Spliceosome Mutations in Uveal Melanoma

 , ,
, , 
Abstract
1. Introduction
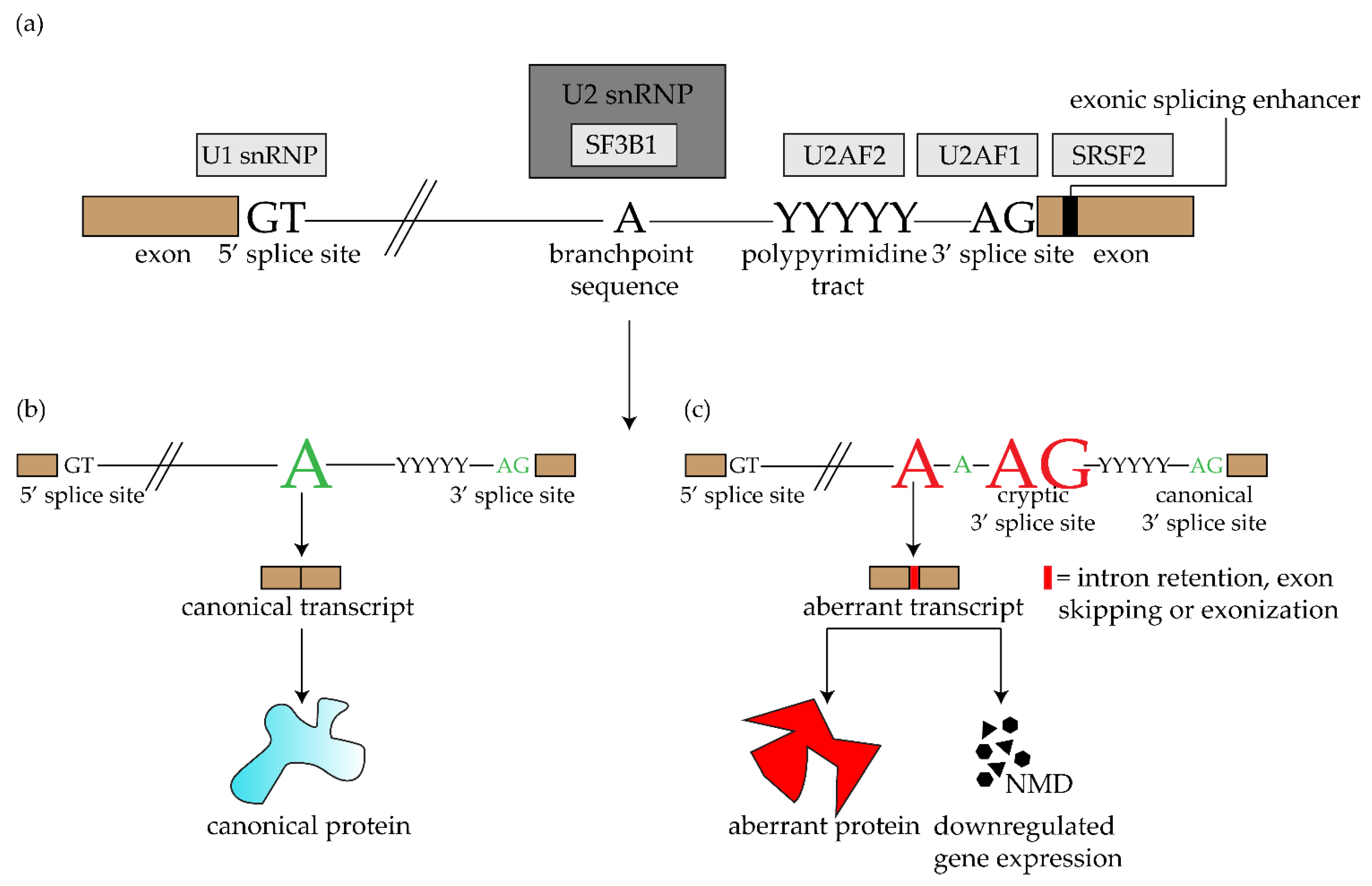
2. SF3B1 and Other Spliceosome Mutations in Uveal Melanoma
3. Chromosomal Anomalies and Epigenetic Changes Unique for SF3B1 UM
4. Telomere Length in SF3B1 Mutated UM
5. Therapeutic Opportunities
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AJCC | American Joint Committee on Cancer |
| BAP1 | BRCA1 associated protein 1 |
| BCL2 | B-cell lymphoma 2 |
| BCL2A1 | BCL2 related protein A1 |
| BCL2L1 | BCL2 like 1 |
| BCLxL | B-cell lymphoma-extra large |
| BPS | Branchpoint sequence |
| BRD9 | Bromodomain containing 9 |
| BUD31 | BUD31 homolog |
| CLL | Chronic Lymphocytic Leukemia |
| CSV | Chromosomal structural variants |
| CYSLTR2 | Cysteinyl Leukotriene Receptor 2 |
| DDX46 | DEAD-Box Helicase 46 |
| DNA-PK | DNA-dependent protein kinase |
| EIF1AX | Eukaryotic Translation Initiation Factor 1A X-Linked |
| EOE | Extraocular tumor extension |
| ESE | Exonic splicing enhancer |
| FAG | Fluorescein Angiography |
| GNAQ | G protein Subunit Alpha Q |
| GNA11 | G protein Subunit Alpha 11 |
| GPCR | G protein-coupled receptors |
| HEAT | Huntington, Elongation factor 3, protein phosphatase 2A, targets of rapamycin 1 |
| HR | Homology recombination |
| K | Lysine |
| M3G8q | Monosomy 3, gain of 8q |
| MCL1 | MCL1 apoptosis regulator |
| MDS | Myelodysplastic syndromes |
| MUT | Mutated |
| MYC | MYC proto-oncogene, BHLH transcription factor |
| ncBAF | Non-canonical BAF |
| NHEJ | Non-homologous end joining |
| NMD | Nonsense mediated decay |
| PLCB4 | Phospholipase C Beta 4 |
| PRMT5 | Protein Arginine Methyltransferase 5 |
| Py | Polypyrimidine |
| R | Arginine |
| RARS | Refractory anemia with ring sideroblasts |
| SF3B1 | Splicing Factor 3b Subunit 1 |
| SRSF2 | Serine- and arginine-rich splicing factor 2 |
| snRNP | Small nuclear ribonucleoprotein particles |
| TERT | Telomerase reverse transcriptase |
| TNM | Tumor-node-metastasis |
| U2AF | U2 small nuclear RNA auxiliary factor |
| U2AF2 | U2 small nuclear RNA auxiliary factor 2 |
| UM | Uveal melanoma |
| US | Ultrasonography |
| WT | Wild type |
References
- Virgili, G.; Gatta, G.; Ciccolallo, L.; Capocaccia, R.; Biggeri, A.; Crocetti, E.; Lutz, J.M.; Paci, E.; Group, E.W. Incidence of uveal melanoma in Europe. Ophthalmology 2007, 114, 2309–2315. [Google Scholar] [CrossRef]
- Shields, C.L.; Furuta, M.; Thangappan, A.; Nagori, S.; Mashayekhi, A.; Lally, D.R.; Kelly, C.C.; Rudich, D.S.; Nagori, A.V.; Wakade, O.A.; et al. Metastasis of uveal melanoma millimeter-by-millimeter in 8033 consecutive eyes. Arch. Ophthalmol. 2009, 127, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Damato, E.M.; Damato, B.E. Detection and time to treatment of uveal melanoma in the United Kingdom: An evaluation of 2384 patients. Ophthalmology 2012, 119, 1582–1589. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, L.E.; McLean, I.W.; Foster, W.D. Does enucleation of the eye containing a malignant melanoma prevent or accelerate the dissemination of tumour cells. Br. J. Ophthalmol. 1978, 62, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Diener-West, M.; Reynolds, S.M.; Agugliaro, D.J.; Caldwell, R.; Cumming, K.; Earle, J.D.; Hawkins, B.S.; Hayman, J.A.; Jaiyesimi, I.; Jampol, L.M.; et al. Development of metastatic disease after enrollment in the COMS trials for treatment of choroidal melanoma: Collaborative Ocular Melanoma Study Group Report No. 26. Arch. Ophthalmol. 2005, 123, 1639–1643. [Google Scholar]
- Eskelin, S.; Pyrhönen, S.; Summanen, P.; Hahka-Kemppinen, M.; Kivelä, T. Tumor doubling times in metastatic malignant melanoma of the uvea: Tumor progression before and after treatment. Ophthalmology 2000, 107, 1443–1449. [Google Scholar] [CrossRef]
- Damato, B. Ocular treatment of choroidal melanoma in relation to the prevention of metastatic death—A personal view. Prog. Retin. Eye Res. 2018, 66, 187–199. [Google Scholar] [CrossRef]
- Amin, M.B.; Edge, S.; Greene, F.; Byrd, D.R.; Brookland, R.K.; Washington, M.K.; Gershenwald, J.E.; Compton, C.C.; Hess, K.R.; Sullivan, D.C.; et al. AJCC Cancer Staging Manual, 8th ed.; Springer International Publishing: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Kujala, E.; Makitie, T.; Kivela, T. Very long-term prognosis of patients with malignant uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4651–4659. [Google Scholar] [CrossRef]
- Coupland, S.E.; Campbell, I.; Damato, B. Routes of extraocular extension of uveal melanoma: Risk factors and influence on survival probability. Ophthalmology 2008, 115, 1778–1785. [Google Scholar] [CrossRef]
- Drabarek, W.; Yavuzyigitoglu, S.; Obulkasim, A.; van Riet, J.; Smit, K.N.; van Poppelen, N.M.; Vaarwater, J.; Brands, T.; Eussen, B.; Verdijk, R.M.; et al. Multi-Modality Analysis Improves Survival Prediction in Enucleated Uveal Melanoma Patients. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3595–3605. [Google Scholar] [CrossRef]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in uveal melanoma. N. Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.I.; Strathmann, M.P.; Gautam, N. Diversity of G proteins in signal transduction. Science 1991, 252, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, A.E.; Vaarwater, J.; Paridaens, D.; Naus, N.C.; Kilic, E.; de Klein, A.; Rotterdam Ocular Melanoma Study group. Patient survival in uveal melanoma is not affected by oncogenic mutations in GNAQ and GNA11. Br. J. Cancer 2013, 109, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009, 457, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.; Kilic, E.; Vaarwater, J.; Bastian, B.C.; Garbe, C.; de Klein, A. Oncogenic GNAQ mutations are not correlated with disease-free survival in uveal melanoma. Br. J. Cancer 2009, 101, 813–815. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.R.; Ceraudo, E.; Sher, J.J.; Guan, Y.; Shoushtari, A.N.; Chang, M.T.; Zhang, J.Q.; Walczak, E.G.; Kazmi, M.A.; Taylor, B.S.; et al. Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat. Genet. 2016, 48, 675–680. [Google Scholar] [CrossRef]
- Johansson, P.; Aoude, L.G.; Wadt, K.; Glasson, W.J.; Warrier, S.K.; Hewitt, A.W.; Kiilgaard, J.F.; Heegaard, S.; Isaacs, T.; Franchina, M.; et al. Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget 2016, 7, 4624–4631. [Google Scholar] [CrossRef]
- Koopmans, A.E.; Verdijk, R.M.; Brouwer, R.W.; van den Bosch, T.P.; van den Berg, M.M.; Vaarwater, J.; Kockx, C.E.; Paridaens, D.; Naus, N.C.; Nellist, M.; et al. Clinical significance of immunohistochemistry for detection of BAP1 mutations in uveal melanoma. Mod. Pathol. 2014, 27, 1321–1330. [Google Scholar] [CrossRef]
- Martin, M.; Masshofer, L.; Temming, P.; Rahmann, S.; Metz, C.; Bornfeld, N.; van de Nes, J.; Klein-Hitpass, L.; Hinnebusch, A.G.; Horsthemke, B.; et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat. Genet. 2013, 45, 933–936. [Google Scholar] [CrossRef]
- Yavuzyigitoglu, S.; Koopmans, A.E.; Verdijk, R.M.; Vaarwater, J.; Eussen, B.; van Bodegom, A.; Paridaens, D.; Kilic, E.; de Klein, A.; Rotterdam Ocular Melanoma Study Group. Uveal Melanomas with SF3B1 Mutations: A Distinct Subclass Associated with Late-Onset Metastases. Ophthalmology 2016, 123, 1118–1128. [Google Scholar] [CrossRef]
- Jensen, D.E.; Proctor, M.; Marquis, S.T.; Gardner, H.P.; Ha, S.I.; Chodosh, L.A.; Ishov, A.M.; Tommerup, N.; Vissing, H.; Sekido, Y.; et al. BAP1: A novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression. Oncogene 1998, 16, 1097–1112. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, J.; Si, K.; Maitra, U. Function of eukaryotic translation initiation factor 1A (eIF1A) (formerly called eIF-4C) in initiation of protein synthesis. J. Biol. Chem. 1997, 272, 7883–7891. [Google Scholar] [CrossRef] [PubMed]
- Furney, S.J.; Pedersen, M.; Gentien, D.; Dumont, A.G.; Rapinat, A.; Desjardins, L.; Turajlic, S.; Piperno-Neumann, S.; de la Grange, P.; Roman-Roman, S.; et al. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov. 2013, 3, 1122–1129. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W.; Roberson, E.D.; Anbunathan, H.; Onken, M.D.; Worley, L.A.; Bowcock, A.M. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat. Genet. 2013, 45, 133–135. [Google Scholar] [CrossRef]
- Seiler, M.; Peng, S.; Agrawal, A.A.; Palacino, J.; Teng, T.; Zhu, P.; Smith, P.G.; Caesar-Johnson, S.J.; Demchok, J.A.; Felau, I.; et al. Somatic Mutational Landscape of Splicing Factor Genes and Their Functional Consequences across 33 Cancer Types. Cell Rep. 2018, 23, 282–296.e4. [Google Scholar] [CrossRef]
- De Conti, L.; Baralle, M.; Buratti, E. Exon and intron definition in pre-mRNA splicing. Wiley Interdiscip. Rev. RNA 2013, 4, 49–60. [Google Scholar] [CrossRef]
- Sibley, C.R.; Blazquez, L.; Ule, J. Lessons from non-canonical splicing. Nat. Rev. Genet. 2016, 17, 407–421. [Google Scholar] [CrossRef]
- Will, C.L.; Luhrmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3, a003707. [Google Scholar] [CrossRef]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef]
- Quesada, V.; Conde, L.; Villamor, N.; Ordonez, G.R.; Jares, P.; Bassaganyas, L.; Ramsay, A.J.; Bea, S.; Pinyol, M.; Martinez-Trillos, A.; et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat. Genet. 2011, 44, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Ellis, M.J.; Ding, L.; Shen, D.; Luo, J.; Suman, V.J.; Wallis, J.W.; Van Tine, B.A.; Hoog, J.; Goiffon, R.J.; Goldstein, T.C.; et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 2012, 486, 353–360. [Google Scholar] [CrossRef]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Hintzsche, J.D.; Gorden, N.T.; Amato, C.M.; Kim, J.; Wuensch, K.E.; Robinson, S.E.; Applegate, A.J.; Couts, K.L.; Medina, T.M.; Wells, K.R.; et al. Whole-exome sequencing identifies recurrent SF3B1 R625 mutation and comutation of NF1 and KIT in mucosal melanoma. Melanoma Res. 2017, 27, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Maguire, S.L.; Leonidou, A.; Wai, P.; Marchio, C.; Ng, C.K.; Sapino, A.; Salomon, A.V.; Reis-Filho, J.S.; Weigelt, B.; Natrajan, R.C. SF3B1 mutations constitute a novel therapeutic target in breast cancer. J. Pathol. 2015, 235, 571–580. [Google Scholar] [CrossRef]
- Alsafadi, S.; Houy, A.; Battistella, A.; Popova, T.; Wassef, M.; Henry, E.; Tirode, F.; Constantinou, A.; Piperno-Neumann, S.; Roman-Roman, S.; et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat. Commun. 2016, 7, 10615. [Google Scholar] [CrossRef]
- Darman, R.B.; Seiler, M.; Agrawal, A.A.; Lim, K.H.; Peng, S.; Aird, D.; Bailey, S.L.; Bhavsar, E.B.; Chan, B.; Colla, S.; et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3′ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015, 13, 1033–1045. [Google Scholar] [CrossRef]
- Paolella, B.R.; Gibson, W.J.; Urbanski, L.M.; Alberta, J.A.; Zack, T.I.; Bandopadhayay, P.; Nichols, C.A.; Agarwalla, P.K.; Brown, M.S.; Lamothe, R.; et al. Copy-number and gene dependency analysis reveals partial copy loss of wild-type SF3B1 as a novel cancer vulnerability. Elife 2017, 6, e23268. [Google Scholar] [CrossRef]
- Tang, Q.; Rodriguez-Santiago, S.; Wang, J.; Pu, J.; Yuste, A.; Gupta, V.; Moldon, A.; Xu, Y.Z.; Query, C.C. SF3B1/Hsh155 HEAT motif mutations affect interaction with the spliceosomal ATPase Prp5, resulting in altered branch site selectivity in pre-mRNA splicing. Genes Dev. 2016, 30, 2710–2723. [Google Scholar] [CrossRef]
- Wang, L.; Lawrence, M.S.; Wan, Y.; Stojanov, P.; Sougnez, C.; Stevenson, K.; Werner, L.; Sivachenko, A.; DeLuca, D.S.; Zhang, L.; et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N. Engl. J. Med. 2011, 365, 2497–2506. [Google Scholar] [CrossRef] [PubMed]
- Malcovati, L.; Karimi, M.; Papaemmanuil, E.; Ambaglio, I.; Jadersten, M.; Jansson, M.; Elena, C.; Galli, A.; Walldin, G.; Della Porta, M.G.; et al. SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood 2015, 126, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Miao, M.; Han, S.; Qi, J.; Wang, H.; Ruan, C.; Wu, D.; Han, Y. Prognostic value and clinical feature of SF3B1 mutations in myelodysplastic syndromes: A meta-analysis. Crit. Rev. Oncol. Hematol. 2019, 133, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Tian, M.; Gu, J.; Cheng, T.; Ma, D.; Feng, L.; Xin, X. SF3B1 mutation is a poor prognostic indicator in luminal B and progesterone receptor-negative breast cancer patients. Oncotarget 2017, 8, 115018–115027. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 2018, 33, 151. [Google Scholar] [CrossRef]
- van Poppelen, N.M.; Drabarek, W.; Smit, K.N.; Vaarwater, J.; Brands, T.; Paridaens, D.; Kilic, E.; de Klein, A. SRSF2 Mutations in Uveal Melanoma: A Preference for In-Frame Deletions? Cancers 2019, 11, 1200. [Google Scholar] [CrossRef]
- Dogrusoz, M.; Jager, M.J. Genetic prognostication in uveal melanoma. Acta Ophthalmol. 2018, 96, 331–347. [Google Scholar] [CrossRef]
- Yavuzyigitoglu, S.; Drabarek, W.; Smit, K.N.; van Poppelen, N.; Koopmans, A.E.; Vaarwater, J.; Brands, T.; Eussen, B.; Dubbink, H.J.; van Riet, J.; et al. Correlation of Gene Mutation Status with Copy Number Profile in Uveal Melanoma. Ophthalmology 2017, 124, 573–575. [Google Scholar] [CrossRef]
- Jeromin, S.; Weissmann, S.; Haferlach, C.; Dicker, F.; Bayer, K.; Grossmann, V.; Alpermann, T.; Roller, A.; Kohlmann, A.; Haferlach, T.; et al. SF3B1 mutations correlated to cytogenetics and mutations in NOTCH1, FBXW7, MYD88, XPO1 and TP53 in 1160 untreated CLL patients. Leukemia 2014, 28, 108–117. [Google Scholar] [CrossRef]
- Smit, K.N.; Boers, R.; Vaarwater, J.; Boers, J.; Brands, T.; Mensink, H.; Verdijk, R.M.; Van Ijcken, W.F.J.; Gribnau, J.; De Klein, A.; et al. Novel DNA-methylation silenced tumor suppressor genes identified for BAP1-mediated uveal melanoma metastasis. 2020, in press. [Google Scholar]
- Naus, N.C.; van Drunen, E.; de Klein, A.; Luyten, G.P.; Paridaens, D.A.; Alers, J.C.; Ksander, B.R.; Beverloo, H.B.; Slater, R.M. Characterization of complex chromosomal abnormalities in uveal melanoma by fluorescence in situ hybridization, spectral karyotyping, and comparative genomic hybridization. Genes Chromosomes Cancer 2001, 30, 267–273. [Google Scholar] [CrossRef]
- Wang, L.; Brooks, A.N.; Fan, J.; Wan, Y.; Gambe, R.; Li, S.; Hergert, S.; Yin, S.; Freeman, S.S.; Levin, J.Z.; et al. Transcriptomic Characterization of SF3B1 Mutation Reveals Its Pleiotropic Effects in Chronic Lymphocytic Leukemia. Cancer Cell 2016, 30, 750–763. [Google Scholar] [CrossRef] [PubMed]
- Maciejowski, J.; de Lange, T. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Bacchetti, S. A survey of telomerase activity in human cancer. Eur. J. Cancer 1997, 33, 787–791. [Google Scholar] [CrossRef]
- Barthel, F.P.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef]
- Koopmans, A.E.; Ober, K.; Dubbink, H.J.; Paridaens, D.; Naus, N.C.; Belunek, S.; Krist, B.; Post, E.; Zwarthoff, E.C.; de Klein, A.; et al. Prevalence and implications of TERT promoter mutation in uveal and conjunctival melanoma and in benign and premalignant conjunctival melanocytic lesions. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6024–6030. [Google Scholar] [CrossRef]
- Doherty, R.E.; Bryant, H.E.; Valluru, M.K.; Rennie, I.G.; Sisley, K. Increased Non-Homologous End Joining Makes DNA-PK a Promising Target for Therapeutic Intervention in Uveal Melanoma. Cancers 2019, 11, 1278. [Google Scholar] [CrossRef]
- Lee, S.C.; Abdel-Wahab, O. Therapeutic targeting of splicing in cancer. Nat. Med. 2016, 22, 976–986. [Google Scholar] [CrossRef]
- Shirai, C.L.; White, B.S.; Tripathi, M.; Tapia, R.; Ley, J.N.; Ndonwi, M.; Kim, S.; Shao, J.; Carver, A.; Saez, B.; et al. Mutant U2AF1-expressing cells are sensitive to pharmacological modulation of the spliceosome. Nat. Commun. 2017, 8, 14060. [Google Scholar] [CrossRef]
- Lagisetti, C.; Yermolina, M.V.; Sharma, L.K.; Palacios, G.; Prigaro, B.J.; Webb, T.R. Pre-mRNA splicing-modulatory pharmacophores: The total synthesis of herboxidiene, a pladienolide-herboxidiene hybrid analog and related derivatives. ACS Chem. Biol. 2014, 9, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Seiler, M.; Yoshimi, A.; Darman, R.; Chan, B.; Keaney, G.; Thomas, M.; Agrawal, A.A.; Caleb, B.; Csibi, A.; Sean, E.; et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med. 2018, 24, 497–504. [Google Scholar] [CrossRef]
- Yokoi, A.; Kotake, Y.; Takahashi, K.; Kadowaki, T.; Matsumoto, Y.; Minoshima, Y.; Sugi, N.H.; Sagane, K.; Hamaguchi, M.; Iwata, M.; et al. Biological validation that SF3b is a target of the antitumor macrolide pladienolide. FEBS J. 2011, 278, 4870–4880. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Dvinge, H.; Kim, E.; Cho, H.; Micol, J.B.; Chung, Y.R.; Durham, B.H.; Yoshimi, A.; Kim, Y.J.; Thomas, M.; et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat. Med. 2016, 22, 672–678. [Google Scholar] [CrossRef]
- Hong, D.S.; Kurzrock, R.; Naing, A.; Wheler, J.J.; Falchook, G.S.; Schiffman, J.S.; Faulkner, N.; Pilat, M.J.; O’Brien, J.; LoRusso, P. A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Investig. New Drugs 2014, 32, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Eskens, F.A.; Ramos, F.J.; Burger, H.; O’Brien, J.P.; Piera, A.; de Jonge, M.J.; Mizui, Y.; Wiemer, E.A.; Carreras, M.J.; Baselga, J.; et al. Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin. Cancer Res. 2013, 19, 6296–6304. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Hori, Y.; Terano, H.; Okuhara, M.; Manda, T.; Matsumoto, S.; Shimomura, K. New antitumor substances, FR901463, FR901464 and FR901465. II. Activities against experimental tumors in mice and mechanism of action. J. Antibiot. 1996, 49, 1204–1211. [Google Scholar] [CrossRef]
- Sakai, Y.; Yoshida, T.; Ochiai, K.; Uosaki, Y.; Saitoh, Y.; Tanaka, F.; Akiyama, T.; Akinaga, S.; Mizukami, T. GEX1 compounds, novel antitumor antibiotics related to herboxidiene, produced by Streptomyces sp. I. Taxonomy, production, isolation, physicochemical properties and biological activities. J. Antibiot. 2002, 55, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Sagane, K.; Owa, T.; Mimori-Kiyosue, Y.; Shimizu, H.; Uesugi, M.; Ishihama, Y.; Iwata, M.; Mizui, Y. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol. 2007, 3, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Lagisetti, C.; Edwards, C.C.; Webb, T.R.; Potter, P.M. Sudemycins, novel small molecule analogues of FR901464, induce alternative gene splicing. ACS Chem. Biol. 2011, 6, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Aird, D.; Teng, T.; Huang, C.L.; Pazolli, E.; Banka, D.; Cheung-Ong, K.; Eifert, C.; Furman, C.; Wu, Z.J.; Seiler, M.; et al. Sensitivity to splicing modulation of BCL2 family genes defines cancer therapeutic strategies for splicing modulators. Nat. Commun. 2019, 10, 137. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.M.; Bezzi, M.; Low, D.H.; Ang, W.X.; Teo, S.X.; Gay, F.P.; Al-Haddawi, M.; Tan, S.Y.; Osato, M.; Sabo, A.; et al. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature 2015, 523, 96–100. [Google Scholar] [CrossRef] [PubMed]
- DePinho, R.; Mitsock, L.; Hatton, K.; Ferrier, P.; Zimmerman, K.; Legouy, E.; Tesfaye, A.; Collum, R.; Yancopoulos, G.; Nisen, P.; et al. Myc family of cellular oncogenes. J. Cell Biochem. 1987, 33, 257–266. [Google Scholar] [CrossRef]
- Neel, B.G.; Jhanwar, S.C.; Chaganti, R.S.; Hayward, W.S. Two human c-onc genes are located on the long arm of chromosome 8. Proc. Natl. Acad. Sci. USA 1982, 79, 7842–7846. [Google Scholar] [CrossRef] [PubMed]
- Anczukow, O.; Krainer, A.R. The spliceosome, a potential Achilles heel of MYC-driven tumors. Genome Med. 2015, 7, 107. [Google Scholar] [CrossRef] [PubMed]
- Inoue, D.; Chew, G.L.; Liu, B.; Michel, B.C.; Pangallo, J.; D’Avino, A.R.; Hitchman, T.; North, K.; Lee, S.C.; Bitner, L.; et al. Spliceosomal disruption of the non-canonical BAF complex in cancer. Nature 2019, 574, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Badran, A.H.; Liu, D.R. CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 2017, 168, 20–36. [Google Scholar] [CrossRef]
- Lentsch, E.; Li, L.; Pfeffer, S.; Ekici, A.B.; Taher, L.; Pilarsky, C.; Grutzmann, R. CRISPR/Cas9-Mediated Knock-Out of Kras(G12D) Mutated Pancreatic Cancer Cell Lines. Int. J. Mol. Sci. 2019, 20, 5706. [Google Scholar] [CrossRef]
- Park, M.Y.; Jung, M.H.; Eo, E.Y.; Kim, S.; Lee, S.H.; Lee, Y.J.; Park, J.S.; Cho, Y.J.; Chung, J.H.; Kim, C.H.; et al. Generation of lung cancer cell lines harboring EGFR T790M mutation by CRISPR/Cas9-mediated genome editing. Oncotarget 2017, 8, 36331–36338. [Google Scholar] [CrossRef]
- Lu, Y.; Xue, J.; Deng, T.; Zhou, X.; Yu, K.; Deng, L.; Huang, M.; Yi, X.; Liang, M.; Wang, Y.; et al. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat. Med. 2020, 26, 732–740. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | Exon | AA Mutation | CDS Mutation | Effect | Frequency (n = 122, %) |
|---|---|---|---|---|---|
| 2: 197402759 | 14 | p.R625H | c.1874G > A | Missense | 61 (50%) |
| 2: 197402760 | 14 | p.R625C | c.1873C > T | Missense | 38 (31%) |
| 2: 197402759 | 14 | p.R625L | c.1874G > T | Missense | 7 (5.7%) |
| 2: 197402760 | 14 | p.R625G | c.1873C > G | Missense | 1 (0.8%) |
| 2: 197402759 | 14 | p.R625P | c.1874G > C | Missense | 1 (0.8%) |
| 2: 197402760 | 14 | p.R625S | c.1873C > A | Missense | 1 (0.8%) |
| 2: 197402775 | 14 | p.M620V | c.1858A > G | Missense | 1 (0.8%) |
| 2: 197402767 | 14 | p.E622D | c.1866G > C | Missense | 1 (0.8%) |
| 2: 197402766 | 14 | p.Y623H | c.1867T > C | Missense | 1 (0.8%) |
| 2: 197402636 | 14 | p.K666T | c.1997A > C | Missense | 2 (1.6%) |
| 2: 197402110 | 15 | p.K700E | c.2098A > G | Missense | 1 (0.8%) |
| 2: 197401765 | 16 | p.E783K | c.2347G > A | Missense | 1 (0.8%) |
| 2: 197401771 | 16 | p.D781N | c.2341G > A | Missense | 1 (0.8%) |
| 2: 197401887 | 16 | p.G742D | c.2225G > A | Missense | 1 (0.8%) |
| 2:197396227 | 23 | p.C1123Y | c.3368G > A | Missense | 1 (0.8%) |
| Splicing Inhibitors | Features | Trials |
|---|---|---|
| FR901464 spliceostatins A-G, meamycins | Targets the U2 snRNP of SF3B1; isolated from fermentation broth of bacterium Pseudomonas sp. No. 2663 [68]. | Preclinical trial in cancer cell lines [68] |
| Herboxidiene | Targets the U2 snRNP of SF3B1; isolated from Streptomyces chromofuscus ATCC 49,982 [69]. | Preclinical trial in cancer cell lines [62] |
| Pladienolide B pladienolides A-G, H3B-8800, E7107 | Targets the U2 snRNP of SF3B1; isolated from bacterium Streptomyces platensis [70]. | Two phase I trials of E7107 on solid tumors, suspended due to side effects [66,67] |
| Sudemycin | Targets SF3B1; pharmacophore between FR901464 and pladienolide B [71]. | Preclinical trial in cancer cell lines and tumor samples [71] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, J.Q.N.; Drabarek, W.; Yavuzyigitoglu, S.; Medico Salsench, E.; Verdijk, R.M.; Naus, N.C.; de Klein, A.; Kiliç, E.; Brosens, E. Spliceosome Mutations in Uveal Melanoma. Int. J. Mol. Sci. 2020, 21, 9546. https://doi.org/10.3390/ijms21249546
Nguyen JQN, Drabarek W, Yavuzyigitoglu S, Medico Salsench E, Verdijk RM, Naus NC, de Klein A, Kiliç E, Brosens E. Spliceosome Mutations in Uveal Melanoma. International Journal of Molecular Sciences. 2020; 21(24):9546. https://doi.org/10.3390/ijms21249546
Chicago/Turabian StyleNguyen, Josephine Q.N., Wojtek Drabarek, Serdar Yavuzyigitoglu, Eva Medico Salsench, Robert M. Verdijk, Nicole C. Naus, Annelies de Klein, Emine Kiliç, and Erwin Brosens. 2020. "Spliceosome Mutations in Uveal Melanoma" International Journal of Molecular Sciences 21, no. 24: 9546. https://doi.org/10.3390/ijms21249546
APA StyleNguyen, J. Q. N., Drabarek, W., Yavuzyigitoglu, S., Medico Salsench, E., Verdijk, R. M., Naus, N. C., de Klein, A., Kiliç, E., & Brosens, E. (2020). Spliceosome Mutations in Uveal Melanoma. International Journal of Molecular Sciences, 21(24), 9546. https://doi.org/10.3390/ijms21249546