Systematic Analysis of the DNA Methylase and Demethylase Gene Families in Rapeseed (Brassica napus L.) and Their Expression Variations After Salt and Heat stresses


Abstract
1. Introduction
2. Results
2.1. Identification of DNA Methyltransferases and Demethylases Genes in Rapeseed Genome
2.2. Multiple Sequences Alignment and Phylogenetic Analysis in Rapeseed, Arabidopsis and Rice
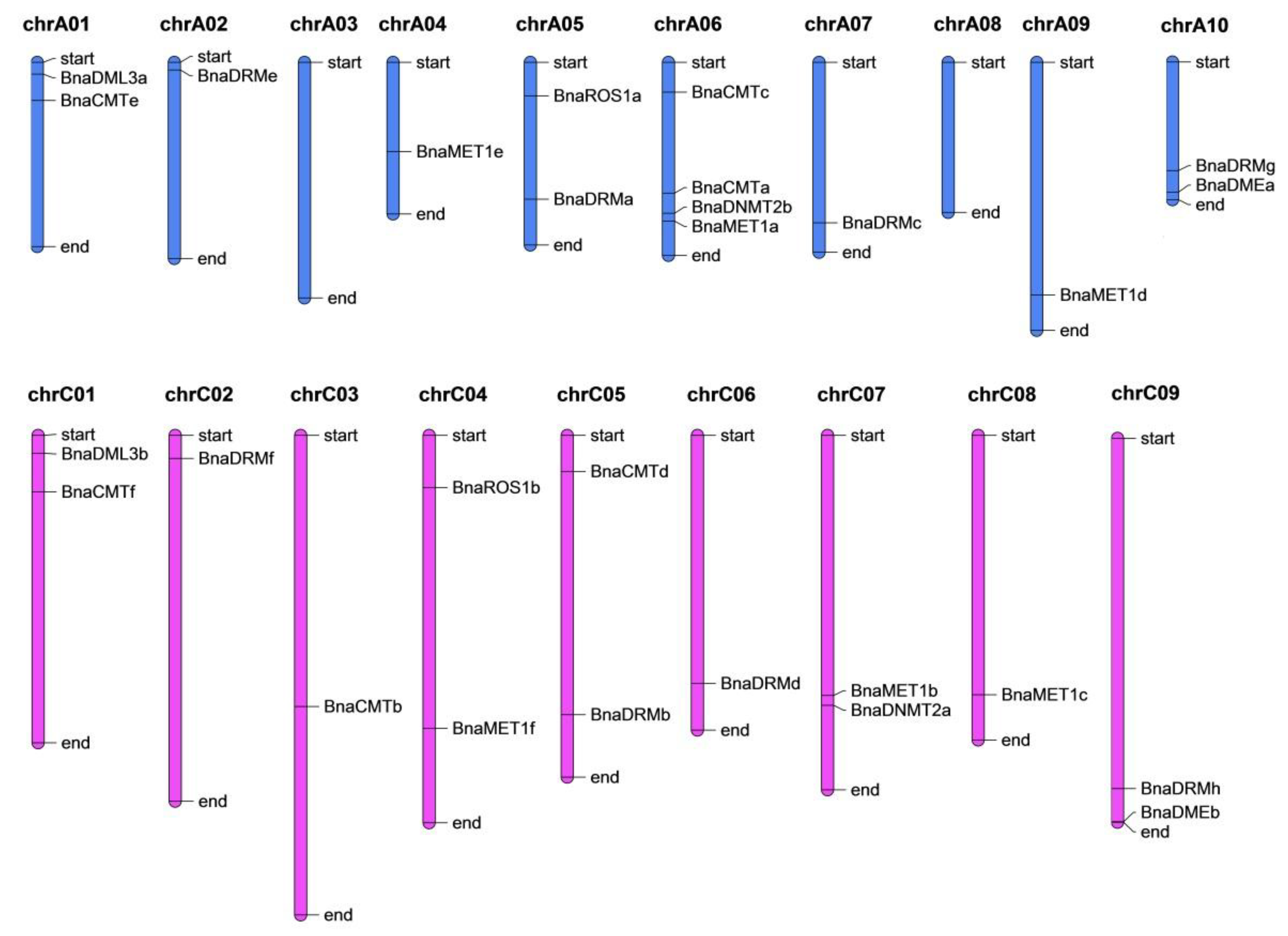
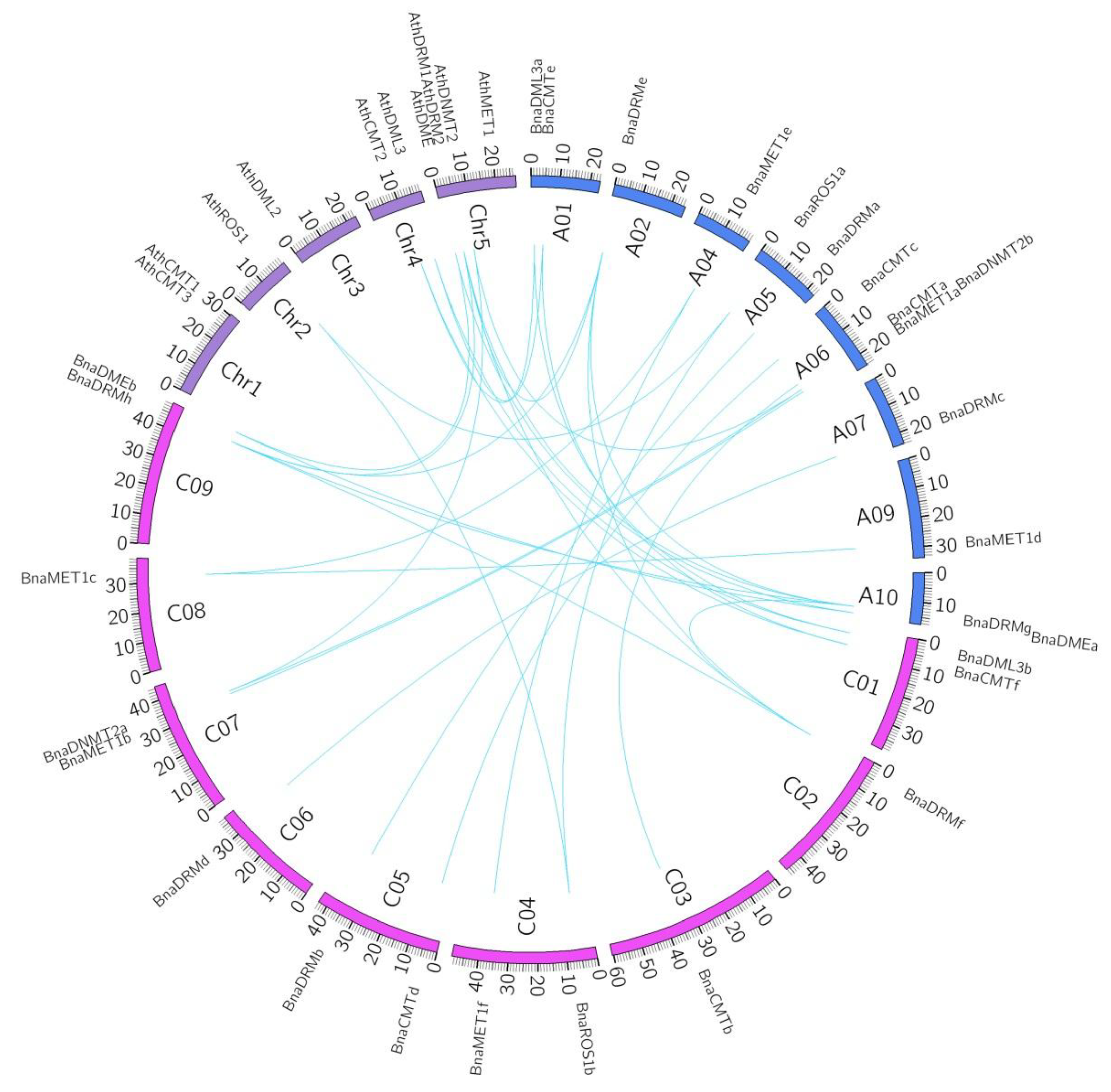
2.3. Chromosomal Location and Synteny Analysis
2.4. Gene Structure and Conserved Motif Distribution Analysis
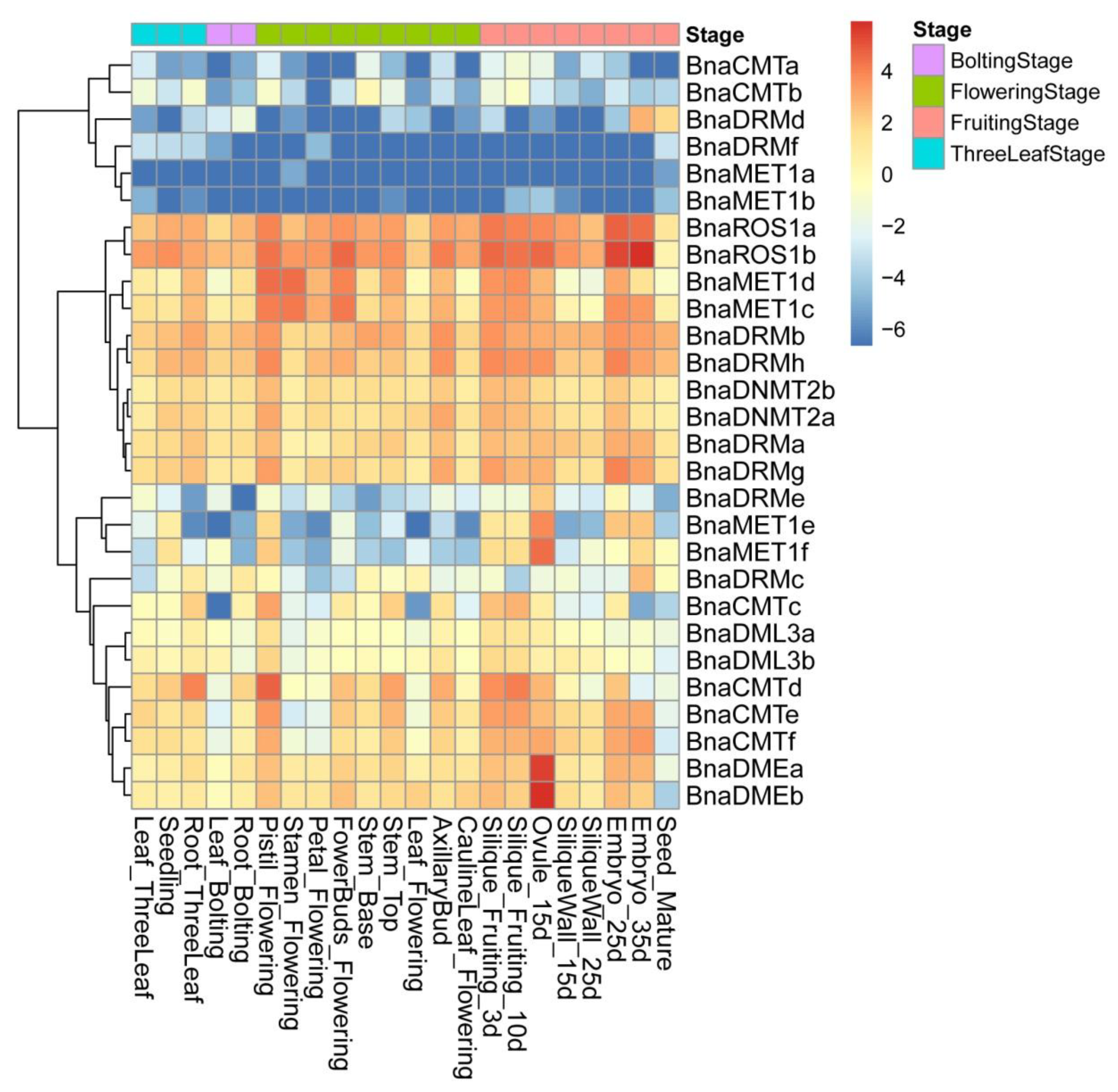
2.5. Expression Patterns of DNA Methylase and Demethylase Genes in Various Tissues of ZS11
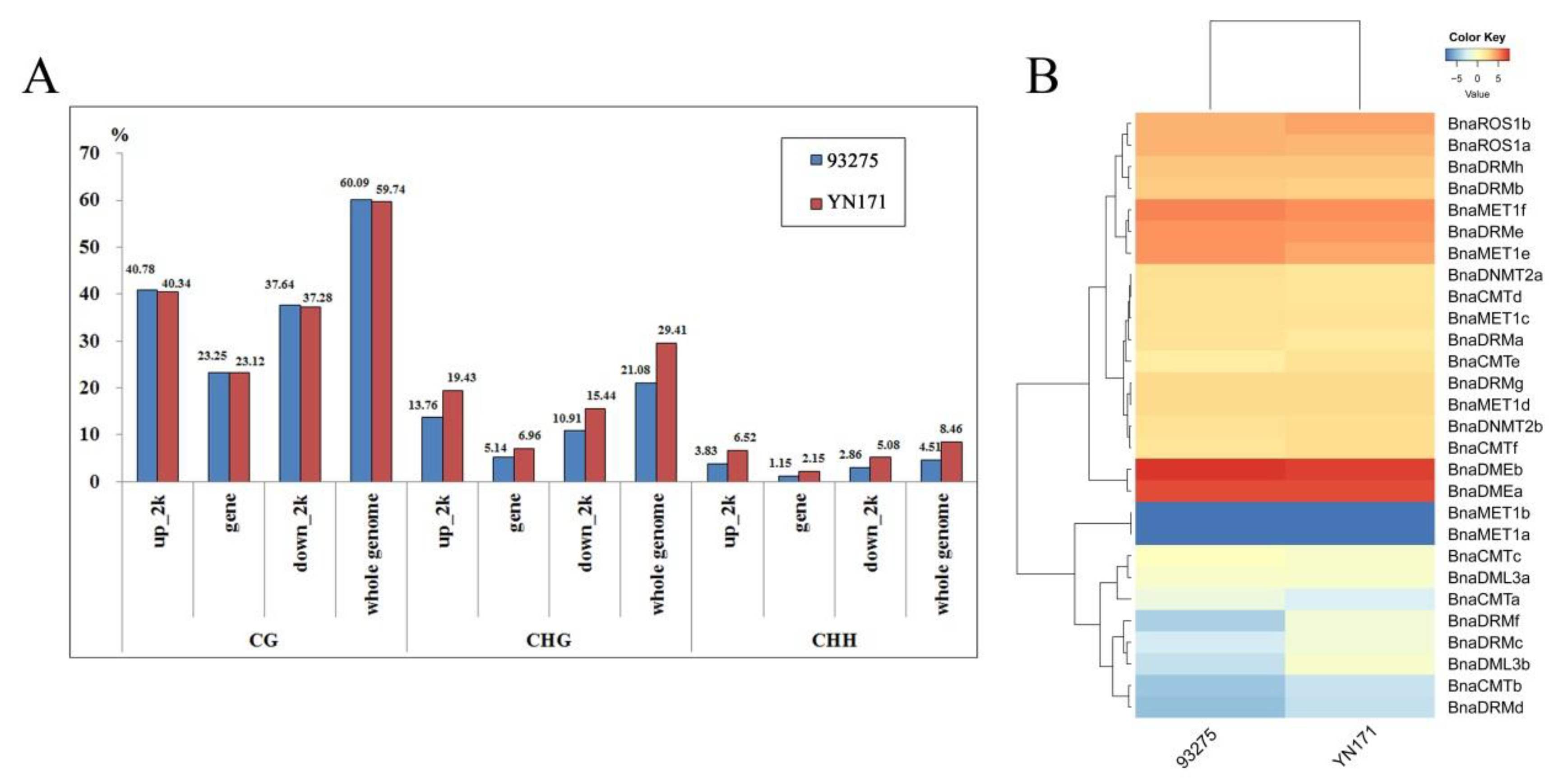
2.6. Correlation between Gene Expression Patterns and Methylation Level in the Endosperm
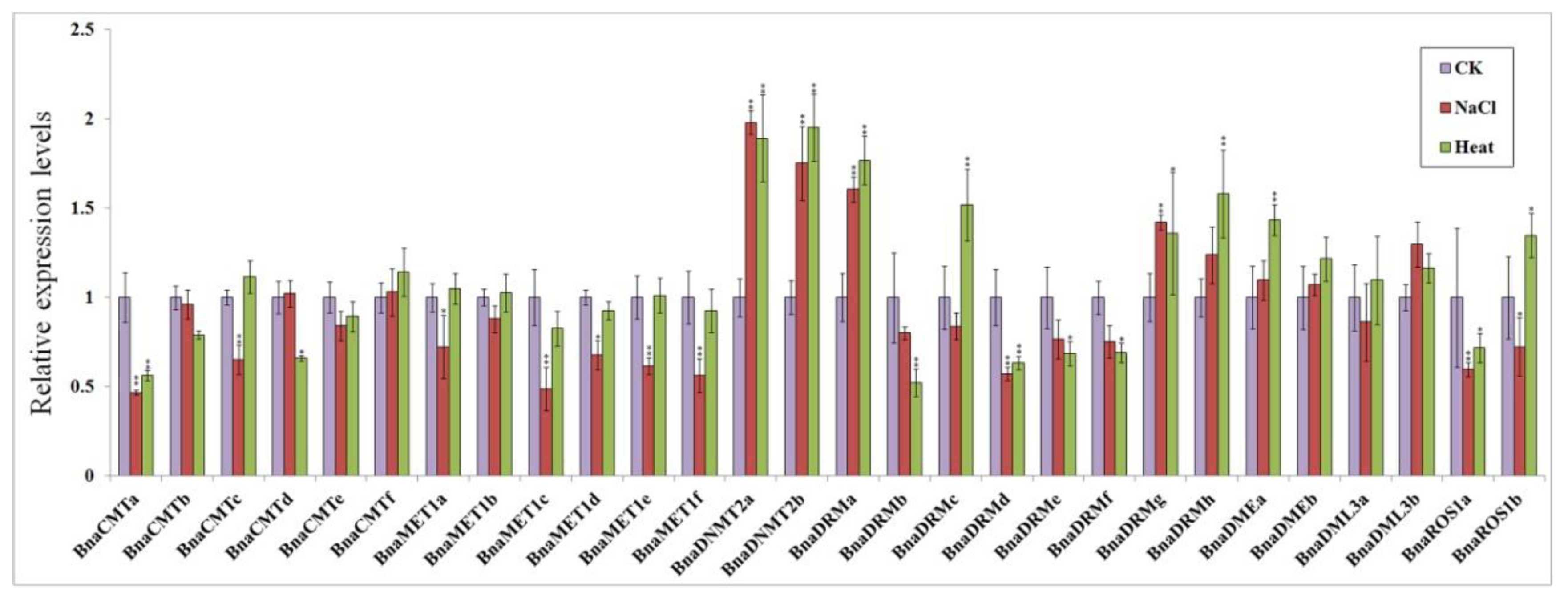
2.7. qPCR Analysis of DNA Methylase and Demethylase Genes in Response to Some Abiotic Stress
3. Discussion
4. Materials and Methods
4.1. Identification of DNA Methylase and Demethylase Proteins in Rapeseed
4.2. Multiple Sequence Alignment and Phylogenetic Analysis
4.3. Chromosome Localization and Collinearity Analysis
4.4. Gene Structure and Conserved Domains Analysis
4.5. Expression Analysis of DNA Methylase and Demethylase Genes
4.6. Plant Material and Treatments
4.7. RNA Extraction and Quantitative Real-Time PCR
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cubas, P.; Vincent, C.; Coen, E. An epigenetic mutation responsible for natural variation in floral symmetry. Nature 1999, 401, 157. [Google Scholar] [CrossRef] [PubMed]
- Manning, K.; Tör, M.; Poole, M.; Hong, Y.; Thompson, A.J.; King, G.J.; Giovannoni, J.J.; Seymour, G.B. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 2006, 38, 948. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.; Zhu, J.; Chen, Q.; Dai, F.; Li, X.; Li, M.; Zhang, H.; Zhang, G.; Li, D.; Dong, Y. Single base–resolution methylome of the silkworm reveals a sparse epigenomic map. Nat. Biotechnol. 2010, 28, 516. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Zhao, X.; Tock, A.J.; Lambing, C.; Underwood, C.J.; Hardcastle, T.J.; Serra, H.; Kim, J.; Cho, H.S.; Kim, J. Nucleosomes and DNA methylation shape meiotic DSB frequency in Arabidopsis thaliana transposons and gene regulatory regions. Genome Res. 2018, 28, 532–546. [Google Scholar] [CrossRef]
- Liu, J.; Li, J.; Liu, H.-F.; Fan, S.-H.; Singh, S.; Zhou, X.-R.; Hu, Z.-Y.; Wang, H.-Z.; Hua, W. Genome-wide screening and analysis of imprinted genes in rapeseed (Brassica napus L.) endosperm. Dna Res. 2018, 25, 629–640. [Google Scholar] [CrossRef]
- Sun, Q.; Qiao, J.; Zhang, S.; He, S.; Shi, Y.; Yuan, Y.; Zhang, X.; Cai, Y. Changes in DNA methylation assessed by genomic bisulfite sequencing suggest a role for DNA methylation in cotton fruiting branch development. PeerJ 2018, 6, e4945. [Google Scholar] [CrossRef]
- Xing, M.-Q.; Zhang, Y.-J.; Zhou, S.-R.; Hu, W.-Y.; Wu, X.-T.; Ye, Y.-J.; Wu, X.-X.; Xiao, Y.-P.; Li, X.; Xue, H.-W. Global analysis reveals the crucial roles of DNA methylation during rice seed development. Plant. Physiol. 2015, 168, 1417–1432. [Google Scholar] [CrossRef]
- Achwal, C.W.; Iyer, C.A.; Chandra, H.S. Immunochemical evidence for the presence of 5mC, 6mA and 7mG in human, Drosophila and mealybug DNA. Febs Lett. 1983, 158, 353–358. [Google Scholar] [CrossRef]
- Klungland, A.; Robertson, A.B. Oxidized C5-methyl cytosine bases in DNA: 5-hydroxymethylcytosine; 5-formylcytosine; and 5-carboxycytosine. Free Radic. Biol. Med. 2017, 107, 62–68. [Google Scholar] [CrossRef]
- Buryanov, Y.I.; Shevchuk, T. DNA methyltransferases and structural-functional specificity of eukaryotic DNA modification. Biochemistry (Mosc.) 2005, 70, 730–742. [Google Scholar] [CrossRef]
- Cao, D.; Ju, Z.; Gao, C.; Mei, X.; Fu, D.; Zhu, H.; Luo, Y.; Zhu, B. Genome-wide identification of cytosine-5 DNA methyltransferases and demethylases in Solanum lycopersicum. Gene 2014, 550, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Dhe-Paganon, S.; Syeda, F.; Park, L. DNA methyl transferase 1: Regulatory mechanisms and implications in health and disease. Int. J. Biochem. Mol. Biol. 2011, 2, 58. [Google Scholar]
- Greb-Markiewicz, B.; Orłowski, M.; Dobrucki, J.; Ożyhar, A. Sequences that direct subcellular traffic of the Drosophila methoprene-tolerant protein (MET) are located predominantly in the PAS domains. Mol. Cell. Endocrinol. 2011, 345, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Clark, T.A.; Murray, I.A.; Morgan, R.D.; Kislyuk, A.O.; Spittle, K.E.; Boitano, M.; Fomenkov, A.; Roberts, R.J.; Korlach, J. Characterization of DNA methyltransferase specificities using single-molecule, real-time DNA sequencing. Nucleic Acids Res. 2011, 40, e29. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Jang, I.-C.; Seo, H.S. COP1 controls abiotic stress responses by modulating AtSIZ1 function through its E3 Ubiquitin ligase activity. Front. Plant Sci. 2016, 7, 1182. [Google Scholar] [CrossRef] [PubMed]
- Ashapkin, V.; Kutueva, L.; Vanyushin, B. Plant DNA methyltransferase genes: Multiplicity, expression, methylation patterns. Biochemistry (Mosc.) 2016, 81, 141–151. [Google Scholar] [CrossRef]
- Gao, Z.; Liu, H.-L.; Daxinger, L.; Pontes, O.; He, X.; Qian, W.; Lin, H.; Xie, M.; Lorkovic, Z.J.; Zhang, S. An RNA polymerase II-and AGO4-associated protein acts in RNA-directed DNA methylation. Nature 2010, 465, 106. [Google Scholar] [CrossRef]
- Eissenberg, J.C. Molecular biology of the chromo domain: An ancient chromatin module comes of age. Gene 2001, 275, 19–29. [Google Scholar] [CrossRef]
- Genger, R.K.; Kovac, K.A.; Dennis, E.S.; Peacock, W.J.; Finnegan, E.J. Multiple DNA methyltransferase genes in Arabidopsis thaliana. Plant. Mol. Biol. 1999, 41, 269–278. [Google Scholar] [CrossRef]
- Ahmad, F.; Huang, X.; Lan, H.; Huma, T.; Bao, Y.; Huang, J.; Zhang, H. Comprehensive gene expression analysis of the DNA (cytosine-5) methyltransferase family in rice (Oryza sativa L.). Genet. Mol. Res. 2014, 13, 5159–5172. [Google Scholar] [CrossRef]
- Palmgren, G.; Mattsson, O.; Okkels, F.T. Specific levels of DNA methylation in various tissues, cell lines, and cell types of Daucus carota. Plant. Physiol. 1991, 95, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Ruffini Castiglione, M.; Venora, G.; Ravalli, C.; Stoilov, L.; Gecheff, K.; Cremonini, R. DNA methylation and chromosomal rearrangements in reconstructed karyotypes of Hordeum vulgare L. Protoplasma 2008, 232, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Xi, Y.; Cheng, B.; Zhu, S. Genome-wide identification and expression profiling of DNA methyltransferase gene family in maize. Plant. Cell Rep. 2014, 33, 1661–1672. [Google Scholar] [CrossRef]
- Teyssier, E.; Bernacchia, G.; Maury, S.; Kit, A.H.; Stammitti-Bert, L.; Rolin, D.; Gallusci, P. Tissue dependent variations of DNA methylation and endoreduplication levels during tomato fruit development and ripening. Planta 2008, 228, 391. [Google Scholar] [CrossRef]
- Zhu, J.-K. Active DNA demethylation mediated by DNA glycosylases. Annu. Rev. Genet. 2009, 43, 143–166. [Google Scholar] [CrossRef]
- Gong, Z.; Morales-Ruiz, T.; Ariza, R.R.; Roldán-Arjona, T.; David, L.; Zhu, J.-K. ROS1, a repressor of transcriptional gene silencing in Arabidopsis, encodes a DNA glycosylase/lyase. Cell 2002, 111, 803–814. [Google Scholar] [CrossRef]
- Zhu, J.; Kapoor, A.; Sridhar, V.V.; Agius, F.; Zhu, J.-K. The DNA glycosylase/lyase ROS1 functions in pruning DNA methylation patterns in Arabidopsis. Curr. Biol. 2007, 17, 54–59. [Google Scholar] [CrossRef]
- Agius, F.; Kapoor, A.; Zhu, J.-K. Role of the Arabidopsis DNA glycosylase/lyase ROS1 in active DNA demethylation. Proc. Natl. Acad. Sci. USA 2006, 103, 11796–11801. [Google Scholar] [CrossRef]
- Choi, Y.; Gehring, M.; Johnson, L.; Hannon, M.; Harada, J.J.; Goldberg, R.B.; Jacobsen, S.E.; Fischer, R.L. DEMETER, a DNA glycosylase domain protein, is required for endosperm gene imprinting and seed viability in Arabidopsis. Cell 2002, 110, 33–42. [Google Scholar] [CrossRef]
- Penterman, J.; Zilberman, D.; Huh, J.H.; Ballinger, T.; Henikoff, S.; Fischer, R.L. DNA demethylation in the Arabidopsis genome. Proc. Natl. Acad. Sci. USA 2007, 104, 6752–6757. [Google Scholar] [CrossRef]
- Zhang, H.; Lang, Z.; Zhu, J.-K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489. [Google Scholar] [CrossRef] [PubMed]
- Pavet, V.; Quintero, C.; Cecchini, N.M.; Rosa, A.L.; Alvarez, M.E. Arabidopsis displays centromeric DNA hypomethylation and cytological alterations of heterochromatin upon attack by Pseudomonas syringae. Mol. Plant.-Microbe Interact. 2006, 19, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.-S.; Sano, H. Abiotic-stress induces demethylation and transcriptional activation of a gene encoding a glycerophosphodiesterase-like protein in tobacco plants. Mol. Genet. Genom. 2007, 277, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Smýkal, P.; Valledor, L.; Rodriguez, R.; Griga, M. Assessment of genetic and epigenetic stability in long-term in vitro shoot culture of pea (Pisum sativum L.). Plant. Cell Rep. 2007, 26, 1985–1998. [Google Scholar]
- Yang, J.; Liu, L.; Gong, Y.; Huang, D.; Wang, F.; He, L. Analysis of genomic DNA methylation level in radish under cadmium stress by methylation-sensitive amplified polymorphism technique. Zhi Wu Sheng Li Yu Fen Zi Sheng Wu Xue Xue Bao = J. Plant Physiol. Mol. Biol. 2007, 33, 219–226. [Google Scholar]
- Bilichak, A.; Ilnystkyy, Y.; Hollunder, J.; Kovalchuk, I. The progeny of Arabidopsis thaliana plants exposed to salt exhibit changes in DNA methylation, histone modifications and gene expression. PLoS ONE 2012, 7, e30515. [Google Scholar] [CrossRef]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. USA 2012, 109, E2183–E2191. [Google Scholar] [CrossRef]
- Sun, F.; Fan, G.; Hu, Q.; Zhou, Y.; Guan, M.; Tong, C.; Li, J.; Du, D.; Qi, C.; Jiang, L. The high-quality genome of Brassica napus cultivar ‘ZS 11′reveals the introgression history in semi-winter morphotype. Plant. J. 2017, 92, 452–468. [Google Scholar] [CrossRef]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.; Wang, J.; Sun, R.; Wu, J.; Liu, S.; Bai, Y.; Mun, J.-H.; Bancroft, I.; Cheng, F. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 2011, 43, 1035. [Google Scholar] [CrossRef]
- Yang, H.; Liu, J.; Huang, S.; Guo, T.; Deng, L.; Hua, W. Selection and evaluation of novel reference genes for quantitative reverse transcription PCR (qRT-PCR) based on genome and transcriptome data in Brassica napus L. Gene 2014, 538, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Monteuuis, G.; Wong, J.J.; Bailey, C.G.; Schmitz, U.; Rasko, J.E. The changing paradigm of intron retention: Regulation, ramifications and recipes. Nucleic Acids Res. 2019, 47, 11497–11513. [Google Scholar] [CrossRef] [PubMed]
- Vertino, P.M.; Yen, R.-W.; Gao, J.; Baylin, S.B. De novo methylation of CpG island sequences in human fibroblasts overexpressing DNA (cytosine-5-)-methyltransferase. Mol. Cell. Biol. 1996, 16, 4555–4565. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, E.J.; Peacock, W.J.; Dennis, E.S. DNA methylation, a key regulator of plant development and other processes. Curr. Opin. Genet. Dev. 2000, 10, 217–223. [Google Scholar] [CrossRef]
- Salilew-Wondim, D.; Saeed-Zidane, M.; Hoelker, M.; Gebremedhn, S.; Poirier, M.; Pandey, H.O.; Tholen, E.; Neuhoff, C.; Held, E.; Besenfelder, U. Genome-wide DNA methylation patterns of bovine blastocysts derived from in vivo embryos subjected to in vitro culture before, during or after embryonic genome activation. Bmc Genom. 2018, 19, 424. [Google Scholar] [CrossRef]
- Matzke, M.A.; Mosher, R.A. RNA-directed DNA methylation: An epigenetic pathway of increasing complexity. Nat. Rev. Genet. 2014, 15, 394–408. [Google Scholar] [CrossRef]
- Steward, N.; Ito, M.; Yamaguchi, Y.; Koizumi, N.; Sano, H. Periodic DNA methylation in maize nucleosomes and demethylation by environmental stress. J. Biol. Chem. 2002, 277, 37741–37746. [Google Scholar] [CrossRef]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- You, J.; Zhang, L.; Song, B.; Qi, X.; Chan, Z. Systematic analysis and identification of stress-responsive genes of the NAC gene family in Brachypodium distachyon. PLoS ONE 2015, 10, e0122027. [Google Scholar] [CrossRef] [PubMed]
- Kayum, M.A.; Park, J.-I.; Nath, U.K.; Saha, G.; Biswas, M.K.; Kim, H.-T.; Nou, I.-S. Genome-wide characterization and expression profiling of PDI family gene reveals function as abiotic and biotic stress tolerance in Chinese cabbage (Brassica rapa ssp. pekinensis). BMC Genom. 2017, 18, 885. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2016, 45, D200–D203. [Google Scholar] [CrossRef] [PubMed]
- Rombauts, S.; Déhais, P.; Van Montagu, M.; Rouzé, P. PlantCARE, a plant cis-acting regulatory element database. Nucleic Acids Res. 1999, 27, 295–296. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | GENE NAME | Sub Cellular | Direction | Start Pos | End Pos | DNA Length | CDS Length | Exon No. | AA Length | Mol.Wt. (kD) | PI |
|---|---|---|---|---|---|---|---|---|---|---|---|
| BnaA06g23880D | BnaCMTa | Nuclear | + | 16,520,582 | 16,525,017 | 4436 | 2418 | 20 | 805 | 91.94 | 6.57 |
| BnaC03g49350D | BnaCMTb | Nuclear | - | 34,264,132 | 34,268,583 | 4452 | 2409 | 20 | 802 | 91.61 | 7.52 |
| BnaA06g07080D | BnaCMTc | Nuclear | - | 3,771,918 | 3,776,153 | 4563 | 2367 | 21 | 788 | 88.78 | 5.15 |
| BnaC05g08730D | BnaCMTd | Nuclear | - | 4,569,531 | 4,573,838 | 4641 | 2391 | 20 | 796 | 89.4 | 5.06 |
| BnaA01g09820D | BnaCMTe | Nuclear | + | 4,828,328 | 4,834,033 | 5706 | 3297 | 23 | 1,098 | 122.34 | 6.02 |
| BnaC01g11520D | BnaCMTf | Nuclear | + | 7,147,733 | 7,153,381 | 5843 | 3435 | 22 | 1,144 | 127.61 | 5.7 |
| BnaA06g29420D | BnaMET1a | Nuclear | - | 20,057,862 | 20,064,228 | 6367 | 3099 | 11 | 1032 | 117.19 | 9.01 |
| BnaC07g27300D | BnaMET1b | Nuclear | + | 32,855,407 | 32,862,845 | 7439 | 3297 | 12 | 1098 | 125.27 | 8.82 |
| BnaC08g34700D | BnaMET1c | Nuclear | + | 32,763,560 | 32,769,056 | 5705 | 4296 | 13 | 1431 | 162.1 | 5.92 |
| BnaA09g42250D | BnaMET1d | Nuclear | + | 29,386,022 | 29,391,515 | 5642 | 4299 | 13 | 1432 | 162.15 | 6.07 |
| BnaA04g13390D | BnaMET1e | Nuclear | - | 11,296,866 | 11,302,393 | 5666 | 4299 | 13 | 1432 | 161.45 | 6.05 |
| BnaC04g35500D | BnaMET1f | Nuclear | - | 37,010,080 | 37,015,638 | 5700 | 4326 | 13 | 1441 | 162.14 | 6.07 |
| BnaC07g29260D | BnaDNMT2a | Nuclear | + | 34,107,074 | 34,109,326 | 2468 | 1164 | 10 | 387 | 43.97 | 5.49 |
| BnaA06g27730D | BnaDNMT2b | Nuclear | - | 19,065,967 | 19,068,137 | 2342 | 1128 | 10 | 375 | 42.49 | 5.68 |
| BnaA05g22820D | BnaDRMa | Extracellular | + | 17,302,556 | 17,308,756 | 6464 | 2655 | 14 | 884 | 96.78 | 5.01 |
| BnaC05g36090D | BnaDRMb | Extracellular | + | 35,279,390 | 35,282,899 | 3889 | 2124 | 9 | 707 | 78.88 | 4.87 |
| BnaA07g28030D | BnaDRMc | Nuclear | + | 20,274,800 | 20,280,859 | 6187 | 2823 | 19 | 940 | 106.79 | 6.47 |
| BnaC06g30710D | BnaDRMd | Nuclear | + | 31,350,535 | 31,353,717 | 3183 | 1911 | 11 | 636 | 72.2 | 5.1 |
| BnaA02g02230D | BnaDRMe | Nuclear | - | 970,840 | 973,703 | 3262 | 1677 | 10 | 558 | 62.91 | 5.31 |
| BnaC02g05620D | BnaDRMf | Nuclear | - | 2,918,874 | 2,921,525 | 2652 | 1668 | 9 | 555 | 62.39 | 5.17 |
| BnaA10g19200D | BnaDRMg | Nuclear | + | 13,731,342 | 13,734,360 | 3780 | 1671 | 11 | 441 | 49.53 | 7.01 |
| BnaC09g42890D | BnaDRMh | Nuclear | + | 44,252,075 | 44,255,166 | 4126 | 1671 | 11 | 556 | 62.09 | 5.26 |
| BnaA10g25630D | BnaDMEa | Nuclear | - | 16,440,585 | 16,454,849 | 15,293 | 5607 | 23 | 1,868 | 207.48 | 8.3 |
| BnaC09g50670D | BnaDMEb | Nuclear | + | 48,435,123 | 48,442,674 | 7929 | 5133 | 20 | 1,710 | 189.21 | 7.98 |
| BnaA01g03070D | BnaDML3a | Nuclear | - | 1,479,536 | 1,485,590 | 6369 | 3243 | 24 | 1080 | 124.52 | 8.59 |
| BnaC01g04300D | BnaDML3b | Nuclear | - | 2,253,312 | 2,260,155 | 6948 | 3549 | 24 | 1182 | 135.52 | 8.37 |
| BnaA05g07800D | BnaROS1a | Nuclear | + | 4,240,657 | 4,246,594 | 6512 | 3903 | 24 | 1300 | 146.68 | 6.27 |
| BnaC04g08810D | BnaROS1b | Nuclear | + | 6,603,988 | 6,609,839 | 6583 | 3975 | 22 | 1324 | 148.73 | 6.08 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, S.; Liu, H.; Liu, J.; Hua, W.; Xu, S.; Li, J. Systematic Analysis of the DNA Methylase and Demethylase Gene Families in Rapeseed (Brassica napus L.) and Their Expression Variations After Salt and Heat stresses. Int. J. Mol. Sci. 2020, 21, 953. https://doi.org/10.3390/ijms21030953
Fan S, Liu H, Liu J, Hua W, Xu S, Li J. Systematic Analysis of the DNA Methylase and Demethylase Gene Families in Rapeseed (Brassica napus L.) and Their Expression Variations After Salt and Heat stresses. International Journal of Molecular Sciences. 2020; 21(3):953. https://doi.org/10.3390/ijms21030953
Chicago/Turabian StyleFan, Shihang, Hongfang Liu, Jing Liu, Wei Hua, Shouming Xu, and Jun Li. 2020. "Systematic Analysis of the DNA Methylase and Demethylase Gene Families in Rapeseed (Brassica napus L.) and Their Expression Variations After Salt and Heat stresses" International Journal of Molecular Sciences 21, no. 3: 953. https://doi.org/10.3390/ijms21030953
APA StyleFan, S., Liu, H., Liu, J., Hua, W., Xu, S., & Li, J. (2020). Systematic Analysis of the DNA Methylase and Demethylase Gene Families in Rapeseed (Brassica napus L.) and Their Expression Variations After Salt and Heat stresses. International Journal of Molecular Sciences, 21(3), 953. https://doi.org/10.3390/ijms21030953