ER–Mitochondria Contacts and Insulin Resistance Modulation through Exercise Intervention

{kind=link}
{kind=link}
Abstract
1. Introduction
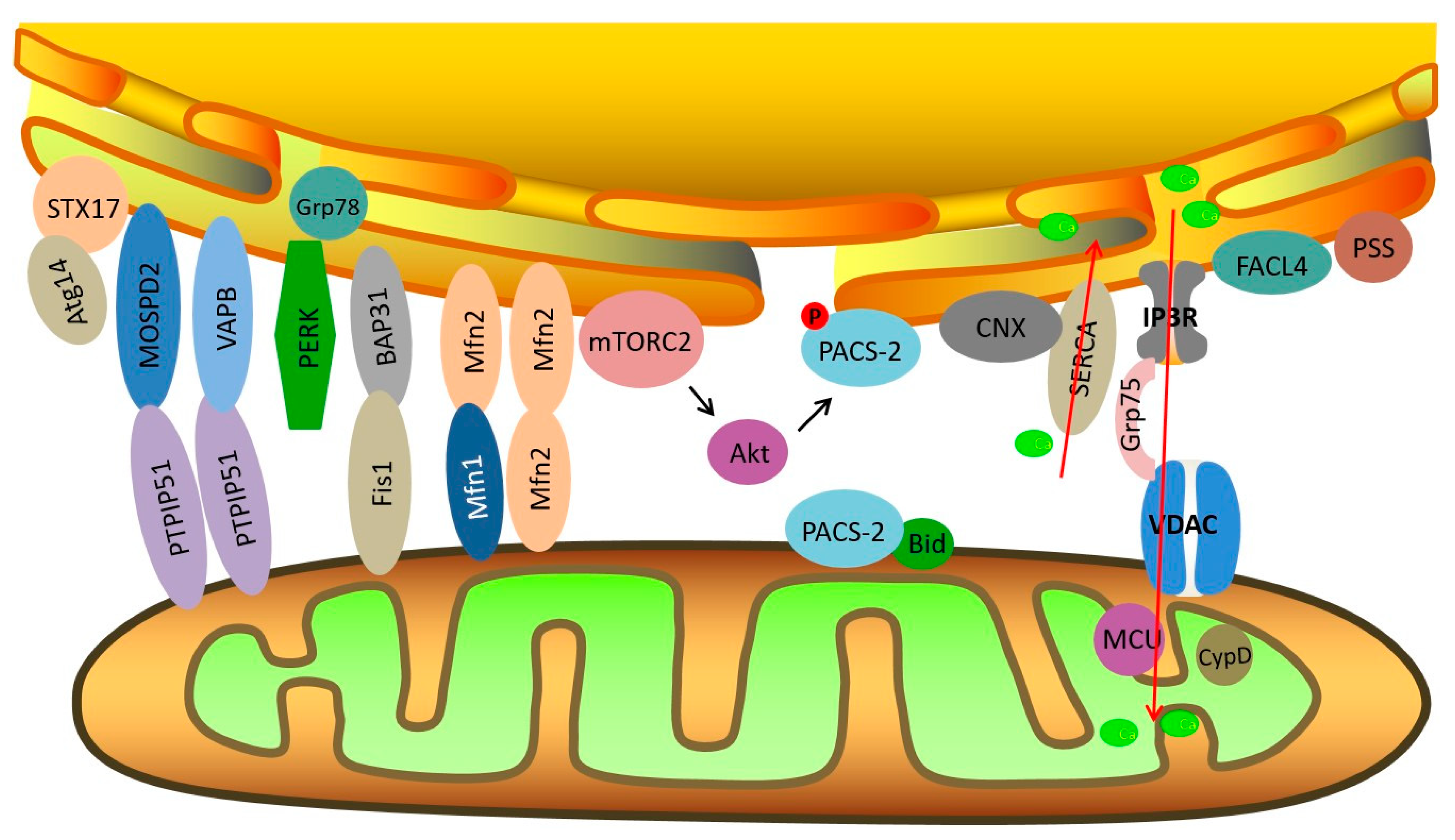
2. The Structure of MAMs
2.1. The IP3R-Grp75-VDAC Complex
2.2. Mfn2
2.3. CypD
2.4. PACS-2
3. The Main Functions of MAMs
3.1. Lipid Exchange
3.2. Ca2+ Homeostasis
3.3. Apoptosis
3.4. Autophagy
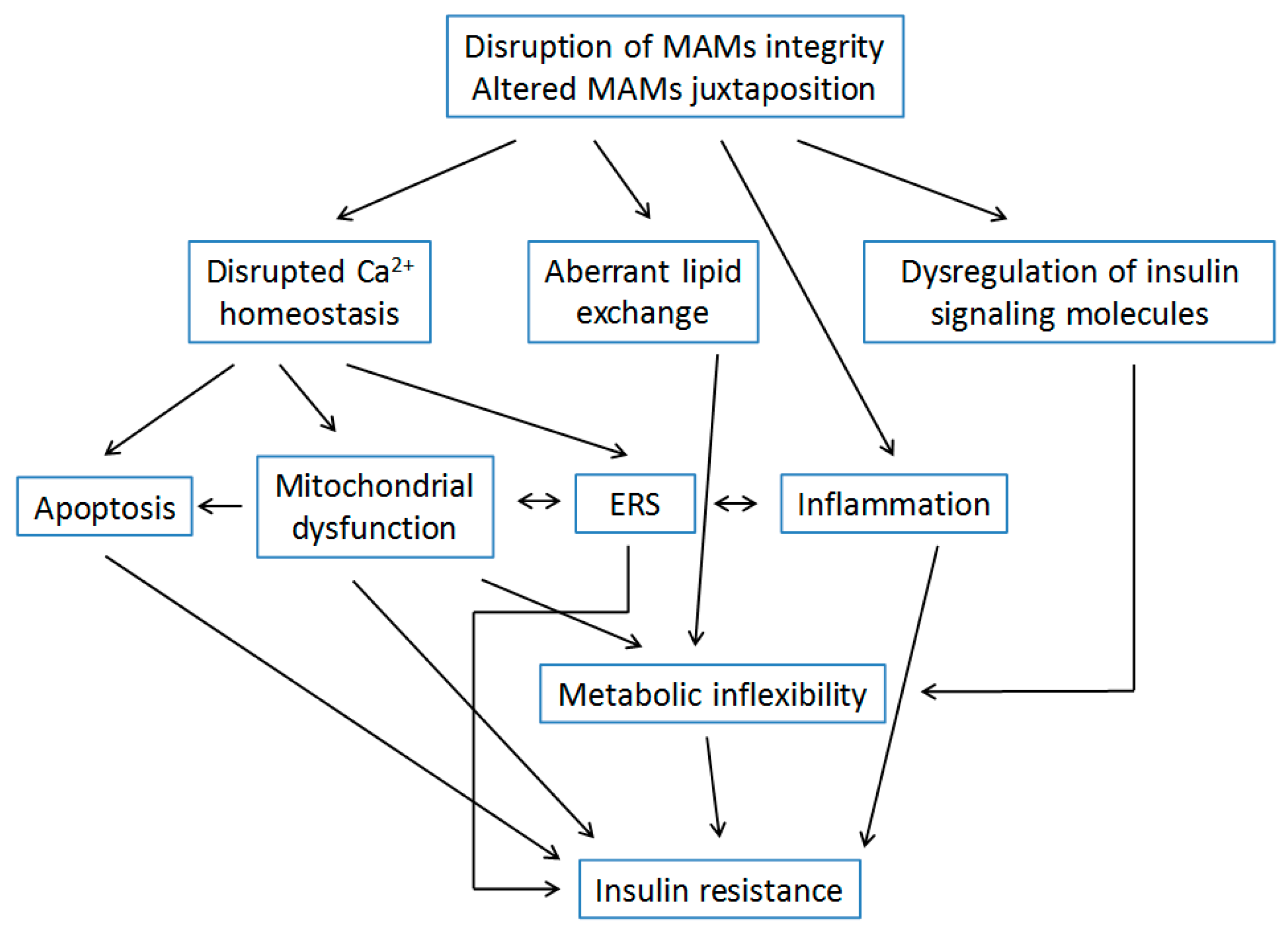
4. Dysfunction of MAMs and Insulin Resistance
4.1. MAMs and Insulin Signaling
4.1.1. MAMs Juxtaposition and Integrity
4.1.2. Insulin Signaling Molecules at MAMs
4.2. Ca2+ Homeostasis and Insulin Resistance
4.3. Metabolic Homeostasis and Insulin Resistance
5. MAMs, Insulin Resistance, and Exercise Intervention
5.1. Exercise Intervention for Mitochondrial Quality Control and Insulin Resistance
5.2. Exercise Intervention for ERS and Insulin Resistance
5.3. The Effect of Exercise Intervention on MAMs and Insulin Resistance
5.3.1. Exercise, Ca2+ Homeostasis, and Insulin Resistance
5.3.2. Exercise, MAMs Components, and Insulin Resistance
5.3.3. Exercise, MAMs-Located Proteins, and Insulin Resistance
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ER | Endoplasmic reticulum |
| MAMs | Mitochondria-associated ER membranes |
| NAFLD | nonalcoholic fatty liver disease |
| PACS-2 | phosphofurin acidic cluster sorting protein 2 |
| Mfn2 | mitofusin 2 |
| IP3R | inositol 1, 4, 5-triphosphate receptor |
| VAPB | vesicle-associated membrane-protein-associated protein B |
| PTPIP51 | protein tyrosine phosphatase-interacting protein 51 |
| CNX | calnexin |
| SERCA | sarco/ER Ca2+ ATPase |
| Sig1R | sigma-1 |
| Ero1α | endoplasmic reticulum oxidoreductin-1α |
| PSS | phosphatidylserine synthase |
| CypD | cyclophilin D |
| mTORC2 | mammalian TOR complex 2 |
| OMM | outer mitochondrial membrane |
| TpMs | trichoplein/mitostatin |
| mPTP | mitochondrial permeability transition pore |
| PE | phosphatidylethanolamine |
| PC | phosphatidylcholine |
| PMCA | plasma membrane Ca2+ ATPases |
| RyR | ryanodine receptor |
| MCU | mitochondrial Ca2+ uniporter |
| PC2 | polycystin 2 |
| PLA | proximity ligation assay |
| HFD | high-fat diet |
| Giamp5 | GTPase of immune-associated protein 5 |
| IRSs | insulin receptor substrates |
| sGC/PKG | soluble guanylate cyclase/protein kinase G |
| ERS | ER stress |
| PDK4 | pyruvate dehydrogenase kinase 4 |
| PML | promyelocytic leukemia |
| UPR | unfolded protein response |
| T2DM | type 2 diabetes |
| PINK1 | PTEN-induced putative kinase 1 |
| Atg7 | autophagy related gene 7 |
| Fundc1 | FUN14 domain-containing 1 |
| HIIT | high-intensity interval training |
| ATF6 | activating transcription factor 6 |
| IRE1α | inositol-requiring enzyme 1α |
| PERK | protein kinase R-like ER protein kinase |
| JNK | c-jun N-terminal kinase |
| IKKβ | I kappa β kinase |
| GLP-1 | glucagon-like peptide-1 |
| REDD1 | regulated in development and DNA damage responses 1 |
| FAS | fatty acid synthase |
References
- Lopez-Crisosto, C.; Bravo-Sagua, R.; Rodriguez-Pena, M.; Mera, C.; Castro, P.F.; Quest, A.F.; Rothermel, B.A.; Cifuentes, M.; Lavandero, S. ER-to-mitochondria miscommunication and metabolic diseases. Biochim. Biophys. Acta 2015, 1852 Pt A, 2096–2105. [Google Scholar] [CrossRef]
- Costello, J.L.; Passmore, J.B.; Islinger, M.; Schrader, M. Multi-localized Proteins: The Peroxisome-Mitochondria Connection. Subcell. Biochem. 2018, 89, 383–415. [Google Scholar] [PubMed]
- Benador, I.Y.; Veliova, M.; Liesa, M.; Shirihai, O.S. Mitochondria Bound to Lipid Droplets: Where Mitochondrial Dynamics Regulate Lipid Storage and Utilization. Cell Metab. 2019, 29, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Missiroli, S.; Patergnani, S.; Duszynski, J.; Wieckowski, M.R.; Pinton, P. Mitochondria-associated membranes: Composition, molecular mechanisms, and physiopathological implications. Antioxid. Redox. Signal. 2015, 22, 995–1019. [Google Scholar] [CrossRef] [PubMed]
- Sasi, U.; Ganapathy, S.; Palayyan, S.R.; Gopal, R.K. Mitochondria Associated Membranes (MAMs): Emerging Drug Targets for Diabetes. Curr. Med. Chem. 2020, 27, 3362–3385. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, C.; Sun, L. Mitochondria-Associated Membranes (MAMs): A Novel Therapeutic Target for Treating Metabolic Syndrome. Curr. Med. Chem. 2020. [CrossRef] [PubMed]
- Wang, J.; He, W.; Tsai, P.J.; Chen, P.H.; Ye, M.; Guo, J.; Su, Z. Mutual interaction between endoplasmic reticulum and mitochondria in nonalcoholic fatty liver disease. Lipids Health Dis. 2020, 19, 72. [Google Scholar] [CrossRef]
- Danese, A.; Patergnani, S.; Bonora, M.; Wieckowski, M.R.; Previati, M.; Giorgi, C.; Pinton, P. Calcium regulates cell death in cancer: Roles of the mitochondria and mitochondria-associated membranes (MAMs). Biochim. Biophys. Acta Bioenergy 2017, 1858, 615–627. [Google Scholar] [CrossRef]
- Delprat, B.; Maurice, T.; Delettre, C. Wolfram syndrome: MAMs’ connection? Cell Death Dis. 2018, 9, 364. [Google Scholar] [CrossRef]
- Rodriguez-Arribas, M.; Yakhine-Diop, S.; Pedro, J.; Gomez-Suaga, P.; Gomez-Sanchez, R.; Martinez-Chacon, G.; Fuentes, J.M.; Gonzalez-Polo, R.A.; Niso-Santano, M. Mitochondria-Associated Membranes (MAMs): Overview and Its Role in Parkinson’s Disease. Mol. Neurobiol. 2017, 54, 6287–6303. [Google Scholar] [CrossRef]
- Li, C.; Li, L.; Yang, M.; Zeng, L.; Sun, L. PACS-2: A key regulator of mitochondria-associated membranes (MAMs). Pharmacol. Res. 2020, 160, 105080. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, D.; Hernandez-Alvarez, M.I.; Segales, J.; Sorianello, E.; Munoz, J.P.; Sala, D.; Waget, A.; Liesa, M.; Paz, J.C.; Gopalacharyulu, P.; et al. Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 5523–5528. [Google Scholar] [CrossRef] [PubMed]
- Iwasawa, R.; Mahul-Mellier, A.L.; Datler, C.; Pazarentzos, E.; Grimm, S. Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J. 2011, 30, 556–568. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Suaga, P.; Paillusson, S.; Stoica, R.; Noble, W.; Hanger, D.P.; Miller, C. The ER-Mitochondria Tethering Complex VAPB-PTPIP51 Regulates Autophagy. Curr. Biol. 2017, 27, 371–385. [Google Scholar] [CrossRef]
- Di Mattia, T.; Wilhelm, L.P.; Ikhlef, S.; Wendling, C.; Spehner, D.; Nomine, Y.; Giordano, F.; Mathelin, C.; Drin, G.; Tomasetto, C.; et al. Identification of MOSPD2, a novel scaffold for endoplasmic reticulum membrane contact sites. EMBO Rep. 2018, 19, 7. [Google Scholar] [CrossRef]
- Myhill, N.; Lynes, E.M.; Nanji, J.A.; Blagoveshchenskaya, A.D.; Fei, H.; Carmine, S.K.; Cooper, T.J.; Thomas, G.; Simmen, T. The subcellular distribution of calnexin is mediated by PACS-2. Mol. Biol. Cell. 2008, 19, 2777–2788. [Google Scholar] [CrossRef]
- Weng, T.Y.; Tsai, S.A.; Su, T.P. Roles of sigma-1 receptors on mitochondrial functions relevant to neurodegenerative diseases. J. Biomed. Sci. 2017, 24, 74. [Google Scholar] [CrossRef]
- Gilady, S.Y.; Bui, M.; Lynes, E.M.; Benson, M.D.; Watts, R.; Vance, J.E.; Simmen, T. Ero1alpha requires oxidizing and normoxic conditions to localize to the mitochondria-associated membrane (MAM). Cell Stress Chaperones 2010, 15, 619–629. [Google Scholar] [CrossRef]
- Lewin, T.M.; Kim, J.H.; Granger, D.A.; Vance, J.E.; Coleman, R.A. Acyl-CoA synthetase isoforms 1, 4, and 5 are present in different subcellular membranes in rat liver and can be inhibited independently. J. Biol. Chem. 2001, 276, 24674–24679. [Google Scholar] [CrossRef]
- Stone, S.J.; Vance, J.E. Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J. Boil. Chem. 2000, 275, 34534–34540. [Google Scholar] [CrossRef]
- Yang, M.; Li, C.; Yang, S.; Xiao, Y.; Xiong, X.; Chen, W.; Zhao, H.; Zhang, Q.; Han, Y.; Sun, L. Mitochondria-Associated ER Membranes—The Origin Site of Autophagy. Front. Cell Dev. Biol. 2020, 8, 595. [Google Scholar] [CrossRef] [PubMed]
- Paillard, M.; Tubbs, E.; Thiebaut, P.A.; Gomez, L.; Fauconnier, J.; Da, S.C.; Teixeira, G.; Mewton, N.; Belaidi, E.; Durand, A.; et al. Depressing mitochondria-reticulum interactions protects cardiomyocytes from lethal hypoxia-reoxygenation injury. Circulation 2013, 128, 1555–1565. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, E.; Theurey, P.; Vial, G.; Bendridi, N.; Bravard, A.; Chauvin, M.A.; Ji-Cao, J.; Zoulim, F.; Bartosch, B.; Ovize, M.; et al. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 2014, 63, 3279–3294. [Google Scholar] [CrossRef] [PubMed]
- Csordas, G.; Renken, C.; Varnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnoczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Rossi, A.E.; Boncompagni, S.; Dirksen, R.T. Sarcoplasmic reticulum-mitochondrial symbiosis: Bidirectional signaling in skeletal muscle. Exerc. Sport Sci. Rev. 2009, 37, 29–35. [Google Scholar] [CrossRef]
- Szabadkai, G.; Bianchi, K.; Varnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef]
- Helle, S.C.; Kanfer, G.; Kolar, K.; Lang, A.; Michel, A.H.; Kornmann, B. Organization and function of membrane contact sites. Biochim. Biophys. Acta 2013, 1833, 2526–2541. [Google Scholar] [CrossRef]
- de Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Cerqua, C.; Anesti, V.; Pyakurel, A.; Liu, D.; Naon, D.; Wiche, G.; Baffa, R.; Dimmer, K.S.; Scorrano, L. Trichoplein/mitostatin regulates endoplasmic reticulum-mitochondria juxtaposition. EMBO Rep. 2010, 11, 854–860. [Google Scholar] [CrossRef]
- Sugiura, A.; Nagashima, S.; Tokuyama, T.; Amo, T.; Matsuki, Y.; Ishido, S.; Kudo, Y.; McBride, H.M.; Fukuda, T.; Matsushita, N.; et al. Mitol regulates endoplasmic reticulum-mitochondria contacts via Mitofusin2. Mol. Cell 2013, 51, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Simmen, T.; Aslan, J.E.; Blagoveshchenskaya, A.D.; Thomas, L.; Wan, L.; Xiang, Y.; Feliciangeli, S.F.; Hung, C.H.; Crump, C.M.; Thomas, G. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J. 2005, 24, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Betz, C.; Stracka, D.; Prescianotto-Baschong, C.; Frieden, M.; Demaurex, N.; Hall, M.N. Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc. Natl. Acad. Sci. USA 2013, 110, 12526–12534. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E. Molecular and cell biology of phosphatidylserine and phosphatidylethanolamine metabolism. Prog. Nucleic Acid. Res. Mol. Biol. 2003, 75, 69–111. [Google Scholar] [PubMed]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Walter, L.; Hajnoczky, G. Mitochondria and endoplasmic reticulum: The lethal interorganelle cross-talk. J. Bioenergy Biomembr. 2005, 37, 191–206. [Google Scholar] [CrossRef]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef]
- Xu, H.; Guan, N.; Ren, Y.L.; Wei, Q.J.; Tao, Y.H.; Yang, G.S.; Liu, X.Y.; Bu, D.F.; Zhang, Y.; Zhu, S.N. IP3R-Grp75-VDAC1-MCU calcium regulation axis antagonists protect podocytes from apoptosis and decrease proteinuria in an Adriamycin nephropathy rat model. BMC Nephrol. 2018, 19, 140. [Google Scholar] [CrossRef]
- Roderick, H.L.; Lechleiter, J.D.; Camacho, P. Cytosolic phosphorylation of calnexin controls intracellular Ca(2+) oscillations via an interaction with SERCA2b. J. Cell. Biol. 2000, 149, 1235–1248. [Google Scholar] [CrossRef]
- Yu, S.; Zhang, L.; Liu, C.; Yang, J.; Zhang, J.; Huang, L. PACS2 is required for ox-LDL-induced endothelial cell apoptosis by regulating mitochondria-associated ER membrane formation and mitochondrial Ca(2+) elevation. Exp. Cell Res. 2019, 379, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Kuo, I.Y.; Brill, A.L.; Lemos, F.O.; Jiang, J.Y.; Falcone, J.L.; Kimmerling, E.P.; Cai, Y.; Dong, K.; Kaplan, D.L.; Wallace, D.P.; et al. Polycystin 2 regulates mitochondrial Ca(2+) signaling, bioenergetics, and dynamics through mitofusin 2. Sci. Signal. 2019, 12, 580. [Google Scholar] [CrossRef] [PubMed]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proc. Natl. Acad. Sci. USA 2015, 112, E2174–E2181. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Suaga, P.; Paillusson, S.; Miller, C. ER-mitochondria signaling regulates autophagy. Autophagy 2017, 13, 1250–1251. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, S.; Iossa, S.; Venditti, P. Skeletal muscle insulin resistance: Role of mitochondria and other ROS sources. J. Endocrinol. 2017, 233, R15–R42. [Google Scholar] [CrossRef]
- Mannella, C.A.; Buttle, K.; Rath, B.K.; Marko, M. Electron microscopic tomography of rat-liver mitochondria and their interaction with the endoplasmic reticulum. BioFactors 1998, 8, 225–228. [Google Scholar] [CrossRef]
- Theurey, P.; Rieusset, J. Mitochondria-Associated Membranes Response to Nutrient Availability and Role in Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 32–45. [Google Scholar] [CrossRef]
- Rieusset, J. Contribution of mitochondria and endoplasmic reticulum dysfunction in insulin resistance: Distinct or interrelated roles? Diabetes Metab. 2015, 41, 358–368. [Google Scholar] [CrossRef]
- Arruda, A.P.; Pers, B.M.; Parlakgul, G.; Guney, E.; Inouye, K.; Hotamisligil, G.S. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat. Med. 2014, 20, 1427–1435. [Google Scholar] [CrossRef]
- Tubbs, E.; Chanon, S.; Robert, M.; Bendridi, N.; Bidaux, G.; Chauvin, M.A.; Ji-Cao, J.; Durand, C.; Gauvrit-Ramette, D.; Vidal, H.; et al. Disruption of Mitochondria-Associated Endoplasmic Reticulum Membrane (MAM) Integrity Contributes to Muscle Insulin Resistance in Mice and Humans. Diabetes 2018, 67, 636–650. [Google Scholar] [CrossRef]
- Theurey, P.; Tubbs, E.; Vial, G.; Jacquemetton, J.; Bendridi, N.; Chauvin, M.A.; Alam, M.R.; Le Romancer, M.; Vidal, H.; Rieusset, J. Mitochondria-associated endoplasmic reticulum membranes allow adaptation of mitochondrial metabolism to glucose availability in the liver. J. Mol. Cell Biol. 2016, 8, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Gan, K.X.; Wang, C.; Chen, J.H.; Zhu, C.J.; Song, G.Y. Mitofusin-2 ameliorates high-fat diet-induced insulin resistance in liver of rats. World J. Gastroenterol. 2013, 19, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Schneeberger, M.; Dietrich, M.O.; Sebastian, D.; Imbernon, M.; Castano, C.; Garcia, A.; Esteban, Y.; Gonzalez-Franquesa, A.; Rodriguez, I.C.; Bortolozzi, A.; et al. Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell 2013, 155, 172–187. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Chen, Y.F.; Wu, C.Y.; Wu, P.C.; Huang, Y.L.; Kao, C.H.; Lin, C.H.; Kao, L.S.; Tsai, T.F.; Wei, Y.H. Cisd2 modulates the differentiation and functioning of adipocytes by regulating intracellular Ca2+ homeostasis. Hum. Mol. Genet. 2014, 23, 4770–4785. [Google Scholar] [CrossRef]
- Destefano, M.A.; Jacinto, E. Regulation of insulin receptor substrate-1 by mTORC2 (mammalian target of rapamycin complex 2). Biochem. Soc. Trans. 2013, 41, 896–901. [Google Scholar] [CrossRef]
- De Palma, C.; Falcone, S.; Pisoni, S.; Cipolat, S.; Panzeri, C.; Pambianco, S.; Pisconti, A.; Allevi, R.; Bassi, M.T.; Cossu, G.; et al. Nitric oxide inhibition of Drp1-mediated mitochondrial fission is critical for myogenic differentiation. Cell Death Differ. 2010, 17, 1684–1696. [Google Scholar] [CrossRef]
- Nisoli, E.; Falcone, S.; Tonello, C.; Cozzi, V.; Palomba, L.; Fiorani, M.; Pisconti, A.; Brunelli, S.; Cardile, A.; Francolini, M.; et al. Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc. Natl. Acad. Sci. USA 2004, 101, 16507–16512. [Google Scholar] [CrossRef]
- Bassot, A.; Chauvin, M.A.; Bendridi, N.; Ji-Cao, J.; Vial, G.; Monnier, L.; Bartosch, B.; Alves, A.; Cottet-Rousselle, C.; Gouriou, Y.; et al. Regulation of Mitochondria-Associated Membranes (MAMs) by NO/sGC/PKG Participates in the Control of Hepatic Insulin Response. Cells 2019, 8, 1319. [Google Scholar] [CrossRef]
- Lim, J.H.; Lee, H.J.; Ho, J.M.; Song, J. Coupling mitochondrial dysfunction to endoplasmic reticulum stress response: A molecular mechanism leading to hepatic insulin resistance. Cell Signal. 2009, 21, 169–177. [Google Scholar] [CrossRef]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef]
- Szado, T.; Vanderheyden, V.; Parys, J.B.; De Smedt, H.; Rietdorf, K.; Kotelevets, L.; Chastre, E.; Khan, F.; Landegren, U.; Soderberg, O.; et al. Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 2427–2432. [Google Scholar] [CrossRef] [PubMed]
- Jeoung, N.H.; Harris, R.A. Pyruvate dehydrogenase kinase-4 deficiency lowers blood glucose and improves glucose tolerance in diet-induced obese mice. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E46–E54. [Google Scholar] [CrossRef] [PubMed]
- Thoudam, T.; Ha, C.M.; Leem, J.; Chanda, D.; Park, J.S.; Kim, H.J.; Jeon, J.H.; Choi, Y.K.; Liangpunsakul, S.; Huh, Y.H.; et al. PDK4 Augments ER-Mitochondria Contact to Dampen Skeletal Muscle Insulin Signaling during Obesity. Diabetes 2019, 68, 571–586. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Ito, K.; Lin, H.K.; Santangelo, C.; Wieckowski, M.R.; Lebiedzinska, M.; Bononi, A.; Bonora, M.; Duszynski, J.; Bernardi, R.; et al. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science 2010, 330, 1247–1251. [Google Scholar] [CrossRef] [PubMed]
- Filadi, R.; Theurey, P.; Pizzo, P. The endoplasmic reticulum-mitochondria coupling in health and disease: Molecules, functions and significance. Cell Calcium. 2017, 62, 1–15. [Google Scholar] [CrossRef]
- Smith, R.L.; Soeters, M.R.; Wust, R.; Houtkooper, R.H. Metabolic Flexibility as an Adaptation to Energy Resources and Requirements in Health and Disease. Endocr. Rev. 2018, 39, 489–517. [Google Scholar] [CrossRef]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef]
- Ahn, B.H.; Kim, H.S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.X.; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452. [Google Scholar] [CrossRef]
- Jeninga, E.H.; Schoonjans, K.; Auwerx, J. Reversible acetylation of PGC-1: Connecting energy sensors and effectors to guarantee metabolic flexibility. Oncogene 2010, 29, 4617–4624. [Google Scholar] [CrossRef]
- Kim, M.K.; Yang, S.; Lee, K.H.; Um, J.H.; Liu, M.; Kang, H.; Park, S.J.; Chung, J.H. Promyelocytic leukemia inhibits adipogenesis, and loss of promyelocytic leukemia results in fat accumulation in mice. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E1130–E1142. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kitamura, Y.I.; Kitamura, T.; Kruse, J.P.; Raum, J.C.; Stein, R.; Gu, W.; Accili, D. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab. 2005, 2, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Guo, S.; Liu, Y.; Chu, H.; Hakimi, P.; Berger, N.A.; Hanson, R.W.; Kao, H.Y. Ablation of promyelocytic leukemia protein (PML) re-patterns energy balance and protects mice from obesity induced by a Western diet. J. Biol. Chem. 2013, 288, 29746–29759. [Google Scholar] [CrossRef] [PubMed]
- Wojtaszewski, J.F.P.; Hansen, B.F.; Gade, J.; Kiens, B.; Markuns, J.F.; Goodyear, L.J.; Richter, E.A. Insulin signaling and insulin sensitivity after exercise in human skeletal muscle. Diabetes 2000, 49, 325–331. [Google Scholar] [CrossRef]
- Ding, C.; Chooi, Y.; Chan, Z.; Lo, J.; Choo, J.; Ding, B.; Leow, M.K.; Magkos, F. Dose-Dependent Effects of Exercise and Diet on Insulin Sensitivity and Secretion. Med. Sci. Sports Exerc. 2019, 51, 2109–2116. [Google Scholar] [CrossRef] [PubMed]
- Colberg, S.R.; Sigal, R.J.; Fernhall, B.; Regensteiner, J.G.; Blissmer, B.J.; Rubin, R.R.; Chasan-Taber, L.; Albright, A.L.; Braun, B. Exercise and type 2 diabetes: The American College of Sports Medicine and the American Diabetes Association: Joint position statement. Diabetes Care 2010, 33, e147–e167. [Google Scholar] [CrossRef]
- Motahari-Tabari, N.; Ahmad, S.M.; Shirzad-E-Ahoodashty, M.; Yousefi-Abdolmaleki, E.; Teimourzadeh, M. The effect of 8 weeks aerobic exercise on insulin resistance in type 2 diabetes: A randomized clinical trial. Glob. J. Health Sci. 2014, 7, 115–121. [Google Scholar] [CrossRef]
- Marson, E.C.; Delevatti, R.S.; Prado, A.K.; Netto, N.; Kruel, L.F. Effects of aerobic, resistance, and combined exercise training on insulin resistance markers in overweight or obese children and adolescents: A systematic review and meta-analysis. Prev. Med. 2016, 93, 211–218. [Google Scholar] [CrossRef]
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef]
- Scheele, C.; Nielsen, A.R.; Walden, T.B.; Sewell, D.A.; Fischer, C.P.; Brogan, R.J.; Petrovic, N.; Larsson, O.; Tesch, P.A.; Wennmalm, K.; et al. Altered regulation of the PINK1 locus: A link between type 2 diabetes and neurodegeneration? FASEB J. 2007, 21, 3653–3665. [Google Scholar] [CrossRef]
- Kim, K.H.; Jeong, Y.T.; Oh, H.; Kim, S.H.; Cho, J.M.; Kim, Y.N.; Kim, S.S.; Kim, D.H.; Hur, K.Y.; Kim, H.K.; et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat. Med. 2013, 19, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Bishop, D.J.; Granata, C.; Eynon, N. Can we optimise the exercise training prescription to maximise improvements in mitochondria function and content? Biochim. Biophys. Acta 2014, 1840, 1266–1275. [Google Scholar] [CrossRef]
- De Strijcker, D.; Lapauw, B.; Ouwens, D.M.; Van de Velde, D.; Hansen, D.; Petrovic, M.; Cuvelier, C.; Tonoli, C.; Calders, P. High intensity interval training is associated with greater impact on physical fitness, insulin sensitivity and muscle mitochondrial content in males with overweight/obesity, as opposed to continuous endurance training: A randomized controlled trial. J. Musculoskelet. Neuronal. Interact. 2018, 18, 215–226. [Google Scholar] [PubMed]
- Di Meo, S.; Iossa, S.; Venditti, P. Improvement of obesity-linked skeletal muscle insulin resistance by strength and endurance training. J. Endocrinol. 2017, 234, R159–R181. [Google Scholar] [CrossRef] [PubMed]
- Pesta, D.; Hoppel, F.; Macek, C.; Messner, H.; Faulhaber, M.; Kobel, C.; Parson, W.; Burtscher, M.; Schocke, M.; Gnaiger, E. Similar qualitative and quantitative changes of mitochondrial respiration following strength and endurance training in normoxia and hypoxia in sedentary humans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1078–R1087. [Google Scholar] [CrossRef] [PubMed]
- Sparks, L.M.; Johannsen, N.M.; Church, T.S.; Earnest, C.P.; Moonen-Kornips, E.; Moro, C.; Hesselink, M.K.; Smith, S.R.; Schrauwen, P. Nine months of combined training improves ex vivo skeletal muscle metabolism in individuals with type 2 diabetes. J. Clin. Endocrinol. Metab. 2013, 98, 1694–1702. [Google Scholar] [CrossRef]
- Ruegsegger, G.N.; Vanderboom, P.M.; Dasari, S.; Klaus, K.A.; Kabiraj, P.; McCarthy, C.B.; Lucchinetti, C.F.; Nair, K.S. Exercise and metformin counteract altered mitochondrial function in the insulin-resistant brain. JCI Insight 2019, 4, 18. [Google Scholar] [CrossRef]
- Zhang, K.; Kaufman, R.J. The unfolded protein response: A stress signaling pathway critical for health and disease. Neurology 2006, 66 (Suppl. 1), S102–S109. [Google Scholar] [CrossRef]
- Szegezdi, E.; Fitzgerald, U.; Samali, A. Caspase-12 and ER-stress-mediated apoptosis: The story so far. Ann. N. Y. Acad. Sci. 2003, 1010, 186–194. [Google Scholar] [CrossRef]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, G.; Zhang, H.; Karin, M.; Bai, H.; Cai, D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 2008, 135, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Da, L.G.; Frederico, M.J.; Da, S.S.; Vitto, M.F.; Cesconetto, P.A.; de Pinho, R.A.; Pauli, J.R.; Silva, A.S.; Cintra, D.E.; Ropelle, E.R.; et al. Endurance exercise training ameliorates insulin resistance and reticulum stress in adipose and hepatic tissue in obese rats. Eur. J. Appl. Physiol. 2011, 111, 2015–2023. [Google Scholar]
- Deldicque, L.; Cani, P.D.; Delzenne, N.M.; Baar, K.; Francaux, M. Endurance training in mice increases the unfolded protein response induced by a high-fat diet. J. Physiol. Biochem. 2013, 69, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Ropelle, E.R.; Flores, M.B.; Cintra, D.E.; Rocha, G.Z.; Pauli, J.R.; Morari, J.; de Souza, C.T.; Moraes, J.C.; Prada, P.O.; Guadagnini, D.; et al. IL-6 and IL-10 anti-inflammatory activity links exercise to hypothalamic insulin and leptin sensitivity through IKKbeta and ER stress inhibition. PLoS Biol. 2010, 8. [Google Scholar] [CrossRef]
- Lee, S.S.; Yoo, J.H.; So, Y.S. Effect of the low- versus high-intensity exercise training on endoplasmic reticulum stress and GLP-1 in adolescents with type 2 diabetes mellitus. J. Phys. Ther. Sci. 2015, 27, 3063–3068. [Google Scholar] [CrossRef]
- Wang, S.; Kaufman, R.J. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012, 197, 857–867. [Google Scholar] [CrossRef]
- Khadir, A.; Kavalakatt, S.; Abubaker, J.; Cherian, P.; Madhu, D.; Al-Khairi, I.; Abu-Farha, M.; Warsame, S.; Elkum, N.; Dehbi, M.; et al. Physical exercise alleviates ER stress in obese humans through reduction in the expression and release of GRP78 chaperone. Metabolism 2016, 65, 1409–1420. [Google Scholar] [CrossRef]
- Merle, A.; Jollet, M.; Britto, F.A.; Goustard, B.; Bendridi, N.; Rieusset, J.; Ollendorff, V.; Favier, F.B. Endurance exercise decreases protein synthesis and ER-mitochondria contacts in mouse skeletal muscle. J. Appl. Physiol. 2019, 127, 1297–1306. [Google Scholar] [CrossRef]
- Flis, D.J.; Dzik, K.; Kaczor, J.J.; Halon-Golabek, M.; Antosiewicz, J.; Wieckowski, M.R.; Ziolkowski, W. Swim Training Modulates Skeletal Muscle Energy Metabolism, Oxidative Stress, and Mitochondrial Cholesterol Content in Amyotrophic Lateral Sclerosis Mice. Oxid. Med. Cell. Longev. 2018, 2018, 5940748. [Google Scholar] [CrossRef]
- Mooren, F.C.; Lechtermann, A.; Fromme, A.; Thorwesten, L.; Volker, K. Alterations in intracellular calcium signaling of lymphocytes after exhaustive exercise. Med. Sci. Sports Exerc. 2001, 33, 242–248. [Google Scholar] [CrossRef]
- Cheng, K.K.Y.; Lam, K.S.L.; Wang, B.; Xu, A. Signaling mechanisms underlying the insulin-sensitizing effects of adiponectin. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Fan, W.; Kruger, K.; Xiao, Y.U.; Pilat, C.; Seimetz, M.; Ringseis, R.; Baumgart-Vogt, E.; Eder, K.; Weissmann, N.; et al. Exercise Affects T-Cell Function by Modifying Intracellular Calcium Homeostasis. Med. Sci. Sports Exerc. 2017, 49, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Palee, S.; Minta, W.; Mantor, D.; Sutham, W.; Jaiwongkam, T.; Kerdphoo, S.; Pratchayasakul, W.; Chattipakorn, S.C.; Chattipakorn, N. Combination of exercise and calorie restriction exerts greater efficacy on cardioprotection than monotherapy in obese-insulin resistant rats through the improvement of cardiac calcium regulation. Metabolism 2019, 94, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Funai, K.; Lodhi, I.J.; Spears, L.D.; Yin, L.; Song, H.; Klein, S.; Semenkovich, C.F. Skeletal Muscle Phospholipid Metabolism Regulates Insulin Sensitivity and Contractile Function. Diabetes 2016, 65, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Venojarvi, M.; Aunola, S.; Puhke, R.; Marniemi, J.; Hamalainen, H.; Halonen, J.P.; Lindstrom, J.; Rastas, M.; Hallsten, K.; Nuutila, P.; et al. Exercise training with dietary counselling increases mitochondrial chaperone expression in middle-aged subjects with impaired glucose tolerance. BMC Endocr. Disord. 2008, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Lappalainen, Z.; Lappalainen, J.; Oksala, N.K.; Laaksonen, D.E.; Khanna, S.; Sen, C.K.; Atalay, M. Exercise training and experimental diabetes modulate heat shock protein response in brain. Scand. J. Med. Sci. Sports 2010, 20, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Veeranki, S.; Givvimani, S.; Kundu, S.; Metreveli, N.; Pushpakumar, S.; Tyagi, S.C. Moderate intensity exercise prevents diabetic cardiomyopathy associated contractile dysfunction through restoration of mitochondrial function and connexin 43 levels in db/db mice. J. Mol. Cell. Cardiol. 2016, 92, 163–173. [Google Scholar] [CrossRef]
- Heo, J.W.; No, M.H.; Park, D.H.; Kang, J.H.; Seo, D.Y.; Han, J.; Neufer, P.D.; Kwak, H.B. Effects of exercise on obesity-induced mitochondrial dysfunction in skeletal muscle. Korean J. Physiol. Pharmacol. 2017, 21, 567–577. [Google Scholar] [CrossRef]
- McLelland, G.L.; Goiran, T.; Yi, W.; Dorval, G.; Chen, C.X.; Lauinger, N.D.; Krahn, A.I.; Valimehr, S.; Rakovic, A.; Rouiller, I.; et al. Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. eLife 2018, 7, e32866. [Google Scholar] [CrossRef]
- Greene, N.P.; Lee, D.E.; Brown, J.L.; Rosa, M.E.; Brown, L.A.; Perry, R.A.; Henry, J.N.; Washington, T.A. Mitochondrial quality control, promoted by PGC-1alpha, is dysregulated by Western diet-induced obesity and partially restored by moderate physical activity in mice. Physiol. Rep. 2015, 3, e12470. [Google Scholar] [CrossRef]
- Yan, Z.; Lira, V.A.; Greene, N.P. Exercise training-induced regulation of mitochondrial quality. Exerc. Sport Sci. Rev. 2012, 40, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Yang, B.; Ren, C.; Fu, J.; Zhang, J. Swimming Exercise Alleviated Insulin Resistance by Regulating Tripartite Motif Family Protein 72 Expression and AKT Signal Pathway in Sprague-Dawley Rats Fed with High-Fat Diet. J. Diabetes Res. 2016, 2016, 1564386. [Google Scholar] [CrossRef]
- Kido, K.; Ato, S.; Yokokawa, T.; Sato, K.; Fujita, S. Resistance training recovers attenuated APPL1 expression and improves insulin-induced Akt signal activation in skeletal muscle of type 2 diabetic rats. Am. J. Physiol. Metab. 2018, 314, E564–E571. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, T.; Shinozaki, S.; Nakamoto, H.; Kaneki, M.; Goto, S.; Shimokado, K.; Kobayashi, H.; Naito, H. Voluntary Exercise Can Ameliorate Insulin Resistance by Reducing iNOS-Mediated S-Nitrosylation of Akt in the Liver in Obese Rats. PLoS ONE 2015, 10, e0132029. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Herzig, S.; Kulkarni, R.N.; Montminy, M. TRB3: A tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science 2003, 300, 1574–1577. [Google Scholar] [CrossRef]
- Koh, H.J.; Toyoda, T.; Didesch, M.M.; Lee, M.Y.; Sleeman, M.W.; Kulkarni, R.N.; Musi, N.; Hirshman, M.F.; Goodyear, L.J. Tribbles 3 mediates endoplasmic reticulum stress-induced insulin resistance in skeletal muscle. Nat. Commun. 2013, 4, 1871. [Google Scholar] [CrossRef] [PubMed]
- Marinho, R.; Mekary, R.A.; Munoz, V.R.; Gomes, R.J.; Pauli, J.R.; de Moura, L.P. Regulation of hepatic TRB3/Akt interaction induced by physical exercise and its effect on the hepatic glucose production in an insulin resistance state. Diabetol. Metab. Syndr. 2015, 7, 67. [Google Scholar] [CrossRef]
- Marcotte, G.R.; West, D.W.; Baar, K. The molecular basis for load-induced skeletal muscle hypertrophy. Calcif. Tissue Int. 2015, 96, 196–210. [Google Scholar] [CrossRef]
- Kleinert, M.; Parker, B.L.; Fritzen, A.M.; Knudsen, J.R.; Jensen, T.E.; Kjobsted, R.; Sylow, L.; Ruegg, M.; James, D.E.; Richter, E.A. Mammalian target of rapamycin complex 2 regulates muscle glucose uptake during exercise in mice. J. Physiol. 2017, 595, 4845–4855. [Google Scholar] [CrossRef]
- Bae, J.Y.; Shin, K.O.; Woo, J.; Woo, S.H.; Jang, K.S.; Lee, Y.H.; Kang, S. Exercise and dietary change ameliorate high fat diet induced obesity and insulin resistance via mTOR signaling pathway. J. Exerc. Nutr. Biochem. 2016, 20, 28–33. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; Ding, S. ER–Mitochondria Contacts and Insulin Resistance Modulation through Exercise Intervention. Int. J. Mol. Sci. 2020, 21, 9587. https://doi.org/10.3390/ijms21249587
Sun Y, Ding S. ER–Mitochondria Contacts and Insulin Resistance Modulation through Exercise Intervention. International Journal of Molecular Sciences. 2020; 21(24):9587. https://doi.org/10.3390/ijms21249587
Chicago/Turabian StyleSun, Yi, and Shuzhe Ding. 2020. "ER–Mitochondria Contacts and Insulin Resistance Modulation through Exercise Intervention" International Journal of Molecular Sciences 21, no. 24: 9587. https://doi.org/10.3390/ijms21249587
APA StyleSun, Y., & Ding, S. (2020). ER–Mitochondria Contacts and Insulin Resistance Modulation through Exercise Intervention. International Journal of Molecular Sciences, 21(24), 9587. https://doi.org/10.3390/ijms21249587