IL-33/IL-31 Axis in Osteoporosis




Abstract
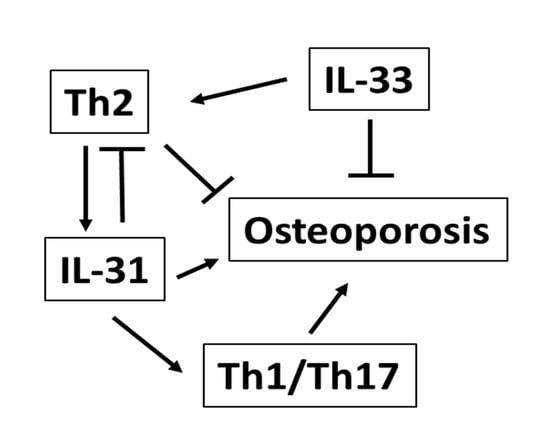
1. Osteoporosis and the Cytokine Regulation of Bone Remodeling
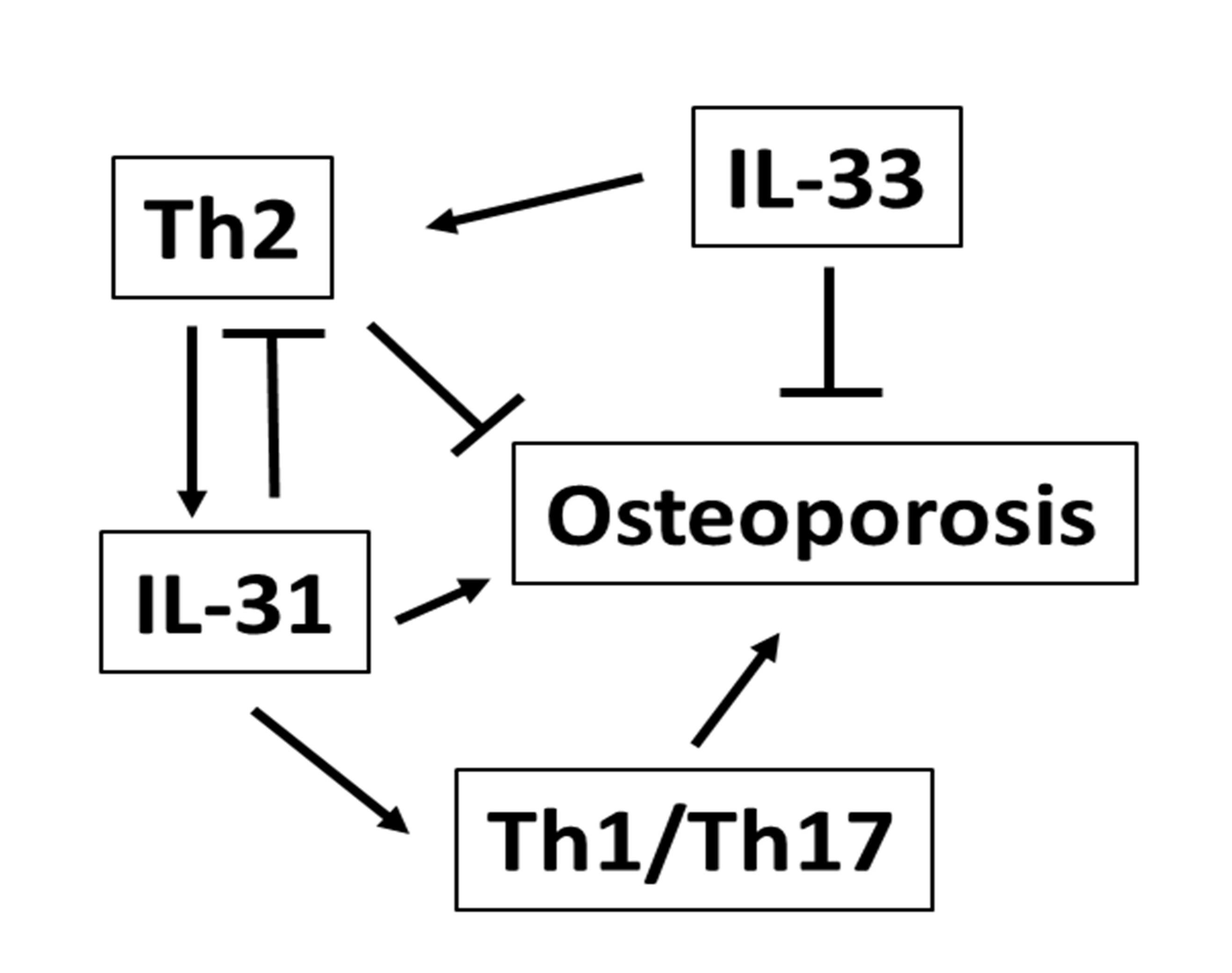
2. The Role of IL-31 in the Immunoskeletal Interface
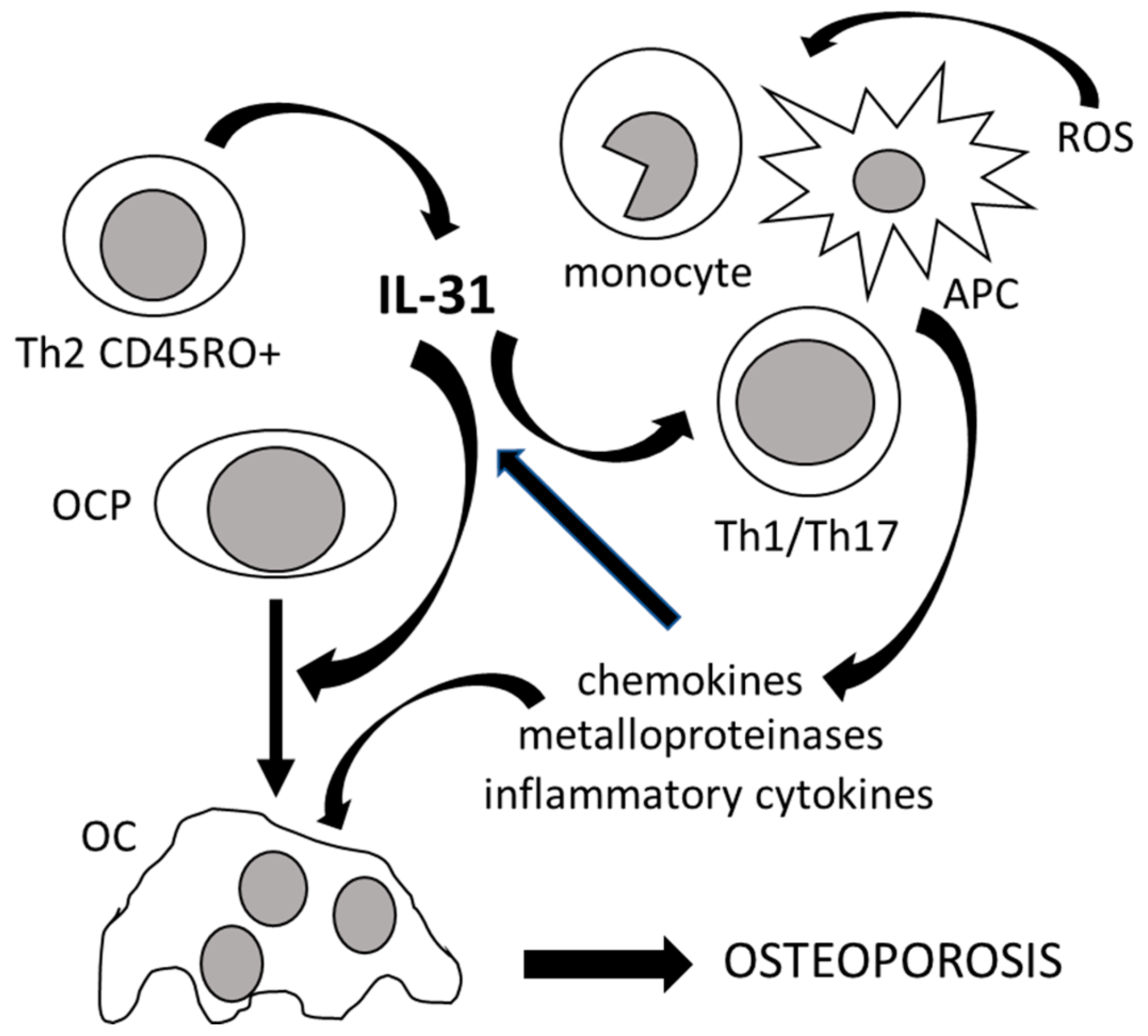
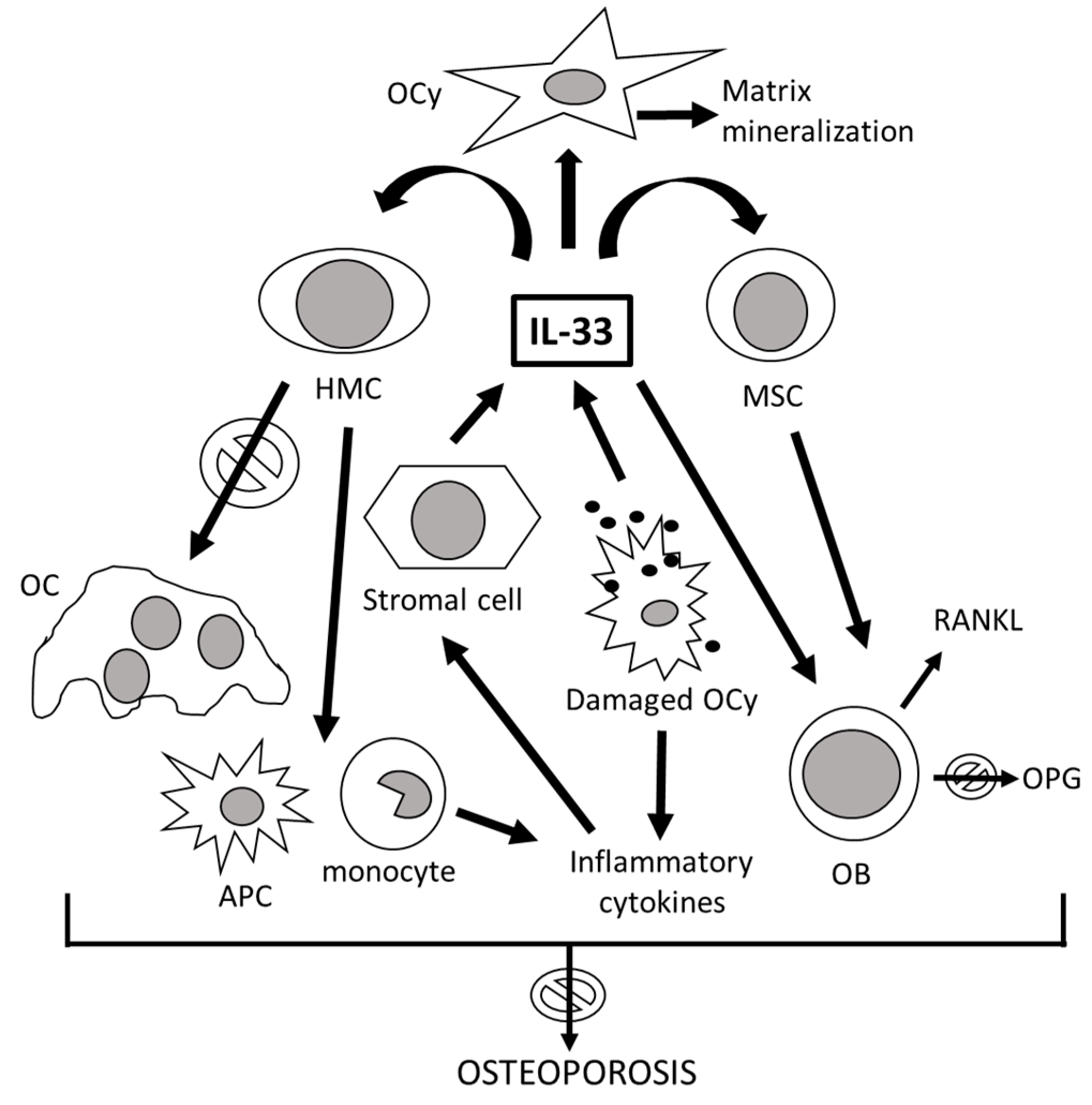
3. The Role of IL-33 in the Immunoskeletal Interface
4. Clinical Significance of Interleukin-31 and Interleukin-33 in Osteoporosis
5. Future Perspectives and Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Akkawi, I.; Zmerly, H. Osteoporosis: Current Concepts. Joints 2018, 6, 122–127. [Google Scholar] [CrossRef]
- De Martinis, M.; Sirufo, M.M.; Ginaldi, L. Osteoporosis: Current and Emerging Therapies Targeted to Immunological Checkpoints. Curr. Med. Chem. 2019, 26, 1–16. [Google Scholar] [CrossRef] [PubMed]
- De Martinis, M.; Di Benedetto, M.C.; Mengoli, L.P.; Ginaldi, L. Senile osteoporosis: Is it an immune-mediated disease? Inflamm. Res. 2006, 55, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Ciccarelli, F.; De Martinis, M.; Ginaldi, L. Glucocorticoids in patients with rheumatic diseases: Friends or enemies of bone? Curr. Med. Chem. 2015, 22, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Güler-Yüksel, M.; Hoes, J.N.; Bultink, I.E.; Lems, W.F. Glucocorticoids, Inflammation and Bone. Calcif. Tissue Int. 2018, 102, 592–606. [Google Scholar] [CrossRef] [PubMed]
- Pacifici, R. T cells, osteoblasts, and osteocytes: Interacting lineages key for the bone anabolic and catabolic activities of parathyroid hormone. Ann. N. Y. Acad. Sci. 2016, 1364, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Kenkre, J.; Bassett, J. The bone remodelling cycle. Ann. Clin. Biochem. Int. J. Lab. Med. 2018, 55, 308–327. [Google Scholar] [CrossRef]
- Awasthi, H.; Mani, D.; Singh, D.; Gupta, A. The underlying pathophysiology and therapeutic approaches for osteoporosis. Med. Res. Rev. 2018, 38, 2024–2057. [Google Scholar] [CrossRef]
- Ginaldi, L.; De Martinis, M. Osteoimmunology and Beyond. Curr. Med. Chem. 2016, 23, 3754–3774. [Google Scholar] [CrossRef]
- Nakamura, M.C. Osteoimmunology: Entwined regulation of integrated systems. Semin. Immunopathol. 2019, 41, 547–549. [Google Scholar] [CrossRef]
- Weitzmann, M.N. Bone and the Immune System. Toxicol. Pathol. 2017, 45, 911–924. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.; Roger, P.-M. From Crosstalk between Immune and Bone Cells to Bone Erosion in Infection. Int. J. Mol. Sci. 2019, 20, 5154. [Google Scholar] [CrossRef] [PubMed]
- Ponzetti, M.; Rucci, N. Updates on Osteoimmunology: What’s New on the Cross-Talk Between Bone and Immune System. Front. Endocrinol. 2019, 10, 236. [Google Scholar] [CrossRef] [PubMed]
- Bultink, I.E.M. Bone Disease in Connective Tissue Disease/Systemic Lupus Erythematosus. Calcif. Tissue Int. 2017, 102, 575–591. [Google Scholar] [CrossRef]
- De Martinis, M.; Ciccarelli, F.; Sirufo, M.M.; Ginaldi, L. An overview of environmental risk factors in systemic sclerosis. Expert. Rev. Clin. Immunol. 2016, 12, 465–478. [Google Scholar] [CrossRef]
- Coury, F.; Peyruchaud, O.; Machuca-Gayet, I. Osteoimmunology of Bone Loss in Inflammatory Rheumatic Diseases. Front. Immunol. 2019, 10, 679. [Google Scholar] [CrossRef]
- Ralston, S.T.; Schett, G. Osteoimmunology. Calcif. Tissue Int. 2018, 102, 501–502. [Google Scholar] [CrossRef]
- Terashima, A.; Takayanagi, H. Overview of Osteoimmunology. Calcif. Tissue Int. 2018, 102, 503–511. [Google Scholar] [CrossRef]
- Giovos, G.; Yavropoulou, M.P.; Yovos, J.G. The role of cellular senescence in diabetes mellitus and osteoporosis: Molecular pathways and potential interventions. Hormones 2019, 18, 339–351. [Google Scholar] [CrossRef]
- Irelli, A.; Sirufo, M.M.; Scipioni, T.; De Pietro, F.; Pancotti, A.; Ginaldi, L.; De Martinis, M. mTOR Links Tumor Immunity and Bone Metabolism: What are the Clinical Implications? Int. J. Mol. Sci. 2019, 20, 5841. [Google Scholar] [CrossRef]
- Xiong, J.; Onal, M.; Jilka, R.L.; Weinstein, R.S.; Manolagas, S.C.; O’Brien, C.A. Matrix-embedded cells control osteoclast formation. Nat. Med. 2011, 17, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; O’Brien, C.A. Osteocyte RANKL: New insights into the control of bone remodeling. J. Bone Miner. Res. 2012, 27, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhang, L.; Xiong, Q.; Gao, Y.; Ge, W.; Tang, P. Bench-to-bedside strategies for osteoporotic fracture: From osteoimmunology to mechanosensation. Bone Res. 2019, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.C.; Takegahara, N.; Kim, H.; Choi, Y. Updating osteoimmunology: Regulation of bone cells by innate and adaptive immunity. Nat. Rev. Rheumatol. 2018, 14, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Amjadi-Moheb, F.; Akhavan-Niaki, H. Wnt signaling pathway in osteoporosis: Epigenetic regulation, interaction with other signaling pathways, and therapeutic promises. J. Cell. Physiol. 2019, 234, 14641–14650. [Google Scholar] [CrossRef]
- Azizieh, F.; Raghupathy, R.; Shehab, D.; Al-Jarallah, K.; Gupta, R. Cytokine profiles in osteoporosis suggest a proresorptive bias. Menopause 2017, 24, 1057–1064. [Google Scholar] [CrossRef]
- Zhang, J.; Fu, Q.; Ren, Z.; Wang, Y.; Wang, C.; Shen, T.; Wang, G.; Wu, L. Changes of serum cytokines-related Th1/Th2/Th17 concentration in patients with postmenopausal osteoporosis. Gynecol. Endocrinol. 2015, 31, 183–190. [Google Scholar] [CrossRef]
- Scheidt-Nave, C.; Bismar, H.; Leidig-Bruckner, G.; Woitge, H.; Seibel, M.J.; Ziegler, R.; Pfeilschifter, J. Serum interleukin 6 is a major predictor of bone loss in women specific to the first decade past menopause. J. Clin. Endocrinol. Metab. 2001, 86, 2032–2042. [Google Scholar]
- Edukulla, R.; Singh, B.; Jegga, A.G.; Sontake, V.; Dillon, S.R.; Madala, S.K. Th2 Cytokines Augment IL-31/IL-31RA Interactions via STAT6-dependent IL-31RA Expression. J. Boil. Chem. 2015, 290, 13510–13520. [Google Scholar] [CrossRef]
- Shen, J.; Shang, Q.; Wong, C.-K.; Li, E.K.; Kun, E.W.; Cheng, I.T.; Li, M.; Li, T.K.; Zhu, T.Y.; Yu, C.-M.; et al. Carotid plaque and bone density and microarchitecture in psoriatic arthritis: The correlation with soluble ST2. Sci. Rep. 2016, 6, 32116. [Google Scholar] [CrossRef]
- Tang, M.; Tian, L.; Luo, G.; Yu, X. Interferon-Gamma-Mediated Osteoimmunology. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.K.; Dar, H.Y.; Mishra, P.K. Immunoporosis: Immunology of Osteoporosis—Role of T Cells. Front. Immunol. 2018, 9, 657. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Yang, C.; Dai, H.; Li, J.; Liu, W.; Luo, Y.; Zhang, X.; Wang, Q. Crocin inhibits titanium particle-induced inflammation and promotes osteogenesis by regulating macrophage polarization. Int. Immunopharmacol. 2019, 76, 105865. [Google Scholar] [CrossRef] [PubMed]
- Catalan-Dibene, J.; McIntyre, L.L.; Zlotnik, A. Interleukin 30 to Interleukin 40. J. Interf. Cytokine Res. 2018, 38, 423–439. [Google Scholar] [CrossRef]
- Ginaldi, L.; De Martinis, M.; Ciccarelli, F.; Saitta, S.; Imbesi, S.; Mannucci, C.; Gangemi, S. Increased levels of interleukin 31 (IL-31) in osteoporosis. BMC Immunol. 2015, 16, 60. [Google Scholar] [CrossRef]
- Ginaldi, L.; De Martinis, M.; Saitta, S.; Sirufo, M.M.; Mannucci, C.; Casciaro, M.; Ciccarelli, F.; Gangemi, S. Interleukin-33 serum levels in postmenopausal women with osteoporosis. Sci. Rep. 2019, 9, 3786. [Google Scholar] [CrossRef]
- Imai, Y. Interleukin-33 in atopic dermatitis. J. Dermatol. Sci. 2019, 96, 2–7. [Google Scholar] [CrossRef]
- Vocca, L.; Di Sano, C.; Uasuf, C.G.; Sala, A.; Riccobono, L.; Gangemi, S.; Albano, G.D.; Bonanno, A.; Gagliardo, R.; Profita, M. IL-33/ST2 axis controls Th2/IL-31 and Th17 immune response in allergic airway diseases. Immunobiology 2015, 220, 954–963. [Google Scholar] [CrossRef]
- Di Salvo, E.; Ventura-Spagnolo, E.; Casciaro, M.; Navarra, M.; Gangemi, S. IL-33/IL-31 axis: A potential inflammatory pathway. Mediat. Inflamm. 2018. [Google Scholar] [CrossRef]
- Petra, A.I.; Tsilioni, I.; Taracanova, A.; Katsarou-Katsari, A.; Theoharides, T.C. Interleukin 33 and interleukin 4 regulate interleukin 31 gene expression and secretion from human laboratory of allergic diseases 2 mast cells stimulated by substance P and/or immunoglobulin E. Allergy Asthma Proc. 2018, 39, 153–160. [Google Scholar] [CrossRef]
- Stott, B.; Lavender, P.; Lehmann, S.; Pennino, D.; Durham, S.; Schmidt-Weber, C.B. Human IL-31 is induced by IL-4 and promotes TH2-driven inflammation. J. Allergy Clin. Immunol. 2013, 132, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Guarneri, F.; Minciullo, P.L.; Mannucci, C.; Calapai, F.; Saitta, S.; Cannavò, S.P.; Gangemi, S. IL-31 and IL-33 circulating levels in allergic contact dermatitis. Eur. Ann. Allergy Clin. Immunol. 2015, 47, 156–158. [Google Scholar]
- Murdaca, G.; Greco, M.; Tonacci, A.; Negrini, S.; Borro, M.; Puppo, F.; Gangemi, S. IL-33/IL-31 Axis in Immune-Mediated and Allergic Diseases. Int. J. Mol. Sci. 2019, 20, 5856. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Zhou, Q.; Liu, C.; Ying, M.; Xu, S. Increased plasma IL-17, IL-31, and IL-33 levels in chronic spontaneous urticaria. Sci. Rep. 2017, 7, 17797. [Google Scholar] [CrossRef] [PubMed]
- Perrigoue, J.G.; Li, J.; Zaph, C.; Goldschmidt, M.; Scott, P.; De Sauvage, F.J.; Pearce, E.J.; Ghilardi, N.; Artis, D. IL-31–IL-31R interactions negatively regulate type 2 inflammation in the lung. J. Exp. Med. 2007, 204, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Broxmeyer, H.E.; Li, J.; Hangoc, G.; Cooper, S.; Tao, W.; Mantel, C.; Graham-Evans, B.; Ghilardi, N.; De Sauvage, F.J. Regulation of myeloid progenitor cell proliferation/survival by IL-31 receptor and IL-31. Exp. Hematol. 2007, 35, 78–86. [Google Scholar] [CrossRef]
- Rosine, N.; Etcheto, A.; Hendel-Chavez, H.; Seror, R.; Briot, K.; Molto, A.; Chanson, P.; Taoufik, Y.; Wendling, D.; Lories, R.; et al. Increase In Il-31 Serum Levels Is Associated With Reduced Structural Damage In Early Axial Spondyloarthritis. Sci. Rep. 2018, 8, 7731. [Google Scholar] [CrossRef]
- Kasraie, S.; Niebuhr, M.; Werfel, T. Interleukin (IL)-31 induces pro-inflammatory cytokines in human monocytes and macrophages following stimulation with staphylococcal exotoxins. Allergy 2010, 65, 712–721. [Google Scholar] [CrossRef]
- Park, K.; Park, J.H.; Yang, W.J.; Lee, J.J.; Song, M.J.; Kim, H.P. Transcriptional activation of the IL31 gene by NFAT and STAT6. J. Leukoc. Biol. 2012, 91, 245–257. [Google Scholar] [CrossRef]
- Hamann, C.R.; Thyssen, J.P. Monoclonal antibodies against interleukin 13 and interleukin 31RA in development for atopic dermatitis. J. Am. Acad. Dermatol. 2018, 78, S37–S42. [Google Scholar] [CrossRef]
- Chen, Z.; Yue, S.X.; Zhou, G.; Greenfield, E.M.; Murakami, S. ERK1 and ERK2 regulate chondrocyte terminal differentiation during endochondral bone formation. J. Bone Miner. Res. 2015, 30, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, C.; Lüscher-Firzlaff, J.; Baron, J.M.; Lüscher, B. Signaling by IL-31 and functional consequences. Eur. J. Cell Boil. 2012, 91, 552–566. [Google Scholar] [CrossRef] [PubMed]
- De Martinis, M.; Franceschi, C.; Monti, D.; Ginaldi, L. Inflamm-ageing and lifelong antigenic load as major determinants of ageing rate and longevity. FEBS Lett. 2005, 579, 2035–2039. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Jenkins, B.J. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat. Rev. Immunol. 2018, 18, 773–789. [Google Scholar] [CrossRef]
- Miake, S.; Tsuji, G.; Takemura, M.; Hashimoto-Hachiya, A.; Vu, Y.H.; Furue, M.; Nakahara, T. IL-4 Augments IL-31/IL-31 Receptor Alpha Interaction Leading to Enhanced Ccl 17 and Ccl 22 Production in Dendritic Cells: Implications for Atopic Dermatitis. Int. J. Mol. Sci. 2019, 20, 4053. [Google Scholar] [CrossRef]
- Wehenkel, M.; Corr, M.; Guy, C.S.; Edwards, B.A.; Castellaw, A.H.; Calabrese, C.; Pagès, G.; Pouysségur, J.; Vogel, P.; McGargill, M.A. Extracellular Signal-Regulated Kinase Signaling in CD4-Expressing Cells Inhibits Osteochondromas. Front. Immunol. 2017, 8, 577. [Google Scholar] [CrossRef]
- Zeng, X.; Zhang, Z.; Gao, Q.-Q.; Wang, Y.-Y.; Yu, X.-Z.; Zhou, B.; Xi, M.-R. Clinical Significance of Serum Interleukin-31 and Interleukin-33 Levels in Patients of Endometrial Cancer: A Case Control Study. Dis. Markers 2016, 2016, 1–7. [Google Scholar] [CrossRef]
- Perrigoue, J.G.; Zaph, C.; Guild, K.; Du, Y.; Artis, D. IL-31-IL-31R interactions limit the magnitude of Th2 cytokine-dependent immunity and inflammation following intestinal helminth infection. J. Immunol. 2009, 182, 6088–6094. [Google Scholar] [CrossRef]
- Schnittker, D.; Kwofie, K.; Ashkar, A.; Trigatti, B.; Richards, C.D. Oncostatin M and TLR-4 Ligand Synergize to Induce MCP-1, IL-6, and VEGF in Human Aortic Adventitial Fibroblasts and Smooth Muscle Cells. Mediat. Inflamm. 2013, 2013, 1–14. [Google Scholar] [CrossRef]
- Cornelissen, C.; Brans, R.; Czaja, K.; Skazik, C.; Marquardt, Y.; Zwadlo-Klarwasser, G.; Kim, A.; Bickers, D.; Luscher-Firzlaff, J.; Lüscher, B.; et al. Ultraviolet B radiation and reactive oxygen species modulate interleukin-31 expression in T lymphocytes, monocytes and dendritic cells. Br. J. Dermatol. 2011, 165, 966–975. [Google Scholar] [CrossRef]
- Azizieh, F.Y.; Shehab, D.; Al Jarallah, K.; Mojiminiyi, O.; Gupta, R.; Raghupathy, R. Circulatory pattern of cytokines, adipokines and bone markers in postmenopausal women with low BMD. J. Inflamm. Res. 2019, 12, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Dattagupta, A.; Immaneni, S.; Sathyamurthy, I. ST2: Current status. Indian Heart J. 2018, 70, S96–S101. [Google Scholar] [PubMed]
- Villarreal, D.O.; Weiner, D.B. Interleukin 33: A switch-hitting cytokine. Curr. Opin. Immunol. 2014, 28, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Macari, S.; Madeira, M.F.M.; Lima, I.L.A.; Pereira, T.S.F.; Dias, G.J.; Cirelli, J.A.; De Molon, R.S.; Fukada, S.Y.; Szawka, R.E.; Garlet, G.P.; et al. ST2 regulates bone loss in a site-dependent and estrogen-dependent manner. J. Cell. Biochem. 2018, 119, 8511–8521. [Google Scholar]
- Miller, A.M. Role of IL-33 in inflammation and disease. J. Inflamm. 2011, 8, 22. [Google Scholar] [CrossRef]
- Ilesanmi-Oyelere, B.L.; Schollum, L.; Kuhn-Sherlock, B.; McConnell, M.; Mros, S.; Coad, J.; Roy, N.C.; Kruger, M.C. Inflammatory markers and bone health in postmenopausal women: A cross-sectional overview. Immun. Ageing 2019, 16, 15. [Google Scholar] [CrossRef]
- Molofsky, A.B.; Savage, A.K.; Locksley, R.M. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity 2015, 42, 1005–1019. [Google Scholar] [CrossRef]
- Laperine, O.; Cloitre, A.; Caillon, J.; Huck, O.; Bugueno, I.M.; Pilet, P.; Sourice, S.; Le Tilly, E.; Palmer, G.; Davideau, J.-L.; et al. Interleukin-33 and RANK-L Interplay in the Alveolar Bone Loss Associated to Periodontitis. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E. HMGB1, IL-1α, IL-33 and S100 proteins: Dual-function alarmins. Cell. Mol. Immunol. 2017, 14, 43–64. [Google Scholar] [CrossRef]
- Schulze, J.; Bickert, T.; Beil, F.T.; Zaiss, M.M.; Albers, J.; Wintges, K.; Streichert, T.; Klaetschke, K.; Keller, J.; Hissnauer, T.-N.; et al. Interleukin-33 is expressed in differentiated osteoblasts and blocks osteoclast formation from bone marrow precursor cells. J. Bone Miner. Res. 2011, 26, 704–717. [Google Scholar]
- Okragly, A.J.; Hamang, M.J.; Pena, E.A.; Baker, H.E.; Bullock, H.A.; Lucchesi, J.; Martín, A.P.; Ma, Y.L.; Benschop, R.J. Elevated levels of Interleukin (IL)-33 induce bone pathology but absence of IL-33 does not negatively impact normal bone homeostasis. Cytokine 2016, 79, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Lima, I.L.; Macari, S.; Madeira, M.F.; Rodrigues, L.F.; Colavite, P.M.; Garlet, G.P.; Soriani, F.M.; Teixeira, M.M.; Fukada, S.Y.; Silva, T.A. Osteoprotective Effects of IL-33/ST2 Link to Osteoclast Apoptosis. Am. J. Pathol. 2015, 185, 3338–3348. [Google Scholar] [CrossRef] [PubMed]
- Omata, Y.; Frech, M.; Primbs, T.; Lucas, S.; Andreev, D.; Scholtysek, C.; Sarter, K.; Kindermann, M.; Yeremenko, N.; Baeten, D.L.; et al. Group 2 Innate Lymphoid Cells Attenuate Inflammatory Arthritis and Protect from Bone Destruction in Mice. Cell Rep. 2018, 24, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Stier, M.T.; Zhang, J.; Goleniewska, K.; Cephus, J.Y.; Rusznak, M.; Wu, L.; Van Kaer, L.; Zhou, B.; Newcomb, D.C.; Peebles, R.S. IL-33 promotes the egress of group 2 innate lymphoid cells from the bone marrow. J. Exp. Med. 2018, 215, 263–281. [Google Scholar] [CrossRef] [PubMed]
- Johnston, L.K.; Hsu, C.-L.; Krier-Burris, R.A.; Chhiba, K.D.; Chien, K.B.; McKenzie, A.; Berdnikovs, S.; Bryce, P.J. IL-33 Precedes IL-5 in Regulating Eosinophil Commitment and Is Required for Eosinophil Homeostasis. J. Immunol. 2016, 197, 3445–3453. [Google Scholar] [CrossRef] [PubMed]
- Sirufo, M.M.; Suppa, M.; Ginaldi, L.; De Martinis, M. Does Allergy Break Bones? Osteoporosis and Its Connection to Allergy. Int. J. Mol. Sci. 2020, 21, 712. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, M.M.; Kurowska-Stolarska, M.; Böhm, C.; Gary, R.; Scholtysek, C.; Stolarski, B.; Reilly, J.; Kerr, S.; Millar, N.L.; Kamradt, T.; et al. IL-33 Shifts the Balance from Osteoclast to Alternatively Activated Macrophage Differentiation and Protects from TNF-α–Mediated Bone Loss. J. Immunol. 2011, 186, 6097–6105. [Google Scholar] [CrossRef]
- Da Luz, F.A.C.; Oliveira, A.P.L.; Borges, D.; Brígido, P.C.; Silva, M.J.B. The Physiopathological Role of IL-33: New Highlights in Bone Biology and a Proposed Role in Periodontal Disease. Mediat. Inflamm. 2014, 2014, 1–8. [Google Scholar] [CrossRef]
- Saleh, H.; Eeles, D.; Hodge, J.M.; Nicholson, G.C.; Gu, R.; Pompolo, S.; Gillespie, M.T.; Quinn, J.M.W. Interleukin-33, a Target of Parathyroid Hormone and Oncostatin M, Increases Osteoblastic Matrix Mineral Deposition and Inhibits Osteoclast Formation in Vitro. Endocrinology 2011, 152, 1911–1922. [Google Scholar] [CrossRef]
- Saidi, S.; Bouri, F.; Lencel, P.; Duplomb, L.; Baud’huin, M.; Delplace, S.; Leterme, D.; Miellot, F.; Heymann, D.; Hardouin, P.; et al. IL-33 is expressed in human osteoblasts, but has no direct effect on bone remodeling. Cytokine 2011, 53, 347–354. [Google Scholar] [CrossRef]
- Keller, J.; Catala-Lehnen, P.; Wintges, K.; Schulze, J.; Bickert, T.; Ito, W.; Horst, A.K.; Amling, M.; Schinke, T. Transgenic over-expression of interleukin-33 in osteoblasts results in decreased osteoclastogenesis. Biochem. Biophys. Res. Commun. 2012, 417, 217–222. [Google Scholar] [CrossRef]
- Mun, S.H.; Ko, N.Y.; Kim, H.S.; Kim, J.W.; Kim, D.K.; Kim, A.R.; Lee, S.H.; Kim, Y.G.; Lee, C.K.; Lee, S.H.; et al. Interleukin-33 stimulates formation of functional osteoclasts from human CD14+ monocytes. Cell. Mol. Life Sci. 2010, 67, 3883–3892. [Google Scholar] [CrossRef] [PubMed]
- Kiyomiya, H.; Ariyoshi, W.; Okinaga, T.; Kaneuji, T.; Mitsugi, S.; Sakurai, T.; Habu, M.; Yoshioka, I.; Tominaga, K.; Nishihara, T. IL-33 inhibits RANKL-induced osteoclast formation through the regulation of Blimp-1 and IRF-8 expression. Biochem. Biophys. Res. Commun. 2015, 460, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhao, Y.; Jiang, Y.; Qin, T.; Chen, J.; Chu, X.; Yi, Q.; Gao, S.; Wang, S. Dectin-1 signaling inhibits osteoclastogenesis via IL-33-induced inhibition of NFATc1. Oncotarget 2017, 8, 53366–53374. [Google Scholar] [CrossRef] [PubMed]
- Velickovic, M.; Pejnovic, N.; Mitrovic, S.; Radosavljevic, G.; Jovanovic, I.; Kanjevac, T.; Jovicic, N.; Lukic, A. ST2 Deletion Increases Inflammatory Bone Destruction in Experimentally Induced Periapical Lesions in Mice. J. Endod. 2015, 41, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Mine, Y.; Makihira, S.; Yamaguchi, Y.; Tanaka, H.; Nikawa, H. Involvement of ERK and p38 MAPK pathways on Interleukin-33-induced RANKL expression in osteoblastic cells. Cell Boil. Int. 2014, 38, 655–662. [Google Scholar] [CrossRef]
- Eeles, D.G.; Hodge, J.M.; Singh, P.P.; Schuijers, J.A.; Grills, B.L.; Gillespie, M.T.; Myers, D.E.; Quinn, J.M. Osteoclast formation elicited by interleukin-33 stimulation is dependent upon the type of osteoclast progenitor. Mol. Cell. Endocrinol. 2015, 399, 259–266. [Google Scholar] [CrossRef]
- Madel, M.-B.; Ibáñez, L.; Wakkach, A.; De Vries, T.J.; Teti, A.; Apparailly, F.; Blin-Wakkach, C. Immune Function and Diversity of Osteoclasts in Normal and Pathological Conditions. Front. Immunol. 2019, 10, 1408. [Google Scholar] [CrossRef]
- Massimini, M.; Palmieri, C.; De Maria, R.; Romanucci, M.; Malatesta, D.; De Martinis, M.; Maniscalco, L.; Ciccarelli, A.; Ginaldi, L.; Buracco, P.; et al. 17-AAG and Apoptosis, Autophagy and Mitophagy in 142 Canine Osteosarcoma Cell Lines. Vet. Pathol. 2017, 54, 405–412. [Google Scholar] [CrossRef]
- Komori, T. Cell Death in Chondrocytes, Osteoblasts, and Osteocytes. Int. J. Mol. Sci. 2016, 17, 2045. [Google Scholar] [CrossRef]
- Xiao, L.; Xiao, Y. The Autophagy in Osteoimmonology: Self-Eating, Maintenance, and Beyond. Front. Endocrinol. 2019, 10, 490. [Google Scholar] [CrossRef] [PubMed]
- Arshad, M.I.; Khan, H.A.; Noel, G.; Piquet-Pellorce, C.; Samson, M. Potential Therapeutic Aspects of Alarmin Cytokine Interleukin 33 or Its Inhibitors in Various Diseases. Clin. Ther. 2016, 38, 1000–1016. [Google Scholar] [CrossRef] [PubMed]
- Mannucci, C.; Calapai, G.; Gangemi, S. Commentary: Circulatory pattern of cytokines, adipokines and bone markers in postmenopausal women with low BMD. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yang, G.; Yang, R.; Peng, X.; Li, J. Interleukin-33 and receptor ST2 as indicators in patients with asthma: A meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 14935–14943. [Google Scholar] [PubMed]
- Xu, W.-D.; Zhang, M.; Zhang, Y.-J.; Ye, N.-Q. IL-33 in rheumatoid arthritis: Potential role in pathogenesis and therapy. Hum. Immunol. 2013, 74, 1057–1060. [Google Scholar] [CrossRef]
- Rostan, O.; Arshad, M.I.; Piquet-Pellorce, C.; Robert-Gangneux, F.; Gangneux, J.-P.; Samson, M. Crucial and Diverse Role of the Interleukin-33/ST2 Axis in Infectious Diseases. Infect. Immun. 2015, 83, 1738–1748. [Google Scholar] [CrossRef]
- López-Mejias, R.; Genre, F.; Remuzgo-Martínez, S.; Robustillo-Villarino, M.; García-Bermúdez, M.; Llorca, J.; Corrales, A.; Gonzalez-Juanatey, C.; Ubilla, B.; Miranda-Filloy, J.A.; et al. Protective Role of the Interleukin 33 rs3939286 Gene Polymorphism in the Development of Subclinical Atherosclerosis in Rheumatoid Arthritis Patients. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Grau-Vorster, M.; Rodríguez, L.; Torrents-Zapata, S.; Vivas, D.; Codinach, M.; Blanco, M.; Oliver-Vila, I.; García-López, J.; Vives, J. Levels of IL-17F and IL-33 correlate with HLA-DR activation in clinical-grade human bone marrow–derived multipotent mesenchymal stromal cell expansion cultures. Cytotherapy 2019, 21, 32–40. [Google Scholar] [CrossRef]
- Fischer, L.; Herkner, C.; Kitte, R.; Dohnke, S.; Riewaldt, J.; Kretschmer, K.; Garbe, A.I. Foxp3+ regulatory T cells in bone and hematopoietic homeostasis. Foxp3+ Regulatory T Cells in Bone and Hematopoietic Homeostasis. Front. Endocrinol. 2019, 10, 578. [Google Scholar] [CrossRef]
- Metzger, C.E.; Narayanan, S.A. The Role of Osteocytes in Inflammatory Bone Loss. Front. Endocrinol. 2019, 10, 285. [Google Scholar] [CrossRef]
- Chicana, B.; Donham, C.; Millan, A.J.; Manilay, J.O. Wnt Antagonists in Hematopoietic and Immune Cell Fate: Implications for Osteoporosis Therapies. Curr. Osteoporos. Rep. 2019, 17, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Takayanagi, H. Osteoimmunology. Cold Spring Harb. Perspect. Med. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Barbarash, O.; Gruzdeva, O.; Uchasova, E.; Dyleva, Y.; Belik, E.; Akbasheva, O.; Karetnikova, V.; Shilov, A. Prognostic Value of Soluble ST2 During Hospitalization for ST-Segment Elevation Myocardial Infarction. Ann. Lab. Med. 2016, 36, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Maupin, K.A.; Himes, E.R.; Plett, A.P.; Chua, H.L.; Singh, P.; Ghosh, J.; Mohamad, S.F.; Abeysekera, I.; Fisher, A.; Sampson, C.; et al. Aging negatively impacts the ability of megakaryocytes to stimulate osteoblast proliferation and bone mass. Bone 2019, 127, 452–459. [Google Scholar] [CrossRef]
- Li, J.; Ayoub, A.; Xiu, Y.; Yin, X.; Sanders, J.O.; Mesfin, A.; Xing, L.; Yao, Z.; Boyce, B.F. TGFβ-induced degradation of TRAF3 in mesenchymal progenitor cells causes age-related osteoporosis. Nat. Commun. 2019, 10, 2795. [Google Scholar] [CrossRef]
- Zupan, J.; Komadina, R.; Marc, J. The relationship between osteoclastogenic and anti-osteoclastogenic pro-inflammatory cytokines differs in human osteoporotic and osteoarthritic bone tissues. J. Biomed. Sci. 2012, 19, 28. [Google Scholar] [CrossRef]
- Mahbub, S.; Deburghgraeve, C.R.; Kovacs, E.J. Advanced Age Impairs Macrophage Polarization. J. Interferon Cytokine Res. 2012, 32, 18–26. [Google Scholar] [CrossRef]
- Menzies, F.M.; Henriquez, F.L.; Alexander, J.; Roberts, C.W. Selective inhibition and augmentation of alternative macrophage activation by progesterone. Immunology 2011, 134, 281–291. [Google Scholar] [CrossRef]
- Cai, S.-Y.; Ge, M.; Mennone, A.; Hoque, R.; Ouyang, X.; Boyer, J.L. Inflammasome Is Activated in the Liver of Cholestatic Patients and Aggravates Hepatic Injury in Bile Duct–Ligated Mouse. Cell. Mol. Gastroenterol. Hepatol. 2019. [Google Scholar] [CrossRef]
- Piehler, D.; Eschke, M.; Schulze, B.; Protschka, M.; Müller, U.; Grahnert, A.; Richter, T.; Heyen, L.; Köhler, G.; Brombacher, F.; et al. The IL-33 receptor (ST2) regulates early IL-13 production in fungus-induced allergic airway inflammation. Mucosal Immunol. 2016, 9, 937–949. [Google Scholar] [CrossRef]
- Engelbertsen, D.; Lichtman, A.H. Innate lymphoid cells in atherosclerosis. Eur. J. Pharmacol. 2017, 816, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tang, J.; Yang, X.; Zanvit, P.; Cui, K.; Ku, W.L.; Jin, W.; Zhang, D.; Goldberg, N.; Cain, A.; et al. TGF-β induces ST2 and programs ILC2 development. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tan, J.; Martino, M.M.; Lui, K.O. Regulatory T-Cells: Potential Regulator of Tissue Repair and Regeneration. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Kuswanto, W.; Burzyn, D.; Panduro, M.; Wang, K.K.; Jang, Y.C.; Wagers, A.J.; Benoist, C.; Mathis, D. Poor Repair of Skeletal Muscle in Aging Mice Reflects a Defect in Local, Interleukin-33-Dependent Accumulation of Regulatory T Cells. Immunity 2016, 44, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Pereira, M.G.; Ciciliot, S.; Rovere-Querini, P. Regulatory T cells and skeletal muscle regeneration. FEBS J. 2016, 284, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhang, L.; Wang, D.; Aiqudsy, L.; Jiang, J.X.; Xu, H.; Shang, P. Muscle-bone crosstalk and potential therapies for sarco-osteoporosis. J. Cell. Biochem. 2019, 120, 14262–14273. [Google Scholar] [CrossRef]
- Kovats, S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell. Immunol. 2015, 294, 63–69. [Google Scholar] [CrossRef]
- Hayashi, M.; Nakashima, T.; Yoshimura, N.; Okamoto, K.; Tanaka, S.; Takayanagi, H. Autoregulation of Osteocyte Sema3A Orchestrates Estrogen Action and Counteracts Bone Aging. Cell Metab. 2019, 29, 627–637. [Google Scholar] [CrossRef]
- Fischer, V.; Kalbitz, M.; Müller-Graf, F.; Gebhard, F.; Ignatius, A.; Liedert, A.; Haffner-Luntzer, M. Influence of Menopause on Inflammatory Cytokines during Murine and Human Bone Fracture Healing. Int. J. Mol. Sci. 2018, 19, 2070. [Google Scholar] [CrossRef]
- Cheloha, R.W.; Gellman, S.H.; Vilardaga, J.-P.; Gardella, T.J. PTH receptor-1 signalling—Mechanistic insights and therapeutic prospects. Nat. Rev. Endocrinol. 2015, 11, 712–724. [Google Scholar] [CrossRef]
- Gao, M.; Yao, X.; Ding, J.; Hong, R.; Wu, Y.; Huang, H.; Zhuang, L.; Li, Z.; Wang, Y.; Zhang, Y.; et al. Low levels of vitamin D and the relationship between vitamin D and Th2 axis-related cytokines in neuromyelitis optica spectrum disorders. J. Clin. Neurosci. 2019, 61, 22–27. [Google Scholar] [CrossRef] [PubMed]
- De Martinis, M.; Ginaldi, L.; Sirufo, M.M.; Pioggia, G.; Calapai, G.; Gangemi, S.; Mannucci, C. Alarmins in osteoporosis, RAGE and IL-33 pathways: A Literature Review. Medicina 2020. under review. [Google Scholar]
- Raggatt, L.J.; Qin, L.; Tamasi, J.; Jefcoat, S.C., Jr.; Shimizu, E.; Selvamurugan, N.; Liew, F.Y.; Bevelock, L.; Feyen, J.H.; Partridge, N.C. Interleukin-18 is regulated by parathyroid hormone and is required for its bone anabolic actions. J. Biol. Chem. 2008, 283, 6790–6798. [Google Scholar] [CrossRef]
- Saluja, R.; Ketelaar, M.E.; Hawro, T.; Church, M.K.; Maurer, M.; Nawijn, M.C. The role of the IL-33/IL-1RL1 axis in mast cell and basophil activation in allergic disorders. Mol. Immunol. 2015, 63, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Griesenauer, B.; Paczesny, S. The ST2/IL-33 Axis in Immune Cells during Inflammatory Diseases. Front. Immunol. 2017, 8, 5207. [Google Scholar] [CrossRef] [PubMed]
- Liew, F.Y. IL-33: A Janus cytokine. Ann. Rheum. Dis. 2012, 71, 101–104. [Google Scholar] [CrossRef]
- Wang, D.; Hu, K.; Gao, N.; Zhang, H.; Jiang, Y.; Liu, C.; Wang, S.; Zhao, Z. High throughput screening of cytokines, chemokines and matrix metalloproteinases in wound fluid induced by mammary surgery. Oncotarget 2015, 6, 29296–29310. [Google Scholar] [CrossRef]
- Brylka, L.J.; Schinke, T. Chemokines in Physiological and Pathological Bone Remodeling. Front. Immunol. 2019, 10, 2182. [Google Scholar] [CrossRef]
- Amarasekara, D.S.; Yun, H.; Kim, S.; Lee, N.; Kim, H.; Rho, J. Regulation of Osteoclast Differentiation by Cytokine Networks. Immune Netw. 2018, 18, 8. [Google Scholar] [CrossRef]
- Xiao, L.; Zhou, Y.; Friis, T.; Beagley, K.; Xiao, Y. S1P-S1PR1 Signaling: The “Sphinx” in Osteoimmunology. Front. Immunol. 2019, 10, 1409. [Google Scholar] [CrossRef]
- Chae, W.-J.; Ehrlich, A.K.; Chan, P.Y.; Teixeira, A.M.; Henegariu, O.; Hao, L.; Shin, J.H.; Park, J.-H.; Tang, W.H.; Kim, S.-T.; et al. The Wnt Antagonist Dickkopf-1 Promotes Pathological Type 2 Cell-Mediated Inflammation. Immunity 2016, 44, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Ciccarelli, F.; De Martinis, M.; Sirufo, M.M.; Ginaldi, L. Psoriasis Induced by Anti-Tumor Necrosis Factor Alpha Agents: A Comprehensive Review of the Literature. Acta Derm. Croat 2016, 24, 169–174. [Google Scholar]
- Folkert, I.W.; Devalaraja, S.; Linette, G.P.; Weber, K.; Haldar, M. Primary Bone Tumors: Challenges and Opportunities for CAR-T Therapies. J. Bone Miner. Res. 2019, 34, 1780–1788. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.T.; Martin, M.U. Interleukin 33 is a guardian of barriers and a localalarmin. Nat. Immunol. 2016, 17, 122–131. [Google Scholar] [CrossRef]
- Lavric, M.; Miranda-García, M.A.; Holzinger, D.; Foell, D.; Wittkowski, H. Alarmins firing arthritis: Helpful diagnostic tools and promising therapeutic targets. Jt. Bone Spine 2017, 84, 401–410. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| IL | Cytokine Family | Expression | Homeostatic Function | Receptors | Effects on Bone |
|---|---|---|---|---|---|
| IL-31 | IL-6 family cytokine | Activated monocytes, myeloid progenitors, macrophages, eosinophils dendritic and CD45RO+ Th2 cells | Multiple regulatory functions on cell cycle and tissue remodeling, tissue damage and repair processes; Th2 response induction; EGF and VEGF upregulation | IL-31RA/OSMR | OPC proliferation; monocyte/macrophage activation; increased proinflammatory cytokine gene transcription, production of osteoclastogenic cytokines, matrix metalloproteinases and chemokines; Th1/Th17 lymphocyte differentiation; APC function regulation; tumorigenesis and bone metastases [29,38,102] |
| IL-33 | IL-1 family cytokine; alarmin | Multiple organs and cell types (mainly bone marrow stromal cells, fibroblasts, epithelial and endothelial cells) following pro-inflammatory stimulation | Alerting for tissue damage; gene transcription control; immune response amplification and tissue repair during cell injury; Th2 cell maturation and chemoattraction; Th2-associated cytokine induction; eosinophil activation and mast cell degranulation; transcription of genes involved in inflammatory responses, including IL-31; Foxp3+ Treg cell induction | IL1RL1/ST2 | Repression of NF-kB transcription; NFATc1 inactivation; inhibition of RANK/RANKL signaling; bone resorption decrease; OC apoptosis induction; OB function stimulation; anti-osteoclastogenic cytokine induction; maintenance of architectural bone structure in response to mechanical stimulations; OC to macrophage switch in myeloid bone marrow progenitors [29,34,108,109] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Martinis, M.; Sirufo, M.M.; Suppa, M.; Ginaldi, L. IL-33/IL-31 Axis in Osteoporosis. Int. J. Mol. Sci. 2020, 21, 1239. https://doi.org/10.3390/ijms21041239
De Martinis M, Sirufo MM, Suppa M, Ginaldi L. IL-33/IL-31 Axis in Osteoporosis. International Journal of Molecular Sciences. 2020; 21(4):1239. https://doi.org/10.3390/ijms21041239
Chicago/Turabian StyleDe Martinis, Massimo, Maria Maddalena Sirufo, Mariano Suppa, and Lia Ginaldi. 2020. "IL-33/IL-31 Axis in Osteoporosis" International Journal of Molecular Sciences 21, no. 4: 1239. https://doi.org/10.3390/ijms21041239
APA StyleDe Martinis, M., Sirufo, M. M., Suppa, M., & Ginaldi, L. (2020). IL-33/IL-31 Axis in Osteoporosis. International Journal of Molecular Sciences, 21(4), 1239. https://doi.org/10.3390/ijms21041239