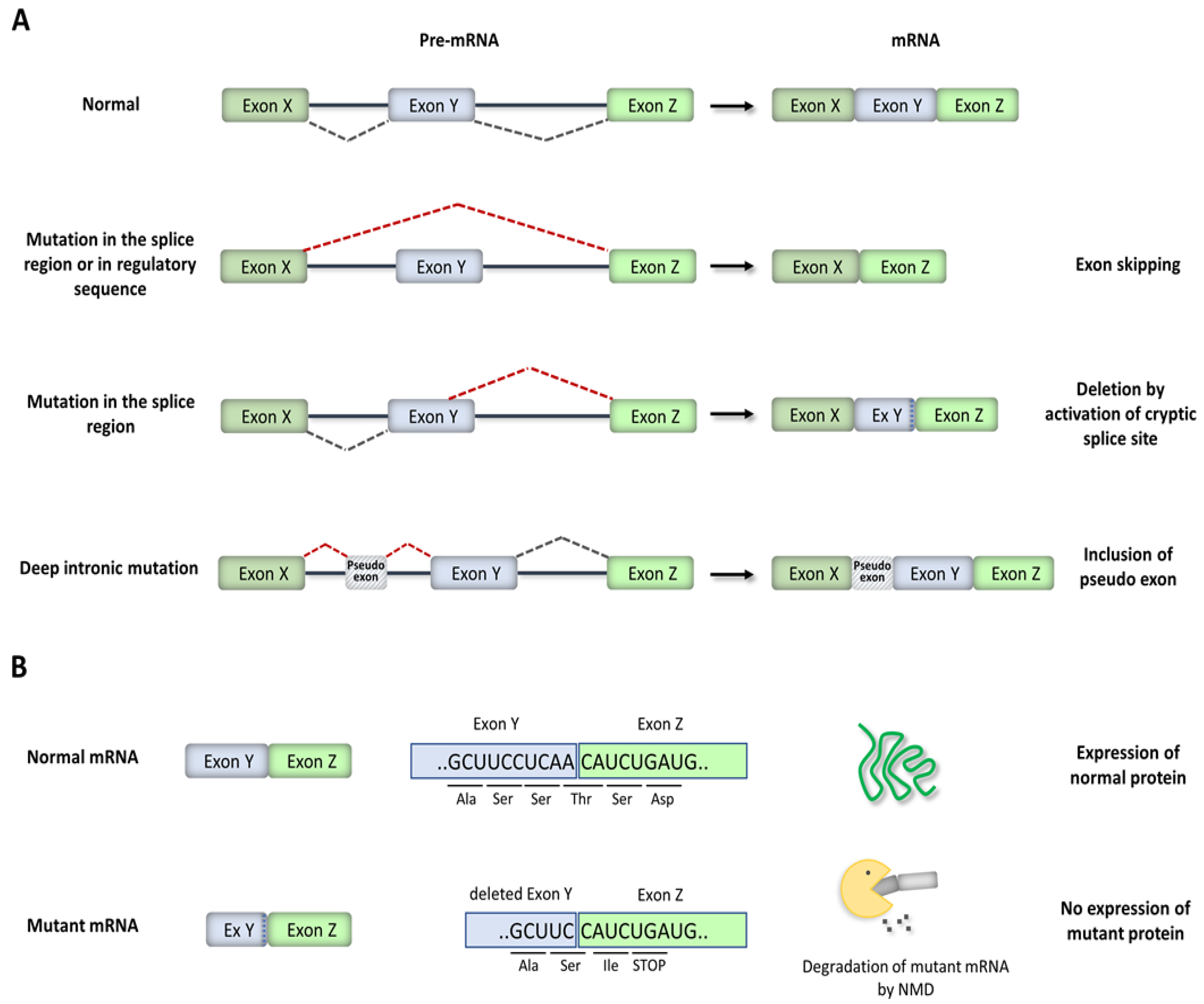
3. Disease-Causing Splicing Mutations
4. HCM-Associated Splicing Mutations
5. RNA Therapeutics for HCM
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maron, B.J.; Gardin, J.M.; Flack, J.M.; Gidding, S.S.; Kurosaki, T.T.; Bild, D.E. Prevalence of hypertrophic cardiomyopathy in a general population of young adults: Echocardiographic analysis of 4111 subjects in the CARDIA study. Circulation 1995, 92, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Semsarian, C.; Ingles, J.; Maron, M.S.; Maron, B.J. New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2015, 65, 1249–1254. [Google Scholar] [CrossRef] [PubMed]
- Klarich, K.W.; Attenhofer Jost, C.H.; Binder, J.; Connolly, H.M.; Scott, C.G.; Freeman, W.K.; Ackerman, M.J.; Nishimura, R.A.; Tajik, A.J.; Ommen, S.R. Risk of death in long-term follow-up of patients with apical hypertrophic cardiomyopathy. Am. J. Cardiol. 2013, 111, 1784–1791. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.J.; Ommen, S.R.; Semsarian, C.; Spirito, P.; Olivotto, I.; Maron, M.S. Hypertrophic cardiomyopathy: Present and future, with translation into contemporary cardiovascular medicine. J. Am. Coll. Cardiol. 2014, 64, 83–99. [Google Scholar] [CrossRef]
- Maron, B.J.; Rowin, E.J.; Casey, S.A.; Link, M.S.; Lesser, J.R.; Chan, R.H.M.; Garberich, R.F.; Udelson, J.E.; Maron, M.S. Hypertrophic cardiomyopathy in adulthood associated with low cardiovascular mortality with contemporary management strategies. J. Am. Coll. Cardiol. 2015, 65, 1915–1928. [Google Scholar] [CrossRef]
- Frey, N.; Luedde, M.; Katus, H.A. Mechanisms of disease: Hypertrophic cardiomyopathy. Nat. Rev. Cardiol. 2012, 9, 91–100. [Google Scholar] [CrossRef]
- Marian, A.J.; Braunwald, E. Hypertrophic cardiomyopathy: Genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef]
- Heitner, S.B. Novel medical therapeutics for hypertrophic cardiomyopathy. In Hypertrophic Cardiomyopathy; Naidu, S.S., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 383–388. ISBN 9783319924236. [Google Scholar]
- Elliott, P.M.; Anastasakis, A.; Michael, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: The task force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar]
- Tejado, B.S.M.; Jou, C. Histopathology in HCM. Glob. Cardiol. Sci. Pract. 2018. [Google Scholar] [CrossRef]
- Grazioli, G.; Usín, D.; Trucco, E.; Sanz, M.; Montserrat, S.; Vidal, B.; Gutierrez, J.; Canal, R.; Brugada, J.; Mont, L.; et al. Differentiating hypertrophic cardiomyopathy from athlete’s heart: An electrocardiographic and echocardiographic approach. J. Electrocardiol. 2016, 49, 539–544. [Google Scholar] [CrossRef]
- Niimura, H.; Bachinski, L.L.; Sangwatanaroj, S.; Watkins, H.; Chudley, A.E.; Mckenna, W.; Kristinsson, A.; Roberts, R.; Sole, M.; Maron, B.J.; et al. Mutations in the gene for cardiac myosin-binding protein C and late-onset familial hypertrophic cardiomyopathy. N. Engl. J. Med. 1998, 338, 1248–1257. [Google Scholar] [CrossRef] [PubMed]
- Niimura, H.; Patton, K.K.; McKenna, W.J.; Soults, J.; Maron, B.J.; Seidman, J.G.; Seidman, C.E. Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly. Circulation 2002, 105, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Gruner, C.; Ivanov, J.; Care, M.; Williams, L.; Moravsky, G.; Yang, H.; Laczay, B.; Siminovitch, K.; Woo, A.; Rakowski, H. Toronto hypertrophic cardiomyopathy genotype score for prediction of a positive genotype in hypertrophic cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.; Thomson, K.L.; Ware, J.S.; Funke, B.H.; Woodley, J.; McGuire, K.J.; Mazzarotto, F.; Blair, E.; Seller, A.; Taylor, J.C.; et al. Reassessment of Mendelian gene pathogenicity using 7855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 2017, 19, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Irimia, M.; Blencowe, B.J. Alternative splicing: Decoding an expansive regulatory layer. Curr. Opin. Cell Biol. 2012, 24, 323–332. [Google Scholar] [CrossRef]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef]
- Weeland, C.J.; van den Hoogenhof, M.M.; Beqqali, A.; Creemers, E.E. Insights into alternative splicing of sarcomeric genes in the heart. J. Mol. Cell. Cardiol. 2015, 81, 107–113. [Google Scholar] [CrossRef]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef]
- Yan, C.; Wan, R.; Shi, Y. Molecular mechanisms of pre-mRNA splicing through structural biology of the spliceosome. Cold Spring Harb. Perspect. Biol. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Kastner, B.; Will, C.L.; Stark, H.; Lührmann, R. Structural insights into nuclear pre-mRNA splicing in higher eukaryotes. Cold Spring Harb. Perspect. Biol. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.A.; Steitz, J.A. Splicing double: Insights from the second spliceosome. Nat. Rev. Mol. Cell Biol. 2003, 4, 960–970. [Google Scholar] [CrossRef]
- Verma, B.; Akinyi, M.V.; Norppa, A.J.; Frilander, M.J. Minor spliceosome and disease. Semin. Cell Dev. Biol. 2018, 79, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Blencowe, B.J. Exonic splicing enhancers: Mechanism of action, diversity and role in human genetic diseases. Trends Biochem. Sci. 2000, 25, 106–110. [Google Scholar] [CrossRef]
- Wang, Z.; Xiao, X.; Van Nostrand, E.; Burge, C.B. General and specific functions of exonic splicing silencers in splicing control. Mol. Cell 2006, 23, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Luco, R.F.; Pan, Q.; Tominaga, K.; Blencowe, B.J.; Pereira-Smith, O.M.; Misteli, T. Regulation of alternative splicing by histone modifications. Science 2010, 327, 996–1000. [Google Scholar] [CrossRef]
- Han, H.; Braunschweig, U.; Gonatopoulos-Pournatzis, T.; Weatheritt, R.J.; Hirsch, C.L.; Ha, K.C.H.; Radovani, E.; Nabeel-Shah, S.; Sterne-Weiler, T.; Wang, J.; et al. Multilayered control of alternative splicing regulatory networks by transcription factors. Mol. Cell 2017, 65, 539–553. [Google Scholar] [CrossRef]
- Grosso, A.R.; Gomes, A.Q.; Barbosa-Morais, N.L.; Caldeira, S.; Thorne, N.P.; Grech, G.; von Lindern, M.; Carmo-Fonseca, M. Tissue-specific splicing factor gene expression signatures. Nucleic Acids Res. 2008, 36, 4823–4832. [Google Scholar] [CrossRef]
- Faustino, N.A.; Cooper, T.A. Pre-mRNA splicing and human disease. Genes Dev. 2003, 17, 419–437. [Google Scholar] [CrossRef] [PubMed]
- Abramowicz, A.; Gos, M. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Rivas, M.A.; Pirinen, M.; Conrad, D.F.; Lek, M.; Tsang, E.K.; Karczewski, K.J.; Maller, J.B.; Kukurba, K.R.; DeLuca, D.S.; Fromer, M.; et al. Effect of predicted protein-truncating genetic variants on the human transcriptome. Science 2015, 348, 666–669. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]
- Zhang, S.; Samocha, K.E.; Rivas, M.A.; Karczewski, K.J.; Daly, E.; Schmandt, B.; Neale, B.M.; MacArthur, D.G.; Daly, M.J. Base-specific mutational intolerance near splice sites clarifies the role of nonessential splice nucleotides. Genome Res. 2018, 28, 968–974. [Google Scholar] [CrossRef]
- Chang, Y.-F.; Imam, J.S.; Wilkinson, M.F. The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 2007, 76, 51–74. [Google Scholar] [CrossRef]
- Wang, Z.; Rolish, M.E.; Yeo, G.; Tung, V.; Mawson, M.; Burge, C.B. Systematic identification and analysis of exonic splicing silencers. Cell 2004, 119, 831–845. [Google Scholar] [CrossRef]
- Wang, Z.; Burge, C.B. Splicing regulation: From a parts list of regulatory elements to an integrated splicing code. RNA 2008, 14, 802–813. [Google Scholar] [CrossRef]
- Cartegni, L.; Chew, S.L.; Krainer, A.R. Listening to silence and understanding nonsense: Exonic mutations that affect splicing. Nat. Rev. Genet. 2002, 3, 285–298. [Google Scholar] [CrossRef]
- Vaz-Drago, R.; Custódio, N.; Carmo-Fonseca, M. Deep intronic mutations and human disease. Hum. Genet. 2017, 136, 1093–1111. [Google Scholar] [CrossRef] [PubMed]
- Vithana, E.N.; Abu-Safieh, L.; Allen, M.J.; Carey, A.; Papaioannou, M.; Chakarova, C.; Al-Maghtheh, M.; Ebenezer, N.D.; Willis, C.; Moore, A.T.; et al. A human homolog of yeast pre-mRNA splicing gene, PRP31, underlies autosomal dominant retinitis pigmentosa on chromosome 19q13.4 (RP11). Mol. Cell 2001, 8, 375–381. [Google Scholar] [CrossRef]
- Chakarova, C.F. Mutations in HPRP3, a third member of pre-mRNA splicing factor genes, implicated in autosomal dominant retinitis pigmentosa. Hum. Mol. Genet. 2002, 11, 87–92. [Google Scholar] [CrossRef] [PubMed]
- McKie, A.B. Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13). Hum. Mol. Genet. 2001, 10, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Paushkin, S.; Gubitz, A.K.; Massenet, S.; Dreyfuss, G. The SMN complex, an assemblyosome of ribonucleoproteins. Curr. Opin. Cell Biol. 2002, 14, 305–312. [Google Scholar] [CrossRef]
- Greaves, S.C.; Roche, A.H.G.; Neutze, J.M.; Whitlock, R.M.L.; Veale, A.M.O. Inheritance of hypertrophic cardiomyopathy: A cross sectional and M mode echocardiographic study of 50 families. Heart 1987, 58, 259–266. [Google Scholar] [CrossRef]
- Watkins, H.; Ashrafian, H.; Redwood, C. Inherited cardiomyopathies. N. Engl. J. Med. 2011, 364, 1643–1656. [Google Scholar] [CrossRef]
- Konno, T.; Chang, S.; Seidman, J.G.; Seidman, C.E. Genetics of hypertrophic cardiomyopathy. Curr. Opin. Cardiol. 2010, 25, 205–209. [Google Scholar] [CrossRef]
- Walsh, R.; Buchan, R.; Wilk, A.; John, S.; Felkin, L.E.; Thomson, K.L.; Chiaw, T.H.; Loong, C.C.W.; Pua, C.J.; Raphael, C.; et al. Defining the genetic architecture of hypertrophic cardiomyopathy: Re-evaluating the role of non-sarcomeric genes. Eur. Heart J. 2017, 38, 3461–3468. [Google Scholar] [CrossRef]
- Tyska, M.J.; Hayes, E.; Giewat, M.; Seidman, C.E.; Seidman, J.G.; Warshaw, D.M. Single-molecule mechanics of R403Q cardiac myosin isolated from the mouse model of familial hypertrophic cardiomyopathy. Circ. Res. 2000, 86, 737–744. [Google Scholar] [CrossRef]
- Marston, S.; Copeland, O.; Jacques, A.; Livesey, K.; Tsang, V.; McKenna, W.J.; Jalilzadeh, S.; Carballo, S.; Redwood, C.; Watkins, H. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ. Res. 2009, 105, 219–222. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, S.J.; Dooijes, D.; dos Remedios, C.; Michels, M.; Lamers, J.M.J.; Winegrad, S.; Schlossarek, S.; Carrier, L.; ten Cate, F.J.; Stienen, G.J.M.; et al. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: Haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation 2009, 119, 1473–1483. [Google Scholar] [CrossRef] [PubMed]
- Barash, Y.; Calarco, J.A.; Gao, W.; Pan, Q.; Wang, X.; Shai, O.; Blencowe, B.J.; Frey, B.J. Deciphering the splicing code. Nature 2010, 465, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Di Giacomo, D.; Gaildrat, P.; Abuli, A.; Abdat, J.; Frébourg, T.; Tosi, M.; Martins, A. Functional analysis of a large set of brca2 exon 7 variants highlights the predictive value of hexamer scores in detecting alterations of exonic splicing regulatory elements. Hum. Mutat. 2013, 34, 1547–1557. [Google Scholar] [CrossRef]
- Erkelenz, S.; Theiss, S.; Otte, M.; Widera, M.; Peter, J.O.; Schaal, H. Genomic HEXploring allows landscaping of novel potential splicing regulatory elements. Nucleic Acids Res. 2014, 42, 10681–10697. [Google Scholar] [CrossRef]
- Rosenberg, A.B.; Patwardhan, R.P.; Shendure, J.; Seelig, G. Learning the sequence determinants of alternative splicing from millions of random sequences. Cell 2015, 163, 698–711. [Google Scholar] [CrossRef]
- Xiong, H.Y.; Alipanahi, B.; Lee, L.J.; Bretschneider, H.; Merico, D.; Yuen, R.K.C.; Hua, Y.; Gueroussov, S.; Najafabadi, H.S.; Hughes, T.R.; et al. The human splicing code reveals new insights into the genetic determinants of disease. Science 2015, 347, 1254806. [Google Scholar] [CrossRef]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting splicing from primary sequence with deep learning. Cell 2019, 176, 535–548. [Google Scholar] [CrossRef]
- Ito, K.; Patel, P.N.; Gorham, J.M.; McDonough, B.; DePalma, S.R.; Adler, E.E.; Lam, L.; MacRae, C.A.; Mohiuddin, S.M.; Fatkin, D.; et al. Identification of pathogenic gene mutations in LMNA and MYBPC3 that alter RNA splicing. Proc. Natl. Acad. Sci. USA 2017, 114, 7689–7694. [Google Scholar] [CrossRef]
- Helms, A.S.; Davis, F.M.; Coleman, D.; Bartolone, S.N.; Glazier, A.A.; Pagani, F.; Yob, J.M.; Sadayappan, S.; Pedersen, E.; Lyons, R.; et al. Sarcomere mutation-specific expression patterns in human hypertrophic cardiomyopathy. Circ. Cardiovasc. Genet. 2014, 7, 434–443. [Google Scholar] [CrossRef]
- Bagnall, R.D.; Ingles, J.; Dinger, M.E.; Cowley, M.J.; Ross, S.B.; Minoche, A.E.; Lal, S.; Turner, C.; Colley, A.; Rajagopalan, S.; et al. Whole Genome Sequencing Improves Outcomes of Genetic Testing in Patients With Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2018, 72, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Singer, E.S.; Ingles, J.; Semsarian, C.; Bagnall, R.D. Key value of RNA analysis of MYBPC3 splice-site variants in hypertrophic cardiomyopathy. Circ. Genom. Precis. Med. 2019, 12, e002368. [Google Scholar] [CrossRef] [PubMed]
- Lan, F.; Lee, A.S.; Liang, P.; Sanchez-Freire, V.; Nguyen, P.K.; Wang, L.; Han, L.; Yen, M.; Wang, Y.; Sun, N.; et al. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell 2013, 12, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.B.; Fraser, S.T.; Bagnall, R.D.; Semsarian, C. Peripheral blood derived induced pluripotent stem cells (iPSCs) from a female with familial hypertrophic cardiomyopathy. Stem Cell Res. 2017, 20, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Zhang, J.Z.; Itzhaki, I.; Zhang, S.L.; Chen, H.; Haddad, F.; Kitani, T.; Wilson, K.D.; Tian, L.; Shrestha, R.; et al. Determining the pathogenicity of a genomic variant of uncertain significance using CRISPR/Cas9 and human-induced pluripotent stem cells. Circulation 2018, 138, 2666–2681. [Google Scholar] [CrossRef] [PubMed]
- Charleston, J.S.; Schnell, F.J.; Dworzak, J.; Donoghue, C.; Lewis, S.; Chen, L.; David Young, G.; Milici, A.J.; Voss, J.; Dealwis, U.; et al. Eteplirsen treatment for Duchenne muscular dystrophy. Neurology 2018, 90, e2135–e2145. [Google Scholar] [CrossRef]
- Michelson, D.; Ciafaloni, E.; Ashwal, S.; Lewis, E.; Narayanaswami, P.; Oskoui, M.; Armstrong, M.J. Evidence in focus: Nusinersen use in spinal muscular atrophy report of the guideline development, dissemination, and implementation subcommittee of the American academy of neurology. Neurology 2018, 91, 923–933. [Google Scholar] [CrossRef]
- Gedicke-Hornung, C.; Behrens-Gawlik, V.; Reischmann, S.; Geertz, B.; Stimpel, D.; Weinberger, F.; Schlossarek, S.; Précigout, G.; Braren, I.; Eschenhagen, T.; et al. Rescue of cardiomyopathy through U7snRNA-mediated exon skipping in Mybpc3-targeted knock-in mice. EMBO Mol. Med. 2013, 5, 1060–1077. [Google Scholar] [CrossRef]
- Setten, R.L.; Rossi, J.J.; Han, S. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019, 18, 421–446. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Montgomery, M.K.; Xu, S.; Fire, A. RNA as a target of double-stranded RNA-mediated genetic interference in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 1998, 95, 15502–15507. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Yang, J. Patisiran for the treatment of hereditary transthyretin-mediated amyloidosis. Expert Rev. Clin. Pharmacol. 2019, 12, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Turanov, A.A.; Lo, A.; Hassler, M.R.; Makris, A.; Ashar-Patel, A.; Alterman, J.F.; Coles, A.H.; Haraszti, R.A.; Roux, L.; Godinho, B.M.D.C.; et al. RNAi modulation of placental sFLT1 for the treatment of preeclampsia. Nat. Biotechnol. 2018, 36, 1164–1173. [Google Scholar] [CrossRef]
- Jiang, J.; Wakimoto, H.; Seidman, J.G.; Seidman, C.E. Allele-specific silencing of mutant Myh6 transcripts in mice suppresses hypertrophic cardiomyopathy. Science 2013, 342, 111–114. [Google Scholar] [CrossRef]
- Berger, A.; Maire, S.; Gaillard, M.C.; Sahel, J.A.; Hantraye, P.; Bemelmans, A.P. mRNA trans-splicing in gene therapy for genetic diseases. Wiley Interdiscip. Rev. RNA 2016, 7, 487–498. [Google Scholar] [CrossRef]
- Mearini, G.; Stimpel, D.; Krämer, E.; Geertz, B.; Braren, I.; Gedicke-Hornung, C.; Précigout, G.; Müller, O.J.; Katus, H.A.; Eschenhagen, T.; et al. Repair of Mybpc3 mRNA by 5′-trans-splicing in a mouse model of hypertrophic cardiomyopathy. Mol. Ther. Nucleic Acids 2013, 2, e102. [Google Scholar] [CrossRef]
- Prondzynski, M.; Krämer, E.; Laufer, S.D.; Shibamiya, A.; Pless, O.; Flenner, F.; Müller, O.J.; Münch, J.; Redwood, C.; Hansen, A.; et al. Evaluation of MYBPC3 trans-splicing and gene replacement as therapeutic options in human ipsc-derived cardiomyocytes. Mol. Ther. Nucleic Acids 2017, 7, 475–486. [Google Scholar] [CrossRef]
- Sayed, D.; Hong, C.; Chen, I.-Y.; Lypowy, J.; Abdellatif, M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ. Res. 2007, 100, 416–424. [Google Scholar] [CrossRef]
- Roncarati, R.; Viviani Anselmi, C.; Losi, M.A.; Papa, L.; Cavarretta, E.; Da Costa Martins, P.; Contaldi, C.; Saccani Jotti, G.; Franzone, A.; Galastri, L.; et al. Circulating miR-29a, among other up-regulated microRNAs, is the only biomarker for both hypertrophy and fibrosis in patients with hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2014, 63, 920–927. [Google Scholar] [CrossRef]
- Song, L.; Su, M.; Wang, S.; Zou, Y.; Wang, X.; Wang, Y.; Cui, H.; Zhao, P.; Hui, R.; Wang, J. MiR-451 is decreased in hypertrophic cardiomyopathy and regulates autophagy by targeting TSC1. J. Cell. Mol. Med. 2014, 18, 2266–2274. [Google Scholar] [CrossRef]
- Su, M.; Wang, S.; Qiu, W.; Li, J.; Hui, R.; Song, L.; Jia, M.; Wang, H.; Wang, J. MIR-139-5p inhibits isoproterenol-induced cardiac hypertrophy by targetting c-Jun. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef]
- Van Rooij, E.; Sutherland, L.B.; Liu, N.; Williams, A.H.; McAnally, J.; Gerard, R.D.; Richardson, J.A.; Olson, E.N. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc. Natl. Acad. Sci. USA 2006, 103, 18255–18260. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro, M.; Furtado, M.; Martins, S.; Carvalho, T.; Carmo-Fonseca, M. RNA Splicing Defects in Hypertrophic Cardiomyopathy: Implications for Diagnosis and Therapy. Int. J. Mol. Sci. 2020, 21, 1329. https://doi.org/10.3390/ijms21041329
Ribeiro M, Furtado M, Martins S, Carvalho T, Carmo-Fonseca M. RNA Splicing Defects in Hypertrophic Cardiomyopathy: Implications for Diagnosis and Therapy. International Journal of Molecular Sciences. 2020; 21(4):1329. https://doi.org/10.3390/ijms21041329
Chicago/Turabian StyleRibeiro, Marta, Marta Furtado, Sandra Martins, Teresa Carvalho, and Maria Carmo-Fonseca. 2020. "RNA Splicing Defects in Hypertrophic Cardiomyopathy: Implications for Diagnosis and Therapy" International Journal of Molecular Sciences 21, no. 4: 1329. https://doi.org/10.3390/ijms21041329
APA StyleRibeiro, M., Furtado, M., Martins, S., Carvalho, T., & Carmo-Fonseca, M. (2020). RNA Splicing Defects in Hypertrophic Cardiomyopathy: Implications for Diagnosis and Therapy. International Journal of Molecular Sciences, 21(4), 1329. https://doi.org/10.3390/ijms21041329