Molecular Features and Clinical Management of Hereditary Gynecological Cancers



Abstract
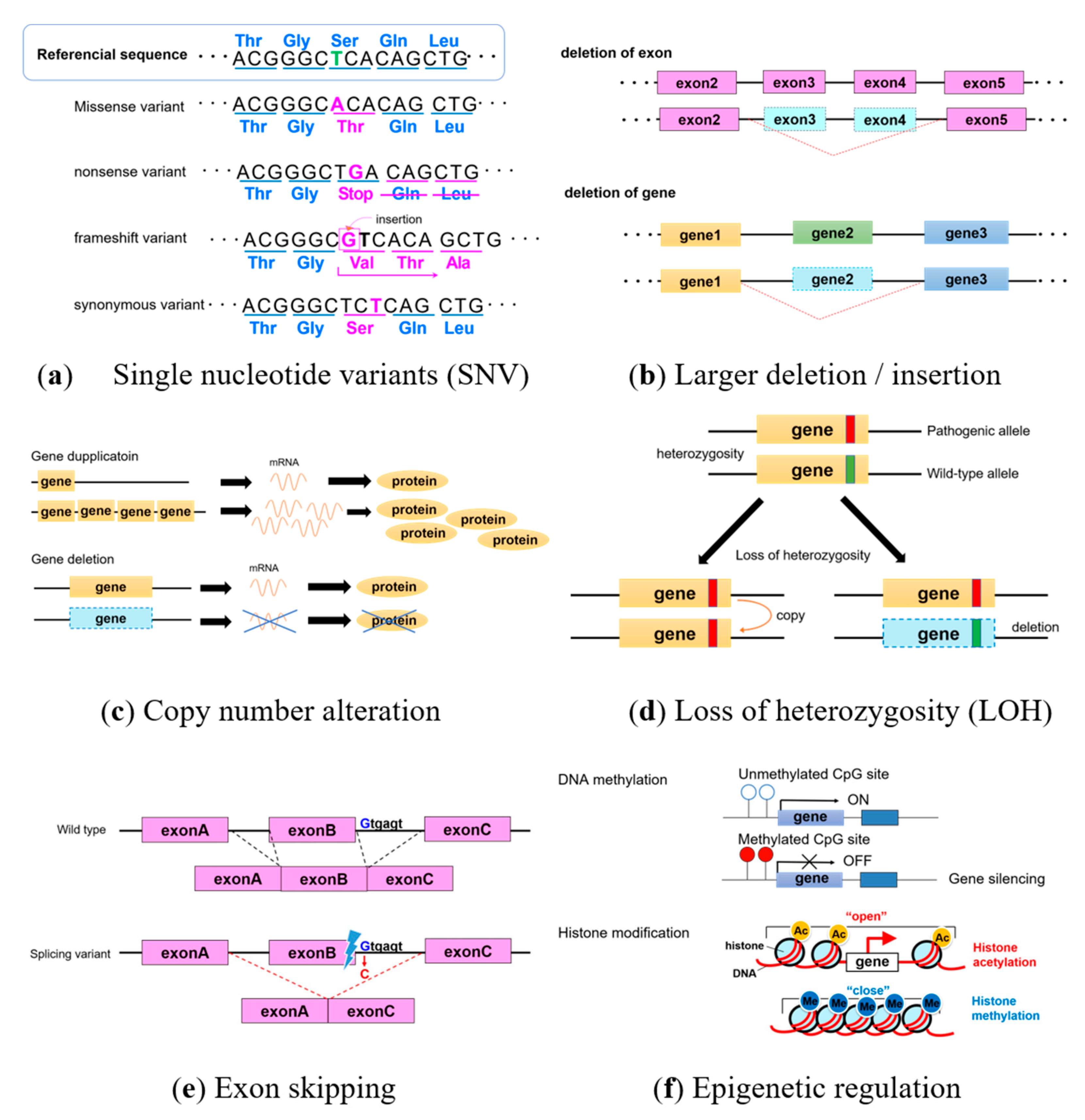
1. Introduction
2. Hereditary Gynecological Cancers
2.1. Hereditary Breast and Ovarian Cancer Syndrome
2.2. Lynch Syndrome
2.3. Li–Fraumeni Syndrome
2.4. Cowden Syndrome
2.5. Peutz–Jeghers Syndrome (PJS)
2.6. Other Cancer-Susceptible Genes
3. Genetic Testing for Hereditary Cancers
3.1. Genetic Tests for Diagnosing Hereditary Cancers
3.2. Genetic Tests for Pharmacogenetics and Personalized Therapy
3.3. Tumor Profiling for Precision Medicine
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACMG | American College of Medical Genetics and Genomics |
| DICER1 | Ribonuclease III; double-stranded RNA-specific endoribonuclease |
| EMA | European Medicines Agency |
| FDA | Food and Drug Administration |
| HBOC | hereditary breast and ovarian cancer syndrome |
| HRD | homologous recombination deficiency |
| HRR | homologous recombination repair |
| ICI | immune checkpoint inhibitor |
| LEGH | lobular endocervical glandular hyperplasia |
| MDA | minimal deviation adenocarcinoma |
| MGP | multi-gene panel |
| MMR | mismatch repair |
| MRI | magnetic resonance imaging |
| MSI | microsatellite instability |
| MSI-H | microsatellite instability-high |
| MSI-L | microsatellite instability-low |
| MSS | microsatellite stable |
| NCCN | National Comprehensive Cancer Network |
| PARP | poly ADP ribose polymerase |
| PJS | Peutz–Jeghers syndrome |
| RRSO | risk-reducing salpingo-oophorectomy |
| SCAT | sex-cord tumor with annular tubules |
| VUS | variant of uncertain significance |
References
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef]
- Fournier, D.M.; Bazzell, A.F.; Dains, J.E. Comparing Outcomes of Genetic Counseling Options in Breast and Ovarian Cancer: An Integrative Review. Oncol. Nurs. Forum 2018, 45, 96–105. [Google Scholar] [CrossRef]
- Hampel, H. Genetic counseling and cascade genetic testing in Lynch syndrome. Fam. Cancer 2016, 15, 423–427. [Google Scholar] [CrossRef]
- Hampel, H.; Bennett, R.L.; Buchanan, A.; Pearlman, R.; Wiesner, G.L. A practice guideline from the American College of Medical Genetics and Genomics and the National Society of Genetic Counselors: Referral indications for cancer predisposition assessment. Genet. Med. 2015, 17, 70–87. [Google Scholar] [CrossRef]
- Hooker, G.W.; Clemens, K.R.; Quillin, J.; Vogel Postula, K.J.; Summerour, P.; Nagy, R.; Buchanan, A.H. Cancer Genetic Counseling and Testing in an Era of Rapid Change. J. Genet. Couns. 2017, 26, 1244–1253. [Google Scholar] [CrossRef]
- NCCN Clinical Practice Guidelines in Oncology. Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. Version1, 2021. Available online: https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf (accessed on 19 November 2020).
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef]
- Hirasawa, A.; Imoto, I.; Naruto, T.; Akahane, T.; Yamagami, W.; Nomura, H.; Masuda, K.; Susumu, N.; Tsuda, H.; Aoki, D. Prevalence of pathogenic germline variants detected by multigene sequencing in unselected Japanese patients with ovarian cancer. Oncotarget 2017, 8, 112258–112267. [Google Scholar] [CrossRef]
- Bolton, K.L.; Chenevix-Trench, G.; Goh, C.; Sadetzki, S.; Ramus, S.J.; Karlan, B.Y.; Lambrechts, D.; Despierre, E.; Barrowdale, D.; McGuffog, L.; et al. Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. JAMA 2012, 307, 382–390. [Google Scholar] [CrossRef]
- Enomoto, T.; Aoki, D.; Hattori, K.; Jinushi, M.; Kigawa, J.; Takeshima, N.; Tsuda, H.; Watanabe, Y.; Yoshihara, K.; Sugiyama, T. The first Japanese nationwide multicenter study of BRCA mutation testing in ovarian cancer: CHARacterizing the cross-sectionaL approach to Ovarian cancer geneTic TEsting of BRCA (CHARLOTTE). Int. J. Gynecol. Cancer 2019, 29, 1043–1049. [Google Scholar] [CrossRef]
- Norquist, B.M.; Harrell, M.I.; Brady, M.F.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Yi, Q.; Burger, R.A.; et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol. 2016, 2, 482–490. [Google Scholar] [CrossRef]
- Yang, D.; Khan, S.; Sun, Y.; Hess, K.; Shmulevich, I.; Sood, A.K.; Zhang, W. Association of BRCA1 and BRCA2 mutations with survival, chemotherapy sensitivity, and gene mutator phenotype in patients with ovarian cancer. JAMA 2011, 306, 1557–1565. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Norquist, B.; Lacchetti, C.; Armstrong, D.; Grisham, R.N.; Goodfellow, P.J.; Kohn, E.C.; Levine, D.A.; Liu, J.F.; Lu, K.H.; et al. Germline and Somatic Tumor Testing in Epithelial Ovarian Cancer: ASCO Guideline. J. Clin. Oncol. 2020, 38, 1222–1245. [Google Scholar] [CrossRef]
- NICE Guideline: Familial Breast Cancer: Classification, Care and Managing Breast Cancer and Related Risks in People with a Family History of Breast Cancer. Available online: www.nice.org.uk/guidance/cg164 (accessed on 19 November 2020).
- Owens, D.K.; Davidson, K.W.; Krist, A.H.; Barry, M.J.; Cabana, M.; Caughey, A.B.; Doubeni, C.A.; Epling, J.W., Jr.; Kubik, M.; Landefeld, C.S.; et al. Risk Assessment, Genetic Counseling, and Genetic Testing for BRCA-Related Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 2019, 322, 652–665. [Google Scholar] [CrossRef]
- Paluch-Shimon, S.; Cardoso, F.; Sessa, C.; Balmana, J.; Cardoso, M.J.; Gilbert, F.; Senkus, E.; Committee, E.G. Prevention and screening in BRCA mutation carriers and other breast/ovarian hereditary cancer syndromes: ESMO Clinical Practice Guidelines for cancer prevention and screening. Ann. Oncol. 2016, 27, v103–v110. [Google Scholar] [CrossRef]
- Rebbeck, T.R.; Kauff, N.D.; Domchek, S.M. Meta-analysis of risk reduction estimates associated with risk-reducing salpingo-oophorectomy in BRCA1 or BRCA2 mutation carriers. J. Natl. Cancer Inst. 2009, 101, 80–87. [Google Scholar] [CrossRef]
- Harmsen, M.G.; Piek, J.M.J.; Bulten, J.; Casey, M.J.; Rebbeck, T.R.; Mourits, M.J.; Greene, M.H.; Slangen, B.F.M.; van Beurden, M.; Massuger, L.; et al. Peritoneal carcinomatosis after risk-reducing surgery in BRCA1/2 mutation carriers. Cancer 2018, 124, 952–959. [Google Scholar] [CrossRef]
- Jacobs, I.J.; Menon, U.; Ryan, A.; Gentry-Maharaj, A.; Burnell, M.; Kalsi, J.K.; Amso, N.N.; Apostolidou, S.; Benjamin, E.; Cruickshank, D.; et al. Ovarian cancer screening and mortality in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): A randomised controlled trial. Lancet 2016, 387, 945–956. [Google Scholar] [CrossRef]
- Iglehart, J.D.; Silver, D.P. Synthetic lethality—A new direction in cancer-drug development. N. Engl. J. Med. 2009, 361, 189–191. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- George, A.; Kaye, S.; Banerjee, S. Delivering widespread BRCA testing and PARP inhibition to patients with ovarian cancer. Nat. Rev. Clin. Oncol. 2017, 14, 284–296. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Oza, A.M.; Cibula, D.; Benzaquen, A.O.; Poole, C.; Mathijssen, R.H.J.; Sonke, G.S.; Colombo, N.; Špaček, J.; Vuylsteke, P.; Hirte, H.; et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: A randomised phase 2 trial. Lancet Oncol. 2015, 16, 87–97. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef]
- Han, H.S.; Diéras, V.; Robson, M.; Palácová, M.; Marcom, P.K.; Jager, A.; Bondarenko, I.; Citrin, D.; Campone, M.; Telli, M.L.; et al. Veliparib with temozolomide or carboplatin/paclitaxel versus placebo with carboplatin/paclitaxel in patients with BRCA1/2 locally recurrent/metastatic breast cancer: Randomized phase II study. Ann. Oncol. 2018, 29, 154–161. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Robson, M.E.; Tung, N.; Conte, P.; Im, S.A.; Senkus, E.; Xu, B.; Masuda, N.; Delaloge, S.; Li, W.; Armstrong, A.; et al. OlympiAD final overall survival and tolerability results: Olaparib versus chemotherapy treatment of physician’s choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann. Oncol. 2019, 30, 558–566. [Google Scholar] [CrossRef]
- Rugo, H.S.; Olopade, O.I.; DeMichele, A.; Yau, C.; van’t Veer, L.J.; Buxton, M.B.; Hogarth, M.; Hylton, N.M.; Paoloni, M.; Perlmutter, J.; et al. Adaptive Randomization of Veliparib-Carboplatin Treatment in Breast Cancer. N. Engl. J. Med. 2016, 375, 23–34. [Google Scholar] [CrossRef]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Hodgson, D.R.; Dougherty, B.A.; Lai, Z.; Fielding, A.; Grinsted, L.; Spencer, S.; O’Connor, M.J.; Ho, T.W.; Robertson, J.D.; Lanchbury, J.S.; et al. Candidate biomarkers of PARP inhibitor sensitivity in ovarian cancer beyond the BRCA genes. Br. J. Cancer 2018, 119, 1401–1409. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Drew, Y.; Kristeleit, R.S. Homologous recombination deficiency and ovarian cancer. Eur. J. Cancer 2016, 60, 49–58. [Google Scholar] [CrossRef]
- Moschetta, M.; George, A.; Kaye, S.B.; Banerjee, S. BRCA somatic mutations and epigenetic BRCA modifications in serous ovarian cancer. Ann. Oncol. 2016, 27, 1449–1455. [Google Scholar] [CrossRef]
- Practice Bulletin No 182: Hereditary Breast and Ovarian Cancer Syndrome. Obstet. Gynecol. 2017, 130, e110–e126. [CrossRef]
- Win, A.K.; Lindor, N.M.; Young, J.P.; Macrae, F.A.; Young, G.P.; Williamson, E.; Parry, S.; Goldblatt, J.; Lipton, L.; Winship, I.; et al. Risks of primary extracolonic cancers following colorectal cancer in lynch syndrome. J. Natl. Cancer Inst. 2012, 104, 1363–1372. [Google Scholar] [CrossRef]
- Sinicrope, F.A. Lynch Syndrome-Associated Colorectal Cancer. N. Engl. J. Med. 2018, 379, 764–773. [Google Scholar] [CrossRef]
- Lu, K.H.; Daniels, M. Endometrial and ovarian cancer in women with Lynch syndrome: Update in screening and prevention. Fam. Cancer 2013, 12, 273–277. [Google Scholar] [CrossRef]
- Lu, K.H.; Dinh, M.; Kohlmann, W.; Watson, P.; Green, J.; Syngal, S.; Bandipalliam, P.; Chen, L.M.; Allen, B.; Conrad, P.; et al. Gynecologic cancer as a “sentinel cancer” for women with hereditary nonpolyposis colorectal cancer syndrome. Obstet. Gynecol. 2005, 105, 569–574. [Google Scholar] [CrossRef]
- NCCN Clinical Practice Guideline in Oncology, Genetic / Familial High-Risk Assessment: Colorectal, Version1. 2020. Available online: https://www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdf (accessed on 19 November 2020).
- Bonadona, V.; Bonaïti, B.; Olschwang, S.; Grandjouan, S.; Huiart, L.; Longy, M.; Guimbaud, R.; Buecher, B.; Bignon, Y.J.; Caron, O.; et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 2011, 305, 2304–2310. [Google Scholar] [CrossRef] [PubMed]
- Ryan, N.A.J.; Evans, D.G.; Green, K.; Crosbie, E.J. Pathological features and clinical behavior of Lynch syndrome-associated ovarian cancer. Gynecol. Oncol. 2017, 144, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.; Bützow, R.; Lynch, H.T.; Mecklin, J.P.; Järvinen, H.J.; Vasen, H.F.; Madlensky, L.; Fidalgo, P.; Bernstein, I. The clinical features of ovarian cancer in hereditary nonpolyposis colorectal cancer. Gynecol. Oncol. 2001, 82, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.; Vasen, H.F.A.; Mecklin, J.P.; Bernstein, I.; Aarnio, M.; Järvinen, H.J.; Myrhøj, T.; Sunde, L.; Wijnen, J.T.; Lynch, H.T. The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. Int. J. Cancer 2008, 123, 444–449. [Google Scholar] [CrossRef]
- Latham, A.; Srinivasan, P.; Kemel, Y.; Shia, J.; Bandlamudi, C.; Mandelker, D.; Middha, S.; Hechtman, J.; Zehir, A.; Dubard-Gault, M.; et al. Microsatellite Instability Is Associated With the Presence of Lynch Syndrome Pan-Cancer. J. Clin. Oncol. 2019, 37, 286–295. [Google Scholar] [CrossRef]
- Vasen, H.F. Clinical diagnosis and management of hereditary colorectal cancer syndromes. J. Clin. Oncol. 2000, 18, 81s–92s. [Google Scholar]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de la Chapelle, A.; Rüschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef]
- Dudley, J.C.; Lin, M.T.; Le, D.T.; Eshleman, J.R. Microsatellite Instability as a Biomarker for PD-1 Blockade. Clin. Cancer Res. 2016, 22, 813–820. [Google Scholar] [CrossRef]
- Le, D.T.; Kim, T.W.; Van Cutsem, E.; Geva, R.; Jäger, D.; Hara, H.; Burge, M.; O’Neil, B.; Kavan, P.; Yoshino, T.; et al. Phase II Open-Label Study of Pembrolizumab in Treatment-Refractory, Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: KEYNOTE-164. J. Clin. Oncol. 2020, 38, 11–19. [Google Scholar] [CrossRef]
- Schneider KA, G.J. Li-Fraumeni Syndrome. In GeneReviews. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1311/ (accessed on 19 November 2020).
- Bougeard, G.; Renaux-Petel, M.; Flaman, J.M.; Charbonnier, C.; Fermey, P.; Belotti, M.; Gauthier-Villars, M.; Stoppa-Lyonnet, D.; Consolino, E.; Brugières, L.; et al. Revisiting Li-Fraumeni Syndrome From TP53 Mutation Carriers. J. Clin. Oncol. 2015, 33, 2345–2352. [Google Scholar] [CrossRef]
- Kratz, C.P.; Achatz, M.I.; Brugières, L.; Frebourg, T.; Garber, J.E.; Greer, M.C.; Hansford, J.R.; Janeway, K.A.; Kohlmann, W.K.; McGee, R.; et al. Cancer Screening Recommendations for Individuals with Li-Fraumeni Syndrome. Clin. Cancer Res. 2017, 23, e38–e45. [Google Scholar] [CrossRef] [PubMed]
- McBride, K.A.; Ballinger, M.L.; Killick, E.; Kirk, J.; Tattersall, M.H.; Eeles, R.A.; Thomas, D.M.; Mitchell, G. Li-Fraumeni syndrome: Cancer risk assessment and clinical management. Nat. Rev. Clin. Oncol. 2014, 11, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Mai, P.L.; Best, A.F.; Peters, J.A.; DeCastro, R.M.; Khincha, P.P.; Loud, J.T.; Bremer, R.C.; Rosenberg, P.S.; Savage, S.A. Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li-Fraumeni syndrome cohort. Cancer 2016, 122, 3673–3681. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, K.D.; Noltner, K.A.; Buzin, C.H.; Gu, D.; Wen-Fong, C.Y.; Nguyen, V.Q.; Han, J.H.; Lowstuter, K.; Longmate, J.; Sommer, S.S.; et al. Beyond Li Fraumeni Syndrome: Clinical characteristics of families with p53 germline mutations. J. Clin. Oncol. 2009, 27, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Lalloo, F.; Varley, J.; Moran, A.; Ellis, D.; O’Dair, L.; Pharoah, P.; Antoniou, A.; Hartley, R.; Shenton, A.; Seal, S.; et al. BRCA1, BRCA2 and TP53 mutations in very early-onset breast cancer with associated risks to relatives. Eur. J. Cancer 2006, 42, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- McCuaig, J.M.; Armel, S.R.; Novokmet, A.; Ginsburg, O.M.; Demsky, R.; Narod, S.A.; Malkin, D. Routine TP53 testing for breast cancer under age 30: Ready for prime time? Fam. Cancer 2012, 11, 607–613. [Google Scholar] [CrossRef]
- Mouchawar, J.; Korch, C.; Byers, T.; Pitts, T.M.; Li, E.; McCredie, M.R.; Giles, G.G.; Hopper, J.L.; Southey, M.C. Population-based estimate of the contribution of TP53 mutations to subgroups of early-onset breast cancer: Australian Breast Cancer Family Study. Cancer Res. 2010, 70, 4795–4800. [Google Scholar] [CrossRef]
- Kamihara, J.; Rana, H.Q.; Garber, J.E. Germline TP53 mutations and the changing landscape of Li-Fraumeni syndrome. Hum. Mutat. 2014, 35, 654–662. [Google Scholar] [CrossRef]
- Valdez, J.M.; Nichols, K.E.; Kesserwan, C. Li-Fraumeni syndrome: A paradigm for the understanding of hereditary cancer predisposition. Br. J. Haematol. 2017, 176, 539–552. [Google Scholar] [CrossRef]
- Mandelker, D.; Donoghue, M.; Talukdar, S.; Bandlamudi, C.; Srinivasan, P.; Vivek, M.; Jezdic, S.; Hanson, H.; Snape, K.; Kulkarni, A.; et al. Germline-focussed analysis of tumour-only sequencing: Recommendations from the ESMO Precision Medicine Working Group. Ann. Oncol. 2019, 30, 1221–1231. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Bubien, V.; Bonnet, F.; Brouste, V.; Hoppe, S.; Barouk-Simonet, E.; David, A.; Edery, P.; Bottani, A.; Layet, V.; Caron, O.; et al. High cumulative risks of cancer in patients with PTEN hamartoma tumour syndrome. J. Med. Genet. 2013, 50, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Pilarski, R.; Burt, R.; Kohlman, W.; Pho, L.; Shannon, K.M.; Swisher, E. Cowden syndrome and the PTEN hamartoma tumor syndrome: Systematic review and revised diagnostic criteria. J. Natl. Cancer Inst. 2013, 105, 1607–1616. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.H.; Mester, J.L.; Ngeow, J.; Rybicki, L.A.; Orloff, M.S.; Eng, C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin. Cancer Res. 2012, 18, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Mandelker, D.; Zhang, L.; Kemel, Y.; Stadler, Z.K.; Joseph, V.; Zehir, A.; Pradhan, N.; Arnold, A.; Walsh, M.F.; Li, Y.; et al. Mutation Detection in Patients With Advanced Cancer by Universal Sequencing of Cancer-Related Genes in Tumor and Normal DNA vs Guideline-Based Germline Testing. JAMA 2017, 318, 825–835. [Google Scholar] [CrossRef] [PubMed]
- Hollander, M.C.; Blumenthal, G.M.; Dennis, P.A. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat. Rev. Cancer 2011, 11, 289–301. [Google Scholar] [CrossRef]
- Komiya, T.; Blumenthal, G.M.; DeChowdhury, R.; Fioravanti, S.; Ballas, M.S.; Morris, J.; Hornyak, T.J.; Wank, S.; Hewitt, S.M.; Morrow, B.; et al. A Pilot Study of Sirolimus in Subjects with Cowden Syndrome or Other Syndromes Characterized by Germline Mutations in PTEN. Oncologist 2019, 24, 1510-e1265. [Google Scholar] [CrossRef]
- Marsh, D.J.; Trahair, T.N.; Martin, J.L.; Chee, W.Y.; Walker, J.; Kirk, E.P.; Baxter, R.C.; Marshall, G.M. Rapamycin treatment for a child with germline PTEN mutation. Nat. Clin. Pract. Oncol. 2008, 5, 357–361. [Google Scholar] [CrossRef]
- Schmid, G.L.; Kässner, F.; Uhlig, H.H.; Körner, A.; Kratzsch, J.; Händel, N.; Zepp, F.P.; Kowalzik, F.; Laner, A.; Starke, S.; et al. Sirolimus treatment of severe PTEN hamartoma tumor syndrome: Case report and in vitro studies. Pediatr. Res. 2014, 75, 527–534. [Google Scholar] [CrossRef]
- Tomlinson, I.P.; Houlston, R.S. Peutz-Jeghers syndrome. J. Med. Genet. 1997, 34, 1007–1011. [Google Scholar] [CrossRef]
- Hemminki, A.; Markie, D.; Tomlinson, I.; Avizienyte, E.; Roth, S.; Loukola, A.; Bignell, G.; Warren, W.; Aminoff, M.; Höglund, P.; et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998, 391, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Jenne, D.E.; Reimann, H.; Nezu, J.; Friedel, W.; Loff, S.; Jeschke, R.; Müller, O.; Back, W.; Zimmer, M. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat. Genet. 1998, 18, 38–43. [Google Scholar] [CrossRef] [PubMed]
- McGarrity, T.J.; Amos, C.I.; Frazier, M.L.; Wei, C. Peutz-Jeghers Syndrome. In GeneReviews. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1266/ (accessed on 10 November 2020).
- Giardiello, F.M.; Trimbath, J.D. Peutz-Jeghers syndrome and management recommendations. Clin. Gastroenterol. Hepatol. 2006, 4, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, A.; Akahane, T.; Tsuruta, T.; Kobayashi, Y.; Masuda, K.; Banno, K.; Fujii, T.; Susumu, N.; Itsubo, T.; Kameyama, K.; et al. Lobular endocervical glandular hyperplasia and peritoneal pigmentation associated with Peutz-Jeghers syndrome due to a germline mutation of STK11. Ann. Oncol. 2012, 23, 2990–2992. [Google Scholar] [CrossRef]
- Karamurzin, Y.S.; Kiyokawa, T.; Parkash, V.; Jotwani, A.R.; Patel, P.; Pike, M.C.; Soslow, R.A.; Park, K.J. Gastric-type Endocervical Adenocarcinoma: An Aggressive Tumor With Unusual Metastatic Patterns and Poor Prognosis. Am. J. Surg. Pathol. 2015, 39, 1449–1457. [Google Scholar] [CrossRef]
- Syngal, S.; Brand, R.E.; Church, J.M.; Giardiello, F.M.; Hampel, H.L.; Burt, R.W. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am. J. Gastroenterol. 2015, 110, 223–262. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Xu, L.; Seki, M.; Yokoyama, T.T.; Kasahara, M.; Kashima, Y.; Ohashi, A.; Shimada, Y.; Motoi, N.; Tsuchihara, K.; et al. Long-read sequencing for non-small-cell lung cancer genomes. Genome Res. 2020, 30, 1243–1257. [Google Scholar] [CrossRef]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef]
- Tsaousis, G.N.; Papadopoulou, E.; Apessos, A.; Agiannitopoulos, K.; Pepe, G.; Kampouri, S.; Diamantopoulos, N.; Floros, T.; Iosifidou, R.; Katopodi, O.; et al. Analysis of hereditary cancer syndromes by using a panel of genes: Novel and multiple pathogenic mutations. BMC Cancer 2019, 19, 535. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Allen, B.; Kaldate, R.R.; Bowles, K.R.; Judkins, T.; Kaushik, P.; Roa, B.B.; Wenstrup, R.J.; Hartman, A.R.; Syngal, S. Identification of a Variety of Mutations in Cancer Predisposition Genes in Patients With Suspected Lynch Syndrome. Gastroenterology 2015, 149, 604–613.e620. [Google Scholar] [CrossRef]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-McCune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef] [PubMed]
- Schultz, K.A.P.; Williams, G.M.; Kamihara, J.; Stewart, D.R.; Harris, A.K.; Bauer, A.J.; Turner, J.; Shah, R.; Schneider, K.; Schneider, K.W.; et al. DICER1 and Associated Conditions: Identification of At-risk Individuals and Recommended Surveillance Strategies. Clin. Cancer Res. 2018, 24, 2251–2261. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.R.; Best, A.F.; Williams, G.M.; Harney, L.A.; Carr, A.G.; Harris, A.K.; Kratz, C.P.; Dehner, L.P.; Messinger, Y.H.; Rosenberg, P.S.; et al. Neoplasm Risk Among Individuals With a Pathogenic Germline Variant in DICER1. J. Clin. Oncol. 2019, 37, 668–676. [Google Scholar] [CrossRef]
- Bailey, K.M.; Jacobs, M.F.; Anderson, B.; Rabah, R.; Wu, Y.M.; Else, T.; Mody, R.J. DICER1 Mutations in the Era of Expanding Integrative Clinical Sequencing in Pediatric Oncology. JCO Precis. Oncol. 2019, 3. [Google Scholar] [CrossRef] [PubMed]
- Kurian, A.W.; Ward, K.C.; Hamilton, A.S.; Deapen, D.M.; Abrahamse, P.; Bondarenko, I.; Li, Y.; Hawley, S.T.; Morrow, M.; Jagsi, R.; et al. Uptake, Results, and Outcomes of Germline Multiple-Gene Sequencing After Diagnosis of Breast Cancer. JAMA Oncol. 2018, 4, 1066–1072. [Google Scholar] [CrossRef]
- LaDuca, H.; Polley, E.C.; Yussuf, A.; Hoang, L.; Gutierrez, S.; Hart, S.N.; Yadav, S.; Hu, C.; Na, J.; Goldgar, D.E.; et al. A clinical guide to hereditary cancer panel testing: Evaluation of gene-specific cancer associations and sensitivity of genetic testing criteria in a cohort of 165,000 high-risk patients. Genet. Med. 2020, 22, 407–415. [Google Scholar] [CrossRef]
- Conley, J.M.; Cook-Deegan, R.; Lázaro-Muñoz, G. MYRIAD AFTER MYRIAD: THE PROPRIETARY DATA DILEMMA. North Carol. J. Law Technol. 2014, 15, 597–637. [Google Scholar]
- Cook-Deegan, R.; Niehaus, A. After Myriad: Genetic Testing in the Wake of Recent Supreme Court Decisions about Gene Patents. Curr. Genet. Med. Rep. 2014, 2, 223–241. [Google Scholar] [CrossRef]
- Daly, M.B.; Pilarski, R.; Yurgelun, M.B.; Berry, M.P.; Buys, S.S.; Dickson, P.; Domchek, S.M.; Elkhanany, A.; Friedman, S.; Garber, J.E.; et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 1.2020. J. Natl. Compr. Cancer Netw. 2020, 18, 380–391. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Seng, S.; Yoo, J.; Equivel, P.; Lum, S.S. Clinical Management of Patients at Risk for Hereditary Breast Cancer with Variants of Uncertain Significance in the Era of Multigene Panel Testing. Ann. Surg. Oncol. 2019, 26, 3389–3396. [Google Scholar] [CrossRef] [PubMed]
- Ndugga-Kabuye, M.K.; Issaka, R.B. Inequities in multi-gene hereditary cancer testing: Lower diagnostic yield and higher VUS rate in individuals who identify as Hispanic, African or Asian and Pacific Islander as compared to European. Fam. Cancer 2019, 18, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Timms, K.M.; Abkevich, V.; Hughes, E.; Neff, C.; Reid, J.; Morris, B.; Kalva, S.; Potter, J.; Tran, T.V.; Chen, J.; et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast. Cancer Res. 2014, 16, 475. [Google Scholar] [CrossRef]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kübler, K.; Mouw, K.W.; et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 2017, 49, 1476–1486. [Google Scholar] [CrossRef]
- Maxwell, K.N.; Wubbenhorst, B.; Wenz, B.M.; De Sloover, D.; Pluta, J.; Emery, L.; Barrett, A.; Kraya, A.A.; Anastopoulos, I.N.; Yu, S.; et al. BRCA locus-specific loss of heterozygosity in germline BRCA1 and BRCA2 carriers. Nat. Commun. 2017, 8, 319. [Google Scholar] [CrossRef]
- Moore, K.N.; Secord, A.A.; Geller, M.A.; Miller, D.S.; Cloven, N.; Fleming, G.F.; Wahner Hendrickson, A.E.; Azodi, M.; DiSilvestro, P.; Oza, A.M.; et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 636–648. [Google Scholar] [CrossRef]
- Byrum, A.K.; Vindigni, A.; Mosammaparast, N. Defining and Modulating ‘BRCAness’. Trends Cell Biol. 2019, 29, 740–751. [Google Scholar] [CrossRef]
- Hoppe, M.M.; Sundar, R.; Tan, D.S.P.; Jeyasekharan, A.D. Biomarkers for Homologous Recombination Deficiency in Cancer. J. Natl. Cancer Inst. 2018, 110, 704–713. [Google Scholar] [CrossRef]
- Jiang, X.; Li, X.; Li, W.; Bai, H.; Zhang, Z. PARP inhibitors in ovarian cancer: Sensitivity prediction and resistance mechanisms. J. Cell Mol. Med. 2019, 23, 2303–2313. [Google Scholar] [CrossRef] [PubMed]
- Pilié, P.G.; Gay, C.M.; Byers, L.A.; O’Connor, M.J.; Yap, T.A. PARP Inhibitors: Extending Benefit Beyond BRCA-Mutant Cancers. Clin. Cancer Res. 2019, 25, 3759–3771. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.H.; Chen, M.; Tsai, L.W.; Lo, C.; Yen, T.C.; Huang, T.Y.; Chen, C.K.; Fan, S.C.; Kuo, S.H.; Huang, C.S. Using next-generation sequencing to redefine BRCAness in triple-negative breast cancer. Cancer Sci. 2020, 111, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Slavin, T.P.; Coffee, B.; Bernhisel, R.; Logan, J.; Cox, H.C.; Marcucci, G.; Weitzel, J.; Neuhausen, S.L.; Mancini-DiNardo, D. Prevalence and characteristics of likely-somatic variants in cancer susceptibility genes among individuals who had hereditary pan-cancer panel testing. Cancer Genet. 2019, 235–236, 31–38. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Li, M.M.; Chao, E.; Esplin, E.D.; Miller, D.T.; Nathanson, K.L.; Plon, S.E.; Scheuner, M.T.; Stewart, D.R. Points to consider for reporting of germline variation in patients undergoing tumor testing: A statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2020, 22, 1142–1148. [Google Scholar] [CrossRef]
- Pujol, P.; Vande Perre, P.; Faivre, L.; Sanlaville, D.; Corsini, C.; Baertschi, B.; Anahory, M.; Vaur, D.; Olschwang, S.; Soufir, N.; et al. Guidelines for reporting secondary findings of genome sequencing in cancer genes: The SFMPP recommendations. Eur. J. Hum. Genet. 2018, 26, 1732–1742. [Google Scholar] [CrossRef]
- Schrader, K.A.; Cheng, D.T.; Joseph, V.; Prasad, M.; Walsh, M.; Zehir, A.; Ni, A.; Thomas, T.; Benayed, R.; Ashraf, A.; et al. Germline Variants in Targeted Tumor Sequencing Using Matched Normal DNA. JAMA Oncol. 2016, 2, 104–111. [Google Scholar] [CrossRef]
- Seifert, B.A.; O’Daniel, J.M.; Amin, K.; Marchuk, D.S.; Patel, N.M.; Parker, J.S.; Hoyle, A.P.; Mose, L.E.; Marron, A.; Hayward, M.C.; et al. Germline Analysis from Tumor-Germline Sequencing Dyads to Identify Clinically Actionable Secondary Findings. Clin. Cancer Res. 2016, 22, 4087–4094. [Google Scholar] [CrossRef]


{kind=link}
| Hereditary Gynecological Cancer | Causative Genes | Related Gynecological Cancers | Related Non-Gynecological Cancers | Drug Sensitivity |
|---|---|---|---|---|
| HBOC | BRCA1, BRCA2 | Ovarian cancer | Breast cancer Prostate cancer Pancreatic cancer | PARP inhibitors |
| Lynch syndrome | MLH1, MSH2, MSH6, PMS2, EPCAM | Endometrial cancer Ovarian cancer | Colorectal cancer Small intestine cancer Ureteral cancer Renal pelvis cancer | ICI |
| Li–Fraumeni syndrome | TP53 | Insufficient evidence of ovarian cancer | Soft tissue sarcoma Osteosarcoma Premenopausal breast cancer Colorectal cancer Gastric cancer Adrenocortical carcinoma Brain tumor | - |
| Cowden syndrome | PTEN | Endometrial cancer | Breast cancer Epithelial thyroid cancer 1 Gastrointestinal hamartoma | - |
| Peutz–Jeghers syndrome | STK11 | Ovarian tumor (SCAT) Cervical tumor (MDA, LEGH) | Colorectal cancer Gastric cancer Pancreatic cancer Breast cancer | - |
| Other cancer-susceptible genes | RAD51C, RAD51D Etc. | Ovarian cancer | Various cancers | HRD status→PARP inhibitors |
| DICER1 | Sertoli–Leydig cell tumor (SLCT) of the ovary | Pleuropulmonary blastoma | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ueki, A.; Hirasawa, A. Molecular Features and Clinical Management of Hereditary Gynecological Cancers. Int. J. Mol. Sci. 2020, 21, 9504. https://doi.org/10.3390/ijms21249504
Ueki A, Hirasawa A. Molecular Features and Clinical Management of Hereditary Gynecological Cancers. International Journal of Molecular Sciences. 2020; 21(24):9504. https://doi.org/10.3390/ijms21249504
Chicago/Turabian StyleUeki, Arisa, and Akira Hirasawa. 2020. "Molecular Features and Clinical Management of Hereditary Gynecological Cancers" International Journal of Molecular Sciences 21, no. 24: 9504. https://doi.org/10.3390/ijms21249504
APA StyleUeki, A., & Hirasawa, A. (2020). Molecular Features and Clinical Management of Hereditary Gynecological Cancers. International Journal of Molecular Sciences, 21(24), 9504. https://doi.org/10.3390/ijms21249504