Albanol B from Mulberries Exerts Anti-Cancer Effect through Mitochondria ROS Production in Lung Cancer Cells and Suppresses In Vivo Tumor Growth

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
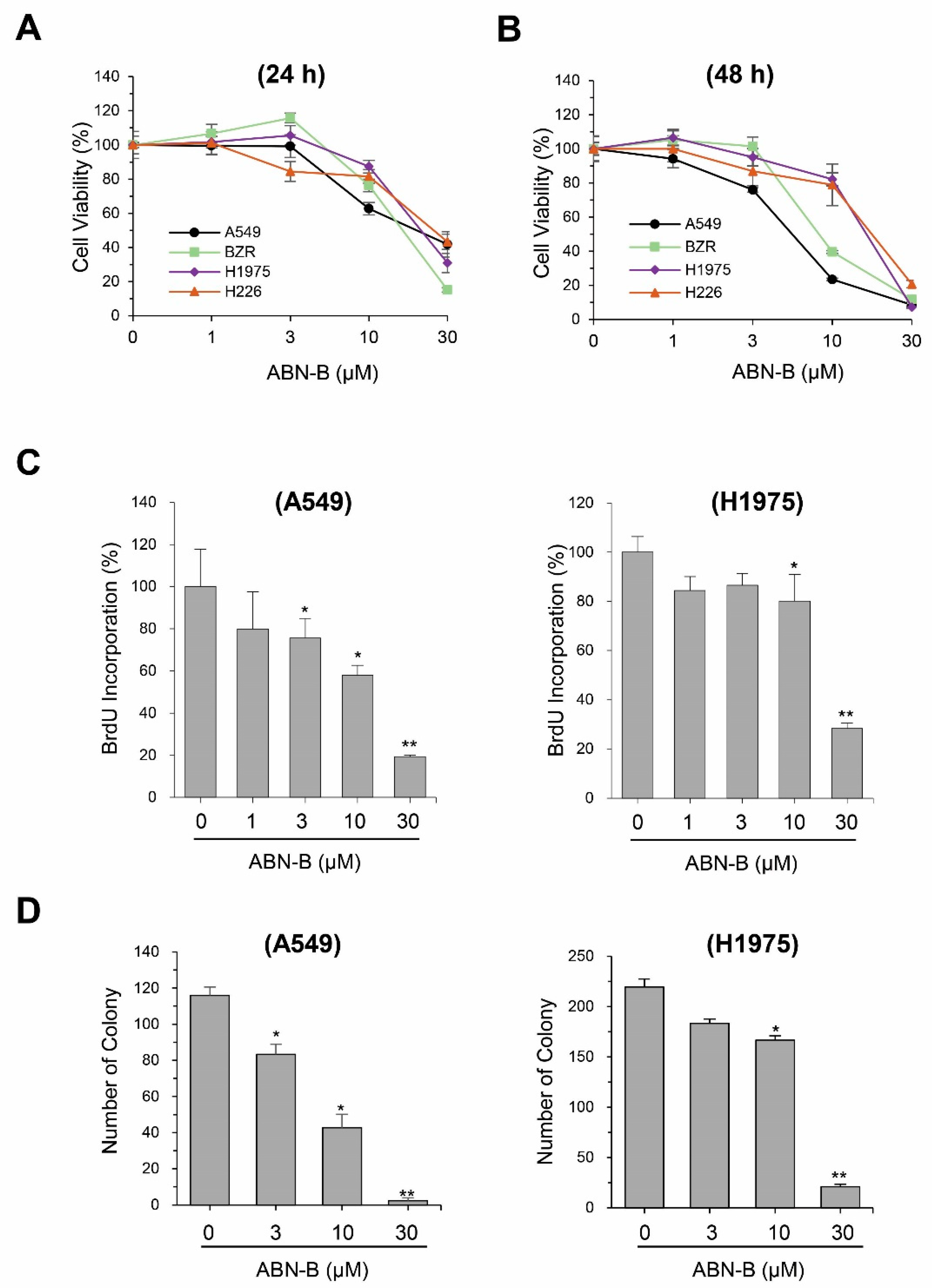
2.1. ABN-B Inhibits the Proliferation of Human Lung Cancer Cells
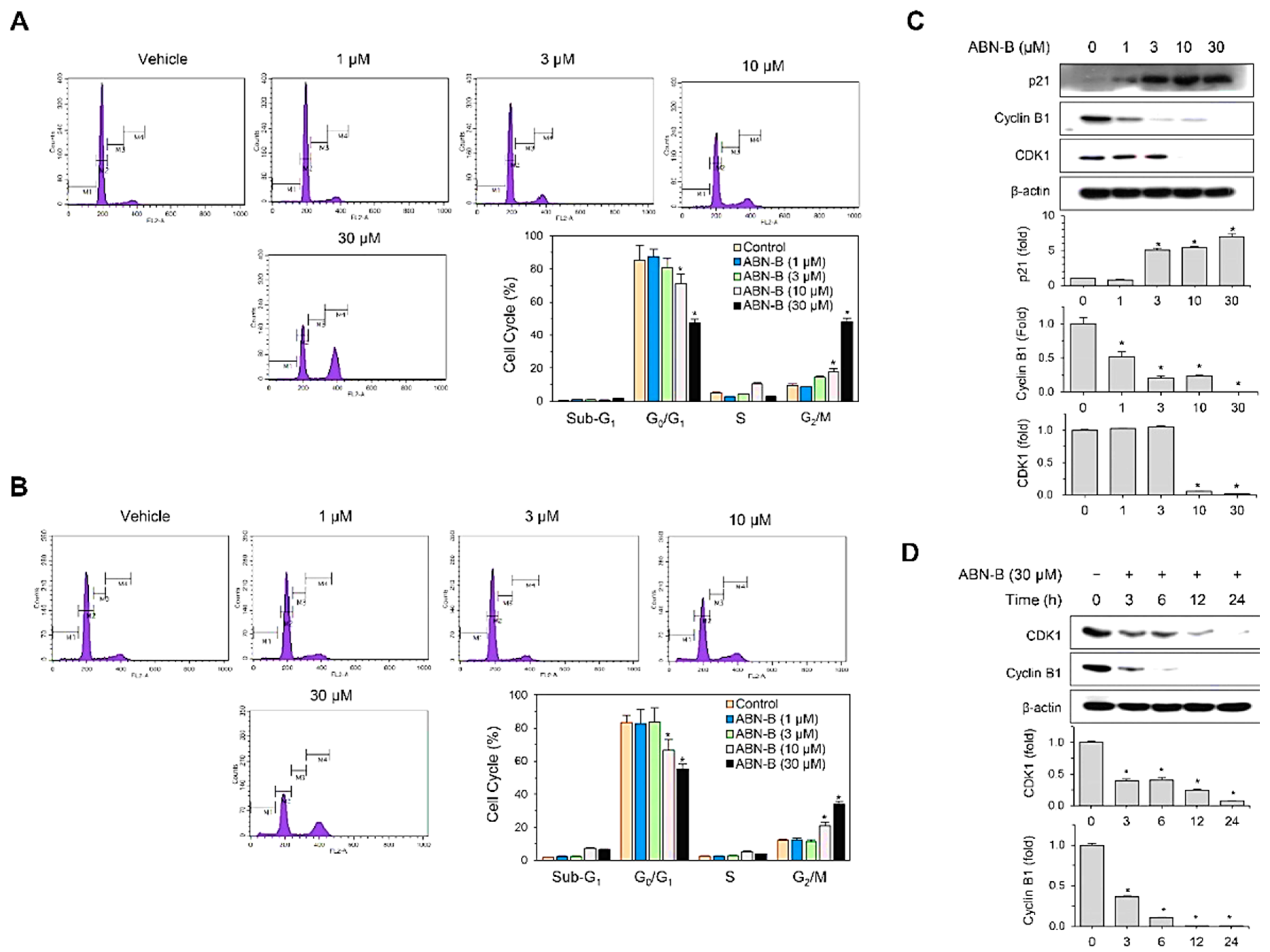
2.2. ABN-B Induces Cell Cycle Arrest at G2/M in A549 and NCI-H1975 Cells by Down-Regulating CDK1 and Cyclin B1, but Up-Regulating p21 Protein Expression
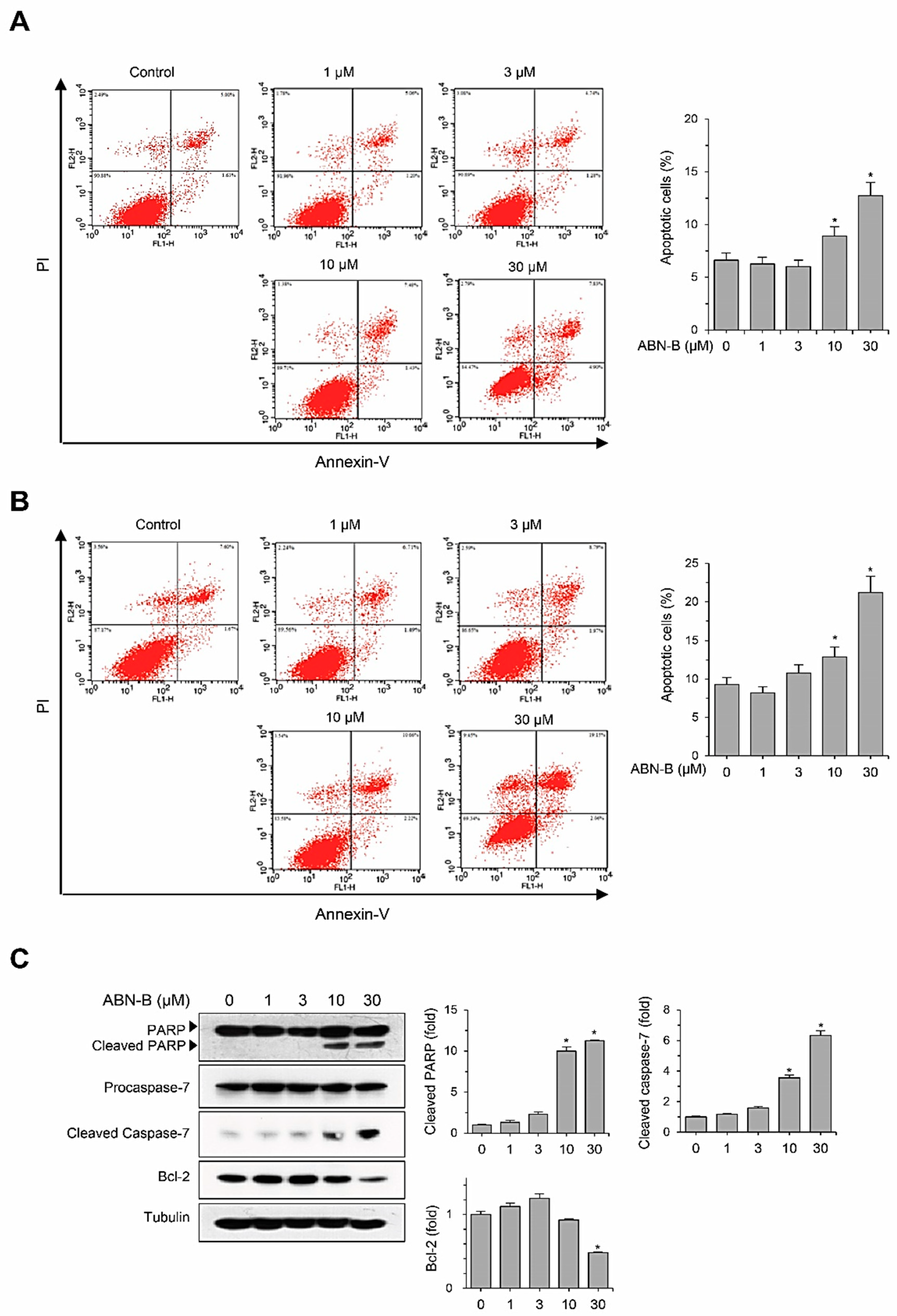
2.3. ABN-B Induces Apoptosis in A549 Cells
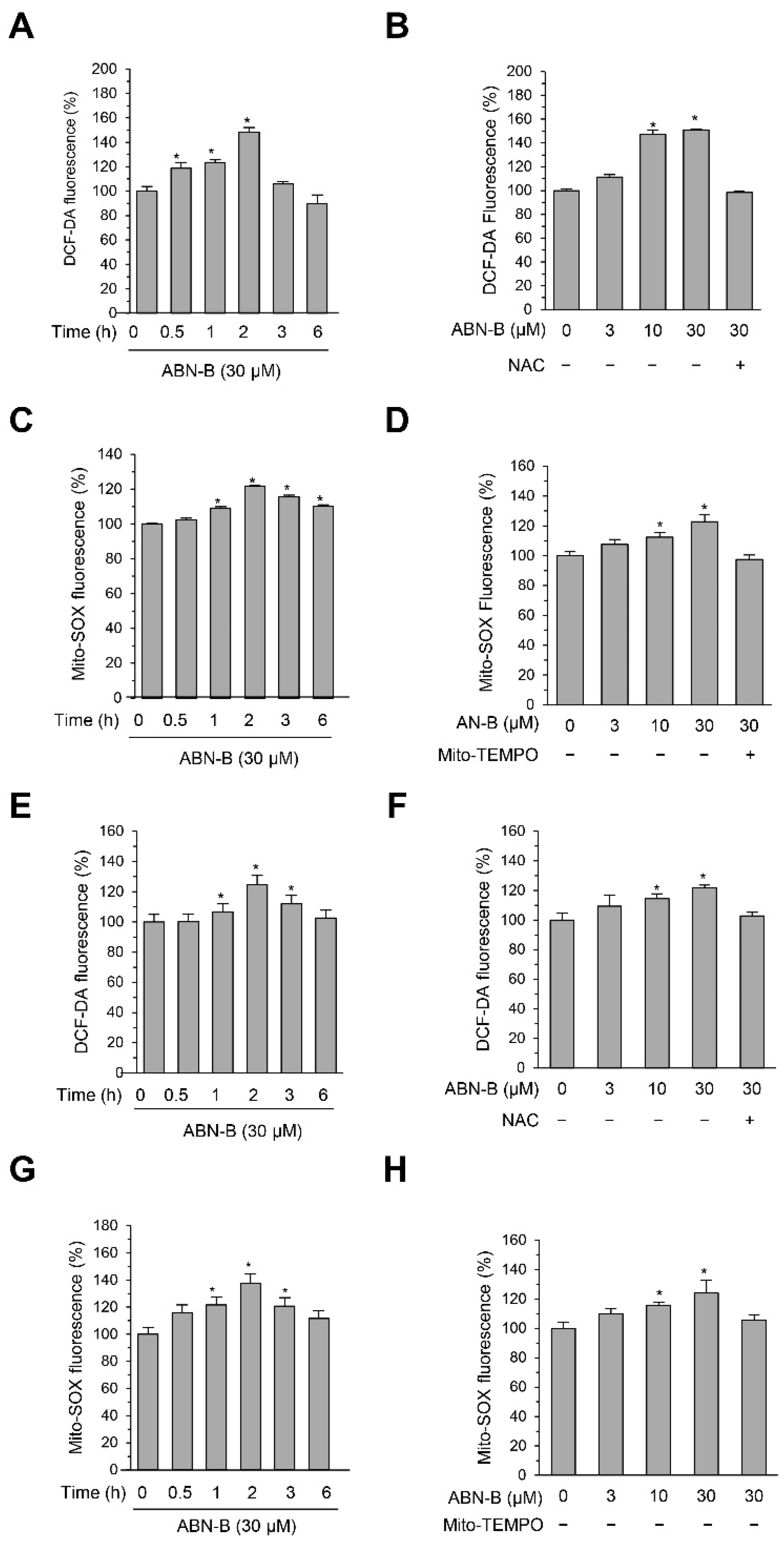
2.4. ABN-B Induced Mitochondrial ROS Production in Both A549 and H1975 Cells
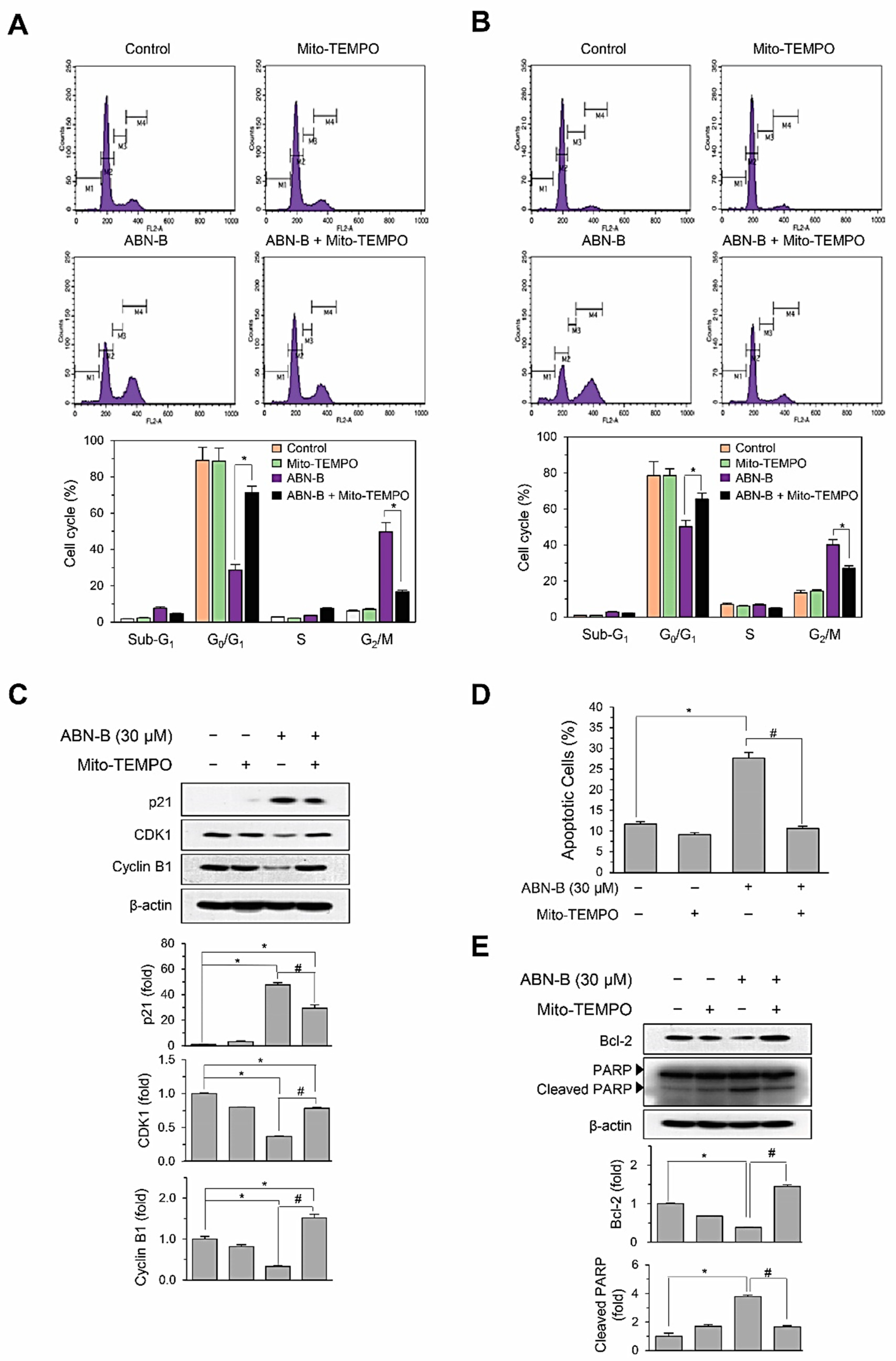
2.5. Mito-TEMPO Attenuated ABN-B-Induced Apoptosis and G2/M Phase Cell Cycle Arrest in Human Lung Cancer Cells
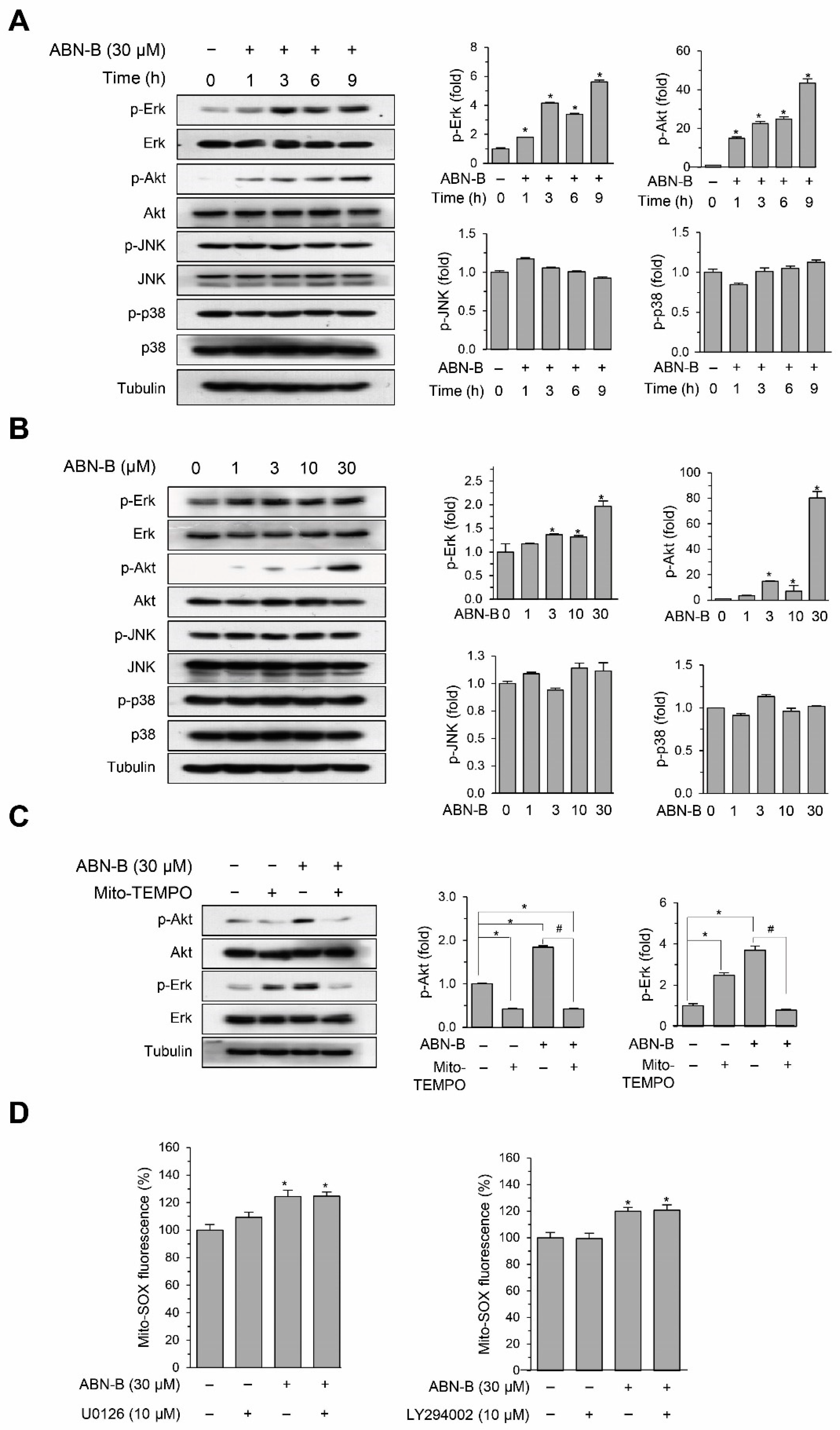
2.6. ABN-B Induced the Phosphorylation of ERK1/2 and AKT via the Mitochondrial ROS Production
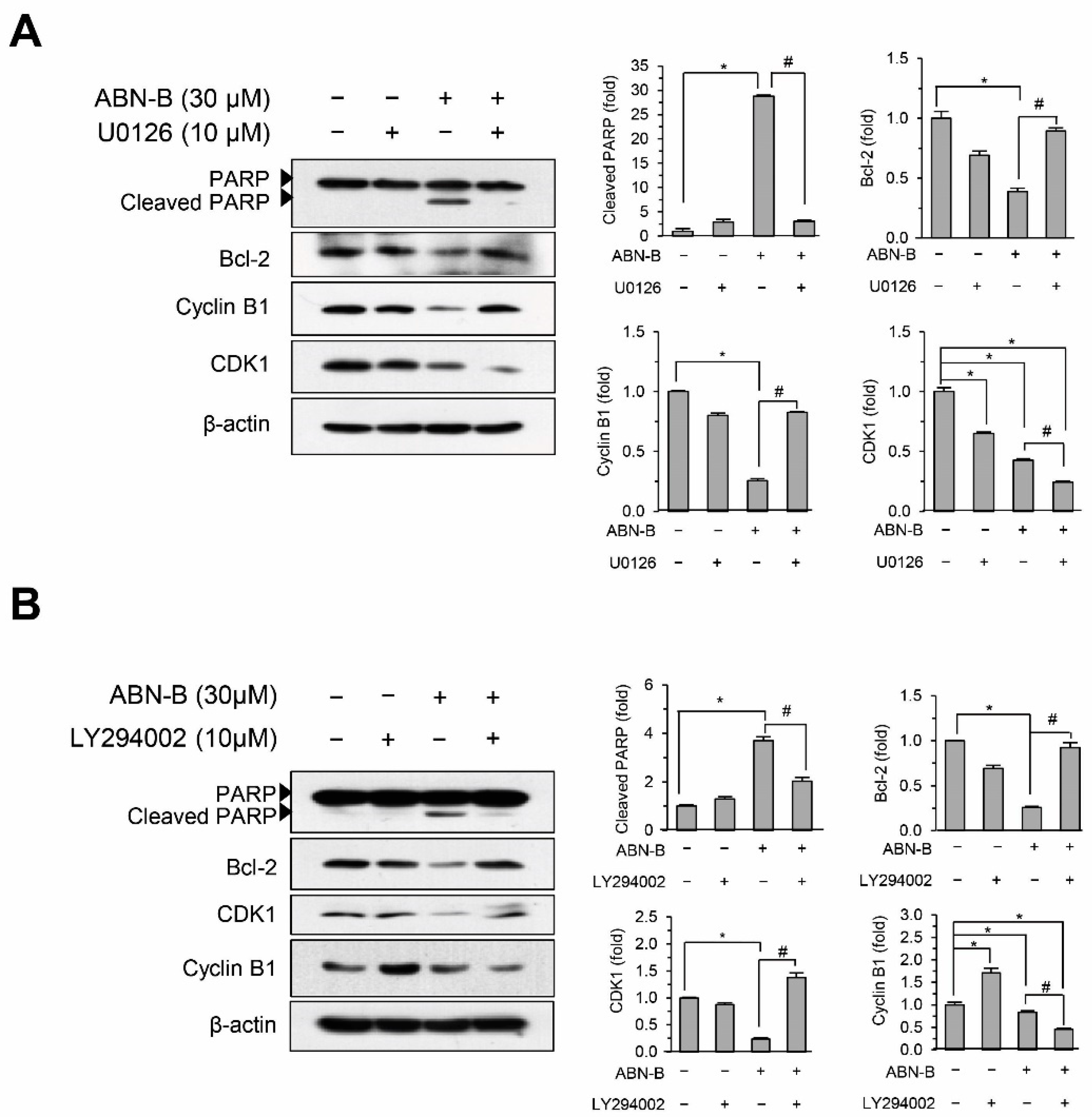
2.7. Inhibition of ERK1/2 and AKT Attenuated ABN-B-Induced Apoptosis and G2/M Arrest
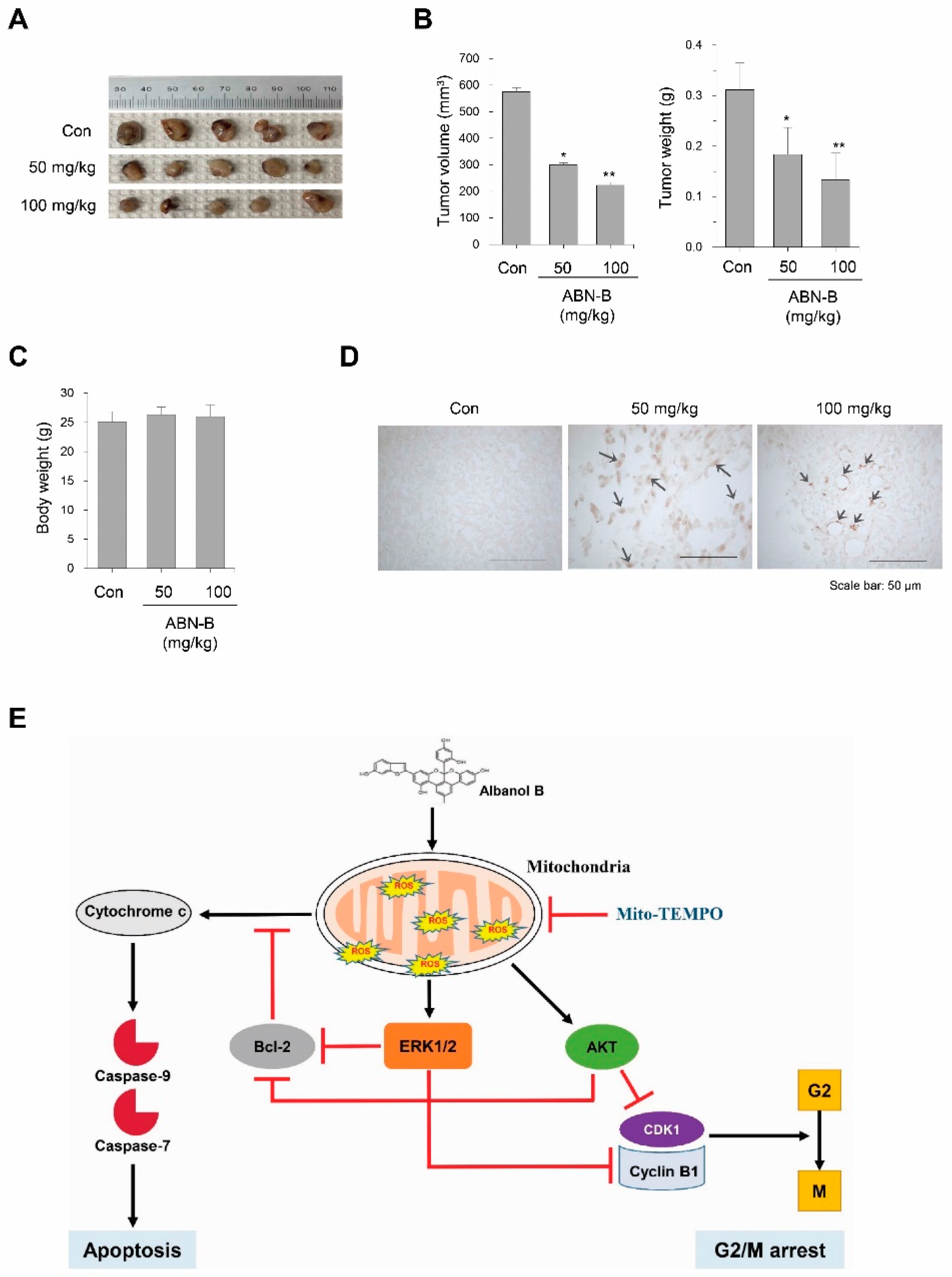
2.8. ABN-B Suppressed Tumor Growth in Ex-3LL Tumor-Bearing Mice
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Chemicals and Reagents
4.3. Isolation of ABN-B
4.4. Cell Viability and Proliferation Assays
4.5. Colony Formation Assay
4.6. Annexin V/PI Double Staining
4.7. Western Blot Analysis
4.8. Cell Cycle Distribution Analysis
4.9. Measurement of Intracellular and Mitochondrial ROS Level
4.10. In Vivo Tumor Growth Assay and Detection of Apoptosis in Tumor Tissues
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABN-B | Albanol B |
| ATF3 | Cyclic AMP-dependent transcription factor 3 |
| Bcl-2 | B-cell lymphoma 2 |
| BrdU | Bromodeoxyuridine |
| CC | Column chromatography |
| CDK1 | Cyclin-dependent kinase 1 |
| DCF-DA | 2′,7′-Dichlorofluorescin diacetate |
| DUSP1 | Dual specificity protein phosphatase 1 |
| DUSP6 | Dual specificity protein phosphatase 6 |
| EDTA | Ethylenediaminetetraacetic acid |
| ERK1/2 | Extracellular signal-regulated kinase 1/2 |
| EGFR | Epidermal growth factor receptor |
| FITC | Fluorescein isothiocyanate |
| FACS | Fluorescence-activated cell sorting |
| HIFs | Hypoxia-inducible factors |
| JCBR | Japanese Collection of Research Biosources |
| JNK | c-Jun n-terminal kinase |
| MAPKs | Mitogen-activated protein kinases |
| MEKK | Mitogen-activated protein kinase kinase |
| MDR1 | P-glycoprotein or ABCB1 |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide |
| NAC | N-Acetylcysteine |
| NSCLC | Non-small cell lung carcinoma |
| P21 | Cyclin-dependent kinase inhibitor 1 |
| PARP | Poly (ADP-ribose) polymerase |
| ROS | Reactive oxygen species |
| PBS | Phosphate-Buffered Saline |
| PI | Propidium iodide |
| PI3K | Phosphatidylinositol 3-kinase |
| STAT | Signal transducers and activators of transcription |
| TUNEL | Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling |
| YB-1 | Y-box binding protein 1 |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.-K. Non-small-cell lung cancers: A heterogeneous set of diseases. Nat. Rev. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef]
- Denisenko, T.V.; Budkevich, I.N.; Zhivotovsky, B. Cell death-based treatment of lung adenocarcinoma. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Dubey, S.; Schiller, J.H. Three Emerging New Drugs for NSCLC: Pemetrexed, Bortezomib, and Cetuximab. Oncologist 2005, 10, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Auten, R.L.; Davis, J.M. Oxygen Toxicity and Reactive Oxygen Species: The Devil Is in the Details. Pediatr. Res. 2009, 66, 121–127. [Google Scholar] [CrossRef]
- Tong, L.; Chuang, C.-C.; Wu, S.; Zuo, L. Reactive oxygen species in redox cancer therapy. Cancer Lett. 2015, 367, 18–25. [Google Scholar] [CrossRef]
- Galadari, S.; Cheratta, A.R.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive Oxygen Species Generated at Mitochondrial Complex III Stabilize Hypoxia-inducible Factor-1α during Hypoxia: A mechanism of o2 sensing. J. Biol. Chem. 2000, 275, 25130–25138. [Google Scholar] [CrossRef]
- Verbon, E.H.; Post, J.A.; Boonstra, J. The influence of reactive oxygen species on cell cycle progression in mammalian cells. Gene 2012, 511, 1–6. [Google Scholar] [CrossRef]
- McCubrey, J.A.; LaHair, M.M.; Franklin, R.A. Reactive Oxygen Species-Induced Activation of the MAP Kinase Signaling Pathways. Antioxid. Redox Signal. 2006, 8, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M.; Jaruga, P. Mechanisms of free radical-induced damage to DNA. Free Radic. Res. 2012, 46, 382–419. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.W.C.; Lye, P.-Y.; Wong, S.-K. Phytochemistry, pharmacology, and clinical trials of Morus alba. Chin. J. Nat. Med. 2016, 14, 17–30. [Google Scholar]
- Wen, P.; Hu, T.-G.; Linhardt, R.J.; Liao, S.; Wu, H.; Zou, Y. Mulberry: A review of bioactive compounds and advanced processing technology. Trends Food Sci. Technol. 2019, 83, 138–158. [Google Scholar] [CrossRef]
- Zhang, Y.; Ren, C.; Lu, G.; Cui, W.; Mu, Z.; Gao, H.; Wang, Y. Purification, characterization and anti-diabetic activity of a polysaccharide from mulberry leaf. Regul. Toxicol. Pharmacol. 2014, 70, 687–695. [Google Scholar] [CrossRef]
- Yuan, Q.; Xie, Y.; Wang, W.; Yan, Y.; Ye, H.; Jabbar, S.; Zeng, X. Extraction optimization, characterization and antioxidant activity in vitro of polysaccharides from mulberry (Morus alba L.) leaves. Carbohydr. Polym. 2015, 128, 52–62. [Google Scholar] [CrossRef]
- Shrikanta, A.; Kumar, A.; Vijayalakshmi, G. Resveratrol content and antioxidant properties of underutilized fruits. J. Food Sci. Technol. 2015, 52, 383–390. [Google Scholar] [CrossRef]
- Liu, C.J.; Lin, J.Y. Anti-inflammatory and anti-apoptotic effects of strawberry and mulberry fruit polysaccharides on lipopolysaccharide-stimulated macrophages through modulating pro-/anti-inflammatory cytokines secretion and Bcl-2/Bak protein ratio. Food Chem. Toxicol. 2012, 50, 3032–3039. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Tien, Y.-J.; Chen, C.-H.; Beltran, F.N.; Amor, E.C.; Wang, R.-J.; Wu, D.-J.; Mettling, C.; Lin, Y.-L.; Yang, W.-C. Morus alba and active compound oxyresveratrol exert anti-inflammatory activity via inhibition of leukocyte migration involving MEK/ERK signaling. BMC Complement. Altern. Med. 2013, 13, 45. [Google Scholar] [CrossRef]
- Kang, T.H.; Hur, J.Y.; Kim, H.B.; Ryu, J.H.; Kim, S.Y. Neuroprotective effects of the cyanidin-3-O-β-d-glucopyranoside isolated from mulberry fruit against cerebral ischemia. Neurosci. Lett. 2006, 391, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-D.; Liu, Z.-H.; Zhang, L.-F.; Yao, J.-N.; Wang, C.-F. Sanggenon C induces apoptosis of colon cancer cells via inhibition of NO production, iNOS expression and ROS activation of the mitochondrial pathway. Oncol. Rep. 2017, 38, 2123–2131. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, D.; Mao, J.; Ke, X.-X.; Zhang, R.; Yin, C.; Gao, N.; Cui, H. Morusin inhibits cell proliferation and tumor growth by down-regulating c-Myc in human gastric cancer. Oncotarget 2017, 8, 57187–57200. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-J.; Min, T.-R.; Chi, G.-Y.; Choi, Y.-H.; Park, S.-H. Induction of apoptosis by morusin in human non-small cell lung cancer cells by suppression of EGFR/STAT3 activation. Biochem. Biophys. Res. Commun. 2018, 505, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Chen, R.-Y.; Yu, D.-Q. Five New Diels-Alder Type Adducts from the Stem and Root Bark of Morus mongolica. Planta Medica 2006, 72, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-X.; Kim, Y.-J.; Kwon, T.-H.; Tan, C.-P.; Son, K.-H.; Kim, T. Anti-inflammatory effects of mulberry (Morus alba L.) root bark and its active compounds. Nat. Prod. Res. 2018, 34, 1786–1790. [Google Scholar] [CrossRef]
- Kuk, E.B.; Jo, A.R.; Oh, S.I.; Sohn, H.S.; Seong, S.H.; Roy, A.; Choi, J.S.; Jung, H.A. Anti-Alzheimer’s disease activity of compounds from the root bark of Morus alba L. Arch. Pharmacal Res. 2017, 40, 338–349. [Google Scholar] [CrossRef]
- Ha, M.T.; Seong, S.H.; Nguyen, T.D.; Cho, W.-K.; Ah, K.J.; Ma, J.Y.; Woo, M.H.; Choi, J.S.; Min, B.S. Chalcone derivatives from the root bark of Morus alba L. act as inhibitors of PTP1B and α-glucosidase. Phytochemistry 2018, 155, 114–125. [Google Scholar] [CrossRef]
- Paudel, P.; Yu, T.; Seong, S.H.; Kuk, E.B.; Jung, H.A.; Choi, J.S. Protein Tyrosine Phosphatase 1B Inhibition and Glucose Uptake Potentials of Mulberrofuran G, Albanol B, and Kuwanon G from Root Bark of Morus alba L. in Insulin-Resistant HepG2 Cells: An In Vitro and In Silico Study. Int. J. Mol. Sci. 2018, 19, 1542. [Google Scholar] [CrossRef]
- Ramalingam, S.; Belani, C.P. Systemic Chemotherapy for Advanced Non-Small Cell Lung Cancer: Recent Advances and Future Directions. Oncologist 2008, 13, 5–13. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, N.; Zhu, M.; Feng, Y.; Li, H.; Tsao, S. Recent progress on anticancer candidates in patents of herbal medicinal products. Recent Patents Food Nutr. Agric. 2011, 3, 30–48. [Google Scholar] [CrossRef] [PubMed]
- Eo, H.J.; Park, J.H.; Park, G.H.; Lee, M.H.; Lee, J.R.; Koo, J.S.; Jeong, J.B. Anti-inflammatory and anti-cancer activity of mulberry (Morus alba L.) root bark. BMC Complement. Altern. Med. 2014, 14, 200. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.K.; Cho, S.G.; Choi, H.S.; Woo, S.M.; Yun, Y.J.; Shin, Y.C.; Ko, S.G. JNK1/2 Activation by an extract from the roots of Morus alba L. reduces the viability of multidrug resistant MCF-7/Dox cells by inhibiting YB-1-Dependent MDR1 expression. Evid. Based Complement Alternat. Med. 2013, 2013, 741985. [Google Scholar] [CrossRef] [PubMed]
- Dat, N.T.; Jin, X.; Lee, K.; Hong, Y.-S.; Kim, Y.H.; Lee, J.J. Hypoxia-Inducible Factor-1 Inhibitory Benzofurans and Chalcone-Derived Diels−Alder Adducts from Morus Species. J. Nat. Prod. 2009, 72, 39–43. [Google Scholar] [CrossRef]
- Kikuchi, T.; Nihei, M.; Nagai, H.; Fukushi, H.; Tabata, K.; Suzuki, T.; Akihisa, T. Albanol A from the root bark of Morus alba L. induces apoptotic cell death in HL60 human leukemia cell line. Chem. Pharm. Bull. 2010, 58, 568–571. [Google Scholar] [CrossRef]
- Sena, L.A.; Chandel, N.S. Physiological Roles of Mitochondrial Reactive Oxygen Species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef]
- Mileo, A.M.; Miccadei, S. Polyphenols as Modulator of Oxidative Stress in Cancer Disease: New Therapeutic Strategies. Oxidative Med. Cell. Longev. 2016, 2016, 1–17. [Google Scholar] [CrossRef]
- González-Vallinas, M.; González-Castejón, M.; Rodríguez-Casado, A.; De Molina, A.R. Dietary phytochemicals in cancer prevention and therapy: A complementary approach with promising perspectives. Nutr. Rev. 2013, 71, 585–599. [Google Scholar] [CrossRef]
- Siddiqui, S.; Ahamad, S.; Jafri, A.; Afzal, M.; Arshad, M. Piperine Triggers Apoptosis of Human Oral Squamous Carcinoma through Cell Cycle Arrest and Mitochondrial Oxidative Stress. Nutr. Cancer 2017, 69, 791–799. [Google Scholar] [CrossRef]
- Cagnol, S.; Chambard, J.-C. ERK and cell death: Mechanisms of ERK-induced cell death-apoptosis, autophagy and senescence. FEBS J. 2009, 277, 2–21. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, Y.-Z.; Kagan, E.; Bonner, J.C. Peroxynitrite Targets the Epidermal Growth Factor Receptor, Raf-1, and MEK Independently to Activate MAPK. J. Biol. Chem. 2000, 275, 22479–22486. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-J.; Cho, H.-N.; Soh, J.-W.; Jhon, G.J.; Cho, C.-K.; Chung, H.-Y.; Bae, S.; Lee, S.-J.; Lee, Y.-S. Oxidative stress-induced apoptosis is mediated by ERK1/2 phosphorylation. Exp. Cell Res. 2003, 291, 251–266. [Google Scholar] [CrossRef]
- Kamata, H.; Honda, S.-I.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive Oxygen Species Promote TNFα-Induced Death and Sustained JNK Activation by Inhibiting MAP Kinase Phosphatases. Cell 2005, 120, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Levinthal, D.J.; DeFranco, D.B. Reversible Oxidation of ERK-directed Protein Phosphatases Drives Oxidative Toxicity in Neurons. J. Biol. Chem. 2004, 280, 5875–5883. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, V.; Park, Y.; Chen, C.-C.; Xu, P.-Z.; Chen, M.-L.; Tonic, I.; Unterman, T.; Hay, N. Akt Determines Replicative Senescence and Oxidative or Oncogenic Premature Senescence and Sensitizes Cells to Oxidative Apoptosis. Cancer Cell 2008, 14, 458–470. [Google Scholar] [CrossRef]
- Chetram, M.A.; Bethea, D.A.; Odero-Marah, V.A.; Don-Salu-Hewage, A.S.; Jones, K.J.; Hinton, C.V. ROS-mediated activation of AKT induces apoptosis via pVHL in prostate cancer cells. Mol. Cell. Biochem. 2013, 376, 63–71. [Google Scholar] [CrossRef]
- Rai, N.; Sarkar, M.; Raha, S. Piroxicam, a traditional non-steroidal anti-inflammatory drug (NSAID) causes apoptosis by ROS mediated Akt activation. Pharmacol. Rep. 2015, 67, 1215–1223. [Google Scholar] [CrossRef]
- Tsai, M.-H.; Liu, J.-F.; Chiang, Y.-C.; Hu, S.C.-S.; Hsu, L.-F.; Lin, Y.-C.; Lin, Z.-C.; Lee, H.-C.; Chen, M.-C.; Huang, C.-L.; et al. Artocarpin, an isoprenyl flavonoid, induces p53-dependent or independent apoptosis via ROS-mediated MAPKs and Akt activation in non-small cell lung cancer cells. Oncotarget 2017, 8, 28342–28358. [Google Scholar] [CrossRef]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; Van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Shin, M.; Lee, B.-M.; Kim, O.; Tran, H.N.K.; Lee, S.; Hwangbo, C.; Min, B.S.; Lee, J.-H. Triterpenoids fromZiziphus jujubainduce apoptotic cell death in human cancer cells through mitochondrial reactive oxygen species production. Food Funct. 2018, 9, 3895–3905. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phan, T.N.; Kim, O.; Ha, M.T.; Hwangbo, C.; Min, B.-S.; Lee, J.-H. Albanol B from Mulberries Exerts Anti-Cancer Effect through Mitochondria ROS Production in Lung Cancer Cells and Suppresses In Vivo Tumor Growth. Int. J. Mol. Sci. 2020, 21, 9502. https://doi.org/10.3390/ijms21249502
Phan TN, Kim O, Ha MT, Hwangbo C, Min B-S, Lee J-H. Albanol B from Mulberries Exerts Anti-Cancer Effect through Mitochondria ROS Production in Lung Cancer Cells and Suppresses In Vivo Tumor Growth. International Journal of Molecular Sciences. 2020; 21(24):9502. https://doi.org/10.3390/ijms21249502
Chicago/Turabian StylePhan, Thanh Nam, Okwha Kim, Manh Tuan Ha, Cheol Hwangbo, Byung-Sun Min, and Jeong-Hyung Lee. 2020. "Albanol B from Mulberries Exerts Anti-Cancer Effect through Mitochondria ROS Production in Lung Cancer Cells and Suppresses In Vivo Tumor Growth" International Journal of Molecular Sciences 21, no. 24: 9502. https://doi.org/10.3390/ijms21249502
APA StylePhan, T. N., Kim, O., Ha, M. T., Hwangbo, C., Min, B.-S., & Lee, J.-H. (2020). Albanol B from Mulberries Exerts Anti-Cancer Effect through Mitochondria ROS Production in Lung Cancer Cells and Suppresses In Vivo Tumor Growth. International Journal of Molecular Sciences, 21(24), 9502. https://doi.org/10.3390/ijms21249502