Tyrosine Phosphorylation of the Kv2.1 Channel Contributes to Injury in Brain Ischemia


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
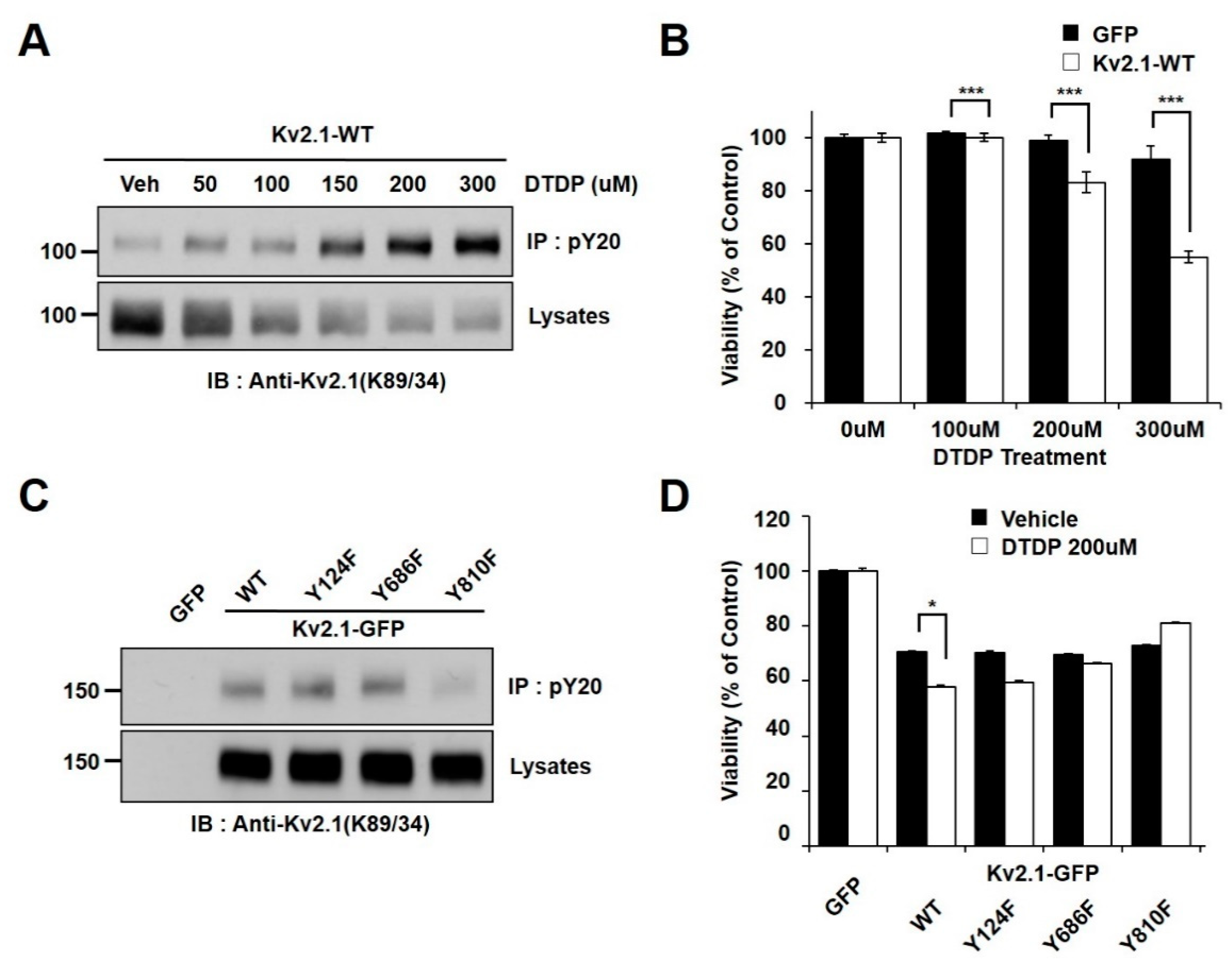
2.1. Oxidative Stress Induces Tyrosine Phosphorylation of Kv2.1
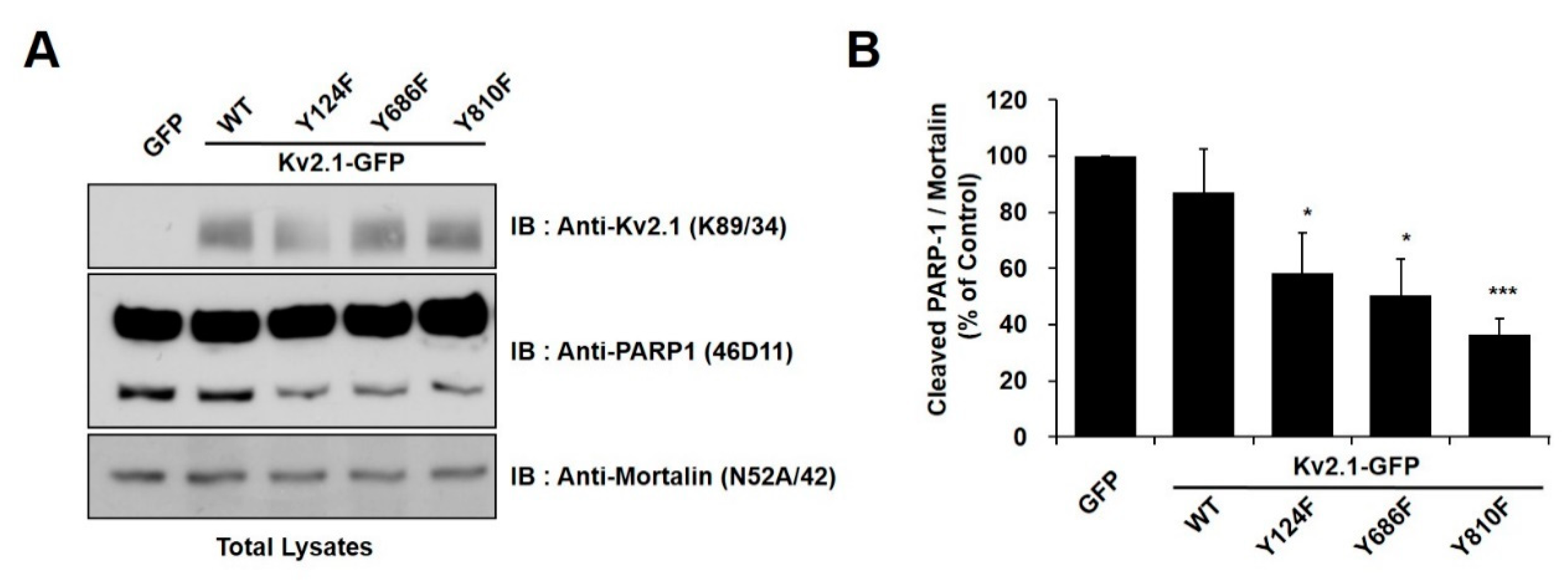
2.2. Regulation of Apoptotic Cell Death by Tyrosine Phosphorylation of Kv2.1
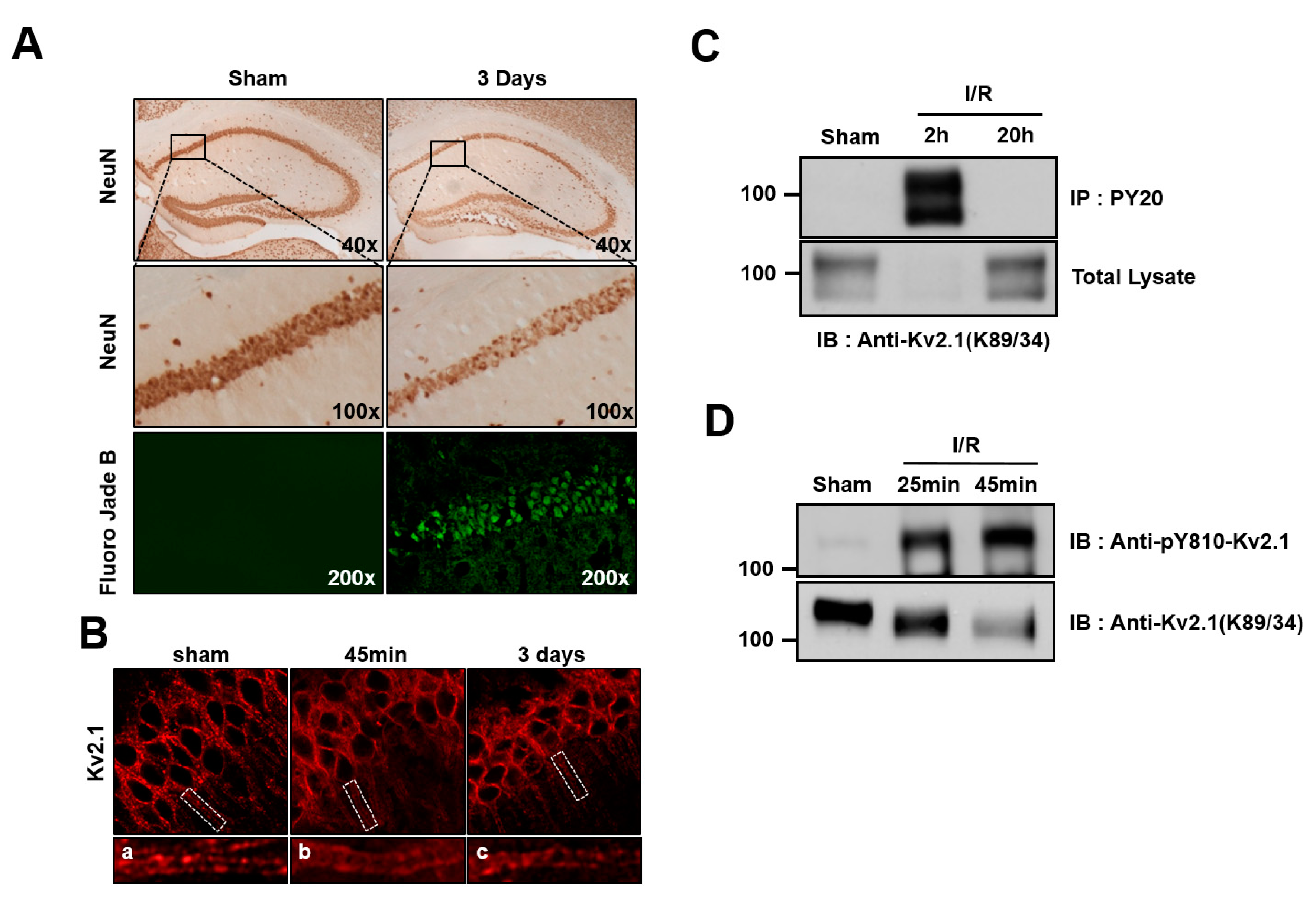
2.3. Brain Ischemia Results in Tyrosine Phosphorylation of the Kv2.1 Channel
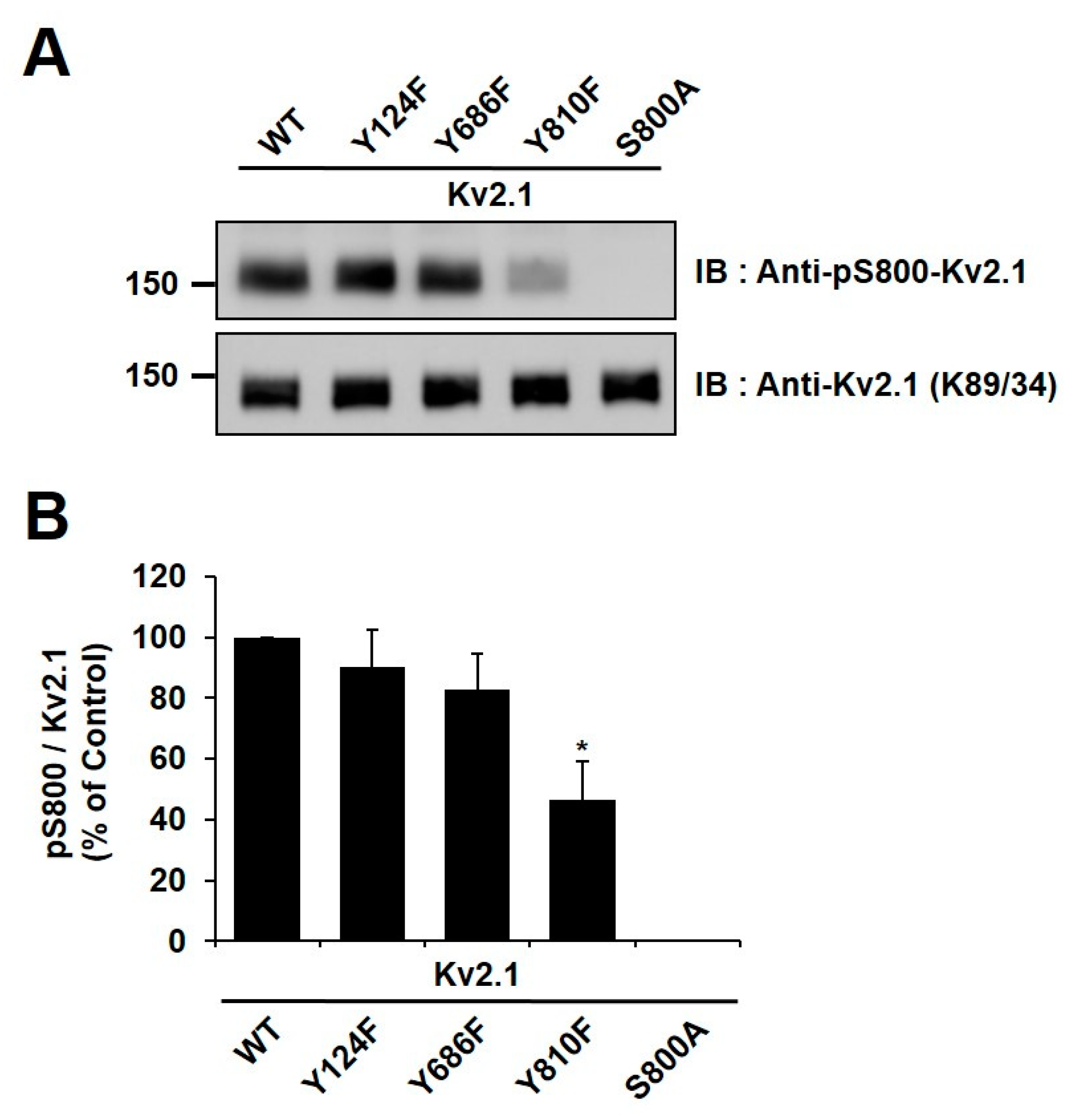
2.4. Y810 Phosphorylation Affects p38-Mediated Phosphorylation of Kv2.1 at S800
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Transient Transfection
4.2. Immunoblotting
4.3. Animals
4.4. Phospho-Specific Antibodies
4.5. Immunoprecipitation
4.6. Two-Vessel Occlusion
4.7. Four-Vessel Occlusion
4.8. Immunohistochemistry
4.9. Cell Viability
4.10. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Kv2.1 | Voltage-dependent potassium channel Kv2.1 |
| mAb | Monoclonal antibody |
| RBM | Rat brain membrane |
| DTDP | 2,2′-dithiodipyridine |
| WST-1 | Ez-Cytox reagent |
| SFK | Src kinase family kinases |
References
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Mehta, S.L.; Kaimal, B.; Lyons, K.; Dempsey, R.J.; Vemuganti, R. Poststroke Induction of alpha-Synuclein Mediates Ischemic Brain Damage. J. Neurosci. 2016, 36, 7055–7065. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhang, H.; Xie, L.; Qian, C.; Ye, Y.; Mao, H.; Wang, B.; Zhang, H.; Zhang, Y.; He, X.; et al. Tristetraprolin destabilizes NOX2 mRNA and protects dopaminergic neurons from oxidative damage in Parkinson’s disease. FASEB J. 2020, 34, 15047–15061. [Google Scholar] [CrossRef] [PubMed]
- Okouchi, M.; Ekshyyan, O.; Maracine, M.; Aw, T.Y. Neuronal apoptosis in neurodegeneration. Antioxid. Redox Signal. 2007, 9, 1059–1096. [Google Scholar] [CrossRef]
- Misonou, H.; Thompson, S.M.; Cai, X. Dynamic regulation of the Kv2. 1 voltage-gated potassium channel during brain ischemia through neuroglial interaction. J. Neurosci. 2008, 28, 8529–8538. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.P. Regulation and critical role of potassium homeostasis in apoptosis. Prog. Neurobiol. 2003, 70, 363–386. [Google Scholar] [CrossRef]
- Al-Owais, M.M.; Scragg, J.L.; Dallas, M.L.; Boycott, H.E.; Warburton, P.; Chakrabarty, A.; Boyle, J.P.; Peers, C. Carbon monoxide mediates the anti-apoptotic effects of heme oxygenase-1 in medulloblastoma DAOY cells via K+ channel inhibition. J. Biol. Chem. 2012, 287, 24754–24764. [Google Scholar] [CrossRef]
- Yu, S.P.; Yeh, C.-H.; Sensi, S.L.; Gwag, B.J.; Canzoniero, L.M.; Farhangrazi, Z.S.; Ying, H.S.; Tian, M.; Dugan, L.L.; Choi, D.W. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science 1997, 278, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Redman, P.T.; He, K.; Hartnett, K.A.; Jefferson, B.S.; Hu, L.; Rosenberg, P.A.; Levitan, E.S.; Aizenman, E. Apoptotic surge of potassium currents is mediated by p38 phosphorylation of Kv2. 1. Proc. Natl. Acad. Sci. USA 2007, 104, 3568–3573. [Google Scholar] [CrossRef]
- Vacher, H.; Mohapatra, D.P.; Trimmer, J.S. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol. Rev. 2008, 88, 1407–1447. [Google Scholar] [CrossRef]
- Sarmiere, P.D.; Weigle, C.M.; Tamkun, M.M. The Kv2. 1 K+ channel targets to the axon initial segment of hippocampal and cortical neurons in culture and in situ. BMC Neurosci. 2008, 9, 112. [Google Scholar] [CrossRef] [PubMed]
- Park, K.-S.; Mohapatra, D.P.; Misonou, H.; Trimmer, J.S. Graded regulation of the Kv2. 1 potassium channel by variable phosphorylation. Science 2006, 313, 976–979. [Google Scholar] [CrossRef]
- Aras, M.A.; Saadi, R.A.; Aizenman, E. Zn2+ regulates Kv2. 1 voltage-dependent gating and localization following ischemia. Eur. J. Neurosci. 2009, 30, 2250–2257. [Google Scholar] [CrossRef] [PubMed]
- Cerda, O.; Trimmer, J.S. Activity-dependent phosphorylation of neuronal Kv2. 1 potassium channels by CDK5. J. Biol. Chem. 2011, 286, 28738–28748. [Google Scholar] [CrossRef] [PubMed]
- Tiran, Z.; Peretz, A.; Attali, B.; Elson, A. Phosphorylation-dependent regulation of Kv2. 1 channel activity at tyrosine 124 by Src and by protein-tyrosine phosphatase ε. J. Biol. Chem. 2003, 278, 17509–17514. [Google Scholar] [CrossRef] [PubMed]
- Song, M.-Y.; Hong, C.; Bae, S.H.; So, I.; Park, K.-S. Dynamic modulation of the Kv2. 1 channel by SRC-dependent tyrosine phosphorylation. J. Proteome Res. 2011, 11, 1018–1026. [Google Scholar] [CrossRef]
- Yeh, C.Y.; Schulien, A.J.; Molyneaux, B.J.; Aizenman, E. Lessons from Recent Advances in Ischemic Stroke Management and Targeting Kv2.1 for Neuroprotection. Int. J. Mol. Sci. 2020, 21, 6107. [Google Scholar] [CrossRef]
- He, K.; McCord, M.C.; Hartnett, K.A.; Aizenman, E. Regulation of Pro-Apoptotic Phosphorylation of Kv2.1 K+ Channels. PLoS ONE 2015, 10, e0129498. [Google Scholar] [CrossRef]
- Wu, X.; Hernandez-Enriquez, B.; Banas, M.; Xu, R.; Sesti, F. Molecular mechanisms underlying the apoptotic effect of KCNB1 K+ channel oxidation. J. Biol Chem. 2013, 288, 4128–4134. [Google Scholar] [CrossRef]
- Song, M.Y.; Moon, Y.J.; Shin, S.K.; Kim, T.Y.; Yune, T.Y.; Park, K.S. Contribution of the delayed-rectifier potassium channel Kv2.1 to acute spinal cord injury in rats. BMB Rep. 2010, 43, 756–760. [Google Scholar] [CrossRef]
- Misonou, H.; Mohapatra, D.P.; Park, E.W.; Leung, V.; Zhen, D.; Misonou, K.; Anderson, A.E.; Trimmer, J.S. Regulation of ion channel localization and phosphorylation by neuronal activity. Nat. Neurosci. 2004, 7, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Redman, P.T.; Hartnett, K.A.; Aras, M.A.; Levitan, E.S.; Aizenman, E. Regulation of apoptotic potassium currents by coordinated zinc-dependent signalling. J. Physiol. 2009, 587, 4393–4404. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.H.; Zhang, R.; Sun, F.Y. Enhancement of ischemia-induced tyrosine phosphorylation of Kv1. 2 by vascular endothelial growth factor via activation of phosphatidylinositol 3-kinase. J. Neurochem. 2003, 87, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- McCord, M.C.; Aizenman, E. Convergent Ca2+ and Zn2+ signaling regulates apoptotic Kv2. 1 K+ currents. Proc. Natl. Acad. Sci. USA 2013, 110, 13988–13993. [Google Scholar] [CrossRef] [PubMed]
- Misonou, H.; Mohapatra, D.P.; Menegola, M.; Trimmer, J.S. Calcium- and metabolic state-dependent modulation of the voltage-dependent Kv2.1 channel regulates neuronal excitability in response to ischemia. J. Neurosci. 2005, 25, 11184–11193. [Google Scholar] [CrossRef] [PubMed]
- Galasso, S.L.; Dyck, R.H. The role of zinc in cerebral ischemia. Mol. Med. 2007, 13, 380–387. [Google Scholar] [CrossRef]
- Zhu, Q.J.; Kong, F.S.; Xu, H.; Wang, Y.; Du, C.P.; Sun, C.C.; Liu, Y.; Li, T.; Hou, X.Y. Tyrosine phosphorylation of GluK2 up-regulates kainate receptor-mediated responses and downstream signaling after brain ischemia. Proc. Natl. Acad. Sci. USA 2014, 111, 13990–13995. [Google Scholar] [CrossRef]
- Du, C.P.; Tan, R.; Hou, X.Y. Fyn Kinases Play a Critical Role in Neuronal Apoptosis Induced by Oxygen and Glucose Deprivation or Amyloid-β Peptide Treatment. CNS Neurosci. Ther. 2012, 18, 754–761. [Google Scholar] [CrossRef]
- Knox, R.; Brennan-Minnella, A.M.; Lu, F.; Yang, D.; Nakazawa, T.; Yamamoto, T.; Swanson, R.A.; Ferriero, D.M.; Jiang, X. NR2B Phosphorylation at Tyrosine 1472 Contributes to Brain Injury in a Rodent Model of Neonatal Hypoxia-Ischemia. Stroke 2014, 45, 3040–3047. [Google Scholar] [CrossRef]
- Wang, W.-W.; Hu, S.-Q.; Li, C.; Zhou, C.; Qi, S.-H.; Zhang, G.-Y. Transduced PDZ1 domain of PSD-95 decreases Src phosphorylation and increases nNOS (Ser847) phosphorylation contributing to neuroprotection after cerebral ischemia. Brain Res. 2010, 1328, 162–170. [Google Scholar] [CrossRef]
- Cao, H.; Sanguinetti, A.R.; Mastick, C.C. Oxidative stress activates both Src-kinases and their negative regulator Csk and induces phosphorylation of two targeting proteins for Csk: Caveolin-1 and paxillin. Exp. Cell Res. 2004, 294, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Martens, J.R.; Navarro-Polanco, R.; Coppock, E.A.; Nishiyama, A.; Parshley, L.; Grobaski, T.D.; Tamkun, M.M. Differential targeting of Shaker-like potassium channels to lipid rafts. J. Biol. Chem. 2000, 275, 7443–7446. [Google Scholar] [CrossRef] [PubMed]
- Aras, M.A.; Aizenman, E. Obligatory role of ASK1 in the apoptotic surge of K+ currents. Neurosci. Lett. 2005, 387, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Schmued, L.C.; Hopkins, K.J. Fluoro-Jade B: A high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000, 874, 123–130. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, M.-Y.; Hwang, J.Y.; Bae, E.J.; Kim, S.; Kang, H.-M.; Kim, Y.J.; Park, C.; Park, K.-S. Tyrosine Phosphorylation of the Kv2.1 Channel Contributes to Injury in Brain Ischemia. Int. J. Mol. Sci. 2020, 21, 9538. https://doi.org/10.3390/ijms21249538
Song M-Y, Hwang JY, Bae EJ, Kim S, Kang H-M, Kim YJ, Park C, Park K-S. Tyrosine Phosphorylation of the Kv2.1 Channel Contributes to Injury in Brain Ischemia. International Journal of Molecular Sciences. 2020; 21(24):9538. https://doi.org/10.3390/ijms21249538
Chicago/Turabian StyleSong, Min-Young, Ji Yeon Hwang, Eun Ji Bae, Saesbyeol Kim, Hye-Min Kang, Yong Jun Kim, Chan Park, and Kang-Sik Park. 2020. "Tyrosine Phosphorylation of the Kv2.1 Channel Contributes to Injury in Brain Ischemia" International Journal of Molecular Sciences 21, no. 24: 9538. https://doi.org/10.3390/ijms21249538
APA StyleSong, M.-Y., Hwang, J. Y., Bae, E. J., Kim, S., Kang, H.-M., Kim, Y. J., Park, C., & Park, K.-S. (2020). Tyrosine Phosphorylation of the Kv2.1 Channel Contributes to Injury in Brain Ischemia. International Journal of Molecular Sciences, 21(24), 9538. https://doi.org/10.3390/ijms21249538