Angiotensin (1-7) Decreases Myostatin-Induced NF-κB Signaling and Skeletal Muscle Atrophy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
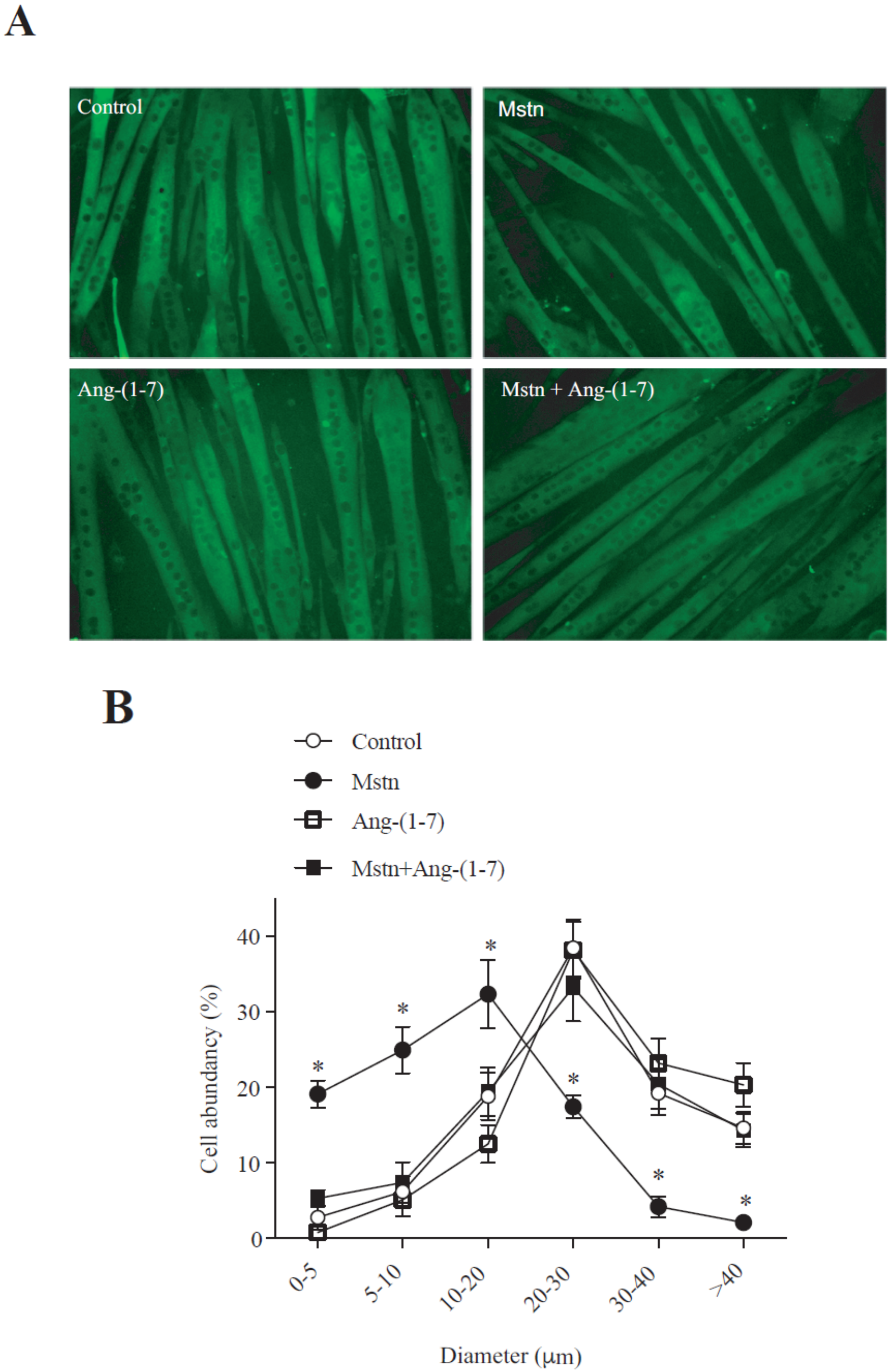
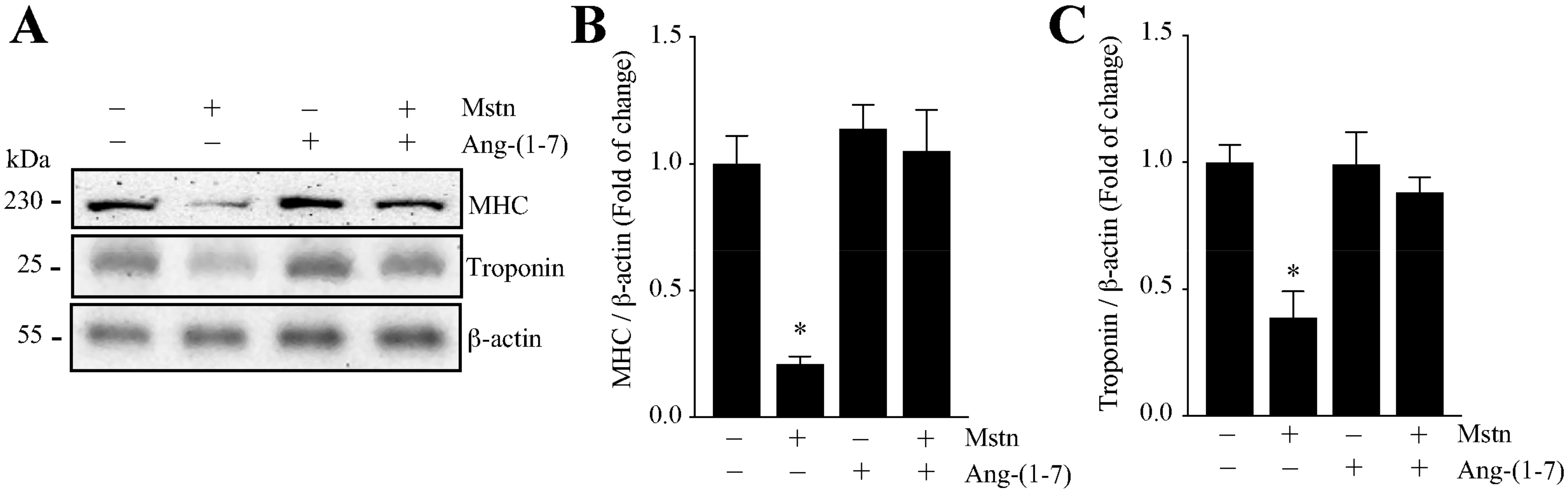
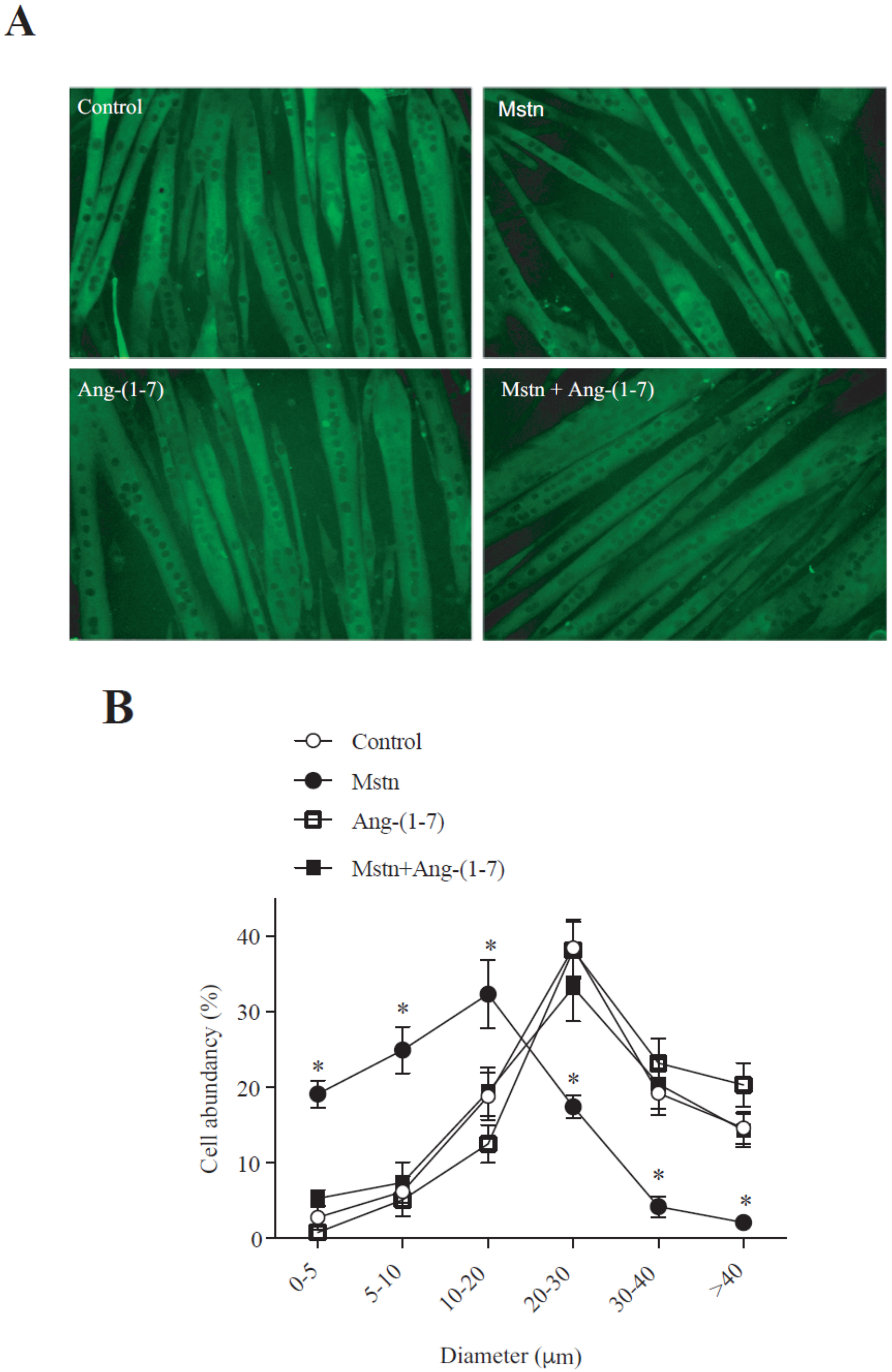
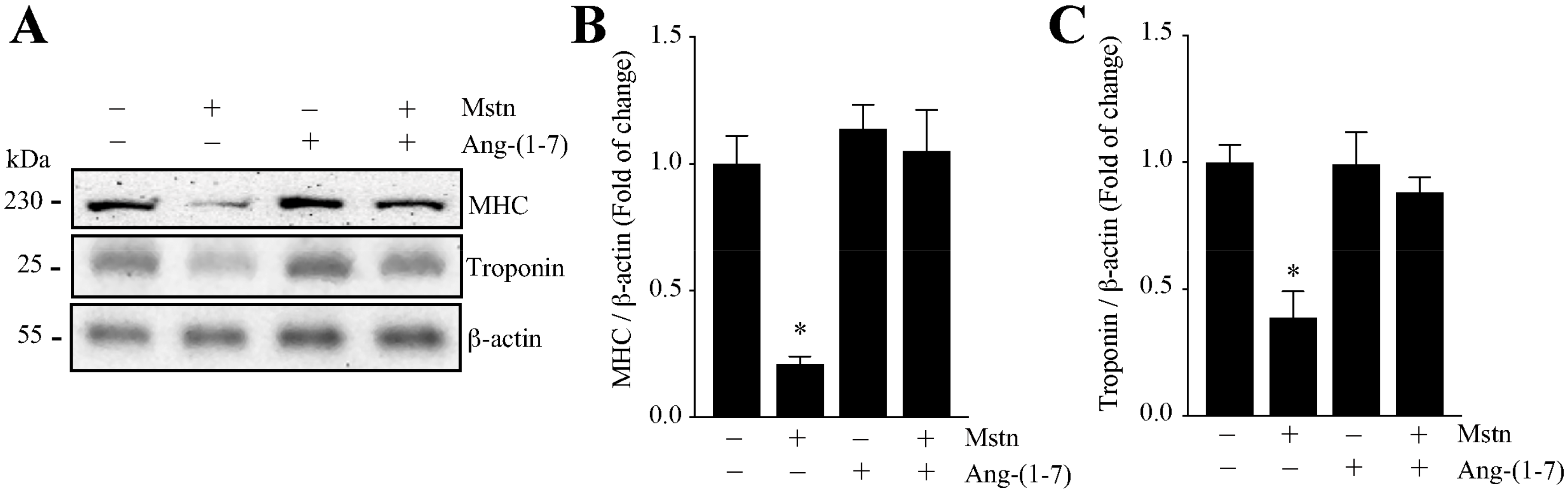
2.1. Ang-(1-7) Prevents the Decrease of the Myotube Diameter and MHC Levels Induced by Myostatin in C2C12 Cells
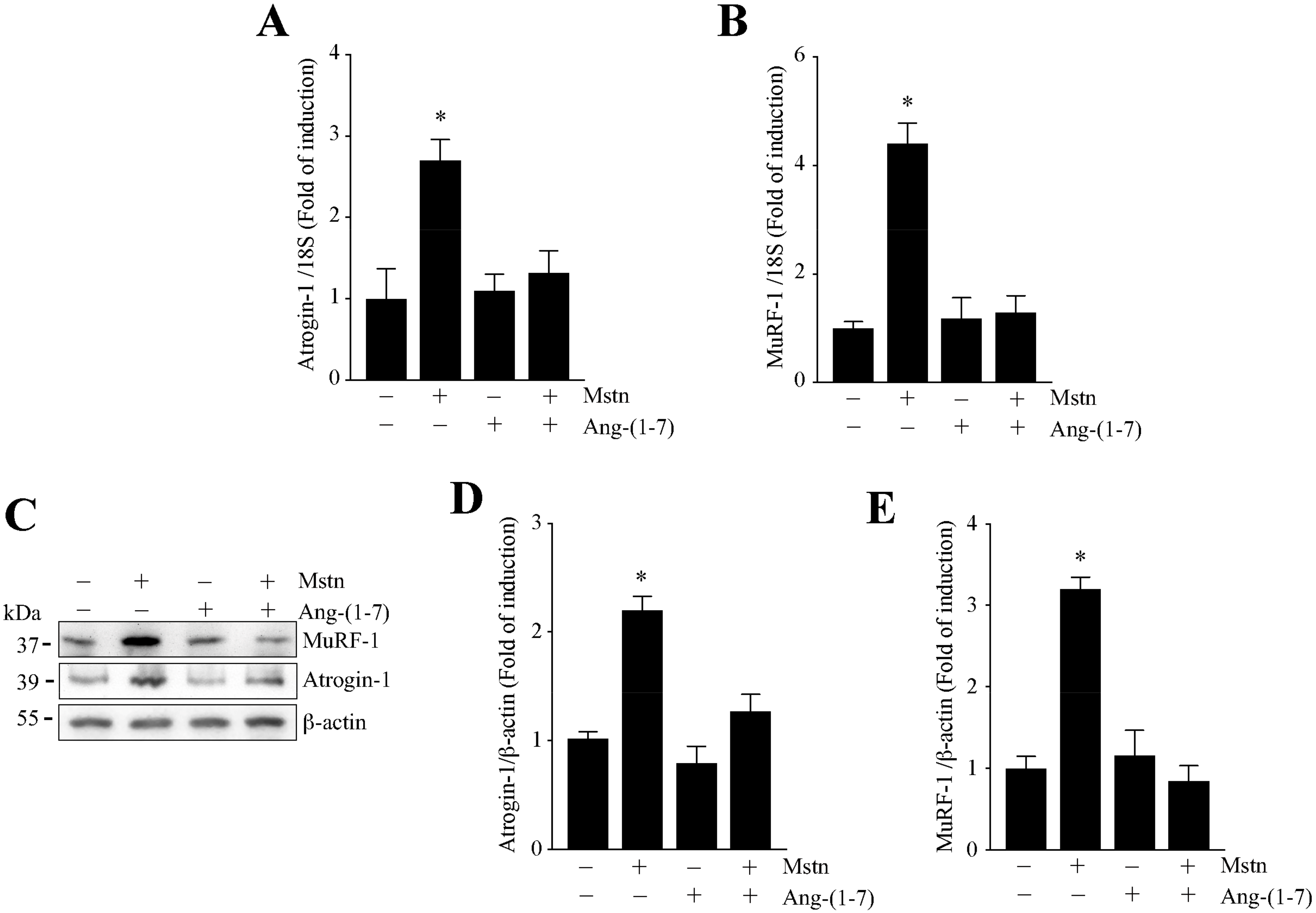
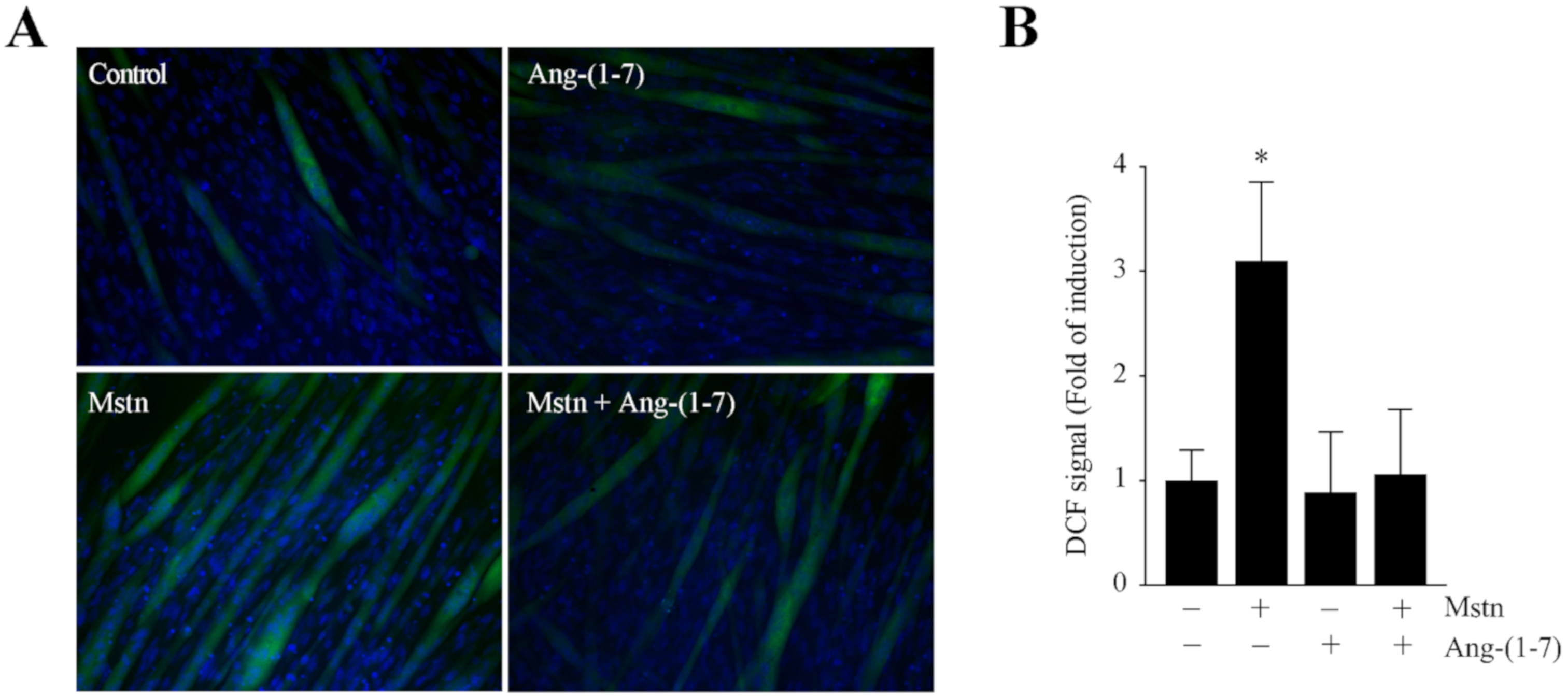
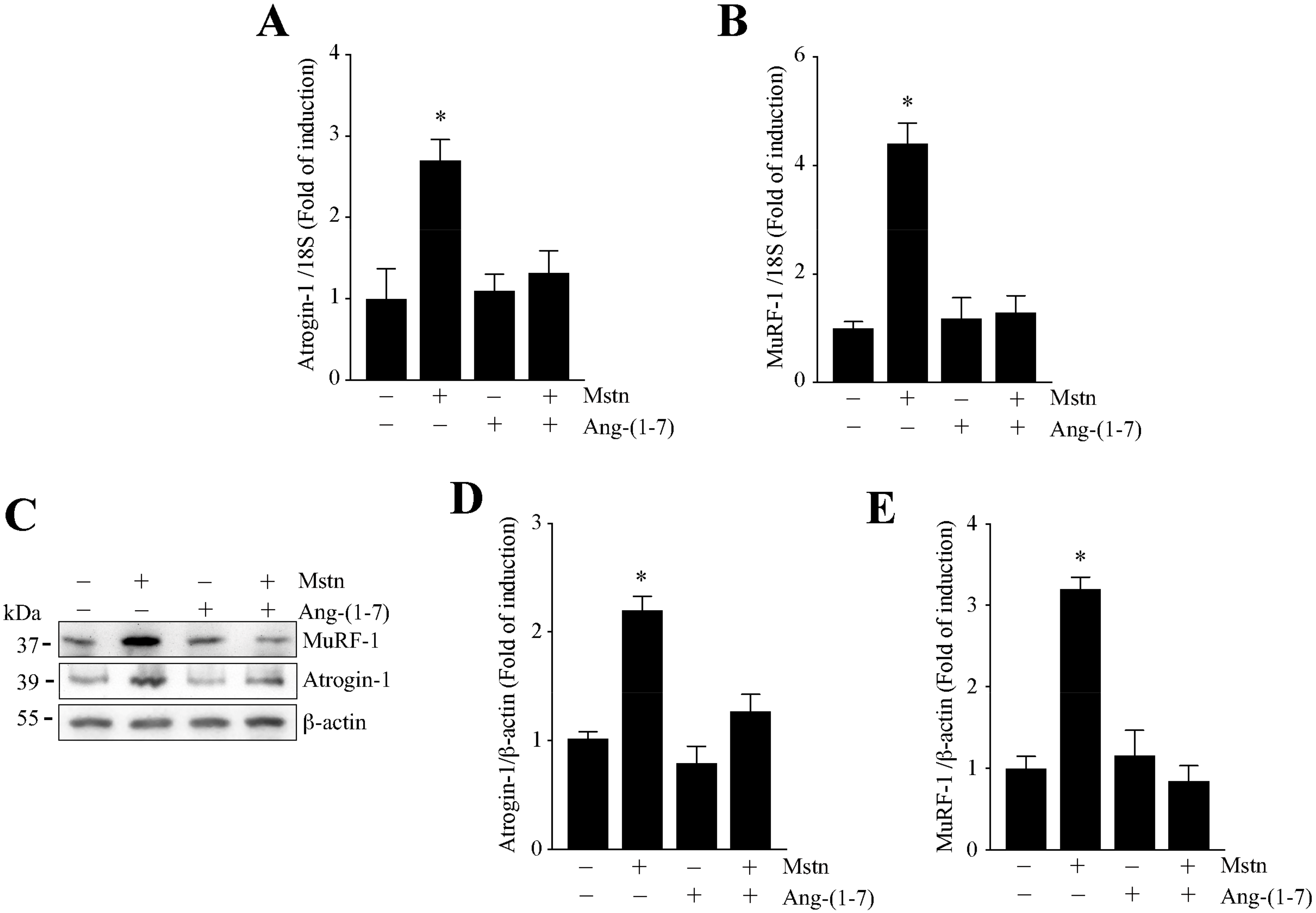
2.2. Ang-(1-7) Decreases the Myostatin-Dependent Increment of E3 Ubiquitin Ligases and ROS in C2C12 Myotubes
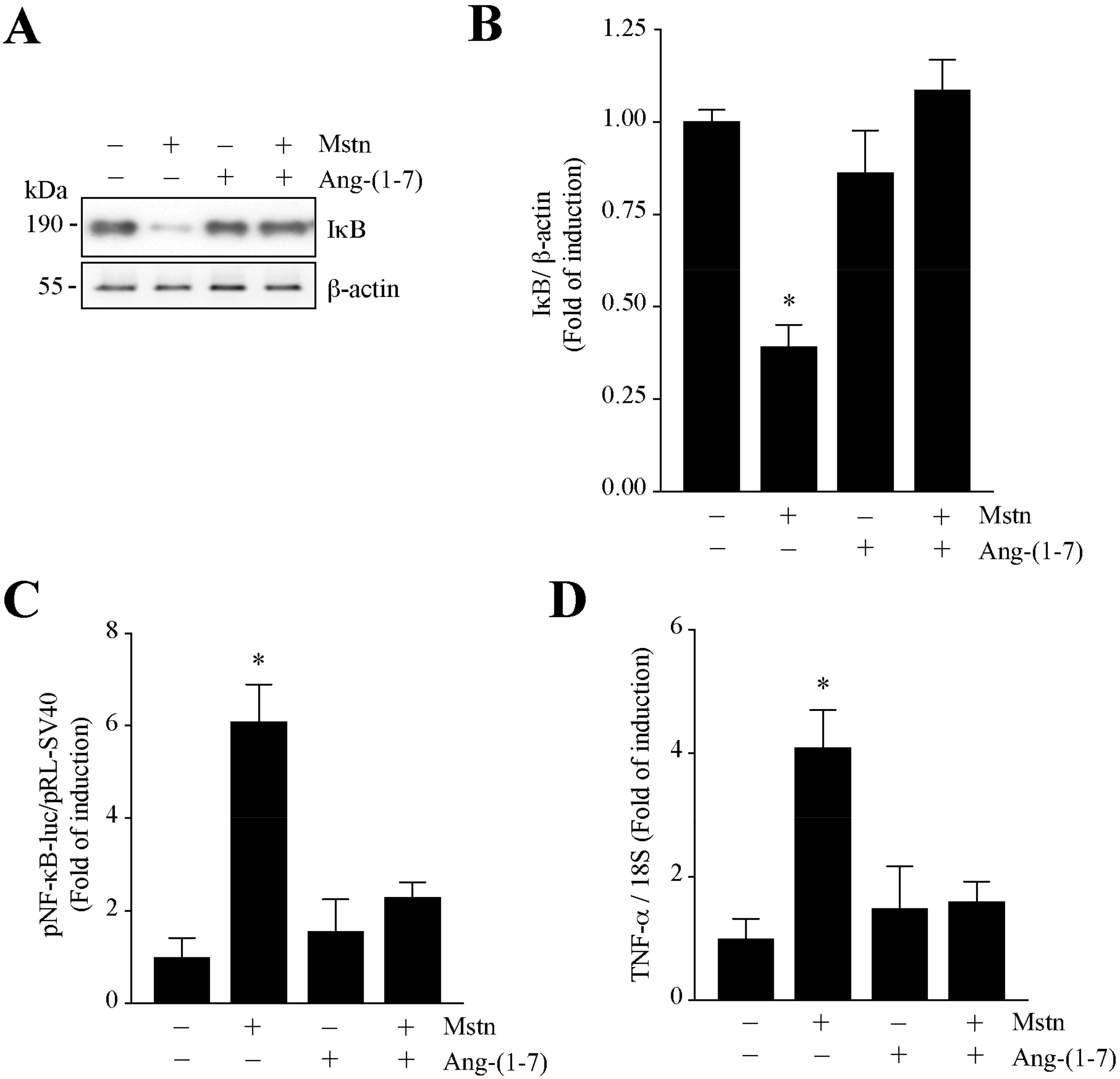
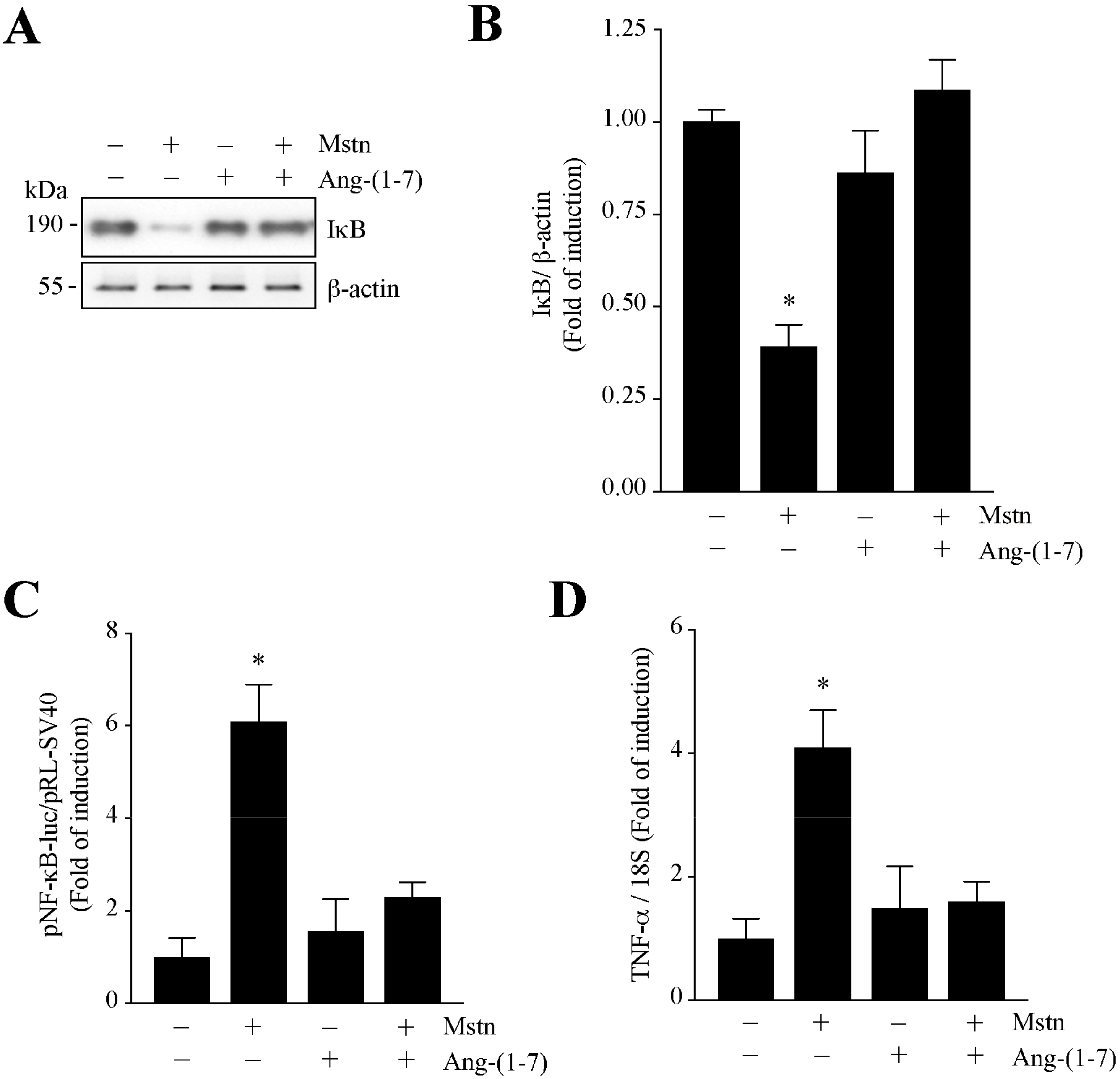
2.3. Ang-(1-7) Decreases the NF-κB Signaling Induced by Myostatin in C2C12 Myotubes
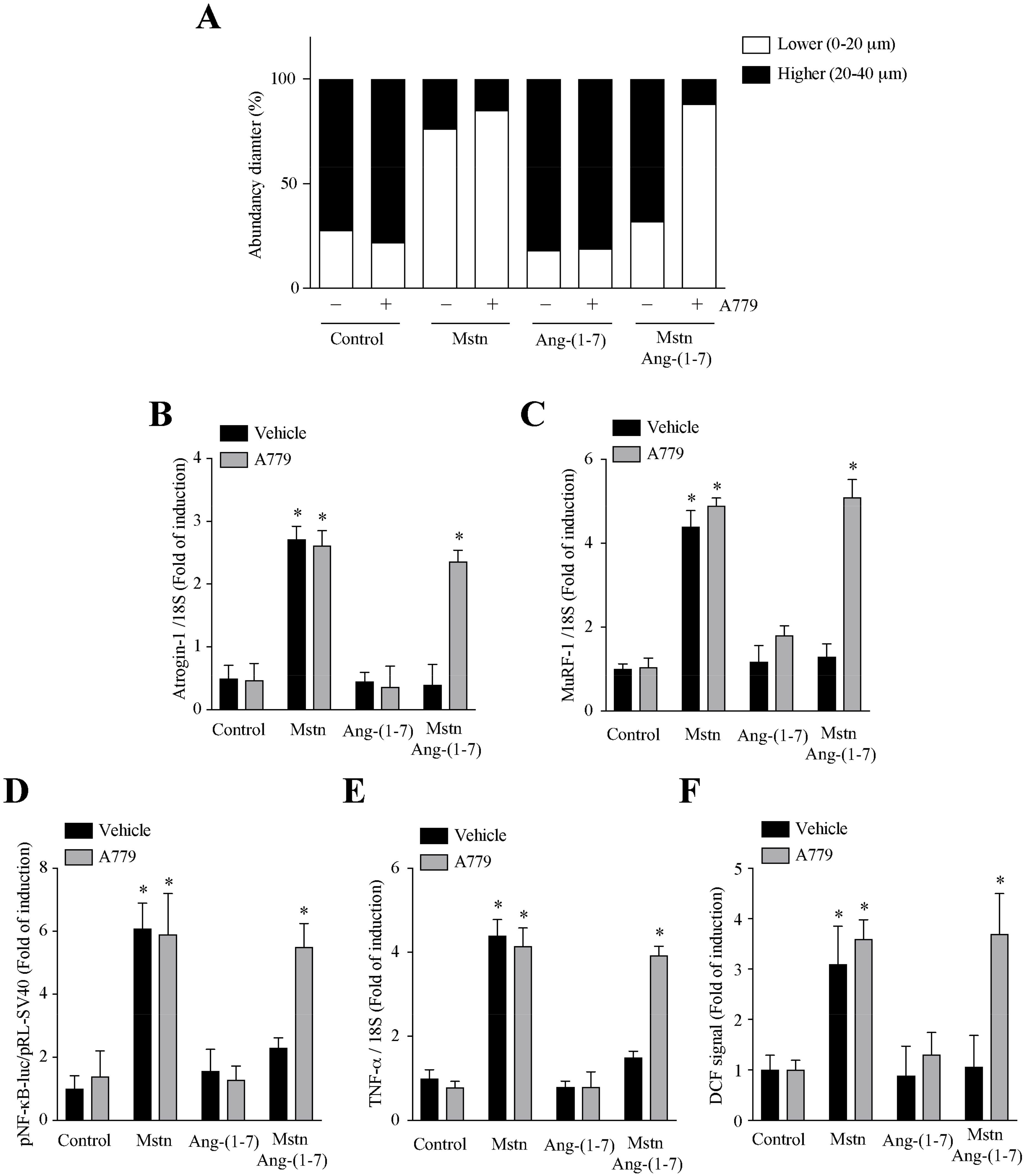
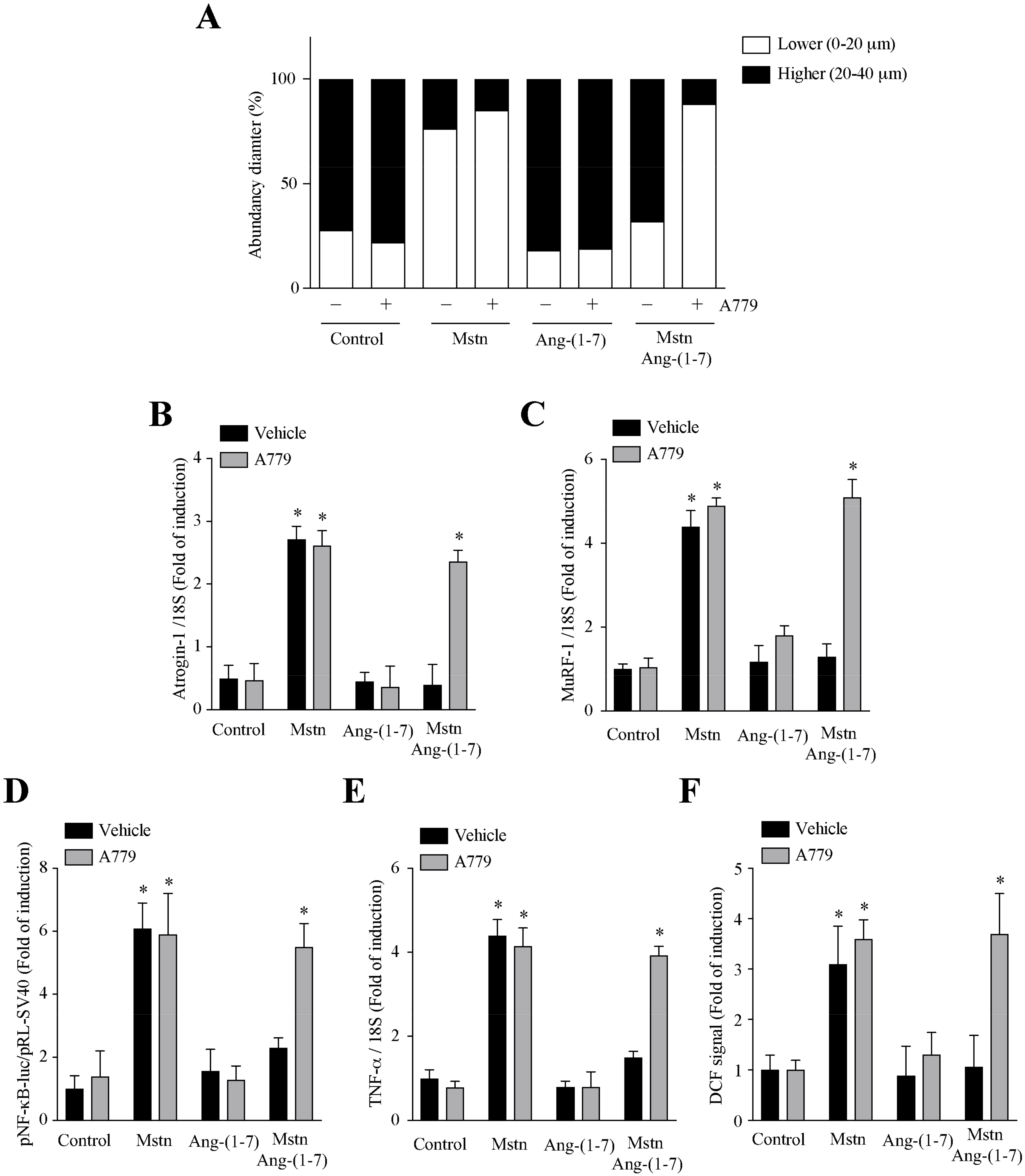
2.4. Ang-(1-7) Decreases the Myostatin-Dependent Activity through the Mas Receptor in C2C12 Myotubes
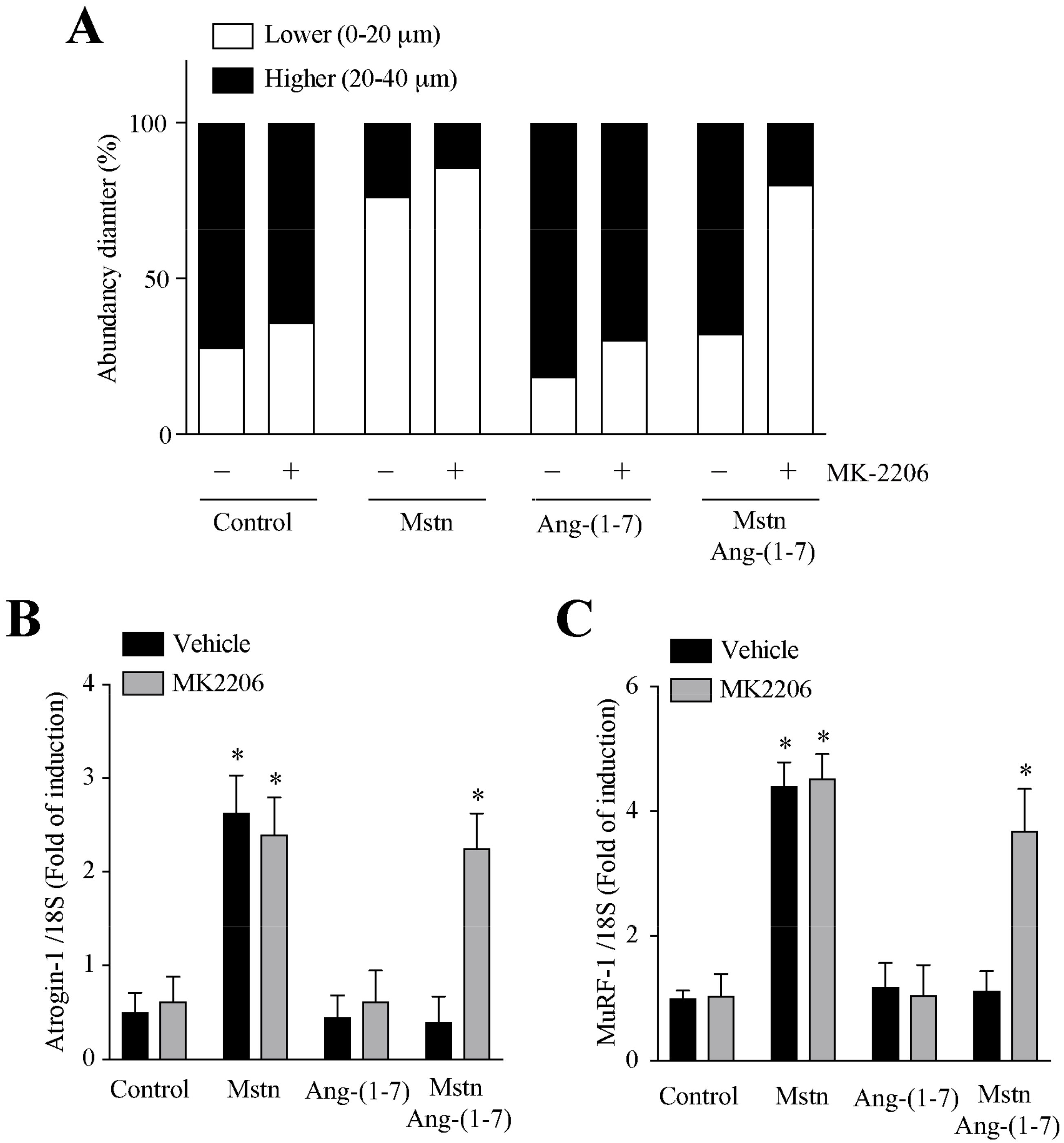
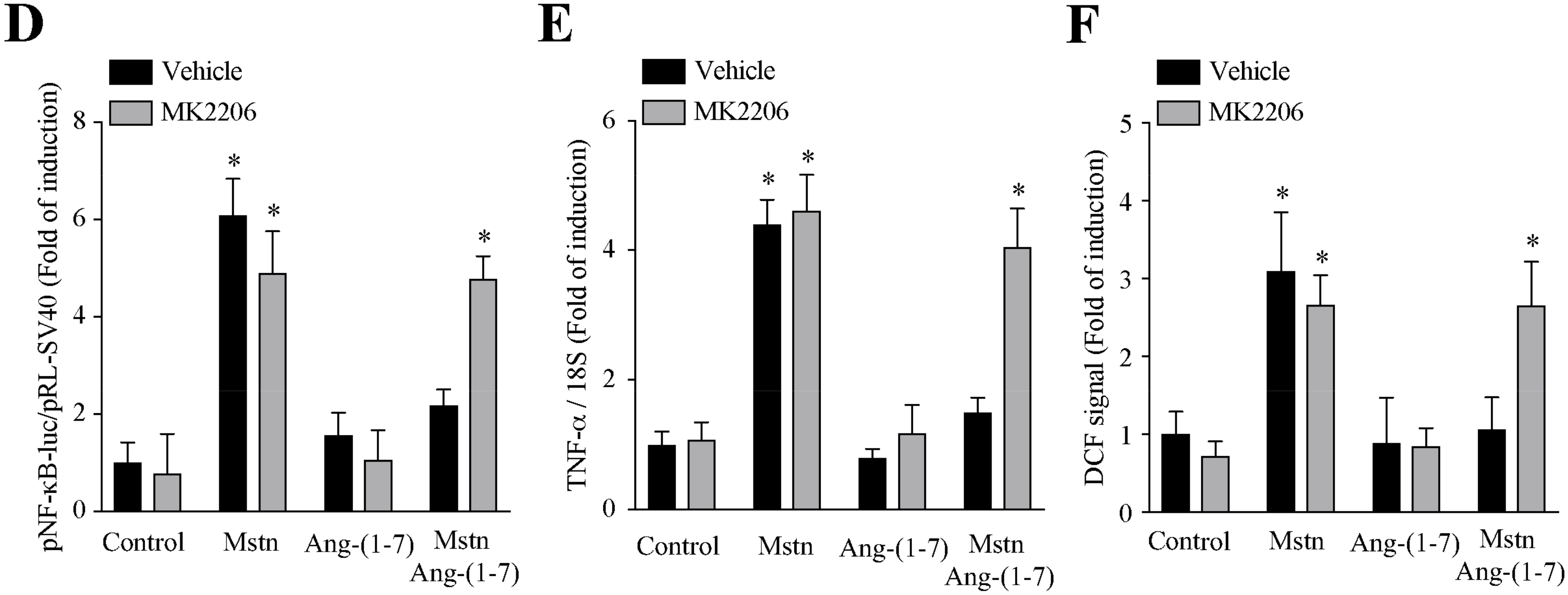
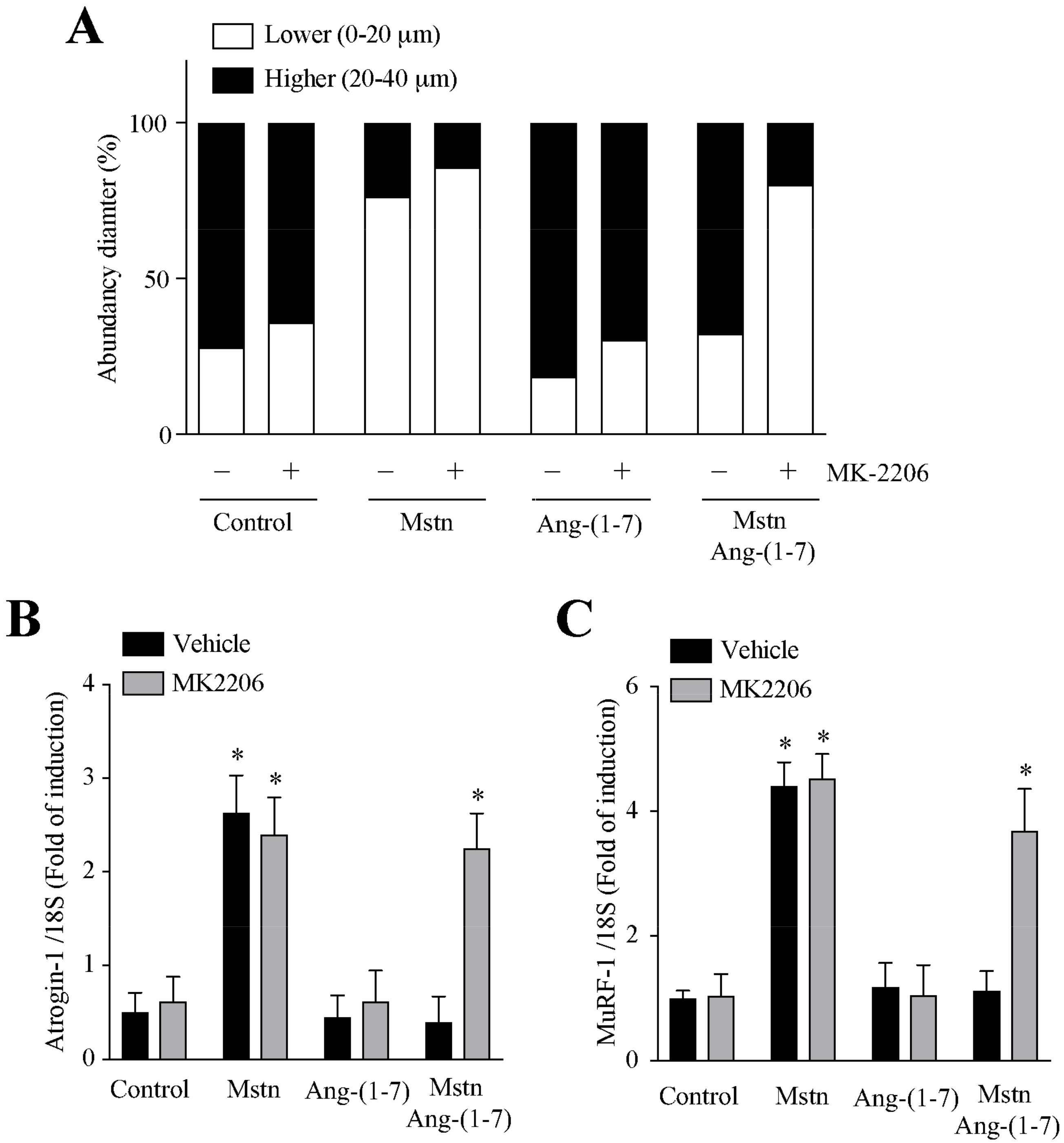
2.5. Ang-(1-7) Decreases the Myostatin-Dependent Activity through the Akt/PKB Activity in C2C12 Myotubes
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Immunoblot Analysis
4.3. Immunofluorescence Microscopy
4.4. Measurement of Myotube Diameter
4.5. Measurement of Intracellular ROS Levels
4.6. Transient Plasmid Transfection
4.7. RNA Isolation, Reverse Transcription, and Quantitative Real-Time PCR
4.8. Statistics
Author Contributions
Funding
Conflicts of Interest
References
- Ding, S.; Dai, Q.; Huang, H.; Xu, Y.; Zhong, C. An Overview of Muscle Atrophy. Adv. Exp. Med. Biol. 2018, 1088, 3–19. [Google Scholar]
- Cao, R.Y.; Li, J.; Dai, Q.; Li, Q.; Yang, J. Muscle Atrophy: Present and Future. Adv. Exp. Med. Biol. 2018, 1088, 605–624. [Google Scholar]
- Abrigo, J.; Simon, F.; Cabrera, D.; Vilos, C.; Cabello-Verrugio, C. Mitochondrial Dysfunction in Skeletal Muscle Pathologies. Curr. Protein Pept. Sci. 2019, 20, 536–546. [Google Scholar]
- Dumitru, A.; Radu, B.M.; Radu, M.; Cretoiu, S.M. Muscle Changes During Atrophy. Adv. Exp. Med. Biol. 2018, 1088, 73–92. [Google Scholar]
- Cabello-Verrugio, C.; Rivera, J.C.; Garcia, D. Skeletal muscle wasting: New role of nonclassical renin-angiotensin system. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 158–163. [Google Scholar]
- Khalil, R. Ubiquitin-Proteasome Pathway and Muscle Atrophy. Adv. Exp. Med. Biol. 2018, 1088, 235–248. [Google Scholar]
- Bilodeau, P.A.; Coyne, E.S.; Wing, S.S. The ubiquitin proteasome system in atrophying skeletal muscle: Roles and regulation. Am. J. Physiol. Cell Physiol. 2016, 311, C392–C403. [Google Scholar]
- Abrigo, J.; Elorza, A.A.; Riedel, C.A.; Vilos, C.; Simon, F.; Cabrera, D.; Estrada, L.; Cabello-Verrugio, C. Role of Oxidative Stress as Key Regulator of Muscle Wasting during Cachexia. Oxid. Med. Cell Longev. 2018, 2018, 2063179. [Google Scholar]
- Bergen, H.R.; Farr, J.N.; Vanderboom, P.M.; Atkinson, E.J.; White, T.A.; Singh, R.J.; Khosla, S.; LeBrasseur, N.K. Myostatin as a mediator of sarcopenia versus homeostatic regulator of muscle mass: Insights using a new mass spectrometry-based assay. Skelet. Muscle 2015, 5, 21. [Google Scholar]
- Hoogaars, W.M.H.; Jaspers, R.T. Past, Present, and Future Perspective of Targeting Myostatin and Related Signaling Pathways to Counteract Muscle Atrophy. Adv. Exp. Med. Biol. 2018, 1088, 153–206. [Google Scholar]
- St Andre, M.; Johnson, M.; Bansal, P.N.; Wellen, J.; Robertson, A.; Opsahl, A.; Burch, P.M.; Bialek, P.; Morris, C.; Owens, J. A mouse anti-myostatin antibody increases muscle mass and improves muscle strength and contractility in the mdx mouse model of Duchenne muscular dystrophy and its humanized equivalent, domagrozumab, (PF-06252616), increases muscle volume in cynomolgus monkeys. Skelet. Muscle 2017, 7, 25. [Google Scholar]
- Desgeorges, M.M.; Devillard, X.; Toutain, J.; Castells, J.; Divoux, D.; Arnould, D.F.; Haqq, C.; Bernaudin, M.; Durieux, A.C.; Touzani, O.; et al. Pharmacological inhibition of myostatin improves skeletal muscle mass and function in a mouse model of stroke. Sci. Rep. 2017, 7, 14000. [Google Scholar]
- Sriram, S.; Subramanian, S.; Juvvuna, P.K.; Ge, X.; Lokireddy, S.; McFarlane, C.D.; Wahli, W.; Kambadur, R.; Sharma, M. Myostatin augments muscle-specific ring finger protein-1 expression through an NF-kB independent mechanism in SMAD3 null muscle. Mol. Endocrinol. 2014, 28, 317–330. [Google Scholar]
- Manfredi, L.H.; Paula-Gomes, S.; Zanon, N.M.; Kettelhut, I.C. Myostatin promotes distinct responses on protein metabolism of skeletal and cardiac muscle fibers of rodents. Braz J. Med. Biol. Res. 2017, 50, e6733. [Google Scholar]
- Wei, Y.; Chen, Y.; Qiu, Y.; Zhao, H.; Liu, G.; Zhang, Y.; Meng, Q.; Wu, G.; Chen, Y.; Cai, X.; et al. Prevention of Muscle Wasting by CRISPR/Cas9-mediated Disruption of Myostatin In Vivo. Mol. Ther. 2016, 24, 1889–1891. [Google Scholar]
- Cabello-Verrugio, C.; Morales, M.G.; Cabrera, D.; Vio, C.P.; Brandan, E. Angiotensin II receptor type 1 blockade decreases CTGF/CCN2-mediated damage and fibrosis in normal and dystrophic skeletal muscles. J. Cell Mol. Med. 2012, 16, 752–764. [Google Scholar]
- Morales, M.G.; Abrigo, J.; Meneses, C.; Simon, F.; Cisternas, F.; Rivera, J.C.; Vazquez, Y.; Cabello-Verrugio, C. The Ang-(1-7)/Mas-1 axis attenuates the expression and signalling of TGF-beta1 induced by AngII in mouse skeletal muscle. Clin. Sci. (Lond.) 2014, 127, 251–264. [Google Scholar]
- Morales, M.G.; Vazquez, Y.; Acuna, M.J.; Rivera, J.C.; Simon, F.; Salas, J.D.; Alvarez Ruf, J.; Brandan, E.; Cabello-Verrugio, C. Angiotensin II-induced pro-fibrotic effects require p38MAPK activity and transforming growth factor beta 1 expression in skeletal muscle cells. Int. J. Biochem. Cell Biol. 2012, 44, 1993–2002. [Google Scholar]
- Russell, S.T.; Sanders, P.M.; Tisdale, M.J. Angiotensin II directly inhibits protein synthesis in murine myotubes. Cancer Lett. 2006, 231, 290–294. [Google Scholar]
- Russell, S.T.; Wyke, S.M.; Tisdale, M.J. Mechanism of induction of muscle protein degradation by angiotensin II. Cell Signal. 2006, 18, 1087–1096. [Google Scholar]
- Cabello-Verrugio, C.; Morales, M.G.; Rivera, J.C.; Cabrera, D.; Simon, F. Renin-angiotensin system: An old player with novel functions in skeletal muscle. Med. Res. Rev. 2015, 35, 437–463. [Google Scholar]
- Da Silveira, K.D.; Coelho, F.M.; Vieira, A.T.; Sachs, D.; Barroso, L.C.; Costa, V.V.; Bretas, T.L.; Bader, M.; de Sousa, L.P.; da Silva, T.A.; et al. Anti-inflammatory effects of the activation of the angiotensin-(1-7) receptor, MAS, in experimental models of arthritis. J. Immunol. 2010, 185, 5569–5576. [Google Scholar]
- Santos, R.A.; Simoes e Silva, A.C.; Maric, C.; Silva, D.M.; Machado, R.P.; de Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.; Lopes, M.T.; Bader, M.; et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263. [Google Scholar]
- Santos, R.A.; Ferreira, A.J.; Simoes, E.S.A.C. Recent advances in the angiotensin-converting enzyme 2-angiotensin(1-7)-Mas axis. Exp. Physiol. 2008, 93, 519–527. [Google Scholar]
- Morales, M.G.; Abrigo, J.; Acuna, M.J.; Santos, R.A.; Bader, M.; Brandan, E.; Simon, F.; Olguin, H.; Cabrera, D.; Cabello-Verrugio, C. Angiotensin-(1-7) attenuates disuse skeletal muscle atrophy in mice via its receptor, Mas. Dis. Models Mech. 2016, 9, 441–449. [Google Scholar]
- Cisternas, F.; Morales, M.G.; Meneses, C.; Simon, F.; Brandan, E.; Abrigo, J.; Vazquez, Y.; Cabello-Verrugio, C. Angiotensin-(1-7) decreases skeletal muscle atrophy induced by angiotensin II through a Mas receptor-dependent mechanism. Clin. Sci. (Lond.) 2015, 128, 307–319. [Google Scholar]
- Marquez-Miranda, V.; Abrigo, J.; Rivera, J.C.; Araya-Duran, I.; Aravena, J.; Simon, F.; Pacheco, N.; Gonzalez-Nilo, F.D.; Cabello-Verrugio, C. The complex of PAMAM-OH dendrimer with Angiotensin, (1-7) prevented the disuse-induced skeletal muscle atrophy in mice. Int J. Nanomed. 2017, 12, 1985–1999. [Google Scholar]
- Abrigo, J.; Simon, F.; Cabrera, D.; Cabello-Verrugio, C. Angiotensin-(1-7) Prevents Skeletal Muscle Atrophy Induced by Transforming Growth Factor Type Beta, (TGF-beta) via Mas Receptor Activation. Cell Physiol. Biochem. 2016, 40, 27–38. [Google Scholar]
- Morales, M.G.; Olguin, H.; Di Capua, G.; Brandan, E.; Simon, F.; Cabello-Verrugio, C. Endotoxin-induced skeletal muscle wasting is prevented by angiotensin-(1-7) through a p38 MAPK-dependent mechanism. Clin. Sci. (Lond.) 2015, 129, 461–476. [Google Scholar]
- Morales, M.G.; Abrigo, J.; Meneses, C.; Cisternas, F.; Simon, F.; Cabello-Verrugio, C. Expression of the Mas receptor is upregulated in skeletal muscle wasting. Histochem. Cell Biol. 2015, 143, 131–141. [Google Scholar]
- Meneses, C.; Morales, M.G.; Abrigo, J.; Simon, F.; Brandan, E.; Cabello-Verrugio, C. The angiotensin-(1-7)/Mas axis reduces myonuclear apoptosis during recovery from angiotensin II-induced skeletal muscle atrophy in mice. Pflug. Arch. 2015, 467, 1975–1984. [Google Scholar]
- Takeshita, H.; Yamamoto, K.; Nozato, S.; Takeda, M.; Fukada, S.I.; Inagaki, T.; Tsuchimochi, H.; Shirai, M.; Nozato, Y.; Fujimoto, T.; et al. Angiotensin-converting enzyme 2 deficiency accelerates and angiotensin 1-7 restores age-related muscle weakness in mice. J. Cachexia Sarcopenia Muscle. 2018, 9, 975–986. [Google Scholar]
- Collins-Hooper, H.; Sartori, R.; Macharia, R.; Visanuvimol, K.; Foster, K.; Matsakas, A.; Flasskamp, H.; Ray, S.; Dash, P.R.; Sandri, M.; et al. Propeptide-mediated inhibition of myostatin increases muscle mass through inhibiting proteolytic pathways in aged mice. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1049–1059. [Google Scholar]
- Liu, D.; Qiao, X.; Ge, Z.; Shang, Y.; Li, Y.; Wang, W.; Chen, M.; Si, S.; Chen, S.Z. IMB0901 inhibits muscle atrophy induced by cancer cachexia through MSTN signaling pathway. Skelet. Muscle 2019, 9, 8. [Google Scholar]
- Trendelenburg, A.U.; Meyer, A.; Rohner, D.; Boyle, J.; Hatakeyama, S.; Glass, D.J. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am. J. Physiol. Cell Physiol. 2009, 296, C1258–C1270. [Google Scholar]
- Yoon, M.S. mTOR as a Key Regulator in Maintaining Skeletal Muscle Mass. Front. Physiol. 2017, 8, 788. [Google Scholar]
- Amirouche, A.; Durieux, A.C.; Banzet, S.; Koulmann, N.; Bonnefoy, R.; Mouret, C.; Bigard, X.; Peinnequin, A.; Freyssenet, D. Down-regulation of Akt/mammalian target of rapamycin signaling pathway in response to myostatin overexpression in skeletal muscle. Endocrinology 2009, 150, 286–294. [Google Scholar]
- Lipina, C.; Kendall, H.; McPherron, A.C.; Taylor, P.M.; Hundal, H.S. Mechanisms involved in the enhancement of mammalian target of rapamycin signalling and hypertrophy in skeletal muscle of myostatin-deficient mice. FEBS Lett. 2010, 584, 2403–2408. [Google Scholar]
- Houddane, A.; Bultot, L.; Novellasdemunt, L.; Johanns, M.; Gueuning, M.A.; Vertommen, D.; Coulie, P.G.; Bartrons, R.; Hue, L.; Rider, M.H. Role of Akt/PKB and PFKFB isoenzymes in the control of glycolysis, cell proliferation and protein synthesis in mitogen-stimulated thymocytes. Cell Signal. 2017, 34, 23–37. [Google Scholar]
- Lai, Y.C.; Liu, Y.; Jacobs, R.; Rider, M.H. A novel PKB/Akt inhibitor, MK-2206, effectively inhibits insulin-stimulated glucose metabolism and protein synthesis in isolated rat skeletal muscle. Biochem. J. 2012, 447, 137–147. [Google Scholar]
- Mirzoev, T.M.; Tyganov, S.A.; Shenkman, B.S. Akt-dependent and Akt-independent pathways are involved in protein synthesis activation during reloading of disused soleus muscle. Muscle Nerve 2017, 55, 393–399. [Google Scholar]
- Abrigo, J.; Rivera, J.C.; Simon, F.; Cabrera, D.; Cabello-Verrugio, C. Transforming growth factor type beta, (TGF-beta) requires reactive oxygen species to induce skeletal muscle atrophy. Cell Signal. 2016, 28, 366–376. [Google Scholar]
- Op den Kamp, C.M.; Langen, R.C.; Snepvangers, F.J.; de Theije, C.C.; Schellekens, J.M.; Laugs, F.; Dingemans, A.M.; Schols, A.M. Nuclear transcription factor kappa B activation and protein turnover adaptations in skeletal muscle of patients with progressive stages of lung cancer cachexia. Am. J. Clin. Nutr. 2013, 98, 738–748. [Google Scholar]
- Ma, J.F.; Sanchez, B.J.; Hall, D.T.; Tremblay, A.K.; Di Marco, S.; Gallouzi, I.E. STAT3 promotes IFNgamma/TNFalpha-induced muscle wasting in an NF-kappaB-dependent and IL-6-independent manner. EMBO Mol. Med. 2017, 9, 622–637. [Google Scholar]
- Thoma, A.; Lightfoot, A.P. NF-kB and Inflammatory Cytokine Signalling: Role in Skeletal Muscle Atrophy. Adv. Exp. Med. Biol. 2018, 1088, 267–279. [Google Scholar]
- Liu, L.; Liu, X.; Bai, Y.; Tang, N.; Li, J.; Zhang, Y.; Wu, J.; Wang, X.; Wei, J. Neuregulin-1beta modulates myogenesis in septic mouse serum-treated C2C12 myotubes in vitro through PPARgamma/NF-kappaB signaling. Mol. Biol. Rep. 2018, 45, 1611–1619. [Google Scholar]
- Lang, C.H.; Frost, R.A.; Nairn, A.C.; MacLean, D.A.; Vary, T.C. TNF-alpha impairs heart and skeletal muscle protein synthesis by altering translation initiation. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E336–E347. [Google Scholar]
- Li, Y.P.; Reid, M.B. NF-kappaB mediates the protein loss induced by TNF-alpha in differentiated skeletal muscle myotubes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R1165–R1170. [Google Scholar]
- Steffen, B.T.; Lees, S.J.; Booth, F.W. Anti-TNF treatment reduces rat skeletal muscle wasting in monocrotaline-induced cardiac cachexia. J. Appl. Physiol. 2008, 105, 1950–1958. [Google Scholar]
- Bihl, J.C.; Zhang, C.; Zhao, Y.; Xiao, X.; Ma, X.; Chen, Y.; Chen, S.; Zhao, B.; Chen, Y. Angiotensin-(1-7) counteracts the effects of Ang II on vascular smooth muscle cells, vascular remodeling and hemorrhagic stroke: Role of the NFsmall ka, CyrillicB inflammatory pathway. Vasc. Pharm. 2015, 73, 115–123. [Google Scholar]
- Yu, X.; Cui, L.; Hou, F.; Liu, X.; Wang, Y.; Wen, Y.; Chi, C.; Li, C.; Liu, R.; Yin, C. Angiotensin-converting enzyme 2-angiotensin, (1-7)-Mas axis prevents pancreatic acinar cell inflammatory response via inhibition of the p38 mitogen-activated protein kinase/nuclear factor-kappaB pathway. Int. J. Mol. Med. 2018, 41, 409–420. [Google Scholar]
- Rabie, M.A.; Abd El Fattah, M.A.; Nassar, N.N.; El-Abhar, H.S.; Abdallah, D.M. Angiotensin 1-7 ameliorates 6-hydroxydopamine lesions in hemiparkinsonian rats through activation of MAS receptor/PI3K/Akt/BDNF pathway and inhibition of angiotensin II type-1 receptor/NF-kappaB axis. Biochem. Pharm. 2018, 151, 126–134. [Google Scholar]
- Martin, A.I.; Gomez-SanMiguel, A.B.; Priego, T.; Lopez-Calderon, A. Formoterol treatment prevents the effects of endotoxin on muscle TNF/NF-kB, Akt/mTOR, and proteolytic pathways in a rat model. Role of IGF-I and miRNA 29b. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E705–E714. [Google Scholar]
- Chacon-Cabrera, A.; Fermoselle, C.; Urtreger, A.J.; Mateu-Jimenez, M.; Diament, M.J.; de Kier Joffe, E.D.; Sandri, M.; Barreiro, E. Pharmacological strategies in lung cancer-induced cachexia: Effects on muscle proteolysis, autophagy, structure, and weakness. J. Cell Physiol. 2014, 229, 1660–1672. [Google Scholar]
- Cabrera, D.; Gutierrez, J.; Cabello-Verrugio, C.; Morales, M.G.; Mezzano, S.; Fadic, R.; Casar, J.C.; Hancke, J.L.; Brandan, E. Andrographolide attenuates skeletal muscle dystrophy in mdx mice and increases efficiency of cell therapy by reducing fibrosis. Skelet. Muscle 2014, 4, 6. [Google Scholar]
- Cabello-Verrugio, C.; Acuna, M.J.; Morales, M.G.; Becerra, A.; Simon, F.; Brandan, E. Fibrotic response induced by angiotensin-II requires NAD(P)H oxidase-induced reactive oxygen species, (ROS) in skeletal muscle cells. Biochem. Biophys. Res. Commun. 2011, 410, 665–670. [Google Scholar]
- Droguett, R.; Cabello-Verrugio, C.; Santander, C.; Brandan, E. TGF-beta receptors, in a Smad-independent manner, are required for terminal skeletal muscle differentiation. Exp. Cell Res. 2010, 316, 2487–2503. [Google Scholar]
- Painemal, P.; Acuna, M.J.; Riquelme, C.; Brandan, E.; Cabello-Verrugio, C. Transforming growth factor type beta 1 increases the expression of angiotensin II receptor type 2 by a SMAD- and p38 MAPK-dependent mechanism in skeletal muscle. Biofactors 2013, 39, 467–475. [Google Scholar]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aravena, J.; Abrigo, J.; Gonzalez, F.; Aguirre, F.; Gonzalez, A.; Simon, F.; Cabello-Verrugio, C. Angiotensin (1-7) Decreases Myostatin-Induced NF-κB Signaling and Skeletal Muscle Atrophy. Int. J. Mol. Sci. 2020, 21, 1167. https://doi.org/10.3390/ijms21031167
Aravena J, Abrigo J, Gonzalez F, Aguirre F, Gonzalez A, Simon F, Cabello-Verrugio C. Angiotensin (1-7) Decreases Myostatin-Induced NF-κB Signaling and Skeletal Muscle Atrophy. International Journal of Molecular Sciences. 2020; 21(3):1167. https://doi.org/10.3390/ijms21031167
Chicago/Turabian StyleAravena, Javier, Johanna Abrigo, Francisco Gonzalez, Francisco Aguirre, Andrea Gonzalez, Felipe Simon, and Claudio Cabello-Verrugio. 2020. "Angiotensin (1-7) Decreases Myostatin-Induced NF-κB Signaling and Skeletal Muscle Atrophy" International Journal of Molecular Sciences 21, no. 3: 1167. https://doi.org/10.3390/ijms21031167
APA StyleAravena, J., Abrigo, J., Gonzalez, F., Aguirre, F., Gonzalez, A., Simon, F., & Cabello-Verrugio, C. (2020). Angiotensin (1-7) Decreases Myostatin-Induced NF-κB Signaling and Skeletal Muscle Atrophy. International Journal of Molecular Sciences, 21(3), 1167. https://doi.org/10.3390/ijms21031167