Mechanical Postconditioning Promotes Glucose Metabolism and AMPK Activity in Parallel with Improved Post-Ischemic Recovery in an Isolated Rat Heart Model of Donation after Circulatory Death

 and
and
Abstract
1. Introduction
2. Results
2.1. Baseline Characteristics
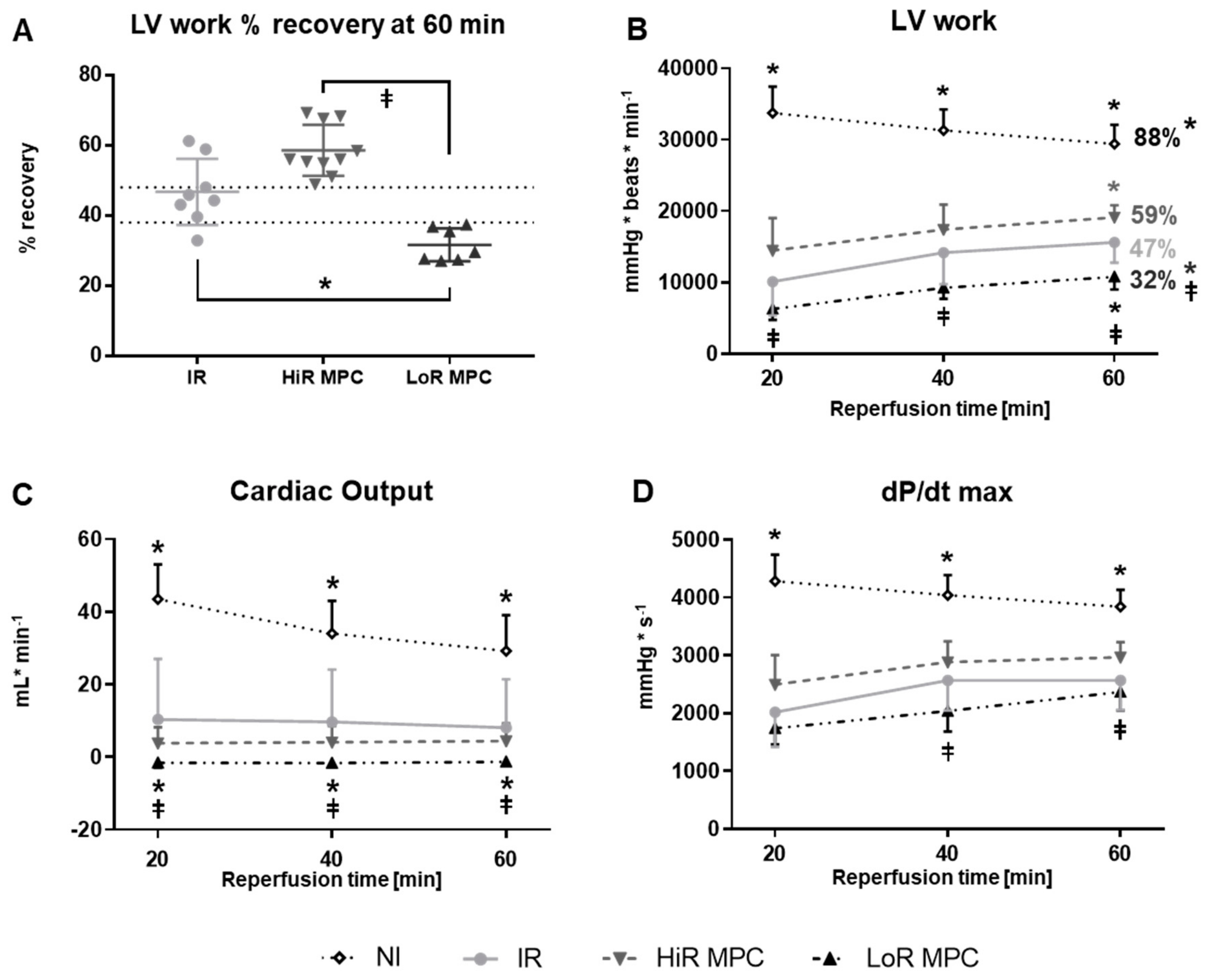
2.2. Post-Ischemic Cardiac Functional Recovery
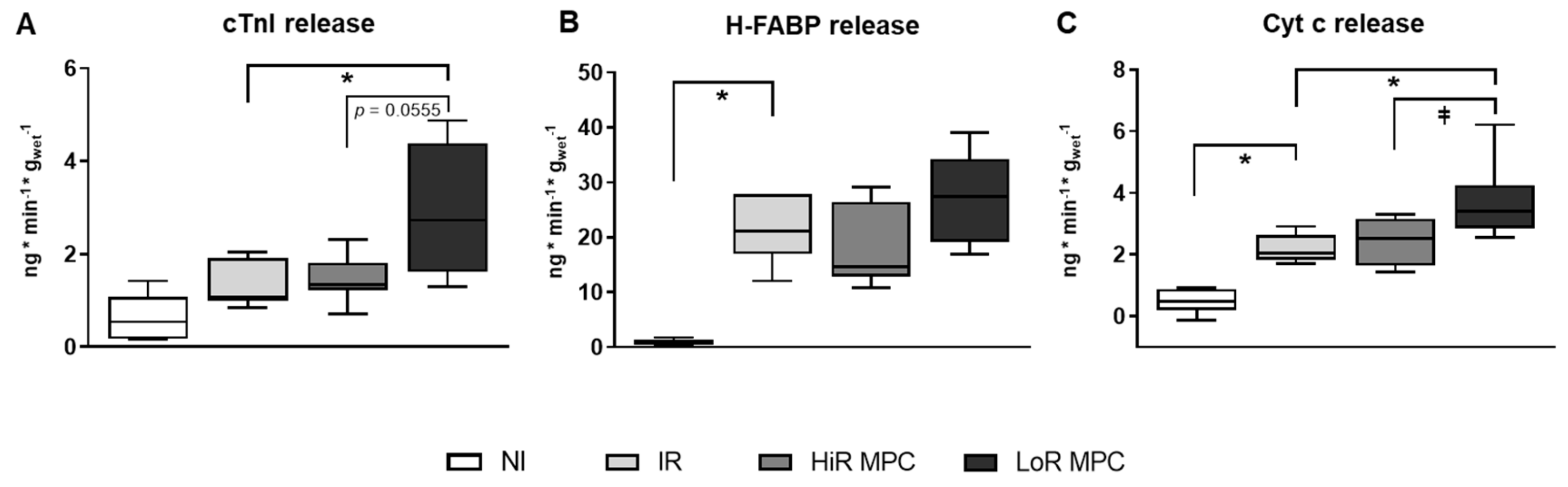
2.3. Markers of Cell Damage
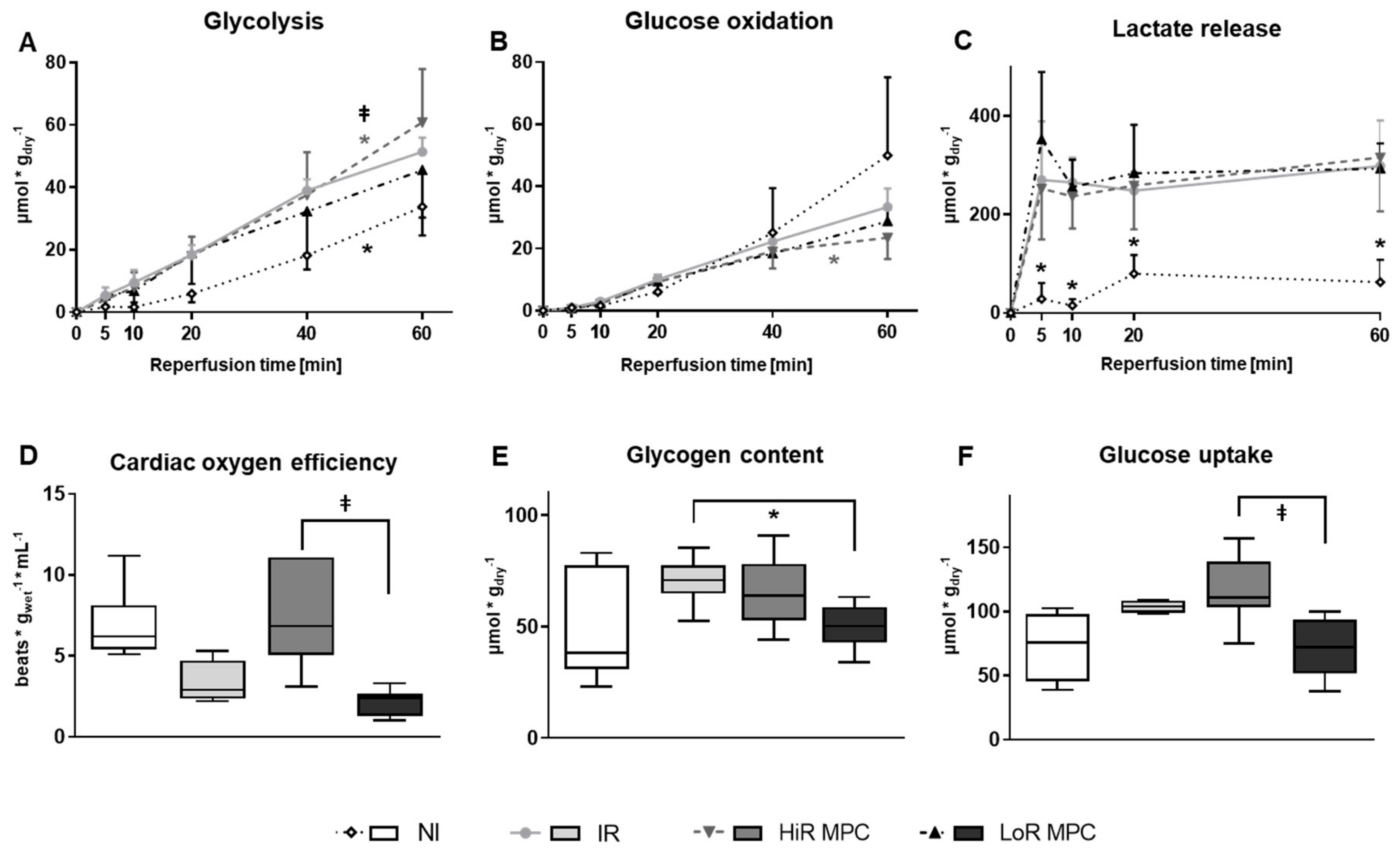
2.4. Post-Ischemic Metabolic Recovery
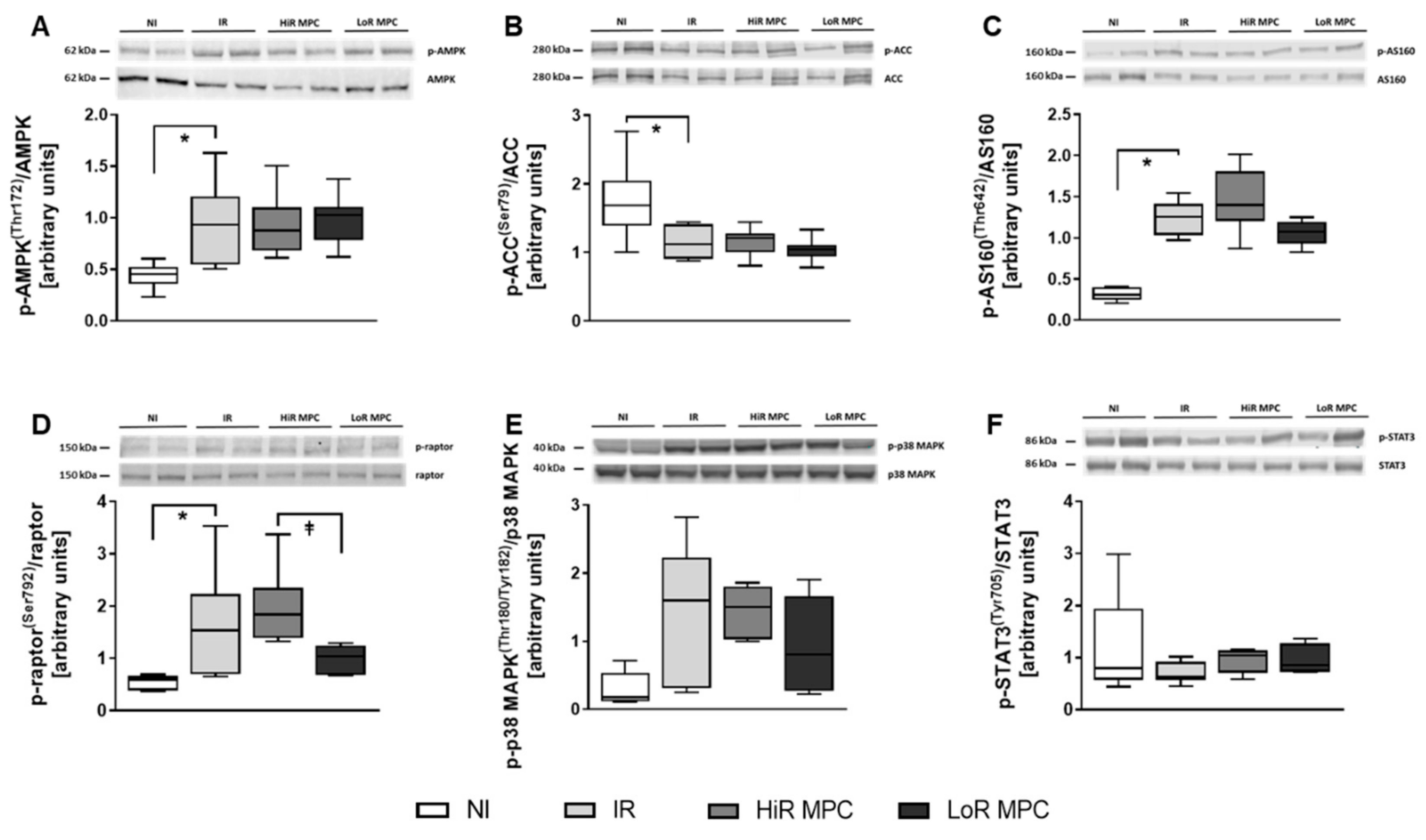
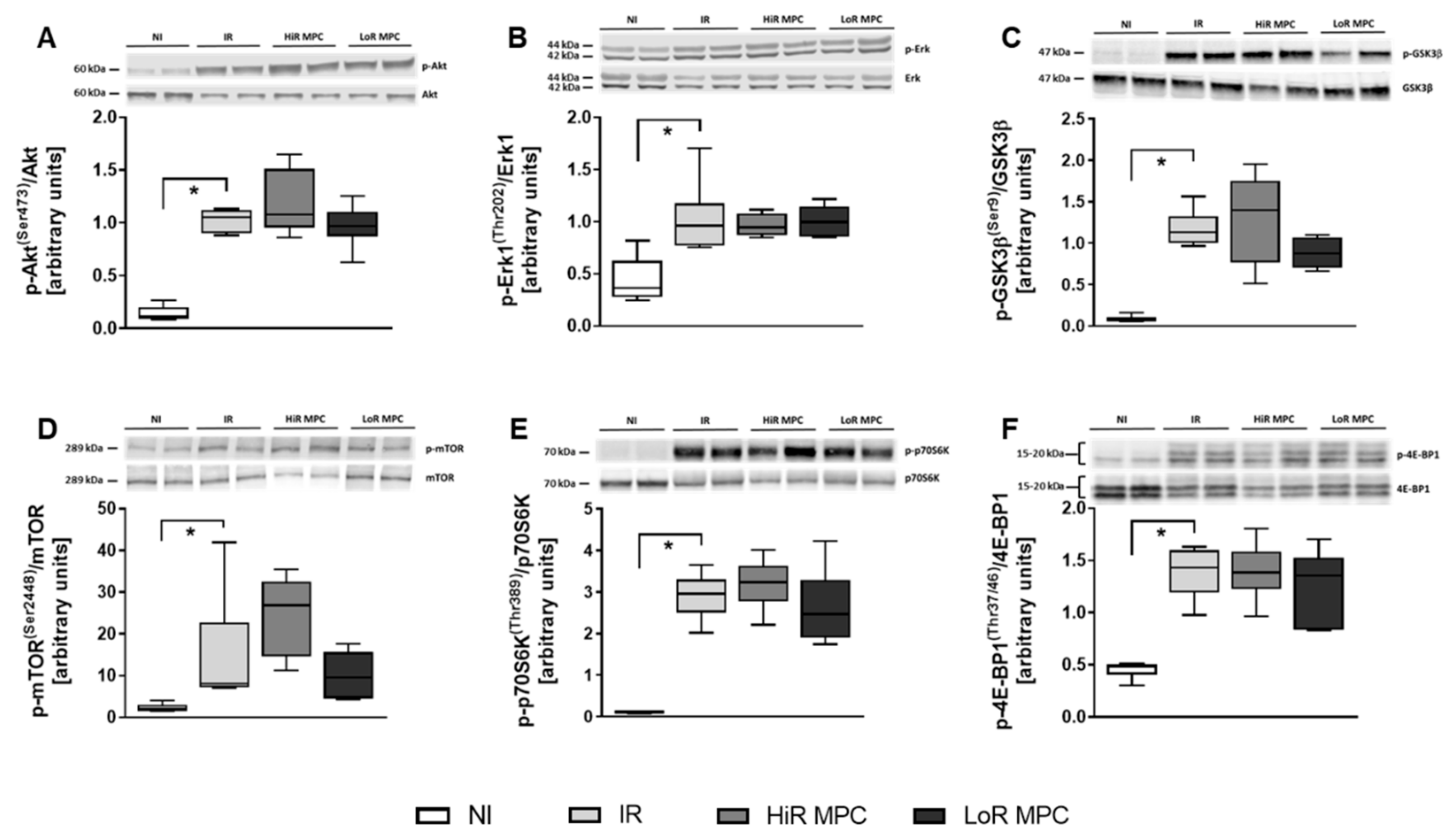
2.5. Intracellular Signaling Pathways
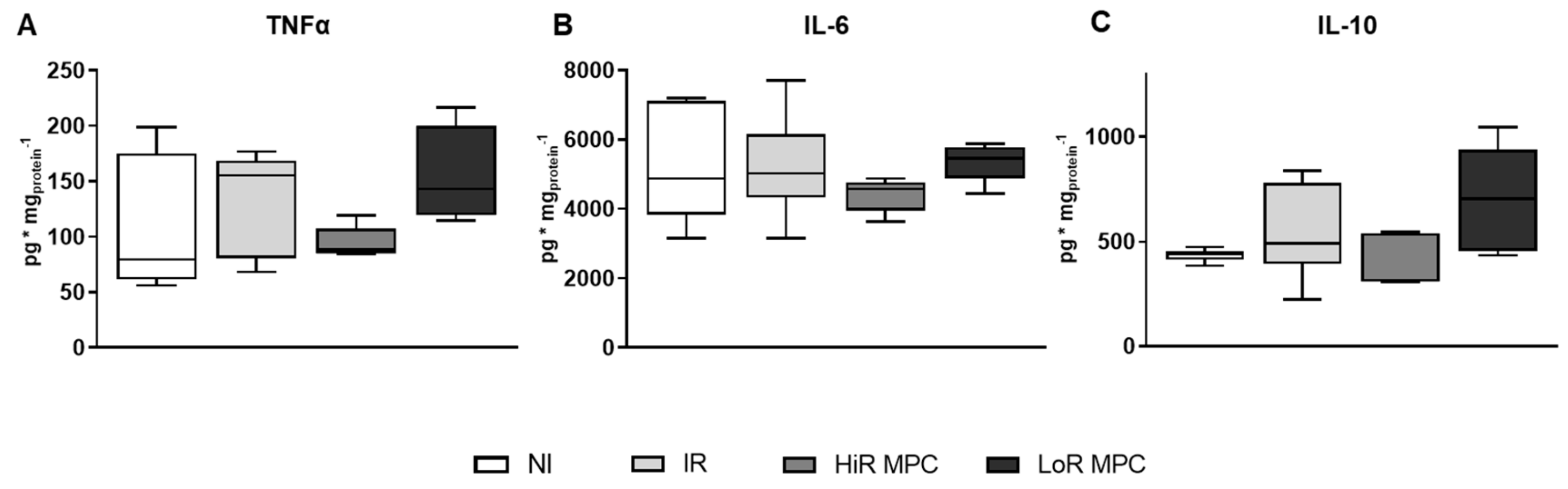
2.6. Cytokine Content
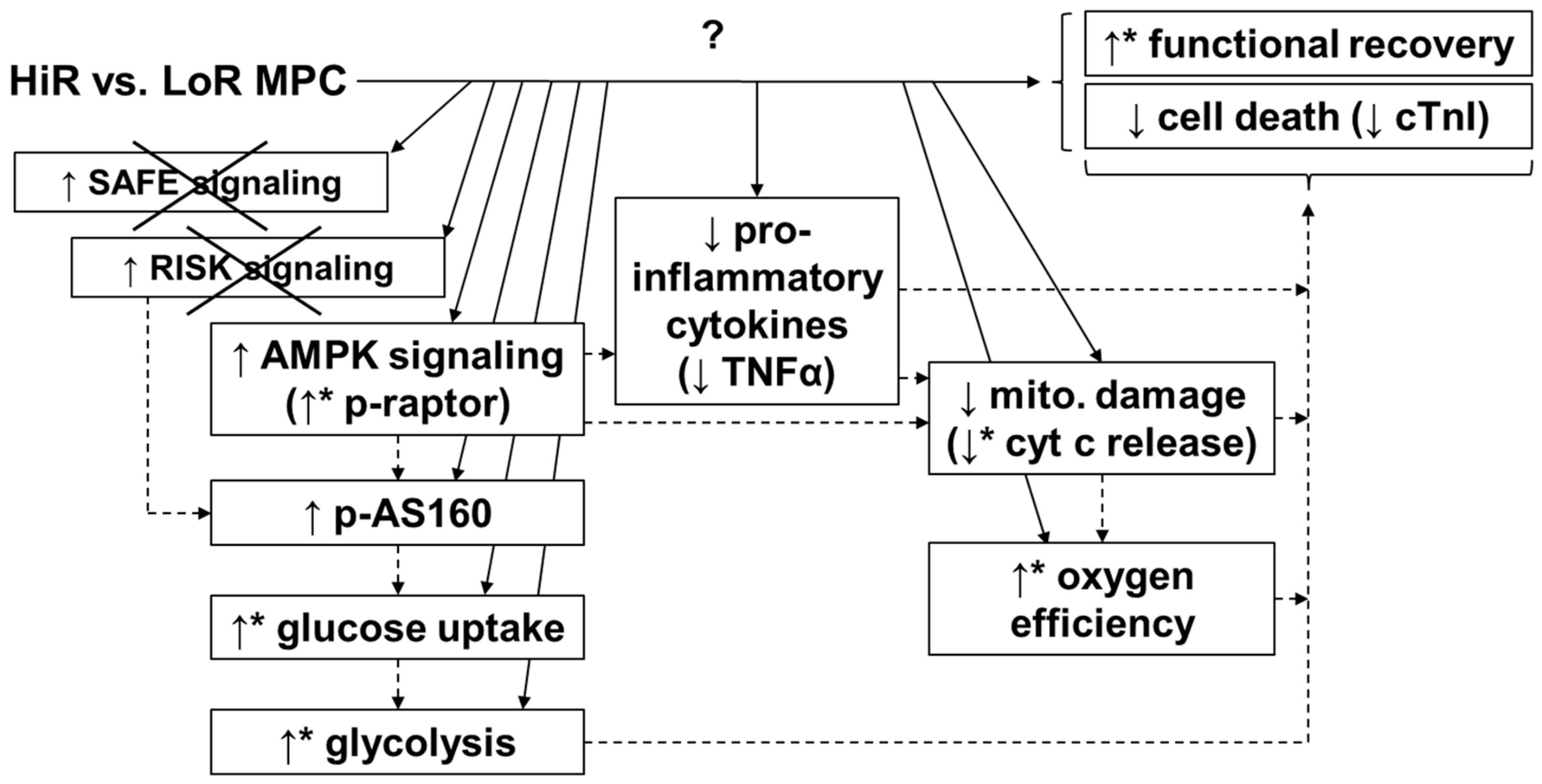
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Isolated Heart Perfusions
4.3. Functional Data Collection
4.4. Rates of Glycolysis and Glucose Oxidation
4.5. Cytochrome c, Cardiac Troponin I and Heart-Type Fatty Acid Binding Protein
4.6. Lactate
4.7. Cardiac Oxygen Consumption and Oxygen Efficiency
4.8. Glycogen Content
4.9. Glucose Uptake
4.10. Phosphorylation of Key Signaling Molecules
4.11. Cytokine Content
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 4E-BP1 | 4E-binding protein 1 |
| ACC | acetyl-CoA carboxylase |
| AMPK | 5′AMP-activated protein kinase |
| AS160 | Akt substrate of 160 kDa |
| cTnI | cardiac troponin I |
| Cyt c | cytochrome c |
| DCD | donation after circulatory death |
| DP | developed pressure |
| dP/dt max | maximum first derivative of left ventricular pressure |
| dP/dt min | minimum first derivative of left ventricular pressure |
| Erk1 | extracellular signal-regulated kinase 1 |
| GSK3β | glycogen synthase kinase 3 beta |
| H-FABP | heart-type fatty acid binding protein |
| HiR | high recovery |
| HR | heart rate |
| IL-6 | interleukin 6 |
| IL-10 | interleukin 10 |
| IRβ | insulin receptor β |
| IRI | ischemia reperfusion injury |
| LoR | low recovery |
| LV work | left ventricular work |
| MPC | mechanical postconditioning |
| mPTP | mitochondrial permeability transition pore |
| mTOR | mammalian target of rapamycin |
| O2C | cardiac oxygen consumption |
| p38 MAPK | p38 mitogen-activated protein kinase |
| p70S6K | ribosomal protein S6 kinase |
| PI3K | phosphoinositide 3-kinase |
| raptor | regulatory associated protein of mTOR |
| RISK | reperfusion injury salvage kinase |
| SAFE | survivor activating factor enhancement |
| STAT3 | signal transducer and activator of transcription 3 |
| TNFα | tumor necrosis factor alpha |
References
- McMurray, J.J.V.; Anker, S.D.; Auricchio, A.; Bohm, M.; Dickstein, K.; Falk, V.; Filippatos, G.; Fonseca, C.; Gomez-Sanchez, M.A.; Jaarsma, T.; et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2012, 33, 1787–1847. [Google Scholar] [PubMed]
- Eurotransplant International Foundation. Eurotransplant Annual Report 2018; Eurotransplant: Leiden, The Netherlands, 2018. [Google Scholar]
- Dhital, K.K.; Iyer, A.; Connellan, M.; Chew, H.C.; Gao, L.; Doyle, A.; Hicks, M.; Kumarasinghe, G.; Soto, C.; Dinale, A.; et al. Adult heart transplantation with distant procurement and ex-vivo preservation of donor hearts after circulatory death: A case series. Lancet 2015, 385, 2585–2591. [Google Scholar] [CrossRef]
- Messer, S.; Large, S. Resuscitating heart transplantation: the donation after circulatory determined death donor. Eur. J. Cardiothorac. Surg. 2016, 49, 1–4. [Google Scholar] [CrossRef] [PubMed]
- García Sáez, D.; Bowles, C.T.; Mohite, P.N.; Zych, B.; Maunz, O.; Popov, A.F.; Hurtado, A.; Raj, B.; Rahman-Haley, S.; Banner, N.; et al. Heart transplantation after donor circulatory death in patients bridged to transplant with implantable left ventricular assist devices. J. Heart Lung Transplant. 2016, 35, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Chew, H.C.; Iyer, A.; Connellan, M.; Scheuer, S.; Villanueva, J.; Gao, L.; Hicks, M.; Harkness, M.; Soto, C.; Dinale, A.; et al. Outcomes of Donation After Circulatory Death Heart Transplantation in Australia. J. Am. Coll. Cardiol. 2019, 73, 1447–1459. [Google Scholar] [CrossRef] [PubMed]
- Messer, S.; Page, A.; Axell, R.; Berman, M.; Hernández-Sánchez, J.; Colah, S.; Parizkova, B.; Valchanov, K.; Dunning, J.; Pavlushkov, E.; et al. Outcome after heart transplantation from donation after circulatory-determined death donors. J. Heart Lung Transplant. 2017, 36, 1311–1318. [Google Scholar] [CrossRef]
- Kin, H. Postconditioning attenuates myocardial ischemia–reperfusion injury by inhibiting events in the early minutes of reperfusion. Cardiovasc. Res. 2004, 62, 74–85. [Google Scholar] [CrossRef]
- Tsang, A.; Hausenloy, D.J.; Mocanu, M.M.; Yellon, D.M. Postconditioning: A Form of “Modified Reperfusion” Protects the Myocardium by Activating the Phosphatidylinositol 3-Kinase-Akt Pathway. Circ. Res. 2004, 95, 230–232. [Google Scholar] [CrossRef]
- Zhao, Z.-Q.; Corvera, J.S.; Halkos, M.E.; Kerendi, F.; Wang, N.-P.; Guyton, R.A.; Vinten-Johansen, J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am. J. Physiol.-Heart Circ. Physiol. 2003, 285, H579–H588. [Google Scholar] [CrossRef]
- Heusch, G. Critical Issues for the Translation of Cardioprotection. Circ. Res. 2017, 120, 1477–1486. [Google Scholar] [CrossRef]
- Bartkevics, M.; Huber, S.; Mathys, V.; Sourdon, J.; Dornbierer, M.; Carmona Mendez, N.; Gahl, B.; Carrel, T.P.; Tevaearai Stahel, H.T.; Longnus, S.L. Efficacy of mechanical postconditioning following warm, global ischaemia depends on circulating fatty acid levels in an isolated, working rat heart model. Eur. J. Cardiothorac. Surg. 2016, 49, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Buechert, A.; Heinen, Y.; Roeskes, C.; Hilfiker-Kleiner, D.; Heusch, G.; Schulz, R. Cardioprotection by Ischemic Postconditioning Is Lost in Aged and STAT3-Deficient Mice. Circ. Res. 2008, 102, 131–135. [Google Scholar] [CrossRef]
- Lee, D.S.; Steinbaugh, G.E.; Quarrie, R.; Yang, F.; Talukder, M.A.H.; Zweier, J.L.; Crestanello, J.A. Ischemic Postconditioning Does Not Provide Cardioprotection from Long-Term Ischemic Injury in Isolated Male or Female Rat Hearts. J. Surg. Res. 2010, 164, 175–181. [Google Scholar] [CrossRef]
- Van Vuuren, D.; Genis, A.; Genade, S.; Lochner, A. Postconditioning the Isolated Working Rat Heart. Cardiovasc. Drugs Ther. 2008, 22, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, B.B.; Fiorelli, A.I.; Gomes, O.M.; Gersak, B. Cardiac Effects of Postconditioning Depend Critically on the Duration of Reperfusion and Reocclusion Episodes. Heart Surg. Forum 2010, 13, 52. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Hausenloy, D.; Duchen, M.R.; Yellon, D.M. Signalling via the reperfusion injury signalling kinase (RISK) pathway links closure of the mitochondrial permeability transition pore to cardioprotection. Int. J. Biochem. Cell Biol. 2006, 38, 414–419. [Google Scholar] [CrossRef]
- Lacerda, L.; Somers, S.; Opie, L.H.; Lecour, S. Ischaemic postconditioning protects against reperfusion injury via the SAFE pathway. Cardiovasc. Res. 2009, 84, 201–208. [Google Scholar] [CrossRef]
- Hermann, R.; Marina Prendes, M.G.; Torresin, M.E.; Vélez, D.; Savino, E.A.; Varela, A. Effects of the AMP-activated protein kinase inhibitor compound C on the postconditioned rat heart. J. Physiol. Sci. 2012, 62, 333–341. [Google Scholar] [CrossRef]
- Granfeldt, A.; Jiang, R.; Wang, N.-P.; Mykytenko, J.; Eldaif, S.; Deneve, J.; Zhao, Z.-Q.; Guyton, R.A.; Tønnesen, E.; Vinten-Johansen, J. Neutrophil inhibition contributes to cardioprotection by postconditioning: Neutrophils in postconditioning cardioprotection. Acta Anaesthesiol. Scand. 2012, 56, 48–56. [Google Scholar] [CrossRef]
- Kin, H.; Wang, N.-P.; Mykytenko, J.; Reeves, J.; Deneve, J.; Jiang, R.; Zatta, A.J.; Guyton, R.A.; Vinten-Johansen, J.; Zhao, Z.-Q. Inhibition of Myocardial Apoptosis By Postconditioning Is Associated With Attenuation Of Oxidative Stress-Mediated Nuclear Factor-κB Translocation And TNFα Release. Shock 2008, 29, 761–768. [Google Scholar] [CrossRef]
- Kewalramani, G.; Puthanveetil, P.; Wang, F.; Kim, M.S.; Deppe, S.; Abrahani, A.; Luciani, D.S.; Johnson, J.D.; Rodrigues, B. AMP-activated protein kinase confers protection against TNF-α-induced cardiac cell death. Cardiovasc. Res. 2009, 84, 42–53. [Google Scholar] [CrossRef]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 545–560. [Google Scholar] [CrossRef]
- Liu, Q.; Docherty, J.C.; Rendell, J.C.T.; Clanachan, A.S.; Lopaschuk, G.D. High levels of fatty acids delay the recovery of intracellular pH and cardiac efficiency inpost-ischemic hearts by inhibiting glucose oxidation. J. Am. Coll. Cardiol. 2002, 39, 718–725. [Google Scholar] [CrossRef]
- Oliver, M.F. Fatty acids and the risk of death during acute myocardial ischaemia. Clin. Sci. 2015, 128, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Niederberger, P.; Farine, E.; Arnold, M.; Wyss, R.K.; Sanz, M.N.; Méndez-Carmona, N.; Gahl, B.; Fiedler, G.M.; Carrel, T.P.; Tevaearai Stahel, H.T.; et al. High pre-ischemic fatty acid levels decrease cardiac recovery in an isolated rat heart model of donation after circulatory death. Metabolism 2017, 71, 107–117. [Google Scholar] [CrossRef]
- Stanley, W.C. Myocardial Substrate Metabolism in the Normal and Failing Heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef]
- Montessuit, C.; Lerch, R. Regulation and dysregulation of glucose transport in cardiomyocytes. Biochim. Biophys. Acta 2013, 1833, 848–856. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Garcia-Dorado, D.; Bøtker, H.E.; Davidson, S.M.; Downey, J.; Engel, F.B.; Jennings, R.; Lecour, S.; Leor, J.; Madonna, R.; et al. Novel targets and future strategies for acute cardioprotection: Position Paper of the European Society of Cardiology Working Group on Cellular Biology of the Heart. Cardiovasc. Res. 2017, 113, 564–585. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Kharbanda, R.K.; Møller, U.K.; Ramlall, M.; Aarøe, J.; Butler, R.; Bulluck, H.; Clayton, T.; Dana, A.; Dodd, M.; et al. Effect of remote ischaemic conditioning on clinical outcomes in patients with acute myocardial infarction (CONDI-2/ERIC-PPCI): A single-blind randomised controlled trial. Lancet 2019, 394, 1415–1424. [Google Scholar] [CrossRef]
- Paiva, M.A.; Rutter-Locher, Z.; Gonçalves, L.M.; Providência, L.A.; Davidson, S.M.; Yellon, D.M.; Mocanu, M.M. Enhancing AMPK activation during ischemia protects the diabetic heart against reperfusion injury. Am. J. Physiol.-Heart Circ. Physiol. 2011, 300, H2123–H2134. [Google Scholar] [CrossRef] [PubMed]
- Perrelli, M.-G. Ischemia/reperfusion injury and cardioprotective mechanisms: Role of mitochondria and reactive oxygen species. World J. Cardiol. 2011, 3, 186. [Google Scholar] [CrossRef] [PubMed]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Large, S.; Tsui, S.; Messer, S. Clinical and ethical challenges in heart transplantation from donation after circulatory determined death donors. Curr. Opin. Organ Transplant. 2017, 22, 251–259. [Google Scholar] [CrossRef]
- Correa, F.; García, N.; Gallardo-Pérez, J.; Carreño-Fuentes, L.; Rodríguez-Enríquez, S.; Marín-Hernández, A.; Zazueta, C. Post-conditioning Preserves Glycolytic ATP During Early Reperfusion: A survival Mechanism for the Reperfused Heart. Cell. Physiol. Biochem. 2008, 22, 635–644. [Google Scholar] [CrossRef]
- Lopaschuk, G.D. AMP-activated protein kinase control of energy metabolism in the ischemic heart. Int. J. Obes. 2008, 32, S29–S35. [Google Scholar] [CrossRef][Green Version]
- Xu, K.Y.; Zweier, J.L.; Becker, L.C. Functional coupling between glycolysis and sarcoplasmic reticulum Ca2+ transport. Circ. Res. 1995, 77, 88–97. [Google Scholar] [CrossRef]
- Dyck, J.R.B.; Lopaschuk, G.D. AMPK alterations in cardiac physiology and pathology: enemy or ally? AMPK alterations in cardiac physiology and pathology. J. Physiol. 2006, 574, 95–112. [Google Scholar] [CrossRef]
- Viollet, B.; Horman, S.; Leclerc, J.; Lantier, L.; Foretz, M.; Billaud, M.; Giri, S.; Andreelli, F. AMPK inhibition in health and disease. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 276–295. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef]
- Larance, M.; Ramm, G.; Stöckli, J.; van Dam, E.M.; Winata, S.; Wasinger, V.; Simpson, F.; Graham, M.; Junutula, J.R.; Guilhaus, M.; et al. Characterization of the Role of the Rab GTPase-activating Protein AS160 in Insulin-regulated GLUT4 Trafficking. J. Biol. Chem. 2005, 280, 37803–37813. [Google Scholar] [CrossRef] [PubMed]
- Dedkova, E.N.; Blatter, L.A. Measuring mitochondrial function in intact cardiac myocytes. J. Mol. Cell. Cardiol. 2012, 52, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Ovize, M.; Baxter, G.F.; Di Lisa, F.; Ferdinandy, P.; Garcia-Dorado, D.; Hausenloy, D.J.; Heusch, G.; Vinten-Johansen, J.; Yellon, D.M.; Schulz, R. Postconditioning and protection from reperfusion injury: Where do we stand? Position Paper from the Working Group of Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc. Res. 2010, 87, 406–423. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.N.; Farine, E.; Niederberger, P.; Méndez-Carmona, N.; Wyss, R.K.; Arnold, M.; Gulac, P.; Fiedler, G.M.; Gressette, M.; Garnier, A.; et al. Cardioprotective reperfusion strategies differentially affect mitochondria: Studies in an isolated rat heart model of donation after circulatory death (DCD). Am. J. Transplant. 2018, 19, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Juhaszova, M.; Zorov, D.B.; Kim, S.-H.; Pepe, S.; Fu, Q.; Fishbein, K.W.; Ziman, B.D.; Wang, S.; Ytrehus, K.; Antos, C.L.; et al. Glycogen synthase kinase-3β mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J. Clin. Investig. 2004, 113, 1535–1549. [Google Scholar] [CrossRef]
- Méndez-Carmona, N.; Wyss, R.K.; Arnold, M.; Joachimbauer, A.; Segiser, A.; Fiedler, G.M.; Carrel, T.P.; Tevaearai Stahel, H.T.; Longnus, S.L. Differential effects of ischemia/reperfusion on endothelial function and contractility in donation after circulatory death. J. Heart Lung Transplant. 2019, 38, 767–777. [Google Scholar] [CrossRef]
- Kleinbongard, P.; Schulz, R.; Heusch, G. TNFα in myocardial ischemia/reperfusion, remodeling and heart failure. Heart Fail. Rev. 2011, 16, 49–69. [Google Scholar] [CrossRef]
- Tafani, M.; Schneider, T.G.; Pastorino, J.G.; Farber, J.L. Cytochrome c-Dependent Activation of Caspase-3 by Tumor Necrosis Factor Requires Induction of the Mitochondrial Permeability Transition. Am. J. Pathol. 2000, 156, 2111–2121. [Google Scholar] [CrossRef]
- Lecour, S. Multiple protective pathways against reperfusion injury: A SAFE path without Aktion? J. Mol. Cell Cardiol. 2009, 46, 607–609. [Google Scholar] [CrossRef]
- Iyer, A.; Chew, H.C.; Gao, L.; Villanueva, J.; Hicks, M.; Doyle, A.; Kumarasinghe, G.; Jabbour, A.; Jansz, P.C.; Feneley, M.P.; et al. Pathophysiological Trends During Withdrawal of Life Support: Implications for Organ Donation After Circulatory Death. Transplantation 2016, 100, 2621–2629. [Google Scholar] [CrossRef]
- White, C.W.; Lillico, R.; Sandha, J.; Hasanally, D.; Wang, F.; Ambrose, E.; Müller, A.; Rachid, O.; Li, Y.; Xiang, B.; et al. Physiologic Changes in the Heart Following Cessation of Mechanical Ventilation in a Porcine Model of Donation After Circulatory Death: Implications for Cardiac Transplantation: Physiologic Response to Donor Extubation. Am. J. Transplant. 2016, 16, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Wyss, R.K.; Méndez-Carmona, N.; Sanz, M.-N.; Arnold, M.; Segiser, A.; Fiedler, G.M.; Carrel, T.P.; Djafarzadeh, S.; Tevaearai Stahel, H.T.; Longnus, S.L. Mitochondrial integrity during early reperfusion in an isolated rat heart model of donation after circulatory death—consequences of ischemic duration. J. Heart Lung Transplant. 2019, 38, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Farine, E.; Niederberger, P.; Wyss, R.K.; Méndez-Carmona, N.; Gahl, B.; Fiedler, G.M.; Carrel, T.P.; Tevaearai Stahel, H.T.; Longnus, S.L. Controlled Reperfusion Strategies Improve Cardiac Hemodynamic Recovery after Warm Global Ischemia in an Isolated, Working Rat Heart Model of Donation after Circulatory Death (DCD). Front. Physiol. 2016, 7, 543. [Google Scholar] [CrossRef] [PubMed]
- Anmann, T.; Varikmaa, M.; Timohhina, N.; Tepp, K.; Shevchuk, I.; Chekulayev, V.; Saks, V.; Kaambre, T. Formation of highly organized intracellular structure and energy metabolism in cardiac muscle cells during postnatal development of rat heart. Biochim. Biophys. Acta 2014, 1837, 1350–1361. [Google Scholar] [CrossRef]
- Barr, R.L.; Lopaschuk, G.D. Methodology for measuring in vitro/ex vivo cardiac energy metabolism. J. Pharmacol. Toxicol. Methods 2000, 43, 141–152. [Google Scholar] [CrossRef]
- Longnus, S.L.; Wambolt, R.B.; Parsons, H.L.; Brownsey, R.W.; Allard, M.F. 5-Aminoimidazole-4-carboxamide 1-β-d-ribofuranoside (AICAR) stimulates myocardial glycogenolysis by allosteric mechanisms. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2003, 284, R936–R944. [Google Scholar] [CrossRef]
- Holland, B.S.; Copenhaver, M.D. An Improved Sequentially Rejective Bonferroni Test Procedure. Biometrics 1987, 43, 417. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Series A | Series B | ||||||
|---|---|---|---|---|---|---|---|---|
| NI | IR | HiR MPC | LoR MPC | NI | IR | HiR MPC | LoR MPC | |
| Number of hearts | 7 | 8 | 11 | 7 | 6 | 6 | 7 | 10 |
| BW (g) | 377 ± 64 | 364 ± 23 | 350 ± 28 | 356 ± 26 | 341 ± 16 | 347 ± 17 | 373 ± 31 | 356 ± 23 |
| HW (g) | 1.69 ± 0.26 | 1.57 ± 0.09 | 1.64 ± 0.15 | 1.58 ± 0.20 | 1.50 ± 0.10 | 1.60 ± 0.20 | 1.75 ± 0.24 | 1.64 ± 0.18 |
| LV work (mmHg ∗ beats ∗ min−1) | 33,507 ± 1895 | 33,673 ± 2677 | 32,760 ± 2468 | 34,036 ± 2007 | 36,990 ± 1917 | 33,625 ± 3674 | 33,573 ± 5043 | 33,727 ± 1674 |
| HR (beats ∗ min−1) | 275 ± 43 | 266 ± 15 | 263 ± 16 | 259 ± 14 | 276 ± 24 | 264 ± 30 | 247 ± 38 | 264 ± 21 |
| DP (mmHg) | 124 ± 15 | 127 ± 10 | 125 ± 9 | 132 ± 6 | 135 ± 9 | 128 ± 12 | 136 ± 11 | 128 ± 8 |
| dP/dt min (mmHg ∗ s−1) | −4313 ± 621 | −4492 ± 991 | −4173 ± 188 | −4598 ± 622 | −4639 ± 356 | −4457 ± 906 | −4797 ± 583 | −5048 ± 1272 |
| dP/dt max (mmHg ∗ s−1) | 4354 ± 526 | 4475 ± 483 | 4361 ± 506 | 4621 ± 244 | 5046 ± 411 | 4246 ± 484 | 4673 ± 556 | 4498 ± 590 |
| CO (mL ∗ min−1) | 64 ± 14 | 61 ± 11 | 61 ± 7 | 63 ± 4 | 66 ± 6 | 64 ± 11 | 68 ± 12 | 65 ± 9 |
| CF (mL ∗ min−1) | 32 ± 5 | 32 ± 5 | 30 ± 3 | 32 ± 4 | 29 ± 4 | 27 ± 8 | 32 ± 6 | 33 ± 4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arnold, M.; Méndez-Carmona, N.; Gulac, P.; Wyss, R.K.; Rutishauser, N.; Segiser, A.; Carrel, T.; Longnus, S. Mechanical Postconditioning Promotes Glucose Metabolism and AMPK Activity in Parallel with Improved Post-Ischemic Recovery in an Isolated Rat Heart Model of Donation after Circulatory Death. Int. J. Mol. Sci. 2020, 21, 964. https://doi.org/10.3390/ijms21030964
Arnold M, Méndez-Carmona N, Gulac P, Wyss RK, Rutishauser N, Segiser A, Carrel T, Longnus S. Mechanical Postconditioning Promotes Glucose Metabolism and AMPK Activity in Parallel with Improved Post-Ischemic Recovery in an Isolated Rat Heart Model of Donation after Circulatory Death. International Journal of Molecular Sciences. 2020; 21(3):964. https://doi.org/10.3390/ijms21030964
Chicago/Turabian StyleArnold, Maria, Natalia Méndez-Carmona, Patrik Gulac, Rahel K Wyss, Nina Rutishauser, Adrian Segiser, Thierry Carrel, and Sarah Longnus. 2020. "Mechanical Postconditioning Promotes Glucose Metabolism and AMPK Activity in Parallel with Improved Post-Ischemic Recovery in an Isolated Rat Heart Model of Donation after Circulatory Death" International Journal of Molecular Sciences 21, no. 3: 964. https://doi.org/10.3390/ijms21030964
APA StyleArnold, M., Méndez-Carmona, N., Gulac, P., Wyss, R. K., Rutishauser, N., Segiser, A., Carrel, T., & Longnus, S. (2020). Mechanical Postconditioning Promotes Glucose Metabolism and AMPK Activity in Parallel with Improved Post-Ischemic Recovery in an Isolated Rat Heart Model of Donation after Circulatory Death. International Journal of Molecular Sciences, 21(3), 964. https://doi.org/10.3390/ijms21030964