Abstract
Proper regulation of the chromatin landscape is essential for maintaining eukaryotic cell identity and diverse cellular processes. The importance of the epigenome comes, in part, from the ability to influence gene expression through patterns in DNA methylation, histone tail modification, and chromatin architecture. Decades of research have associated this process of chromatin regulation and gene expression with human diseased states. With the goal of understanding how chromatin dysregulation contributes to disease, as well as preventing or reversing this type of dysregulation, a multidisciplinary effort has been launched to control the epigenome. Chemicals that alter the epigenome have been used in labs and in clinics since the 1970s, but more recently there has been a shift in this effort towards manipulating the chromatin landscape in a locus-specific manner. This review will provide an overview of chromatin biology to set the stage for the type of control being discussed, evaluate the recent technological advances made in controlling specific regions of chromatin, and consider the translational applications of these works.
1. Introduction
1.1. Chromatin
Differential regulation of the chromatin landscape (composed of DNA wrapped around histone linkers or octamers) allows for genetically identical eukaryotic cells to perform specific, discrete functions [1]. This is achieved by regulating different gene expression profiles, which in turn result in a cell adopting specific proteomes to conduct specialized functions in a multicellular organism. A nucleosome represents the core building block of chromatin and is comprised of 147 DNA base pairs (bp) wrapped around histone protein octamers (two sets of each histone protein H2A, H2B, H3, and H4) [2]. For three billion base pairs of DNA to fit within a 10 μm nucleus while still allowing functional information to be expressed, chromatin needs to be folded in a precise, organized manner [3]. Chromatin is differentially compacted along a spectrum of condensation states, with the term “heterochromatin” referring to areas of tight compaction (associated with low transcriptional activity) and “euchromatin” referring to areas of less compaction (associated with high transcriptional activity) [4].
Chromatin compaction has been identified as a physical determinant of DNA’s accessibility to transcription-initiating proteins, and these processes are highly regulated through deposition of histone protein variants, ATP-dependent nucleosome remodeling, methylation of DNA, and posttranslational modifications of histone tails [5]. These interrelated processes work in concert to control transcriptional activity, DNA replication, damage repair, and nuclear organization [4,6,7,8]. For the purposes of this review, we focus primarily on the contribution of DNA methylation and histone tail modifications to the regulation of gene expression.
Protruding from nucleosomes are N-terminal histone tails, capable of being post- translationally chemically modified, and DNA bases can also be modified at cytosine (C) and adenine (A) nucleotides [9,10,11,12]. Specific combinations of histone and DNA modifications give rise to an epigenetic code, with specific modifications being associated with high or low levels of gene expression [13]. To date, four nucleotide modifications have been observed: methylation, hydroxy-methylation, formylation, and carboxylation [14]. Over ten modifications have been identified for histone tails, with more modifications currently being validated and characterized [15,16].
The Allis group coined the phrase “the histone code” to describe the observation that distinct histone marks function sequentially or in combination to initiate distinct downstream changes [11,13]. Changing modifications of nucleotides or histone tails requires a series of proteins: writers, erasers, and readers. “Writer” enzymes deposit the nucleotide or histone tail modification, “eraser” enzymes remove the modification, and “reader” domains recognize a modification, initiating downstream consequences such as recruitment of transcription factors [5]. Two of the most widely studied nucleotide modifications are methylation (of DNA and of the lysine residue) and acetylation (of the lysine residue) [17]. These chromatin-based mechanisms are commonly used in the field of epigenetic biotechnology to control gene expression. Importantly, there is debate between scientists about correlation versus causation. Are the actual histone marks and DNA modifications important for changes in transcription, or are the modifications passengers? There are examples of evidence for both models [18,19,20]. Fortunately, gene-specific manipulation of chromatin can serve as a new tool to further study these models. The classification and function of the proteins mediating DNA methylation and lysine methylation/acetylation are important for the design of chromatin recruitment tools.
1.2. DNA Methylation
Eukaryotic DNA methylation is most often observed as 5-methylcytosine in the context of a cytosine–guanine (CpG) dinucleotide and is strongly associated with gene repression, though there are some notable exceptions [21,22]. The mark is deposited by DNA methyltransferases (DNMTs), enzymes that transfer a methyl group from S-adenosyl-L-methionine (SAM) to cytosine at the 5′ position [23]. Within the mammalian DNMT family, DNMT1 is considered the maintenance methyltransferase, as it propagates the methylation pattern from parental to daughter strands during cell replication [24,25]. DNMT3A and DNMT3B are both considered the de novo methyltransferases, as they methylate both hemi-methylated and un-methylated DNA. DNMT3A/B are critical for the initiation of DNA methylation during gametogenesis and the early stages of embryogenesis [21,26]. Importantly, these are the DNMT family members that have been used for epigenomic manipulation (discussed in Section 3 of this review), in part due to the stability of DNA methylation to progress through cell replication [27,28]. The DNA methylation “erasers” are part of the ten-eleven translocation (TET) family. It has been shown that this family of enzymes catalyze the stepwise oxidation of methyl groups, ultimately removing the methylation from the cytosine [14] in concert with the TDG DNA glycosylase [29]. The proteins that recognize methylated DNA are the methyl CpG binding proteins (MBPs), including zinc finger (ZnF) domain-, SRA domain-, and methyl CpG binding domain- (MBD-) containing proteins [30]. Following recognition of methylated DNA, compaction and transcriptional silencing is initiated, in part through coordinated recruitment of chromatin remodeling complexes.
1.3. Histone Lysine Methylation
The lysine residues on histone tails can be mono-, di-, or tri-methylated by a set of histone writer proteins, generically referred to as histone lysine methyltransferases (KMTs). Methyl groups are transferred from the methyl donor, adenosyl-methionine. KMTs are split into two families based on the presence (or lack) of a SET domain [17]. Mammalian examples of SET-containing enzymes include H3K4 methyltransferases (such as SET1A/B and SET7 [31,32]), H3K9 methyltransferases (such as EHMT2/G9a and SUV39H1/2) [6,33,34,35], H3K27 methyltransferases (such as EZH1/2) [36,37,38], H3K36 methyltransferases (such as NSD1) [39], and H4K20 methyltransferases (such as SUV4-20H1/H2) [40]. Fewer studies have been done on KMTs lacking the SET domain other than the highly studied H3K79 methyltransferase, DOT1 [41,42,43]. Proteins that recognize histone lysine methylation contain one, or multiple, reader domains: chromodomain [44], chromobarrel domain [45], double chromodomain [46], PWWP domain [47], plant homeodomain [48], WD40 domain [49], malignant brain tumor domain [50], and Tudor domain [17,51]. Some of these reader domains have been discovered in proteins alongside other reader domains (i.e., UHRF1 recognizes hemi-methylated DNA and H3K9me3) [52]. The removal of lysine methylation is performed by one of two ways: lysine-specific demethylase (LSD) members, LSD1/KDM1A and LSD2/KDM1B, contain an amine oxidase (AOD) domain responsible for demethylase activity in a flavin adenine dinucleotide (FAD)-dependent manner [53], whereas Jumonji C (JmjC) domain-containing enzymes [54] utilize the co-substrates 2-oxoglutarate (2OG) and dioxygen with cofactor Fe(II) [55,56,57,58,59]. Unlike DNA methylation, the transcriptional influence of lysine methylation is strongly context dependent [60]. For example, H3K36me3 is observed in the gene body of transcriptionally active genes, whereas H3K9me3 is linked with gene repression when concentrated near transcriptional start sites (TSS) [60,61]. For this reason, the chromodomain, a reader of H3K9me3, has been used for recruitment to induce targeted control of chromatin (in systems discussed in Section 3 of this review).
1.4. Histone Lysine Acetylation
Lysine residues can be acetylated, which typically results in a less condensed chromatin state coupled with an increase in transcriptional activity [62,63]. Histone acetyl transferases (HATs) are the writer enzymes that deposit acetylation marks from acetyl-CoA. HAT families are classified based on cellular location, with Type A HATs functioning on chromatin in the nucleus and Type B HATs acting on newly translated histones in the cytoplasm. The Type A HATs are further classified into five subtypes based on their structural and functional characteristics: GNAT, MYST, p300/CREB, TAF250, and SRC/NCoA [17]. The most commonly used HAT family in the field of epigenetic biotechnology is the p300/CBP family of HATs, KAT3A/CBP and KAT3B/p300, which acetylates all four histone subunits and contains several domains including a KIX domain, a bromodomain, and a HAT domain [64]. Histone deacetylases (HDACs) are the eraser enzymes that remove lysine acetylation, supporting a transcriptionally silenced state. This set of enzymes is subdivided into Class I, IIa, IIb, III, and IV [17,65,66]. HDAC Class I, IIa, IIb, and IV require an interaction with zinc, whereas class III, or sirtuin, requires an HDACs function with NAD+ [17,65,67]. For chromatin manipulation with biotechnology, class I HDACs (HDAC1/2/3/8) are most commonly used, in part because all are observed predominantly in the nucleus and are expressed in most tissue types. In addition, HDAC1/2/3 function in collaboration with repressive complexes, which is important for downstream recruitment and targeted transcriptional repression. The three peptide folds that interact with histone lysine acetylation are the bromodomain (BRD), double PHD finger domain, and Yeats domain [17]. BRD-containing proteins are the most abundant lysine acetylation readers and are sub-classified into eight families based on similarities in structure and sequence [17,68,69] including the BRD II family comprising the Bromodomain and Extra-Terminal Domain (BET) proteins BRD2/3/4 and BRDT. BET proteins recognize H4K12ac, H3K14 ac, H4K5ac, and H4K8ac. Lastly, BRD family III domains are found in many proteins that are ubiquitously expressed across cell types including EP300 and CREBBP [69].
1.5. Chromatin and Transcription
Models that describe the close interplay between specific chromatin environments and their associated transcriptional activities are based on two characteristics: the physical accessibility of the DNA due to the level of chromatin compaction and the recruitment of transcriptional machinery by chromatin modifications. At the eukaryotic promoter, RNA polymerase II (Pol II) binds to DNA, in coordination with other general transcription factors, and forms the preinitiation complex (PIC) [70]. The chromatin environment surrounding the promoter, as well as functionally relevant enhancer regions, is important for Pol II binding, PIC formation, subsequent elongation, and proper termination. For example, variations in nucleosome acetylation levels impact how readily the transcriptional machinery runs along the DNA [71]. Additionally, H4K16ac blocks chromatin compaction and increases overall accessibility of the DNA to the transcriptional machinery [72]. Recruitment of SetD2 by Pol II is an import step that occurs during transcriptional elongation. Upon Set2 recruitment, H3K36me3 is deposited, followed by hypoacetylation of the gene body [73]. Lastly, prolonged recruitment of HP1 (through recognition of H3K9me3) results in the deposition of DNA methylation, an epigenetically stable and transcriptionally repressive modification [74]. While this interplay is more complicated than what has been described above, these examples illustrate how transcriptional regulation and the chromatin environment are related. Given this tight interaction, manipulating the function, recruitment, and/or expression of chromatin modifying proteins can subsequently control the expression of essential genes [75].
2. Global Control of Chromatin
For decades, researchers have sought to understand the mechanisms by which the chromatin regulatory machinery functions to control higher-ordered genomes. Understanding the function and transcriptional effects induced by various DNA and histone writers, readers, and erasers are a major goal for basic scientists and translational scientists. There are several ways in which scientists have worked towards understanding and controlling these pathways by modulating the activity of a chromatin modifying enzymes, either by inhibiting their catalytic domains [76,77,78], causing their degradation [79], or preventing them from binding [20,80].
By directly binding and inhibiting the catalytic activity of chromatin modifying enzymes, effects on transcription and translation have been inferred. For example, the first two DNMT inhibitors (DNMTi) synthesized, Azacytidine (5-azacytidine) and Decitabine (5-aza-2′-deoxycytidine), are classified as “nucleoside” inhibitors. They mimic the structure of cytidine, are incorporated into the DNA, and form irreversible interactions with DNMTs [81]. Several inhibitors of EZH1 and EZH2 (HMTs that catalyze H3K27 methylation) exist and are classified based on their chemical structure into pyridine-indazole scaffolds (EPZ005687, UNC1999, and GSK343), pyridine-phenyl scaffolds (EPZ006088 and EPZ6438), and pyridine-indole scaffolds (GSK126, CP1-1205, and EI1) [82]. To study and control histone acetylation, a potent and selective p300/CBP inhibitor, termed A-485, was synthesized and shown to bind the catalytic active site of p300, competing with acetyl-CoA [83]. These types of chemicals block function by directly targeting the protein-of-interest.
Another means by which scientists have studied and controlled the chromatin regulatory machinery is through targeted degradation with Proteolysis Targeted Chimeras (PROTACs). These bifunctional molecules bind a protein of interest with one portion of the PROTAC, while the other end binds an E3 ubiquitin ligase protein [84]. Through recruitment of E3 ubiquitin ligase, the protein-of-interest is rapidly degraded. For example, Zengerle et al. developed a PROTAC capable of selectively targeting and degrading BRD4 (a member of the BET family of histone acetylation readers) [79].
Lastly, by blocking the chromatin-based target, the function of specific chromatin marks and enzymes can also be inferred. For example, DNMTis have been designed and synthesized in the “non-nucleoside” category. For instance, Procainamide binds CpG regions of DNA to block DNMT-based activity [80]. These types of chemicals do not bind to the actual DNMT protein, but rather the target, CpG regions of DNA. Another way that chromatin regulation has been studied is through histone replacement. In the approach, the genes that code for histones in Drosophila are replaced with histone genes incapable of having certain post-translational modifications. In a seminal paper, McKay et al., mutated the H3K27, H3K36, or H4K20 residue from lysine (K) to Alanine (A), which is incapable of being methylated or acetylated and observed that H3K36 is required for viability and H3K27 is essential for maintenance of cellular identity [20].
In the above examples, global chromatin is being impacted, either through inhibiting or degrading the chromatin modifying machinery or by preventing a DNA or histone modification from being deposited. The disadvantage of chemically inhibiting or degrading chromatin regulatory machinery in the whole cell or organism is that many of the existing chemicals bind off-target proteins. Especially in instances where a conserved domain is being targeting (for example with HDAC class I inhibitors wherein the lysine-binding groove is very similar between all Class I HDACs), specificity is difficult to achieve [85,86,87]. These non-specific binding instances could impact the study’s results or the therapeutic potential. In addition, many of these proteins have multiple substrates, unrelated to chromatin. Downstream effects of the inhibitor could be, at least in part, related to these other substrates [88]. While off-target specificity has been addressed with some of these substrate-blocking methods (CpG binding and histone replacement), the ability to precisely study coordinated pathways with multiple proteins and steps is somewhat limited. In addition, any chromatin-based changes will be epigenome-wide, resulting in direct and indirect changes in gene expression. It is important to recognize that in some disease settings, targeting multiple genes with similar aberrant chromatin modifications is beneficial. When large cohorts of genes are co-repressed by compacted chromatin, targeting multiple genes represents a more efficient solution than targeting one gene at a time [89]. To complement and expand upon previous work with these epigenome-wide approaches, scientists have begun investigating and controlling chromatin environments at a gene-specific level. By targeting defined chromatin modulators to a gene-of-interest, minute mechanistic questions can be answered, and gene-specific transcriptional activity can be controlled.
3. Short-Range Locus-Specific Control of Chromatin
While a lot can be learned through global perturbation of chromatin, site-specific technologies offer an opportunity to examine chromatin regulation in the context of a more physiologic setting without gross changes to the cellular environment. To recruit chromatin effectors to a specific gene, technologies have been developed and utilized to genetically modify a gene-of-interest, insert a Gal4-bind arrays, and recruit Gal4-fused chromatin modifiers [74,90,91,92]. Native to yeast, Gal4 fused to a defined chromatin-modifier would achieve specificity to the mammalian gene of interest. Many other similar proteins with matched DNA binding arrays have also been used (e.g., LexA). The requirement of homologous recombination or other DNA insertion technique to edit genes, in the pre-CRISPR era, made the upfront work for these types of experiments relatively time-consuming and expensive. With the advancements of custom zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and clustered regulatory interspaced short palindromic repeats (CRISPR)/CRISPR-associated (Cas) systems, epigenome editing has been possible without initial modification of mammalian genes [93]. To edit the genome, double-stranded break (DSB) or non-homologues end joining repair pathways are initiated by fusing sequence-specific DNA-binding domains (ZFN or TALEN) to the FokI restriction endonuclease or by creating a sequence-specific single guide RNA (sgRNA) to recruit a Cas endonuclease [94,95,96,97,98]. These techniques were later adapted to serve as a recruitment, rather than DNA-editing, platform and paved the way for targeted chromatin editing, consequently advancing the fields of basic science and translational research. We summarize examples of current technologies to achieve gene-specific control of chromatin in Figure 1.
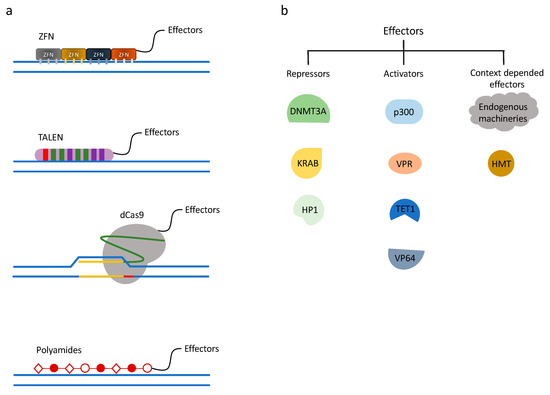
Figure 1.
Chromatin engaging technologies and gene regulation systems. (a) Chromatin engaging technologies. ZF (zinc finger), TALE (transcription activator-like effector), dCas9, and polyamides are chromatin-engaging techniques used for precise gene targeting, often fused directly to effector proteins. (b) Gene regulation systems. Examples of different effectors used in chromatin engaging technologies. By fusing repressive or activating effectors to chromatin engaging platforms, gene regulation can be achieved.
3.1. Zinc Finger Nucleases
ZFNs were the first of these technologies to be developed for editing the mammalian genome (Figure 1a). The Cys2-His2 zinc-finger domain has been the most common type of zinc-finger used for these editing platforms. Each zinc-finger consists of roughly 30 amino acids (aa) to form a ββα conformation, with a α-helix and the adjacent turn designed to bind 3 bps of DNA. The optimized systems often use six zinc finger domains strung together, targeting 18 bps. This 18 bp length provides specificity within 68 million bases [99]. Zinc fingers have been developed to recognize most of the possible nucleotide triplets, such that pre-designed zinc fingers triples can be linked with molecular cloning techniques in a specific order to target a predefined DNA sequence. If fused to the FokI endonuclease, the zinc fingers are able to induce a DSB in a locus-specific manner. More recently, scientists have used these zinc-finger domains to recruit chromatin modifying machinery to a region of DNA, enabling them to study the resulting chromatin and transcriptional changes [92,100,101]. For example, Snowden et al. targeted the endogenous, mammalian VEGF-A locus with a gene-specific zinc finger fused to an HMT. They demonstrated increased H3K9 methylation as well as targeted gene repression [101]. The main disadvantage of the zinc finger technology is that designing and creating zinc fingers is a time-consuming and labor-insensitive process. An alternative way to use this technology is through ordering pre-made constructs to target your gene-of-interest, though they are relatively expensive.
3.2. Transcription Activator-Like Effector Nucleases
Transcription Activator-Like Effector (TALE) proteins are found natively in bacteria and contain a series of 33-35 amino acid repeat domains that recognize a single DNA base pair (Figure 1a). Similar to ZnFs, TALE repeats are strung together to recognize specific DNA sequences [102]. To create a TALE nuclease (TALEN) for the purpose of gene-specific editing, an endonuclease is fused to the TALE. This single-base-pair approach allows for more flexibility in the design than the triple nucleotide design of zinc fingers, however the design and cloning process is more arduous and expensive. Regardless of the upfront efforts, several groups have utilized the TALEs system to study the chromatin-based and transcriptional effects of a given chromatin regulatory protein [92,103,104,105]. For example, Maeder and colleagues demonstrated the ability to actively demethylate human endogenous RHOXF2 with a TALE-TET1 fusion. Upon recruitment of this fusion protein, CpG methylation levels decreased and transcriptional activity increased [105].
3.2.1. Introduction to dCas9
The clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated protein (Cas) system provides a technique for mammalian genome editing. Cas9 is the most widely used CRISPR protein for epigenomic engineering. CRISPR RNA (crRNA) and trans-activating crRNA (tracrRNA) recruit Cas9 to a complementary sequence of DNA and sequence-specific nuclease cleavage is introduced. The CRISPR-Cas system was first discovered in prokaryotes as an adaptive immunity to foreign viruses and plasmid DNA. To apply the CRISPR/Cas9 system to mammalian genome manipulation, a single-chain chimeric RNA was created by fusing crRNA and tracrRNA, termed single guide RNA (sgRNA) [106]. This sgRNA contains a complementary sequence to the genome, typically 20 base pairs in length, providing the design capability to target any gene of interest. The protospacer-adjacent motif (PAM) sequence, a unique sequence downstream of the 20-mer, which the Cas9 protein needs for recognizing a target DNA region, is the only required component at the 3′ end of the DNA complementary region [107]. Therefore, compared to ZFN and TALEN, CRISPR/Cas9 can be customized by a 20 base pair synthetic sequence with relatively little molecular cloning. To repurpose Cas9 for epigenome regulation, instead of genome editing, a nuclease deficient Cas9 (dCas9) was developed by introducing mutations at the RuvC1 and HNH nuclease domains (D10A and H841A, respectively) [108]. The dCas9 retains the recruitment characteristics, such that it can bind to a predefined DNA sequence without cutting (Figure 1a). Soon after the dCas9 platform was introduced, an evolving field has grown around dCas9-related protein engineering and technology development to regulate the mammalian epigenome.
3.2.2. Approaches Using Direct Fusions to Activate or Repress Gene Expression
Shortly after Qi et al. published the design of the deactivated Cas9 (dCas9), several labs began to engineer activating protein domains to dCas9 to increase endogenous gene expression. One of the first works published was the dCas9-VP64 fusion from Meader et al. VP64 is a tetrameric repeat of herpes simplex VP16′s minimal activation domain [109]. Meader et al. linked VP64 to the C-terminus of dCas9 and activated expression of human VEGFA and NTF3. In this work they also created a multiple sgRNA targeting system with which a set of sgRNAs can be co-expressed to target the same gene at several places showing that the activation level of the targeted genes was elevated when compared to using only one sgRNA. A variety of different mammalian genes were activated by other groups by directly fusing activating factors, including VP64 [97,110,111], VP160 (VP16x10) [112], VP192 [113], p65 AD (activation domain) [110], VPR (VP64-Rta-p65) [114,115], Tet1 CD (catalytic domain) [116,117,118], and the p300 core domain [119] (Figure 1b).
Besides exploring activation of a gene by using dCas9 fusion proteins, groups have also demonstrated the possibility of engineered dCas9 proteins to efficiently repress gene expression. Gilbert et al. attached KRAB to dCas9 (Figure 1a,b) and showed repression on a fluorescent reporter gene by 15-fold. Furthermore, RNA-seq analysis indicated high specificity. They next knocked down endogenous gene expression of CXCR4 and CD71 in cell lines stably expressing dCas9-KRAB and transduced with sgRNAs [110]. Vojta et al. applied the dCas9 direct fusion technique into DNA methylation and created dCas9-DNMT3A fusion proteins (Figure 1a,b) to specifically increase CpG methylation at a roughly 35 base pair window at the promoter region of the targeted gene locus using multiplexed sgRNAs. They demonstrated that the expression of the targeted endogenous genes was transcriptionally decreased with a concurrent increase in CpG methylation [27]. It is worth mentioning that Qi lab also demonstrated that dCas9 alone can interfere with transcription when the sgRNA targets the protein-coding region by blocking RNA polymerase and transcript elongation [108]. The dCas9-KRAB fusion protein technology has also recently been used in large genome wide screens to map the impact of enhancer elements on gene expression [120].
In addition to the promoter region of genes, different publications have shown the ability to target dCas9 fusions to non-promoter regulatory elements as a means to control the chromatin environment. Hilton et al. expanded the targeting region to core enhancers (CE), distal regulatory regions (DRR), proximal enhancers (PE), and distal enhancers (DE). Both dCas9-p300 core and dCas9-VP64 were used in their experiments targeting MYOD1. Based on the activation level of MYOD1, sgRNAs targeting the CE, DDR, or promoter region in combination with dCas9-p300 core showed better efficacy than dCas9-VP64. On another gene, the human β-globin locus, by targeting the hypersensitive site 2 (HS2) enhancer region with sgRNAs and dCas9-activator fusion proteins, multiple downstream hemoglobin genes were significantly overexpressed simultaneously [119]. Thakore et al. from the same research group continued working on targeting the same HS2 enhancer with dCas9-KRAB and successfully induced H3K9me3 deposition, decreasing chromatin accessibility [121]. Kearns et al. studied and published that cell type-specific regulatory elements can be controlled through recruitment of histone demethylase activity with dCas9-LSD1 [122]. As more attention was drawn to dCas9 technology, a variety of approaches were introduced into this field. Klann et al. developed CRISPR/Cas9-based epigenomic regulatory element screening (CERES) for high-throughput screening of regulatory element activity in their native genomic context. They used dCas9-p300 or dCas9-KRAB constructs and sgRNA libraries to target DNase I hypersensitive sites (regions of open chromatin) surrounding a gene-of-interest. They demonstrated that CERES can identify both known and undiscovered regulatory elements, like HER2 [123]. However, the off-target effects of CRISPR/dCas9 system still need to be addressed by genome-wide readouts.
3.2.3. Indirect dCas9 Recruitment Strategies
After the development of dCas9 direct fusion technologies, researchers developed dCas9-based indirect effectors. The incorporation of SunTag into the dCas9 system has significantly increased the number of epigenetic modulators being recruited. The SunTag is a repeating peptide array, which can recruit a single chain variable fragment (scFv) antibody fusion. Tenenbaum et al. tested the recruitments of 4, 5, 10, or 24 scFv-VP64 fusions and successfully promoted the regulatory effect by increasing the number of antibody fusions [124]. They continued evolving the dCas9-SunTag system with scFv-TET1 [117] and scFv-DNMT3A [125] and observed significant changes in endogenous gene methylation.
Not only have groups focused on the dCas9 fusion protein engineering, but also on the scaffolding sgRNAs (scRNAs). By engineering the sgRNA with additional stem loops, the recruitment power of the sgRNAs can be increased. Zalatan et al. created three scRNA systems and compared the recruitment efficacy between them. They engineered MS2 stemloops to recruit MS2-capping proteins (MCP), PP7 stemloops to recruit PP7-coat proteins (PCP), and Com stemloops to recruit Com proteins. By fusing MCP, PCP, and Com proteins to VP64, transcriptional activation through stemloop-based recruitment was observed. Furthermore, they developed a two-stemloop version with identical stemloops in the scRNA and the activation was improved [126]. Further optimization of the MS2 system was developed based on the crystal structure of the interaction between dCas9 and sgRNA. Incorporation of the MS2 stemloops in the dCas9-recruiting sgRNA stemloops promoted the recruitment power with MCP-VP64, which was 14-fold more efficient in targeting ASCL1 than placing the addition loops downstream of the sgRNA stemloops. This improvement likely occurred because either the positioning or the degradation protection of the MS2 protein was improved. In the same study, it was demonstrated that engaging different recruitment strategies (dCas9-VP64 and MCP-P65) was more powerful than using the same activator in both dCas9 and MCP fusions. By utilizing this MS2 stemloop sgRNA, MCP-TET1-CD can activate gene expression by DNA demethylation [127]. With the optimizations of the SunTag system and the scRNA, a side-by-side comparison of dCas9-direct fusions, scRNAs, and SunTag activation efficacy was conducted by Chavez et al. They tested on human, mouse, and fly cell lines, and found that the activation level was effector-, cell line-, and sgRNA-dependent [128].
3.2.4. Inducible Systems
While researchers were focusing on controlling gene expression by utilizing synthetic devices, several labs began developing chemically responsive gene expression control constructs. By introducing small molecules into biological systems, gene expression can be controlled in a temporal manner. The Conklin lab took advantage of doxycycline (Dox; Figure 2a) and iPSC’s differentiation mechanism and created a doxycycline-inducible dCas9-KRAB repression domain. By introducing Dox into the system, they demonstrated that dCas9-KRAB repression was highly Dox-dependent and reversible [129]. As an attempt at the other direction of chemical control, the Adli lab developed a dCas9 construct that can be turned off by the addition of a plant-based technology called auxin-induced degron. By tethering the degron peptide to dCas9-p300 fusion protein, adding auxin can induce recognition of dCas9 constructs by SCF-E3 ubiquitin ligase through the TIR1 protein. In this technology, roughly 90% of the degron-dCas9-p300 expression can be reduced in a rapid (within 6 h of auxin treatment) and reversible manner [130].
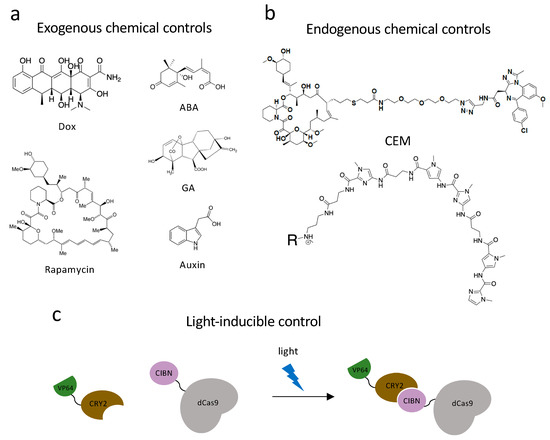
Figure 2.
Dynamic control systems. (a) Exogenous chemical controls. Dox, rapamycin, ABA (abscisic acid), GA (gibberellic acid) and auxin can interact with DNA-binding systems to recruit effectors to chromatin and achieve dynamic chromatin regulation. (b) Endogenous chemical controls. CEMs and Syn-TEFs are synthetic molecules designed to facilitate with chromatin engaging constructs and achieve precise gene regulation by recruiting endogenous protein complexes. (c) Light-inducible control technology. Light-inducible heterodimerizing proteins, CRY2 and CIBN, undergo a conformational change in the presence of a specific fluorescent light and bring effectors to chromatin engaging constructs to regulate gene transcriptions.
Apart from the two chemically dependent systems mentioned above, there is another approach, chemical inducers of proximity (CIP), being applied to both study and control chromatin biology. This system utilizes a class of bifunctional molecules, which can recruit two protein fusions together and initiate a new biological response. The first CIP was a homodimer of FK506 (FK1012) [131]. FK506 is a calcineurin inhibitor, which can bind FK506-binding-protein (FKBP12) from one side and bind calcineurin on the other. By appending two FK506 molecules into one homodimer, FK1012 can bring together two FKBP proteins. Another FKBP-interacting molecule is rapamycin (Figure 2a), which is a macrolide immunosuppressor. Rapamycin is capable of bringing together FKBP12 and the FKBP12-rapamycin-binding (FRB) domain of the TOR kinase [132]. Three other commonly used CIPs are plant-based molecules, which are advantageous because they are non-toxic while in mammalian systems. Abscisic acid (ABA) is a plant hormone, which functions with the Pyl receptor and phosphatase ABI1 (Figure 2a). Gibberellin (GA) is another plant hormone that induces tethering of GID1 and GAI (Figure 2a). The last common plant-based CIP is indole-3-acetic acid, also named auxin, which was previously used in dCas9 proteasomal degradation (Figure 2a). Auxin can recruit the Cul1 complex to degron-labeled proteins. The discovery and characterization of these CIPs encouraged scientists to develop new techniques and utilize these molecules in diverse ways [132].
With the development of CRISPR/Cas9, many labs have incorporated CIPs and dCas9 fusion proteins to control chromatin structure and gene expression. The Crabtree lab developed a dCas9-based rapamycin, FKBP, and FRB system. By creating ms2-FKBP and FRB-chromatin regulator fusions (e.g., FRB-HP1cs and FRB-BAF), they achieved reversible control of the chromatin environment of endogenous mammalian genes and a change in transcription activities [133]. Zetsche et al. combined rapamycin with a different approach by splitting a dCas9-VP64 fusion into dCas9(N)-FRB and dCas9(C)-FKBP-VP64 [134]. Upon treatment with nanomolar amounts of rapamycin, the C-terminal dCas9 fragment and the N-terminal dCas9 fragment interacted and activated ASCL expression at a level comparable to the full-length dCas9-VP64 (57-fold overexpression compared to untreated cells). The Qi lab also developed a highly intricate dCas9 and CIP system. They utilized both ABA and GA to create dCas9-based synthetic gene circuits. They can recruit activating or repressive machinery in a chemically dependent manner by creating dCas9-AB1/Pyl1-fusions and dCas9-GAI/GID1 fusions. The ABA- and GA- inducible recruitments were reversible and orthogonal [135]. Another CIP application was in a split SunTag system whereby the ScFv was fused to FKBP12 and VP64 was fused to a mutated FRB domain (T3089L) [136]. A rapamycin analogue AP21967 that can bind to the mutated FRB selectively over the endogenous TOR protein was developed. This system was used to activate a BFP reporter gene, and 140-fold activation was achieved following treatment with AP21967. For further details of dCas9- and CIP based chromatin regulation please refer to Corson et al. [137] and Stanton et al. [132].
Optogenetic systems have an advantage in precise spatial and temporal controls. Polstein and Gersbach engineered a light-activated dCas9 effector (LACE) system to induce the transcription of endogenous genes by illuminating blue light [138]. They utilized a pair of light-inducible heterodimerizing proteins, CRY2 and CIBN, by fusing CIBN to dCas9 and CRY2 to VP64 (Figure 2c). CIBN-dCas9-CIBN is localized by sgRNA to the targeted gene. In the presence of blue light, CRY2 undergoes a conformational change and heterodimerizes to CIBN, which recruits VP64 to the gene-of-interest and causes transcriptional activation. The Sato group used another pair of light-inducible heterodimerizing domains (positive magnet and negative magnet) and generated photoactivatable dCas9 (padCas9) by splitting dCas9 [139]. They attached the N-terminus of dCas9 to pMag and the C-terminus to nMag and VP64. In the presence of blue light, pMag and nMag will assemble the whole dCas9 protein and targeted endogenous genes will be activated. They successfully induced neuronal differentiation in induced pluripotent stem cells with this approach. Shao et al. incorporated optogenetic far-red light (FRL) to create an activator dCas9 and developed an orthogonal, reversible endogenous gene-activation system (FACE) [140]. FRL has the ability to penetrate deeply into tissues and can activate engineered bacterial photoreceptor BphS, which converts GTP to c-di-GMP. When c-di-GMP concentration is increased, the FRL-dependent transactivator will bind to its chimeric promoter and initiate MS2, P65, and HSF1 transcription. With the expression of MS2 fusions, dCas9 and sgRNA can activate targeted gene expression.
Similar to the split dCas9 protein technology, the Muir lab used another split intein-mediated protein trans-splicing strategy (PTS) and designed dCas9-InN (a genetic fusion of dCas9 and the N-terminal fragment of a split intein) and InC-effector. They tested dCas9-InN and InC-VP64 as a direct fusion, which led to transcriptional activation of targeted genes (IL1RN and NTF3). Next, they expanded the effector into engineered InC-JQ1x4 (small molecule) and InC-UNC3866 (synthetic peptide) and recruited endogenous BRD4 and PRC1 complexes, respectively [141].
Recruiting endogenous chromatin modifying enzymes has a great advantage over exogenous engagement in the simplicity of the technology. With the development of dCas9 approaches to regulate chromatin structure, research groups have expanded the application, utilizing chemical endogenous recruiting warheads. We have developed a bifunctional molecule named chemical epigenetic modifier (CEM), which has two functional components, FK506 and a molecule interacting with cellular epigenetic machinery [142] (Figure 2b). We incorporated dCas9 with MS2 sgRNA stemloops and MCP-FKBPx2 fusion and determined it to be the most effective dCas9 recruiting machinery after screening a series of other recruitment systems including dCas9-FKBP and SunTag system. Different recruiting warheads were tested to target endogenous genes (CXCR4, MYOD1, etc.) in multiple cell lines, and CEM87, which is composed of FK506 and iBet762 (a BRD inhibitor), was determined to be the best-in-class CEM. With different concentrations of CEM treatment, graded activation of targeted genes can be achieved. CEM can also provide reversible gene activation by washing out the molecule. To determine the off-target effects, ChIP-sequencing (ChIP-seq) and RNA-sequencing (RNA-seq) were conducted. With expression of the targeting sgRNA and addition of CEM87, no off-target effects were observed, which is indicative of the specificity of CEM technology.
3.3. Synthetic Transcription Factors
Transcription factors (TFs) are diverse signaling proteins that play important roles in gene expression and cell differentiation in a dynamic manner. During cellular development, transcription factors interpret signals and initiate programmed transcriptional changes. Artificial transcription factors (ATFs) have been designed and customized to reprogram somatic cells into induced pluripotent stem cells (iPSCs) [143]. Repurposing ATFs as a toolkit to regulate transcription provides another category of gene-specific targeting and regulatory technology. ATFs are composed of a DNA Binding Domain (DBD), an Interaction Domain (ID), and an Effector Domain (ED). TALEs, ZnFs, CRISPR-Cas9, and polyamides can serve as the DBD. Pyrrole- and imidazole-based polyamides are a class of synthetic molecules developed by Ansari and colleagues, designed to be sequence-specific synthetic transcription elongation factors (Syn-TEFs), which are programmable DNA-binding ligands that target desired genomic loci and engage the transcription elongation machinery [144] (Figure 2c). The programmed polyamides were designed to bind at the frataxin (FXN) gene and were conjugated to JQ1 (BRD4 ligands). By recruiting BRD4 to FXN, JQ1 can engage the transcription elongation machinery P-TEFb and facilitate transcription elongation. Activation of FXN was observed in lymphoblastoid cell lines, and no off-target effects were seen with RNA-seq and ChIP-seq. With treatment of Syn-TEF to overexpress FXN, they induced iPSCs to differentiate into cardiomyocytes that expressed cardiac-specific markers and were observed to beat rhythmically in culture. Finally, they examined the utility of Syn-TEF in vivo by establishing an immunocompromised mouse model bearing a luciferase reporter fused with FXN, and upon Syn-TEF treatment luciferase expression was stimulated. More ATF technologies can be found in the review written by Heiderscheit et al. [145].
4. Translational Implications
Discoveries over the past few decades have demonstrated the importance of chromatin regulatory pathways and have implicated their dysregulation in several human diseases (Table 1). Technological advances and new ways of thinking have paved the way for understanding how these processes are dysregulated in diseased settings, what the downstream transcriptional consequences are, and how best to correct these changes. Epigenetic-based clinical trials are ongoing and dozens of epigenetic-based drugs have been approved, yet more specific and safer approaches are needed.

Table 1.
Histone and DNA writers, readers, and erasers modified in diseases.
With the understanding that changes in chromatin can contribute to disease initiation, growth, and/or therapeutic resistance, small molecules have been developed to study epigenome modulators and regulate their activity [194]. The idea of using epigenomic agents in combination with the standard-of-care therapy regiment has been around for over 40 years [195]. As previously discussed, targeting chromatin regulators in a cell-wide manner can result in off-target effects. Chromatin modifying enzymes can have non-histone substrates. The inhibition of an acetyl transferase, for example, could result in undesired effects. In addition, dysregulation of these pathways impacts the expression of dozens to thousands of genes, while only a small percentage of these gene expression changes are contributing to the diseased phenotype. Lastly, many of these inhibitors have limited target specificity, resulting in inhibition of other proteins. All of these whole-cell changes and the lack of specificity results in less effective drugs and increased toxicity in patients. For example, high levels of toxicity have been observed in patients treated with HDACi [196], likely due to poor bioavailability and/or off-target binding effects [195,197]. It has been demonstrated that the compounds also inhibit methylation and phosphorylation of non-histone substrates [198]. A trial using Vorinostat in combination with a chemotherapy (docetaxel) was terminated due to toxicities including neutropenia, peripheral neuropathy, and gastrointestinal bleeding [199]. Additional patient toxicities include fatigue, nausea, vomiting, thrombocytopenia, and weight loss [200]. While improvements in development of potent, targeted drugs have been made, the ability to control the chromatin environment and the expression of specific genes and/or target exposure to exclusively cancer cells are areas being actively pursued in research.
In some cases, groups have been able to specify a tumor suppressor or oncogene for a given disease, in which the chromatin environment is altered at the gene locus, thereby affecting the transcriptional activity (Table 2). These examples represent ideal targets for gene-specific control of chromatin environments. In addition to manipulating the chromatin environment at a specific gene, this level of control enables scientists to model mislocalization of chromatin machinery and specific changes in histone or DNA modifications, to then determine the direct downstream recruitment and transcriptional effects.
Table 2.
Genes with modified chromatin environments in a diseased context.
While gene-specific control of chromatin is not currently possible in the clinical setting, several recent advances have brought the field much closer to this being a reality. CRISPR-Cas9-based therapies have already been done in patients ex vivo [214], the dCas9 system has been demonstrated as a therapeutic approach in murine models [215], Syn-TEF has advanced into in vivo studies in immunocompromised mouse models, and delivery systems such as AAV [216] and whole-protein-deliveries [217] are also being optimized. Simultaneously, advancements have been made in decreasing the size of the CRISPR system, providing greater efficiency in delivery systems such as AAV. This has been through the discovery of smaller versions Cas9 (Staphylococcus aureus dCas9) [218,219], smaller Cas9-like CRISPR systems (deactivated CasX [220] and deactivated Cpf1 [221,222]), CRE-inducible CRISPR [223], molecular engineering of artificial split Cas9 genes [136], and sa-Cas9 proteins lacking non-essential domains (mini-Cas9 [224]). The smaller size of these emerging Cas variants will greatly contribute to making gene-specific control of chromatin a clinical reality. The stability of epigenomic modifications must also be addressed. Unless the gene-specific chromatin modifier is constituently expressed in the target tissue many publications have reported reversions back to the initial chromatin state before synthetic perturbation. For instance Kungulovski et al. found that in ovarian cancer cells that changes made by ZF-DNMT or ZF-HKMT fusion proteins were not stably maintained even 10 days after adenoviral treatment [225].
5. Conclusions
Development of new ways to tether proteins and proteins associated with RNA to chromatin has opened up the ability to modulate specific endogenous genes by an assortment of chromatin modifying activities. Using gene-specific targeting technologies in conjunction with chromatin regulators has allowed for directed epigenomic engineering. Furthermore, bringing temporal and spatial control with these technologies has allowed finer governing of chromatin state at specific times and in subsets of cell populations. These technologies have been used to study the mechanisms of chromatin regulation in new ways and across a diverse set of genes. Modern research is also examining the ability of these approaches to be used to treat human diseases, yet most of this is still in the preclinical setting. The overexpression of synthetic effectors is required in most of the technologies summarized in this review, having undesirable impacts to the epigenome. While epigenomic engineering has been empowered by technology, it is likely to be driven further by techniques that are yet to be developed. Expect the future to hold a wider assortment of chromatin regulatory activities to be tethered to chromatin as only a subset of all the wide array of chromatin regulatory enzymes have been tested by these approaches. Additionally, new methods to anchor chromatin-modifying enzymes to target genes will lead to even more effective and specific chromatin modulation than current techniques.
Funding
The Hathaway lab is supported by grants R01GM118653 and R01GM132299 from the US National Institutes of Health, as well as grants from the Eshelman Institute for Innovation.
Acknowledgments
We apologize to the authors we could not cite in this review due to space limits. The authors thank the Reviewers for suggestions that greatly improved the scope and readability of this review. We also thank J. Umana and S. Clinkscales for critical reading of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Han, L.; Su, B.; Li, W.H.; Zhao, Z. CpG island density and its correlations with genomic features in mammalian genomes. Genome Biol. 2008, 9, R79. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Reinberg, D. Chromatin structure and the inheritance of epigenetic information. Nat. Rev. Genet. 2010, 11, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Wallrath, L.L.; Vitalini, M.W.; Elgin, S.C.R. Fundamentals of Chromatin; Springer New York: New York, NY, USA, 2014; pp. 529–552. [Google Scholar]
- Gardner, K.E.; Allis, C.D.; Strahl, B.D. Operating on chromatin, a colorful language where context matters. J. Mol. Biol. 2011, 409, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, I.; Margueron, R.; Shukeir, N.; Eisold, M.; Fritzsch, C.; Richter, F.M.; Mittler, G.; Genoud, C.; Goyama, S.; Kurokawa, M.; et al. Prdm3 and Prdm16 are H3K9me1 methyltransferases required for mammalian heterochromatin integrity. Cell 2012, 150, 948–960. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Sandoval, A.; Towbin, B.D.; Kalck, V.; Cabianca, D.S.; Gaidatzis, D.; Hauer, M.H.; Geng, L.; Wang, L.; Yang, T.; Wang, X.; et al. Perinuclear Anchoring of H3K9-Methylated Chromatin Stabilizes Induced Cell Fate in C. elegans Embryos. Cell 2015, 163, 1333–1347. [Google Scholar] [CrossRef]
- De Koning, L.; Savignoni, A.; Boumendil, C.; Rehman, H.; Asselain, B.; Sastre-Garau, X.; Almouzni, G. Heterochromatin protein 1alpha: a hallmark of cell proliferation relevant to clinical oncology. EMBO Mol. Med. 2009, 1, 178–191. [Google Scholar] [CrossRef]
- Müller-Ott, K.; Erdel, F.; Matveeva, A.; Mallm, J.-P.; Rademacher, A.; Hahn, M.; Bauer, C.; Zhang, Q.; Kaltofen, S.; Schotta, G.; et al. Specificity, propagation, and memory of pericentric heterochromatin. Mol. Syst. Biol. 2014, 10, 746. [Google Scholar] [CrossRef]
- Luger, K.; Richmond, T.J. The histone tails of the nucleosome. Curr. Opin. Genet. Dev. 1998, 8, 140–146. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Jeltsch, A.; Jurkowska, R.Z. New concepts in DNA methylation. Trends Biochem. Sci. 2014, 39, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Allis, C.D. Translating the Histone Code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- Kebede, A.F.; Schneider, R.; Daujat, S. Novel types and sites of histone modifications emerge as players in the transcriptional regulation contest. FEBS J. 2015, 282, 1658–1674. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Biswas, S.; Rao, C.M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef]
- Dorighi, K.M.; Swigut, T.; Henriques, T.; Bhanu, N.V.; Scruggs, B.S.; Nady, N.; Still, C.D.; Garcia, B.A.; Adelman, K.; Wysocka, J. Mll3 and Mll4 Facilitate Enhancer RNA Synthesis and Transcription from Promoters Independently of H3K4 Monomethylation. Mol. Cell 2017, 66, 568–576.e4. [Google Scholar] [CrossRef]
- Rickels, R.; Herz, H.-M.; Sze, C.C.; Cao, K.; Morgan, M.A.; Collings, C.K.; Gause, M.; Takahashi, Y.-H.; Wang, L.; Rendleman, E.J.; et al. Histone H3K4 monomethylation catalyzed by Trr and mammalian COMPASS-like proteins at enhancers is dispensable for development and viability. Nat. Genet. 2017, 49, 1647–1653. [Google Scholar] [CrossRef]
- McKay, D.J.; Klusza, S.; Penke, T.J.R.; Meers, M.P.; Curry, K.P.; McDaniel, S.L.; Malek, P.Y.; Cooper, S.W.; Tatomer, D.C.; Lieb, J.D.; et al. Interrogating the function of metazoan histones using engineered gene clusters. Dev. Cell 2015, 32, 373–386. [Google Scholar] [CrossRef]
- El-Osta, A.; Wolffe, A.P. DNA methylation and histone deacetylation in the control of gene expression: Basic biochemistry to human development and disease. Gene Expr. 2000, 9, 63–75. [Google Scholar] [CrossRef]
- Stewart, M.D.; Li, J.; Wong, J. Relationship between Histone H3 Lysine 9 Methylation, Transcription Repression, and Heterochromatin Protein 1 Recruitment Relationship between Histone H3 Lysine 9 Methylation, Transcription Repression, and Heterochromatin Protein 1 Recruitment. Mol. Cell. Biol. 2005, 25, 2525–2538. [Google Scholar] [CrossRef] [PubMed]
- Santi, D.V.; Norment, A.; Garrett, C.E. Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc. Natl. Acad. Sci. USA 1984, 81, 6993–6997. [Google Scholar] [CrossRef] [PubMed]
- Detich, N.; Ramchandani, S.; Szyf, M. A Conserved 3′-Untranslated Element Mediates Growth Regulation of DNA Methyltransferase 1 and Inhibits Its Transforming Activity. J. Biol. Chem. 2001, 276, 24881–24890. [Google Scholar] [CrossRef] [PubMed]
- Dhe-paganon, S.; Syeda, F.; Park, L. DNA methyl transferase 1: regulatory mechanisms and implications in health and disease. J. Biochem. Mol. Biol. 2011, 2, 58–66. [Google Scholar]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Vojta, A.; Dobrinić, P.; Tadić, V.; Bočkor, L.; Korać, P.; Julg, B.; Klasić, M.; Zoldoš, V. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016, 44, 5615–5628. [Google Scholar] [CrossRef]
- Lin, L.; Liu, Y.; Xu, F.; Huang, J.; Daugaard, T.F.; Petersen, T.S.; Hansen, B.; Ye, L.; Zhou, Q.; Fang, F.; et al. Genome-wide determination of on-target and off-target characteristics for RNA-guided DNA methylation by dCas9 methyltransferases. Gigascience 2018, 7, 1–19. [Google Scholar] [CrossRef]
- He, Y.F.; Li, B.Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, N.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Gu, B.; Lee, M. Histone H3 lysine 4 methyltransferases and demethylases in self-renewal and differentiation of stem cells. Cell Biosci. 2013, 3, 39. [Google Scholar] [CrossRef]
- Development, S. A central role of H3K4me3 extended chromatin domains in gene regulation. Epigenomics 2016, 8, 1011–1014. [Google Scholar]
- Völkel, P.; Angrand, P.-O. The control of histone lysine methylation in epigenetic regulation. Biochimie 2007, 89, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Rauch, T.; Chen, Z.; Szabo, P.E.; Riggs, A.D.; Pfeifer, G.P. The Histone Methyltransferase SETDB1 and the DNA Methyltransferase DNMT3A Interact Directly and Localize to Promoters Silenced in Cancer Cells. J. Biol. Chem. 2006, 281, 19489–19500. [Google Scholar] [CrossRef]
- Keniry, A.; Gearing, L.J.; Jansz, N.; Liu, J.; Holik, A.Z.; Hickey, P.F.; Kinkel, S.A.; Moore, D.L.; Breslin, K.; Chen, K.; et al. Setdb1-mediated H3K9 methylation is enriched on the inactive X and plays a role in its epigenetic silencing. Epigenetics Chromatin 2016, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Trojer, P.; Xu, C.F.; Cheung, P.; Kuo, A.; Drury, W.J.; Qiao, Q.; Neubert, T.A.; Xu, R.M.; Gozani, O.; et al. The Target of the NSD Family of Histone Lysine Methyltransferases Depends on the Nature of the Substrate. J. Biol. Chem. 2009, 284, 34283–34295. [Google Scholar] [CrossRef]
- Margueron, R.; Li, G.; Sarma, K.; Blais, A.; Zavadil, J.; Woodcock, C.L.; Dynlacht, B.D.; Reinberg, D. Ezh1 and Ezh2 Maintain Repressive Chromatin through Different Mechanisms. Mol. Cell 2008, 32, 503–518. [Google Scholar] [CrossRef]
- Hansen, K.H.; Bracken, A.P.; Pasini, D.; Dietrich, N.; Gehani, S.S.; Monrad, A.; Rappsilber, J.; Lerdrup, M.; Helin, K. A model for transmission of the H3K27me3 epigenetic mark. Nat. Cell Biol. 2008, 10, 1291–1300. [Google Scholar] [CrossRef]
- Wagner, E.J.; Carpenter, P.B. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 2012, 13, 115–126. [Google Scholar] [CrossRef]
- Jørgensen, S.; Schotta, G.; Sørensen, C.S. Histone H4 lysine 20 methylation: key player in epigenetic regulation of genomic integrity. Nucleic Acids Res. 2013, 41, 2797–2806. [Google Scholar] [CrossRef]
- Stulemeijer, I.J.E.; De Vos, D.; van Harten, K.; Joshi, O.K.; Blomberg, O.; van Welsem, T.; Terweij, M.; Vlaming, H.; de Graaf, E.L.; Altelaar, A.F.M.; et al. Dot1 histone methyltransferases share a distributive mechanism but have highly diverged catalytic properties. Sci. Rep. 2015, 5, 1–11. [Google Scholar] [CrossRef]
- Vlaming, H.; van Leeuwen, F. The upstreams and downstreams of H3K79 methylation by DOT1L. Chromosoma 2016, 125, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Dindar, G.; Anger, A.M.; Mehlhorn, C.; Hake, S.B.; Janzen, C.J. Structure-guided mutational analysis reveals the functional requirements for product specificity of DOT1 enzymes. Nat. Commun. 2014, 5, 5313. [Google Scholar] [CrossRef]
- Jacobs, S.A.; Khorasanizadeh, S. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science 2002, 295, 2080–2083. [Google Scholar] [CrossRef] [PubMed]
- Yap, K.L.; Zhou, M.-M. Keeping it in the family: diverse histone recognition by conserved structural folds. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 488–505. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Marfella, C.G.A.; Imbalzano, A.N. The Chd family of chromatin remodelers. Mutat. Res. 2007, 618, 30–40. [Google Scholar] [CrossRef]
- Dhayalan, A.; Rajavelu, A.; Rathert, P.; Tamas, R.; Jurkowska, R.Z.; Ragozin, S.; Jeltsch, A. The Dnmt3a PWWP Domain Reads Histone 3 Lysine 36 Trimethylation and Guides DNA Methylation. J. Biol. Chem. 2010, 285, 26114–26120. [Google Scholar] [CrossRef]
- Sanchez, R.; Zhou, M.-M. The PHD finger: A versatile epigenome reader. Trends Biochem. Sci. 2011, 36, 364–372. [Google Scholar] [CrossRef]
- Trievel, R.C.; Shilatifard, A. WDR5, a complexed protein. Nat. Struct. Mol. Biol. 2009, 16, 678–680. [Google Scholar] [CrossRef]
- Trojer, P.; Reinberg, D. Beyond histone methyl-lysine binding: How malignant brain tumor (MBT) protein L3MBTL1 impacts chromatin structure. Cell Cycle 2008, 7, 578–585. [Google Scholar] [CrossRef][Green Version]
- Brien, G.L.; Gambero, G.; O’Connell, D.J.; Jerman, E.; Turner, S.A.; Egan, C.M.; Dunne, E.J.; Jurgens, M.C.; Wynne, K.; Piao, L.; et al. Polycomb PHF19 binds H3K36me3 and recruits PRC2 and demethylase NO66 to embryonic stem cell genes during differentiation. Nat. Struct. Mol. Biol. 2012, 19, 1273–1281. [Google Scholar] [CrossRef]
- Cheng, J.; Yang, Y.; Fang, J.; Xiao, J.; Zhu, T.; Chen, F.; Wang, P.; Li, Z.; Yang, H.; Xu, Y. Structural Insight into Coordinated Recognition of Trimethylated Histone H3 Lysine 9 (H3K9me3) by the Plant Homeodomain (PHD) and Tandem Tudor Domain (TTD) of UHRF1 (Ubiquitin-like, Containing PHD and RING Finger Domains, 1) Protein. J. Biol. Chem. 2013, 288, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Maiques-Diaz, A.; Somervaille, T.C. LSD1: biologic roles and therapeutic targeting. Epigenomics 2016, 8, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Walport, L.J.; Hopkinson, R.J.; Schofield, C.J. Mechanisms of human histone and nucleic acid demethylases. Curr. Opin. Chem. Biol. 2012, 16, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Klein, B.J.; Piao, L.; Xi, Y.; Rincon-Arano, H.; Rothbart, S.B.; Peng, D.; Wen, H.; Larson, C.; Zhang, X.; Zheng, X.; et al. The histone-H3K4-specific demethylase KDM5B Binds to its substrate and product through distinct PHD fingers. Cell Rep. 2014, 6, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Aranda, S.; Mas, G.; Croce, L. Di Regulation of gene transcription by Polycomb proteins. Sci. Adv. 2015, 2, 1–15. [Google Scholar]
- Labbé, R.M.; Holowatyj, A.; Yang, Z.-Q. Histone lysine demethylase (KDM) subfamily 4: structures, functions and therapeutic potential. Am. J. Transl. Res. 2013, 6, 1–15. [Google Scholar]
- Becker, J.S.; Nicetto, D.; Zaret, K.S. H3K9me3-Dependent Heterochromatin: Barrier to Cell Fate Changes. Trends Genet. 2016, 32, 29–41. [Google Scholar] [CrossRef]
- Walport, L.J.; Hopkinson, R.J.; Vollmar, M.; Madden, S.K.; Gileadi, C.; Oppermann, U.; Schofield, C.J.; Johansson, C. Human UTY(KDM6C) Is a Male-specific N ϵ -Methyl Lysyl Demethylase. J. Biol. Chem. 2014, 289, 18302–18313. [Google Scholar] [CrossRef]
- Vermeulen, M.; Eberl, H.C.; Matarese, F.; Marks, H.; Denissov, S.; Butter, F.; Lee, K.K.; Olsen, J.V.; Hyman, A.A.; Stunnenberg, H.G.; et al. Quantitative Interaction Proteomics and Genome-wide Profiling of Epigenetic Histone Marks and Their Readers. Cell 2010, 142, 967–980. [Google Scholar] [CrossRef]
- Ho, J.W.K.; Jung, Y.L.; Liu, T.; Alver, B.H.; Lee, S.; Ikegami, K.; Sohn, K.-A.; Minoda, A.; Tolstorukov, M.Y.; Appert, A.; et al. Comparative analysis of metazoan chromatin organization. Nature 2014, 512, 449–452. [Google Scholar] [CrossRef]
- Katan-Khaykovich, Y.; Struhl, K. Dynamics of global histone acetylation and deacetylation in vivo: rapid restoration of normal histone acetylation status upon removal of activators and repressors. Genes Dev. 2002, 16, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Struhl, K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998, 12, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Dancy, B.M.; Cole, P.A. Protein Lysine Acetylation by p300/CBP. Chem. Rev. 2015, 115, 2419–2452. [Google Scholar] [CrossRef] [PubMed]
- De Ruijter, A.J.M.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B.P. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Livyatan, I.; Meshorer, E. The HDAC interaction network. Mol. Syst. Biol. 2013, 9, 671. [Google Scholar] [CrossRef] [PubMed]
- Schemies, J.; Uciechowska, U.; Sippl, W.; Jung, M. NAD+-dependent histone deacetylases (sirtuins) as novel therapeutic targets. Med. Res. Rev. 2010, 30, 861–889. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, R.; Meslamani, J.; Zhou, M.M. The bromodomain: From epigenome reader to druggable target. Biochim. Biophys. Acta - Gene Regul. Mech. 2014, 1839, 676–685. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.G.; Zhou, M.-M. The Bromodomain: A New Target in Emerging Epigenetic The Bromodomain: A New Target in Emerging Epigenetic Medicine. 2015 ACS Chem. Biol. 2015, 11, 598–608. [Google Scholar] [CrossRef]
- Thomas, M.C.; Chiang, C.M. The General Transcription Machinery and General Cofactors. Crit. Rev. Biochem. Mol. Biol. 2006, 41, 105–178. [Google Scholar] [CrossRef]
- Reinke, H.; Hörz, W. Histones Are First Hyperacetylated and Then Lose Contact with the Activated PHO5 Promoter. Mol. Cell 2003, 11, 1599–1607. [Google Scholar] [CrossRef]
- Shogren-Knaak, M.; Ishii, H.; Sun, J.M.; Pazin, M.J.; Davie, J.R.; Peterson, C.L. Histone H4-K16 Acetylation Controls Chromatin Structure and Protein Interactions. Science 2006, 311, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.A.; Struhl, K. Eaf3 Chromodomain Interaction with Methylated H3-K36 Links Histone Deacetylation to Pol II Elongation. Mol. Cell 2005, 20, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Hathaway, N.A.; Bell, O.; Hodges, C.; Miller, E.L.; Neel, D.S.; Crabtree, G.R. Dynamics and memory of heterochromatin in living cells. Cell 2012, 149, 1447–1460. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Carey, M.; Workman, J.L. The Role of Chromatin during Transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef]
- Herold, J.M.; Wigle, T.J.; Norris, J.L.; Lam, R.; Korboukh, V.K.; Gao, C.; Ingerman, L.A.; Kireev, D.B.; Senisterra, G.; Vedadi, M.; et al. Small Molecule Ligands of Methyl-Lysine Binding Proteins. J. Med. Chem. 2011, 54, 2504–2511. [Google Scholar] [CrossRef]
- Frye, S.V.; Jin, J. Novel Therapeutics Targeting Epigenetics: New Molecules, New Methods. ACS Med. Chem. Lett. 2016, 7, 123. [Google Scholar] [CrossRef][Green Version]
- James, L.I.; Frye, S.V. Targeting chromatin readers. Clin. Pharmacol. Ther. 2013, 93, 312–314. [Google Scholar] [CrossRef]
- Zengerle, M.; Chan, K.H.; Ciulli, A. Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. [Google Scholar] [CrossRef]
- Villar-Garea, A.; Fraga, M.F.; Espada, J.; Esteller, M. Procaine is a DNA-demethylating agent with growth-inhibitory effects in human cancer cells. Cancer Res. 2003, 63, 4984–4989. [Google Scholar]
- Christman, J.K. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483–5495. [Google Scholar] [CrossRef]
- Kuntz, K.W.; Campbell, J.E.; Keilhack, H.; Pollock, R.M.; Knutson, S.K.; Porter-Scott, M.; Richon, V.M.; Sneeringer, C.J.; Wigle, T.J.; Allain, C.J.; et al. The Importance of Being Me: Magic Methyls, Methyltransferase Inhibitors, and the Discovery of Tazemetostat. J. Med. Chem. 2016, 59, 1556–1564. [Google Scholar] [CrossRef] [PubMed]
- Lasko, L.M.; Jakob, C.G.; Edalji, R.P.; Qiu, W.; Montgomery, D.; Digiammarino, E.L.; Hansen, T.M.; Risi, R.M.; Frey, R.; Manaves, V.; et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineagespecific tumours. Nature 2018, 550, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Toure, M.; Crews, C.M. Small-Molecule PROTACS: New Approaches to Protein Degradation. Angew. Chem. Int. Ed. Engl. 2016, 55, 1966–1973. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA Approval Summary: Vorinostat for Treatment of Advanced Primary Cutaneous T-Cell Lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef]
- Jimeno, A.; McDermott, J. Belinostat for the treatment of peripheral T-cell lymphomas. Drugs of Today 2014, 50, 337. [Google Scholar] [CrossRef]
- Stiff, A.; Caserta, E.; Sborov, D.W.; Nuovo, G.J.; Mo, X.; Schlotter, S.Y.; Canella, A.; Smith, E.; Badway, J.; Old, M.; et al. Histone Deacetylase Inhibitors Enhance the Therapeutic Potential of Reovirus in Multiple Myeloma. Mol. Cancer Ther. 2016, 15, 830–841. [Google Scholar] [CrossRef]
- Huang, J.; Berger, S.L. The emerging field of dynamic lysine methylation of non-histone proteins. Curr. Opin. Genet. Dev. 2008, 18, 152–158. [Google Scholar] [CrossRef]
- Baskin, N.L.; Haynes, K.A. Chromatin engineering offers an opportunity to advance epigenetic cancer therapy. Nat. Struct. Mol. Biol. 2019, 26, 842–845. [Google Scholar] [CrossRef]
- Butler, K.V.; Chiarella, A.M.; Jin, J.; Hathaway, N.A. Targeted Gene Repression Using Novel Bifunctional Molecules to Harness Endogenous Histone Deacetylation Activity. ACS Synth. Biol. 2018, 7, 38–45. [Google Scholar] [CrossRef]
- Chiarella, A.M.; Wang, T.A.; Butler, K.V.; Jin, J.; Hathaway, N.A. Repressing Gene Transcription by Redirecting Cellular Machinery with Chemical Epigenetic Modifiers. J. Vis. Exp. 2018, 1, e58222. [Google Scholar] [CrossRef]
- Li, F.; Papworth, M.; Minczuk, M.; Rohde, C.; Zhang, Y.; Ragozin, S.; Jeltsch, A. Chimeric DNA methyltransferases target DNA methylation to specific DNA sequences and repress expression of target genes. Nucleic Acids Res. 2007, 35, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Wyman, C.; Kanaar, R. DNA Double-Strand Break Repair: All’s Well that Ends Well. Annu. Rev. Genet. 2006, 40, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Cermak, T.; Doyle, E.L.; Christian, M.; Wang, L.; Zhang, Y.; Schmidt, C.; Baller, J.A.; Somia, N.V.; Bogdanove, A.J.; Voytas, D.F. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011, 39, e82. [Google Scholar] [CrossRef]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Esvelt, K.M.; Church, G.M. Cas9 as a versatile tool for engineering biology. Nat. Methods 2013, 10, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Liu, Q.; Segal, D.J.; Ghiara, J.B.; Barbas, C.F. Design of polydactyl zinc-finger proteins for unique addressing within complex genomes. Proc. Natl. Acad. Sci. 1997, 94, 5525–5530. [Google Scholar] [CrossRef]
- Rivenbark, A.G.; Stolzenburg, S.; Beltran, A.S.; Yuan, X.; Rots, M.G.; Strahl, B.D.; Blancafort, P. Epigenetic reprogramming of cancer cells via targeted DNA methylation. Epigenetics 2012, 7, 350–360. [Google Scholar] [CrossRef]
- Snowden, A.W.; Gregory, P.D.; Case, C.C.; Pabo, C.O. Gene-Specific Targeting of H3K9 Methylation Is Sufficient for Initiating Repression In Vivo. Curr. Biol. 2002, 12, 2159–2166. [Google Scholar] [CrossRef]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the Code of DNA Binding Specificity of TAL-Type III Effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Lei, Y.; Wong, W.-K.; Liu, S.; Lee, K.-C.; He, X.; You, W.; Zhou, R.; Guo, J.-T.; Chen, X.; et al. Direct activation of human and mouse Oct4 genes using engineered TALE and Cas9 transcription factors. Nucleic Acids Res. 2014, 42, 4375–4390. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, D.L.; Le Lay, J.E.; Ruano, E.G.; Kaestner, K.H. TALE-mediated epigenetic suppression of CDKN2A increases replication in human fibroblasts. J. Clin. Invest. 2015, 125, 1998–2006. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Angstman, J.F.; Richardson, M.E.; Linder, S.J.; Cascio, V.M.; Tsai, S.Q.; Ho, Q.H.; Sander, J.D.; Reyon, D.; Bernstein, B.E.; et al. Targeted DNA demethylation and activation of endogenous genes using programmable TALE-TET1 fusion proteins. Nat. Biotechnol. 2013, 31, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef]
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 2012, 482, 331–338. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W. a Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef]
- Beerli, R.R.; Segal, D.J.; Dreier, B.; Barbas, C.F. Toward controlling gene expression at will: Specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. 1998, 95, 14628-33. Proc. Nati. Acad. Sci. 1998, 95, 14628–14633. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef]
- Jost, M.; Chen, Y.; Gilbert, L.A.; Horlbeck, M.A.; Krenning, L.; Menchon, G.; Rai, A.; Cho, M.Y.; Stern, J.J.; Prota, A.E.; et al. Combined CRISPRi/a-Based Chemical Genetic Screens Reveal that Rigosertib Is a Microtubule-Destabilizing Agent. Mol. Cell 2017, 68, 210–223.e6. [Google Scholar] [CrossRef]
- Cheng, A.W.; Wang, H.; Yang, H.; Shi, L.; Katz, Y.; Theunissen, T.W.; Rangarajan, S.; Shivalila, C.S.; Dadon, D.B.; Jaenisch, R. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013, 23, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Balboa, D.; Weltner, J.; Eurola, S.; Trokovic, R.; Wartiovaara, K.; Otonkoski, T. Conditionally Stabilized dCas9 Activator for Controlling Gene Expression in Human Cell Reprogramming and Differentiation. Stem Cell Reports 2015, 5, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; P R Iyer, E.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [PubMed]
- Le Sage, C.; Lawo, S.; Panicker, P.; Scales, T.M.E.; Rahman, S.A.; Little, A.S.; McCarthy, N.J.; Moore, J.D.; Cross, B.C.S. Dual direction CRISPR transcriptional regulation screening uncovers gene networks driving drug resistance. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Choudhury, S.R.; Cui, Y.; Lubecka, K.; Stefanska, B.; Irudayaraj, J. CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget 2016, 7, 46545–46556. [Google Scholar] [CrossRef]
- Morita, S.; Noguchi, H.; Horii, T.; Nakabayashi, K.; Kimura, M.; Okamura, K.; Sakai, A.; Nakashima, H.; Hata, K.; Nakashima, K.; et al. Targeted DNA demethylation in vivo using dCas9-peptide repeat and scFv-TET1 catalytic domain fusions. Nat. Biotechnol. 2016, 34, 1060–1065. [Google Scholar] [CrossRef]
- Liu, S.; Wu, H.; Ji, X.; Stelzer, Y.; Wu, X.; Czauderna, S.; Shu, J.; Dadon, D.; Young, R.A.; Jaenisch, R. Editing DNA methylation in the mammalian genome. Cell 2016, 167, 233–247. [Google Scholar] [CrossRef]
- Hilton, I.B.; D’Ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C. a Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef]
- Gasperini, M.; Hill, A.J.; McFaline-Figueroa, J.L.; Martin, B.; Kim, S.; Zhang, M.D.; Jackson, D.; Leith, A.; Schreiber, J.; Noble, W.S.; et al. A Genome-wide Framework for Mapping Gene Regulation via Cellular Genetic Screens. Cell 2019, 176, 377–390.e19. [Google Scholar] [CrossRef]
- Thakore, P.I.; D’Ippolito, A.M.; Song, L.; Safi, A.; Shivakumar, N.K.; Kabadi, A.M.; Reddy, T.E.; Crawford, G.E.; Gersbach, C.A. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 2015, 12, 1143–1149. [Google Scholar] [CrossRef]
- Kearns, N.A.; Pham, H.; Tabak, B.; Genga, R.M.; Garber, M.; Maehr, R. Functional annotation of native enhancers with a Cas9 -histone demethylase fusion demethylase fusion. Nat. Methods 2015, 12, 401–403. [Google Scholar] [CrossRef] [PubMed]
- Klann, T.S.; Black, J.B.; Chellappan, M.; Safi, A.; Song, L.; Hilton, I.B.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. CRISPR-Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat. Biotechnol. 2017, 35, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A Protein-Tagging System for Signal Amplification in Gene Expression and Fluorescence Imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Pflueger, C.; Tan, D.; Swain, T.; Nguyen, T.; Pflueger, J.; Nefzger, C.; Polo, J.M.; Ford, E.; Lister, R. A modular dCas9-SunTag DNMT3A epigenome editing system overcomes pervasive off-target activity of direct fusion dCas9-DNMT3A constructs. Genome Res. 2018, 28, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Zalatan, J.G.; Lee, M.E.; Almeida, R.; Gilbert, L.A.; Whitehead, E.H.; La Russa, M.; Tsai, J.C.; Weissman, J.S.; Dueber, J.E.; Qi, L.S.; et al. Engineering complex synthetic transcriptional programs with CRISPR RNA scaffolds. Cell 2015, 160, 339–350. [Google Scholar] [CrossRef]
- Xu, X.; Tao, Y.; Gao, X.; Zhang, L.; Li, X.; Zou, W.; Ruan, K.; Wang, F.; Xu, G.; Hu, R. A CRISPR-based approach for targeted DNA demethylation. Cell Discov. 2016, 2, 16009. [Google Scholar] [CrossRef]
- Chavez, A.; Tuttle, M.; Pruitt, B.W.; Ewen-Campen, B.; Chari, R.; Ter-Ovanesyan, D.; Haque, S.J.; Cecchi, R.J.; Kowal, E.J.K.; Buchthal, J.; et al. Comparison of Cas9 activators in multiple species. Nat. Methods 2016, 13, 7–10. [Google Scholar] [CrossRef]
- Mandegar, M.A.; Huebsch, N.; Frolov, E.B.; Shin, E.; Truong, A.; Olvera, M.P.; Chan, A.H.; Miyaoka, Y.; Holmes, K.; Spencer, C.I.; et al. CRISPR Interference Efficiently Induces Specific and Reversible Gene Silencing in Human iPSCs. Cell Stem Cell 2016, 18, 541–553. [Google Scholar] [CrossRef]
- Kuscu, C.; Mammeadov, R.; Czikora, A.; Unlu, H.; Tufan, T.; Fischer, N.L.; Arslan, S.; Bekiranov, S.; Kanemaki, M.; Adli, M. Temporal and Spatial Epigenome Editing Allows Precise Gene Regulation in Mammalian Cells. J. Mol. Biol. 2018, 1–11. [Google Scholar] [CrossRef]
- Spencer, D.M.; Wandless, T.J.; Schreiber, S.L.; Crabtree, G.R. Controlling signal transduction with synthetic ligands. Science 1993, 262, 1019–1024. [Google Scholar] [CrossRef]
- Stanton, B.Z.; Chory, E.J.; Crabtree, G.R. Chemically induced proximity in biology and medicine. Science 2018, 359. [Google Scholar]
- Braun, S.M.G.; Kirkland, J.G.; Chory, E.J.; Husmann, D.; Calarco, J.P.; Crabtree, G.R. Rapid and reversible epigenome editing by endogenous chromatin regulators. Nat. Commun. 2017, 8, 560. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Volz, S.E.; Zhang, F. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat. Biotechnol. 2015, 33, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Xiong, X.; Wong, S.; Charles, E.J.; Lim, W.A.; Qi, L.S. Complex transcriptional modulation with orthogonal and inducible dCas9 regulators. Nat. Methods 2016, 12, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Peng, S.; Xie, Z. Integration and exchange of split dCas9 domains for transcriptional controls in mammalian cells. Nat. Commun. 2016, 7, 13056. [Google Scholar] [CrossRef]
- Corson, T.W.; Aberle, N.; Crews, C.M. Design and Applications of Bifunctional Small Molecules: Why Two Heads Are Better Than One. ACS Chem. Biol. 2008, 3, 677–692. [Google Scholar] [CrossRef]
- Polstein, L.R.; Gersbach, C.A. A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nat Chem Biol. 2015, 11, 198–200. [Google Scholar] [CrossRef]
- Nihongaki, Y.; Furuhata, Y.; Otabe, T.; Hasegawa, S.; Yoshimoto, K.; Sato, M. CRISPR-Cas9-based photoactivatable transcription systems to induce neuronal differentiation. Nat. Methods 2017, 14, 963–966. [Google Scholar] [CrossRef]
- Shao, J.; Wang, M.; Yu, G.; Zhu, S.; Yu, Y.; Wu, J.; Ye, H. Synthetic far-red light-mediated CRISPR-dCas9 device for inducing functional neuronal differentiation. Proc. Natl. Acad. Sci. USA 2018, 115, 6722–6730. [Google Scholar] [CrossRef]
- Liszczak, G.P.; Brown, Z.Z.; Kim, S.H.; Oslund, R.C.; David, Y.; Muir, T.W. Genomic targeting of epigenetic probes using a chemically tailored Cas9 system. Proc. Natl. Acad. Sci. USA 2017, 114, 681–686. [Google Scholar] [CrossRef]
- Chiarella, A.M.; Butler, K.V.; Gryder, B.E.; Lu, D.; Wang, T.A.; Yu, X.; Pomella, S.; Khan, J.; Jin, J.; Hathaway, N.A. Dose-dependent activation of gene expression is achieved using CRISPR and small molecules that recruit endogenous chromatin machinery. Nat. Biotechnol. 2020, 38, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Cherry, A.B.C.; Daley, G.Q. Reprogramming cellular identity for regenerative medicine. Cell 2012, 148, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Erwin, G.S.; Grieshop, M.P.; Ali, A.; Qi, J.; Lawlor, M.; Kumar, D.; Ahmad, I.; Mcnally, A.; Teider, N.; Worringer, K.; et al. Synthetic transcription elongation factors license transcription across repressive chromatin. Science 2017, 358, 1617–1622. [Google Scholar] [CrossRef] [PubMed]
- Heiderscheit, E.A.; Eguchi, A.; Spurgat, M.C.; Ansari, A.Z. Reprogramming cell fate with artificial transcription factors. FEBS Lett. 2018, 592, 888–900. [Google Scholar] [CrossRef]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. DNMT3A Mutations in Acute Myeloid Leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A.; et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009, 10, 223–232. [Google Scholar] [CrossRef]
- Lorsbach, R.B.; Moore, J.; Mathew, S.; Raimondi, S.C.; Mukatira, S.T.; Downing, J.R. TET1, a member of a novel protein family, is fused to MLL in acute myeloid leukemia containing the t(10;11)(q22;q23). Leukemia 2003, 17, 637. [Google Scholar] [CrossRef]
- Delhommeau, F.; Dupont, S.; Valle, V.D.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in Myeloid Cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Cimmino, L.; Abdel-Wahab, O.; Levine, R.L.; Aifantis, I. TET Family Proteins and Their Role in Stem Cell Differentiation and Transformation. Cell Stem Cell 2011, 9, 193–204. [Google Scholar] [CrossRef]
- Langemeijer, S.M.C.; Kuiper, R.P.; Berends, M.; Knops, R.; Aslanyan, M.G.; Massop, M.; Stevens-Linders, E.; Van Hoogen, P.; Van Kessel, A.G.; Raymakers, R.A.P.; et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat. Genet. 2009, 41, 838–842. [Google Scholar] [CrossRef]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulos, I.; Fioretos, T.; Isaksson, M.; Samuelsson, U.; Billström, R.; Strömbeck, B.; Mitelman, F.; Johansson, B. Fusion of the MORF and CBP genes in acute myeloid leukemia with the t(10;16)(q22;p13). Hum. Mol. Genet. 2001, 10, 395–404. [Google Scholar] [CrossRef]
- Sobulo, O.M.; Borrow, J.; Tomek, R.; Reshmi, S.; Harden, A.; Schlegelberger, B.; Housman, D.; Doggett, N.A.; Rowley, J.D.; Zeleznik-Le, N.J. MLL is fused to CBP, a histone acetyltransferase, in therapy-related acute myeloid leukemia with a t(11;16)(q23;p13.3). Proc. Natl. Acad. Sci. USA 1997, 94, 8732–8737. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Trifonov, V.; Fabbri, G.; Ma, J.; Rossi, D.; Chiarenza, A.; Wells, V.A.; Grunn, A.; Messina, M.; Elliot, O.; et al. Analysis of the Coding Genome of Diffuse Large B-Cell Lymphoma. Nat. Genet. 2011, 43, 830–837. [Google Scholar] [CrossRef]
- Taki, T.; Sako, M.; Tsuchida, M.; Hayashi, Y. The t(11; 16)(q23; p13) Translocation in Myelodysplastic Syndrome Fuses the MLL Gene to the CBP Gene. Blood 1997, 89, 3945–3950. [Google Scholar] [CrossRef] [PubMed]
- Petrif, F.; Giles, R.H.; Dauwerse, H.G.; Saris, J.J.; Hennekam, R.C.M.; Masuno, M.; Tommerup, N.; van Ommen, G.-J.B.; Goodman, R.H.; Peters, D.J.M.; et al. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature 1995, 376, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Laï, J.L.; Jouet, J.P.; Bauters, F.; Deminatti, M. Chronic myelogenous leukemia with translocation (8;22): Report of a new case. Cancer Genet. Cytogenet. 1985, 17, 365–366. [Google Scholar] [CrossRef]
- Gayther, S.A.; Batley, S.J.; Linger, L.; Bannister, A.; Thorpe, K.; Chin, S.-F.; Daigo, Y.; Russell, P.; Wilson, A.; Sowter, H.M.; et al. Mutations truncating the EP300 acetylase in human cancers. Nat. Genet. 2000, 24, 300. [Google Scholar] [CrossRef]
- Kitabayashi, I.; Aikawa, Y.; Yokoyama, A.; Hosoda, F.; Nagai, M.; Kakazu, N.; Abe, T.; Ohki, M. Fusion of MOZ and p300 histone acetyltransferases in acute monocytic leukemia with a t(8;22)(p11;q13) chromosome translocation. Leukemia 2001, 15, 89–94. [Google Scholar] [CrossRef]
- Huntly, B.J.P.; Shigematsu, H.; Deguchi, K.; Lee, B.H.; Mizuno, S.; Duclos, N.; Rowan, R.; Amaral, S.; Curley, D.; Williams, I.R.; et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 2004, 6, 587–596. [Google Scholar] [CrossRef]
- Moore, S.D.P.; Herrick, S.R.; Ince, T.A.; Kleinman, M.S.; Cin, P.D.; Morton, C.C.; Quade, B.J. Uterine Leiomyomata with t(10;17) Disrupt the Histone Acetyltransferase. Cancer Res. 2004, 64, 5570–5577. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Walker, S.R.; Nelson, E.A.; Cerulli, R.; Xiang, M.; Toniolo, P.A.; Qi, J.; Stone, R.M.; Wadleigh, M.; Bradner, J.E.; et al. Targeting STAT5 in Hematologic Malignancies through Inhibition of the Bromodomain and Extra-Terminal (BET) Bromodomain Protein BRD2. Mol. Cancer Ther. 2014, 13, 1194–1205. [Google Scholar] [CrossRef] [PubMed]
- French, C.A.; Ramirez, C.L.; Kolmakova, J.; Hickman, T.T.; Cameron, M.J.; Thyne, M.E.; Kutok, J.L.; Toretsky, J.A.; Tadavarthy, A.K.; Kees, U.R.; et al. BRD–NUT oncoproteins: a family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene 2007, 27, 2237–2242. [Google Scholar] [CrossRef] [PubMed]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the Hallmarks of Lung Adenocarcinoma with Massively Parallel Sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- French, C.A.; Kutok, J.L.; Faquin, W.C.; Toretsky, J.A.; Antonescu, C.R.; Griffin, C.A.; Nose, V.; Vargas, S.O.; Moschovi, M.; Tzortzatou-Stathopoulou, F.; et al. Midline carcinoma of children and young adults with NUT rearrangement. J. Clin. Oncol. 2004, 22, 4135–4139. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Wang, L.; Hasanovic, A.; Suehara, Y.; Lipson, D.; Stephens, P.; Ross, J.; Miller, V.; Ginsberg, M.; Zakowski, M.F.; et al. Response to Cabozantinib in Patients with RET Fusion-Positive Lung Adenocarcinomas. Cancer Discov. 2013, 3, 630–635. [Google Scholar] [CrossRef]
- Elzamly, S.; Chavali, S.; Tonk, V.; Tonk, S.; Gaur, S.; Tarango, D.; Torabi, A. Acute myeloid leukemia with KMT2A-SEPT5 translocation: A case report and review of the literature. SAGE Open Med. Case Reports 2018, 6, 2050313X17750334. [Google Scholar] [CrossRef]
- Winters, A.C.; Bernt, K.M. MLL-Rearranged Leukemias—An Update on Science and Clinical Approaches. Front. Pediatr. 2017, 5, 4. [Google Scholar] [CrossRef]
- Parsons, D.W.; Li, M.; Zhang, X.; Jones, S.; Leary, R.J.; Lin, J.C.-H.; Boca, S.M.; Carter, H.; Samayoa, J.; Bettegowda, C.; et al. The Genetic Landscape of the Childhood Cancer Medulloblastoma. Science 2011, 331, 435–439. [Google Scholar] [CrossRef]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Van Loo, P.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef]
- Dalgliesh, G.L.; Furge, K.; Greenman, C.; Chen, L.; Bignell, G.; Butler, A.; Davies, H.; Edkins, S.; Hardy, C.; Latimer, C.; et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 2010, 463, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef]
- Hollink, I.H.I.M.; van den Heuvel-Eibrink, M.M.; Arentsen-Peters, S.T.C.J.M.; Pratcorona, M.; Abbas, S.; Kuipers, J.E.; van Galen, J.F.; Beverloo, H.B.; Sonneveld, E.; Kaspers, G.-J.J.L.; et al. NUP98/NSD1 characterizes a novel poor prognostic group in acute myeloid leukemia with a distinct HOX gene expression pattern. Blood 2011, 118, 3645–3656. [Google Scholar] [CrossRef]
- Stec, I.; Wright, T.J.; Van Ommen, G.J.B.; De Boer, P.A.J.; Van Haeringen, A.; Moorman, A.F.M.; Altherr, M.R.; Den Dunnen, J.T. WHSC1, a 90 kb SET domain-containing gene, expressed in early development and homologous to a Drosophila dysmorphy gene maps in the Wolf-Hirschhorn syndrome critical region and is fused to IgH in t(4;14) multiple myeloma. Hum. Mol. Genet. 1998, 7, 1071–1082. [Google Scholar] [CrossRef]
- Jaffe, J.D.; Wang, Y.; Chan, H.M.; Zhang, J.; Huether, R.; Kryukov, G.V.; Bhang, H.C.; Taylor, J.E.; Hu, M.; Englund, N.P.; et al. Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 1386–1391. [Google Scholar] [CrossRef]
- Rosati, R.; La Starza, R.; Veronese, A.; Aventin, A.; Schwienbacher, C.; Vallespi, T.; Negrini, M.; Martelli, M.F.; Mecucci, C. NUP98 is fused to the NSD3 gene in acute myeloid leukemia associated with t(8;11)(p11.2;p15). Blood 2002, 99, 3857–3860. [Google Scholar] [CrossRef]
- Dubois, S.; Mareschal, S.; Cornic, M.; Picquenot, J.M.; Bertrand, P.; Bohers, E.; Maingonnat, C.; Viailly, P.J.; Ruminy, P.; Alcantara, M.; et al. Targeted EZH2 Inhibitors in Diffuse Large B-Cell Lymphoma (DLBCL): Immunohistochemical and Mutational Profiles of Patients May Determine Candidates for Treatment. Blood 2014, 124, 1656. [Google Scholar] [CrossRef]
- Shih, A.H.; Abdel-Wahab, O.; Patel, J.P.; Levine, R.L. The role of mutations in epigenetic regulators in myeloid malignancies. Nat. Rev. Cancer 2012, 12, 599–612. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical Effect of Point Mutations in Myelodysplastic Syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [PubMed]
- Gough, S.M.; Slape, C.I.; Aplan, P.D. NUP98 gene fusions and hematopoietic malignancies: common themes and new biologic insights. Blood 2011, 118, 6247–6257. [Google Scholar] [CrossRef] [PubMed]
- Van Haaften, G.; Dalgliesh, G.L.; Davies, H.; Chen, L.; Bignell, G.; Greenman, C.; Edkins, S.; Hardy, C.; O’Meara, S.; Teague, J.; et al. Somatic mutations of the histone H3K27 demethylase, UTX, in human cancer. Nat. Genet. 2009, 41, 521–523. [Google Scholar] [CrossRef] [PubMed]
- Van Vlierberghe, P.; Palomero, T.; Khiabanian, H.; Van der Meulen, J.; Castillo, M.; Van Roy, N.; De Moerloose, B.; Philippé, J.; González-García, S.; Toribio, M.L.; et al. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat. Genet. 2010, 42, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Van Vlierberghe, P.; Patel, J.; Abdel-Wahab, O.; Lobry, C.; Hedvat, C.V.; Balbin, M.; Nicolas, C.; Payer, A.R.; Fernandez, H.F.; Tallman, M.S.; et al. PHF6 mutations in adult acute myeloid leukemia. Leukemia 2010, 25, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Totoki, Y.; Abe, T.; Boroevich, K.A.; Hosoda, F.; Nguyen, H.H.; Aoki, M.; Hosono, N.; Kubo, M.; Miya, F.; et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012, 44, 760–764. [Google Scholar] [CrossRef]
- Kanai, Y.; Ushijima, S.; Nakanishi, Y.; Sakamoto, M.; Hirohashi, S. Mutation of the DNA methyltransferase (DNMT) 1 gene in human colorectal cancers. Cancer Lett. 2003, 192, 75–82. [Google Scholar] [CrossRef]
- Ropero, S.; Fraga, M.F.; Ballestar, E.; Hamelin, R.; Yamamoto, H.; Boix-Chornet, M.; Caballero, R.; Alaminos, M.; Setien, F.; Paz, M.F.; et al. A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat. Genet. 2006, 38, 566. [Google Scholar] [CrossRef]
- Berger, M.F.; Lawrence, M.S.; Demichelis, F.; Drier, Y.; Cibulskis, K.; Sivachenko, A.Y.; Sboner, A.; Esgueva, R.; Pflueger, D.; Sougnez, C.; et al. The genomic complexity of primary human prostate cancer. Nature 2011, 470, 214–220. [Google Scholar] [CrossRef]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.; Sougnez, C.; McKenna, A.; et al. The Mutational Landscape of Head and Neck Squamous Cell Carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef]
- Fontebasso, A.M.; Schwartzentruber, J.; Khuong-Quang, D.A.; Liu, X.Y.; Sturm, D.; Korshunov, A.; Jones, D.T.W.; Witt, H.; Kool, M.; Albrecht, S.; et al. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol. 2013, 125, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Piva, F.; Santoni, M.; Matrana, M.R.; Satti, S.; Giulietti, M.; Occhipinti, G.; Massari, F.; Cheng, L.; Lopez-Beltran, A.; Scarpelli, M.; et al. BAP1, PBRM1 and SETD2 in clear-cell renal cell carcinoma: molecular diagnostics and possible targets for personalized therapies. Expert Rev. Mol. Diagn. 2015, 15, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Haynes, K.A. Chromatin research and biological engineering: an evolving relationship poised for new biomedical impacts. Curr. Opin. Syst. Biol. 2019, 14, 73–81. [Google Scholar] [CrossRef]
- Göttlicher, M.; Minucci, S.; Zhu, P.; Krämer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef] [PubMed]
- Warrell, R.P.; He, L.Z.; Richon, V.; Calleja, E.; Pandolfi, P.P. Therapeutic Targeting of Transcription in Acute Promyelocytic Leukemia by Use of an Inhibitor of Histone Deacetylase. JNCI J. Natl. Cancer Inst. 1998, 90, 1621–1625. [Google Scholar] [CrossRef]
- Kijima, M.; Yoshida, M.; Sugita, K.; Horinouchi, S.; Beppu, T. Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase. J. Biol. Chem. 1993, 268, 22429–22435. [Google Scholar]
- Newmark, H.L.; Young, C.W. Butyrate and phenylacetate as differentiating agents: practical problems and opportunities. J. Cell. Biochem. Suppl. 1995, 22, 247–253. [Google Scholar] [CrossRef]
- Schneider, B.J.; Kalemkerian, G.P.; Bradley, D.; Smith, D.C.; Egorin, M.J.; Daignault, S.; Dunn, R.; Hussain, M. Phase I study of vorinostat (suberoylanilide hydroxamic acid, NSC 701852) in combination with docetaxel in patients with advanced and relapsed solid malignancies. Invest. New Drugs 2012, 30, 249–257. [Google Scholar] [CrossRef]
- Rathkopf, D.E.; Picus, J.; Hussain, A.; Ellard, S.; Chi, K.N.; Nydam, T.; Allen-Freda, E.; Mishra, K.K.; Porro, M.G.; Scher, H.I.; et al. A phase 2 study of intravenous panobinostat in patients with castration-resistant prostate cancer. Cancer Chemother. Pharmacol. 2013, 72, 537–544. [Google Scholar] [CrossRef]
- Jesionek-Kupnicka, D.; Szybka, M.; Malachowska, B.; Fendler, W.; Potemski, P.; Piaskowski, S.; Jaskolski, D.; Papierz, W.; Skowronski, W.; Och, W.; et al. TP53 Promoter Methylation in Primary Glioblastoma: Relationship with TP53 mRNA and Protein Expression and Mutation Status. DNA Cell Biol. 2014, 33, 217–226. [Google Scholar] [CrossRef]
- Robaina, M.C.S.; Faccion, R.S.; Arruda, V.O.; de Rezende, L.M.M.; Vasconcelos, G.M.; Apa, A.G.; Bacchi, C.E.; Klumb, C.E. Quantitative analysis of CDKN2A methylation, mRNA, and p16INK4a protein expression in children and adolescents with Burkitt lymphoma: Biological and clinical implications. Leuk. Res. 2015, 39, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Shen, L.; Suzuki, S.; Kurokawa, T.; Masuko, K.; Tanaka, Y.; Kato, H.; Mizuno, Y.; Yokoe, M.; Sugauchi, F.; et al. Alterations of DNA methylation and histone modifications contribute to gene silencing in hepatocellular carcinomas. Hepatol. Res. 2007, 37, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, Y.; Lv, Y.; Meng, G.; Guo, Q.N. Epigenetic regulation of the pro-apoptosis gene TSSC3 in human osteosarcoma cells. Biomed. Pharmacother. 2014, 68, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Cabianca, D.S.; Casa, V.; Bodega, B.; Xynos, A.; Ginelli, E.; Tanaka, Y.; Gabellini, D. A Long ncRNA Links Copy Number Variation to a Polycomb/Trithorax Epigenetic Switch in FSHD Muscular Dystrophy. Cell 2012, 149, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Ortega, E.; Arzate-Mejía, R.; Pérez-Molina, R.; González-Buendía, E.; Meier, K.; Guerrero, G.; Recillas-Targa, F. Epigenetic silencing of miR-181c by DNA methylation in glioblastoma cell lines. BMC Cancer 2016, 16, 226. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391. [Google Scholar] [CrossRef]
- Richon, V.M.; Sandhoff, T.W.; Rifkind, R.A.; Marks, P.A. Histone deacetylase inhibitor selectively induces p21(WAF1) expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. USA 2000, 97, 10014–10019. [Google Scholar] [CrossRef]
- Miao, F.; Smith, D.D.; Zhang, L.; Min, A.; Feng, W.; Natarajan, R. Lymphocytes From Patients With Type 1 Diabetes Display a Distinct Profile of Chromatin Histone H3 Lysine 9 Dimethylation: An Epigenetic Study in Diabetes. Diabetes 2008, 57, 3189–3198. [Google Scholar] [CrossRef]
- Navarro, J.M.; Touzart, A.; Pradel, L.C.; Loosveld, M.; Koubi, M.; Fenouil, R.; Le Noir, S.; Maqbool, M.A.; Morgado, E.; Gregoire, C.; et al. Site- and allele-specific polycomb dysregulation in T-cell leukaemia. Nat. Commun. 2015, 6, 6094. [Google Scholar] [CrossRef]
- Villalba, M.; DiazLlagares, A.; Redrado, M.; De, A.L.; Freire, J.; Gomez-roman, J.; Montuenga, L.M.; Esteller, M. Epigenetic alterations leading to TMPRSS4 promoter hypomethylation and protein overexpression predict poor prognosis in squamous lung cancer patients. Oncotarget 2016, 7, 22752–22769. [Google Scholar] [CrossRef]
- Strunnikova, M.; Schagdarsurengin, U.; Garbe, J.C.; Stampfer, M.R.; Kehlen, A.; Dammann, R. Chromatin Inactivation Precedes De Novo DNA Methylation during the Progressive Epigenetic Silencing of the RASSF1A Promoter Chromatin Inactivation Precedes De Novo DNA Methylation during the Progressive Epigenetic Silencing of the RASSF1A Promoter. Mol.Cell.Biol. 2005, 25, 3923–3933. [Google Scholar] [CrossRef] [PubMed]
- Jancar, S.; Serezani, C.H. Imbalance between HDAC and HAT activities drives aberrant STAT1/MyD88 expression in macrophages from type 1 diabetic mice. 2018, 31, 334–339.
- Thulin, L. Four U.S. CRISPR Trials Editing Human DNA to Research New Treatments. S. CRISPR Trials Editing Human DNA to Research New Treatments. Smithsonian.com 2019. [Google Scholar]
- Liu, Q.; Zhao, K.; Wang, C.; Zhang, Z.; Zheng, C.; Zhao, Y.; Zheng, Y.; Liu, C.; An, Y.; Shi, L.; et al. Multistage Delivery Nanoparticle Facilitates Efficient CRISPR/dCas9 Activation and Tumor Growth Suppression In Vivo. Adv. Sci. 2018, 6, 1801423. [Google Scholar] [CrossRef] [PubMed]
- Lykken, E.A.; Shyng, C.; Edwards, R.J.; Rozenberg, A.; Gray, S.J. Recent progress and considerations for AAV gene therapies targeting the central nervous system. J. Neurodev. Disord. 2018, 10, 16. [Google Scholar] [CrossRef]
- Stanislawska, I.; Liwinska, W.; Lyp, M.; Stojek, Z.; Zabost, E. Recent Advances in Degradable Hybrids of Biomolecules and NGs for Targeted Delivery. Molecules 2019, 24, 1873. [Google Scholar] [CrossRef]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef]
- Thakore, P.I.; Kwon, J.B.; Nelson, C.E.; Rouse, D.C.; Gemberling, M.P.; Oliver, M.L.; Gersbach, C.A. RNA-guided transcriptional silencing in vivo with S. aureus CRISPR-Cas9 repressors. Nat. Commun. 2018, 9, 1674. [Google Scholar] [CrossRef]
- Liu, J.J.; Orlova, N.; Oakes, B.L.; Ma, E.; Spinner, H.B.; Baney, K.L.M.; Chuck, J.; Tan, D.; Knott, G.J.; Harrington, L.B.; et al. CasX enzymes comprise a distinct family of RNA-guided genome editors. Nature 2019, 566, 218–233. [Google Scholar] [CrossRef]
- Nihongaki, Y.; Otabe, T.; Ueda, Y.; Sato, M. A split CRISPR-Cpf1 platform for inducible genome editing and gene activation. Nat. Chem. Biol. 2019, 15, 882–888. [Google Scholar] [CrossRef]
- Kim, S.K.; Kim, H.; Ahn, W.C.; Park, K.H.; Woo, E.; Lee, D.H.; Lee, S.G. Efficient Transcriptional Gene Repression by Type V-A CRISPR-Cpf1 from Eubacterium eligens. ACS Synth. Biol. 2017, 6, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Chylinski, K.; Hubmann, M.; Hanna, R.E.; Yanchus, C.; Michlits, G.; Uijttewaal, E.C.H.; Doench, J.; Schramek, D.; Elling, U. CRISPR-Switch regulates sgRNA activity by Cre recombination for sequential editing of two loci. Nat. Commun. 2019, 10, 5454. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Peng, S.; Huang, W.; Cai, Z.; Xie, Z. Rational Design of Mini-Cas9 for Transcriptional Activation. ACS Synth. Biol. 2018, 7, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Kungulovski, G.; Nunna, S.; Thomas, M.; Zanger, U.M.; Reinhardt, R.; Jeltsch, A. Targeted epigenome editing of an endogenous locus with chromatin modifiers is not stably maintained. Epigenetics and Chromatin 2015, 8, 12. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).