Peptide-Based Identification of Phytophthora Isolates and Phytophthora Detection in Planta

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
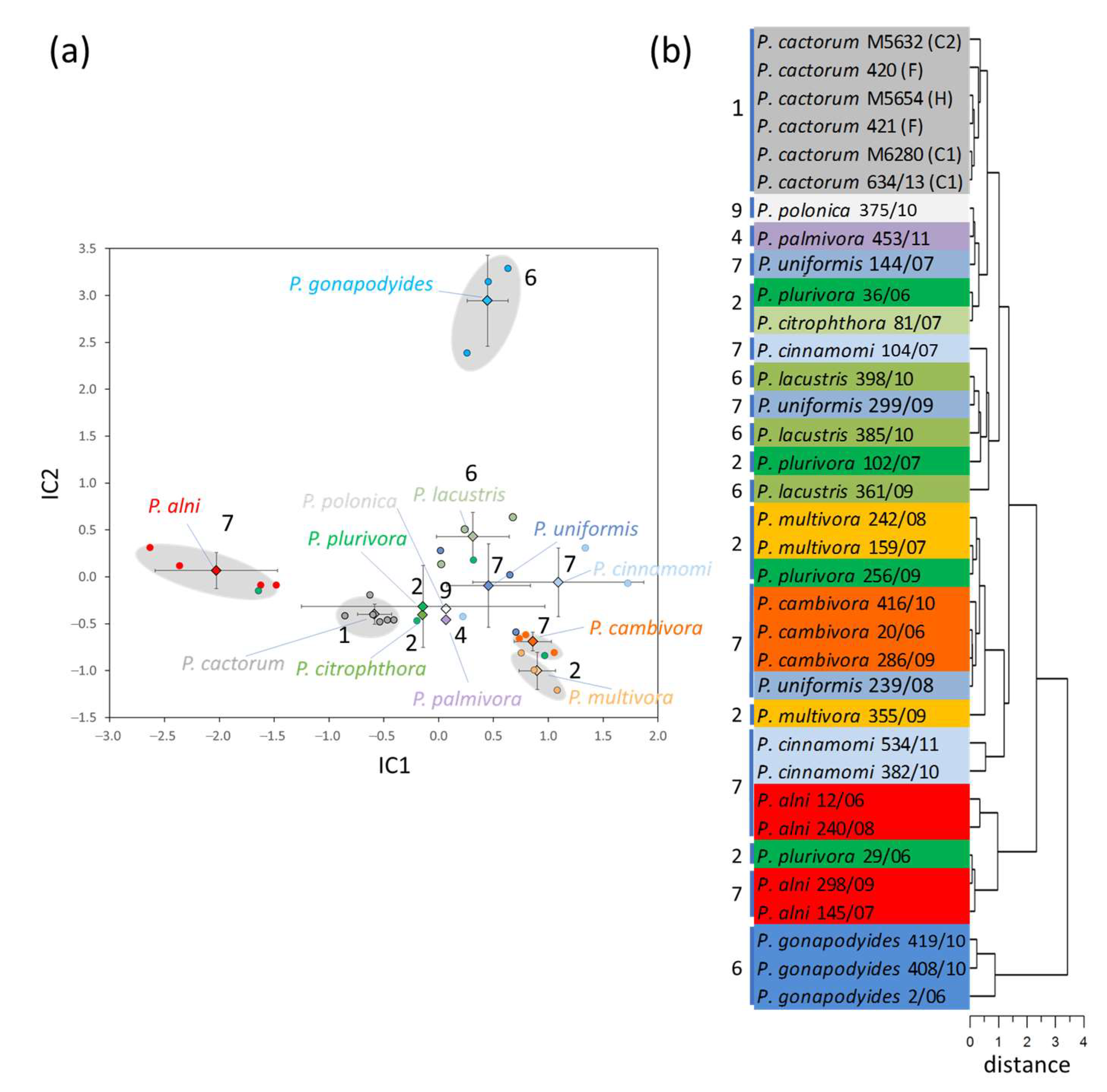
2.1. Peptide-Based Identification of Phytophthora Isolates
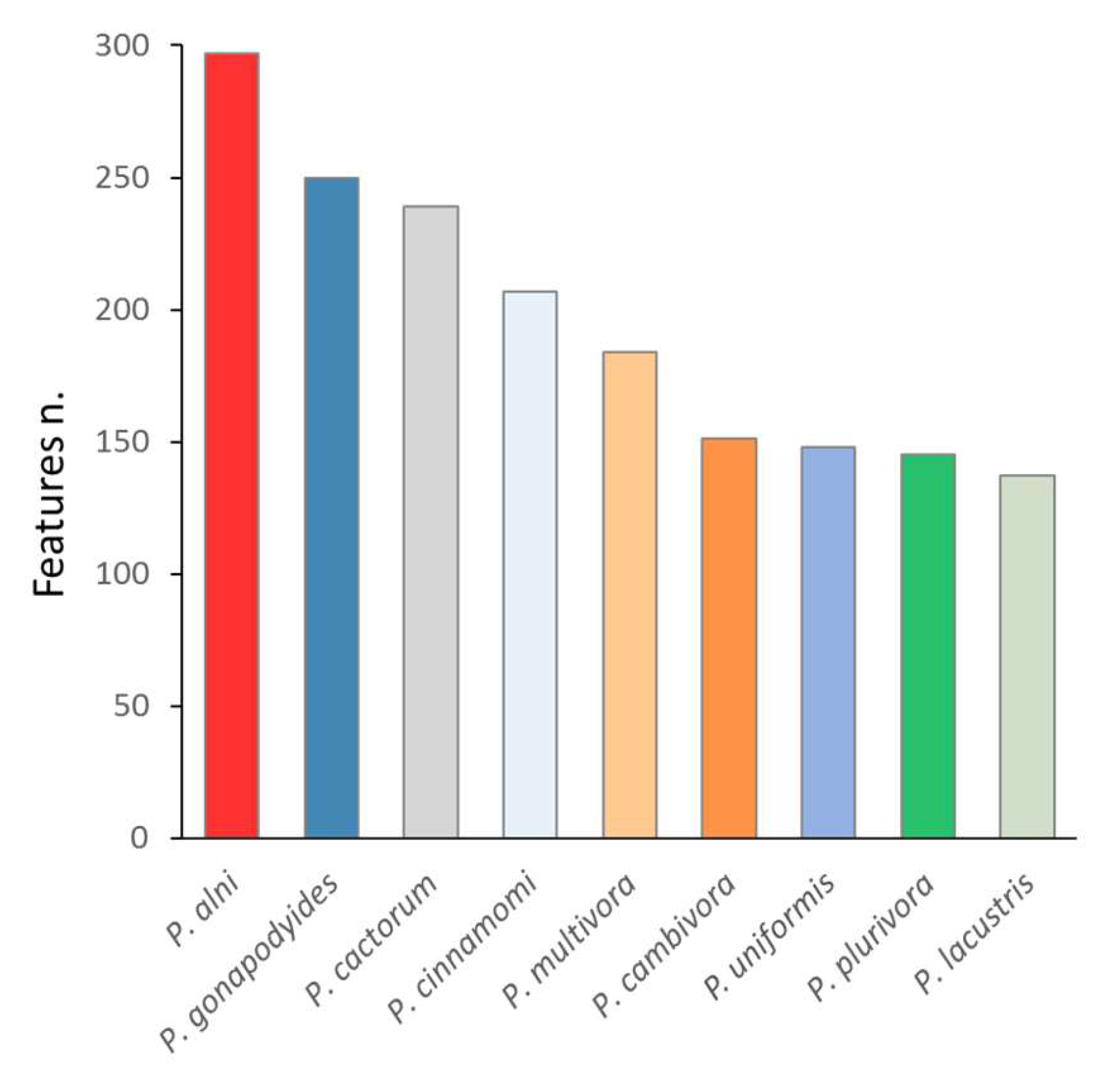
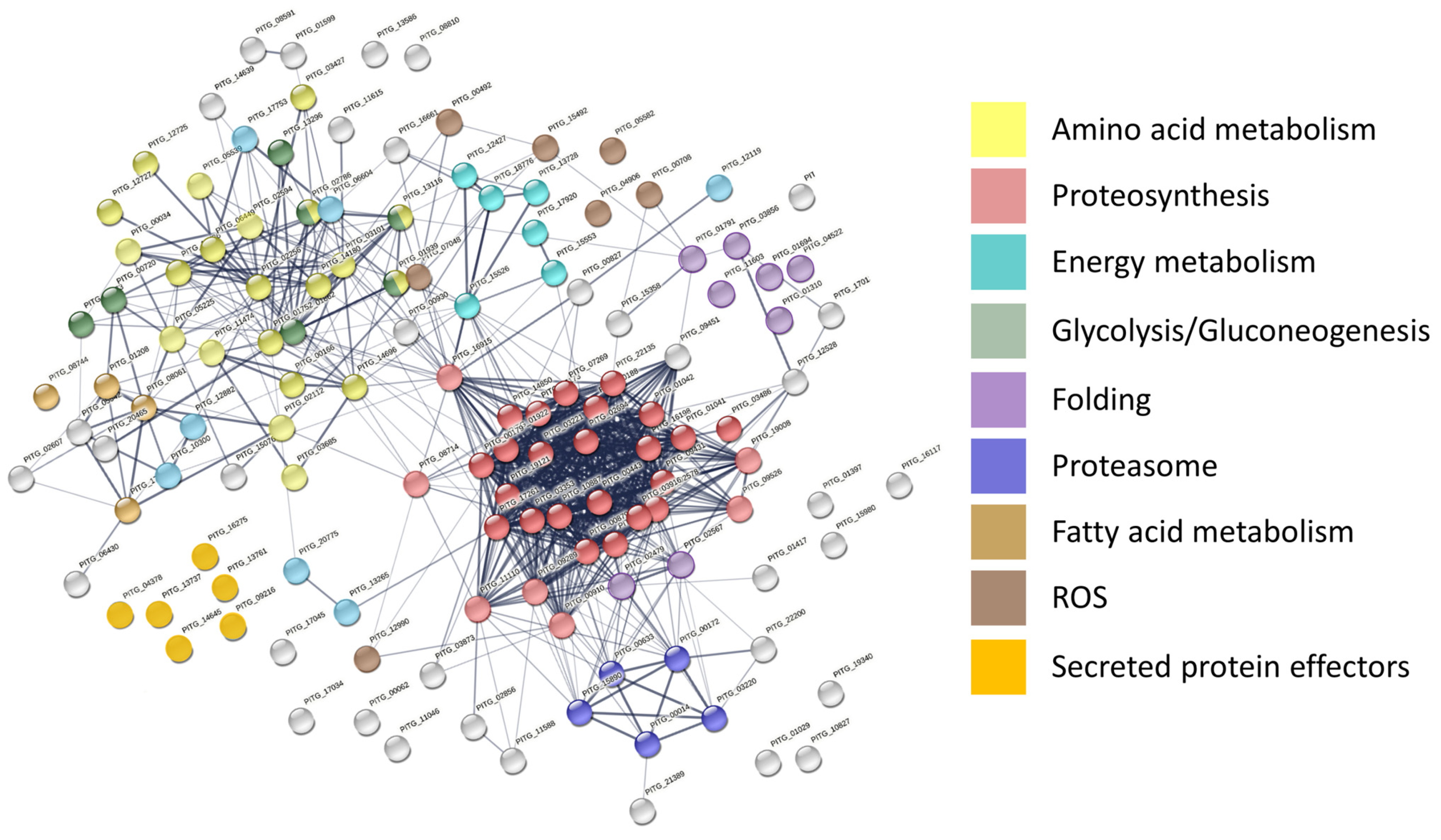
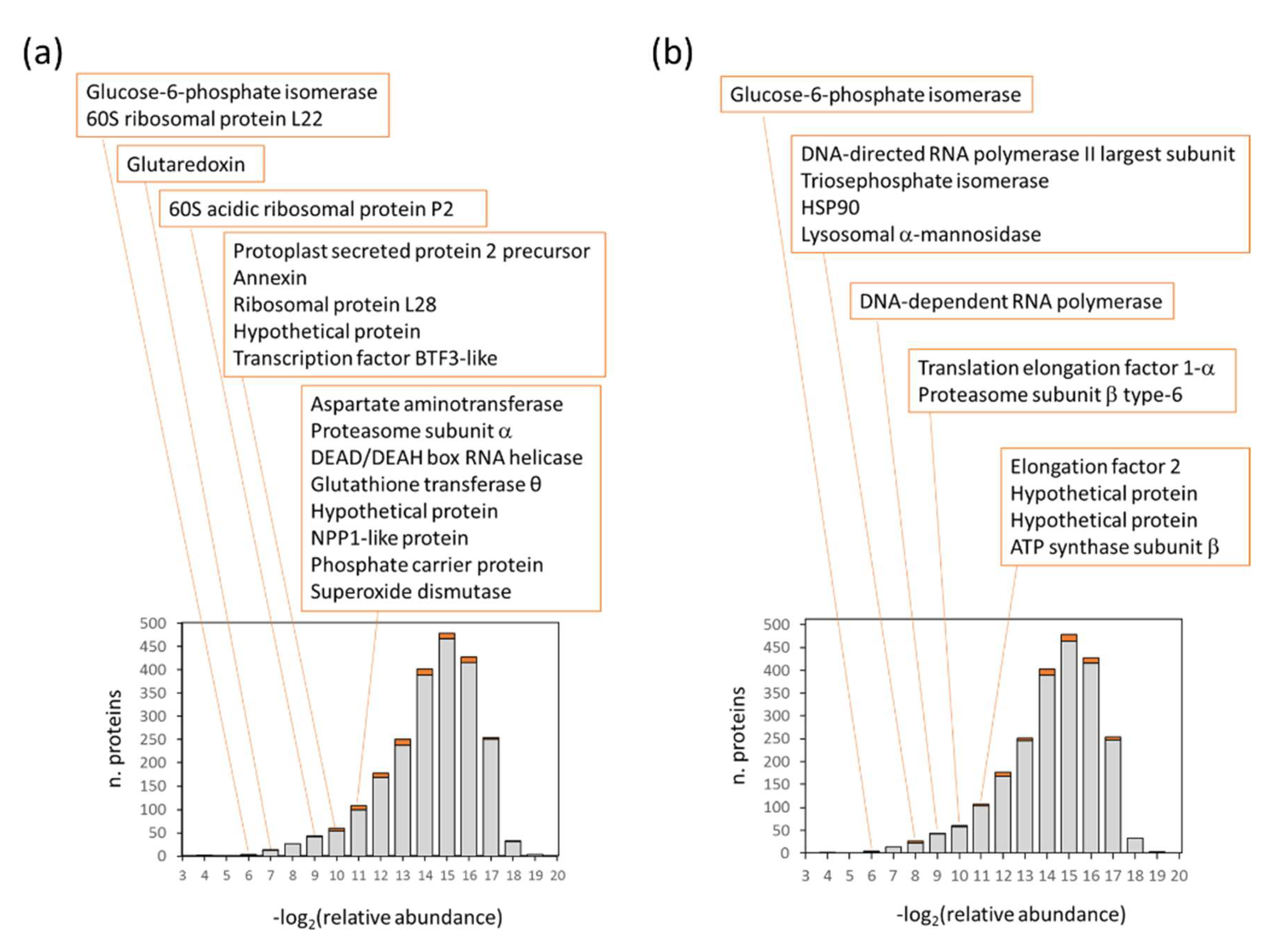
2.2. The Most Abundant Species-Specific Peptides Predominantly Originated from Enzymes of Major Metabolic Pathways
2.3. Phytophthora Detection in Planta
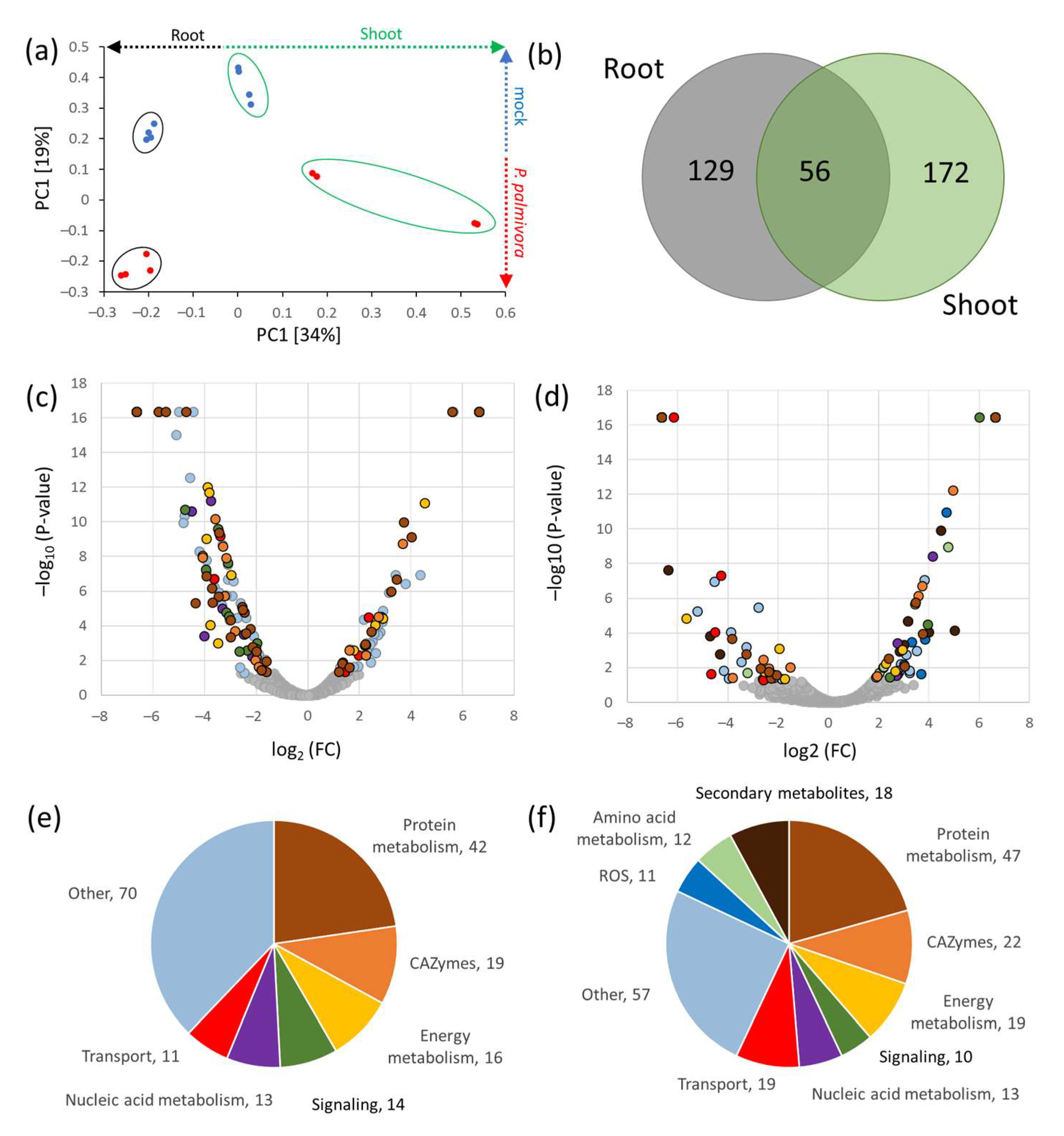
2.3.1. Detection of P. palmivora Proteins in Infected Seedlings of H. vulgare
2.3.2. P. palmivora Altered Barley Metabolism and Stress Signaling
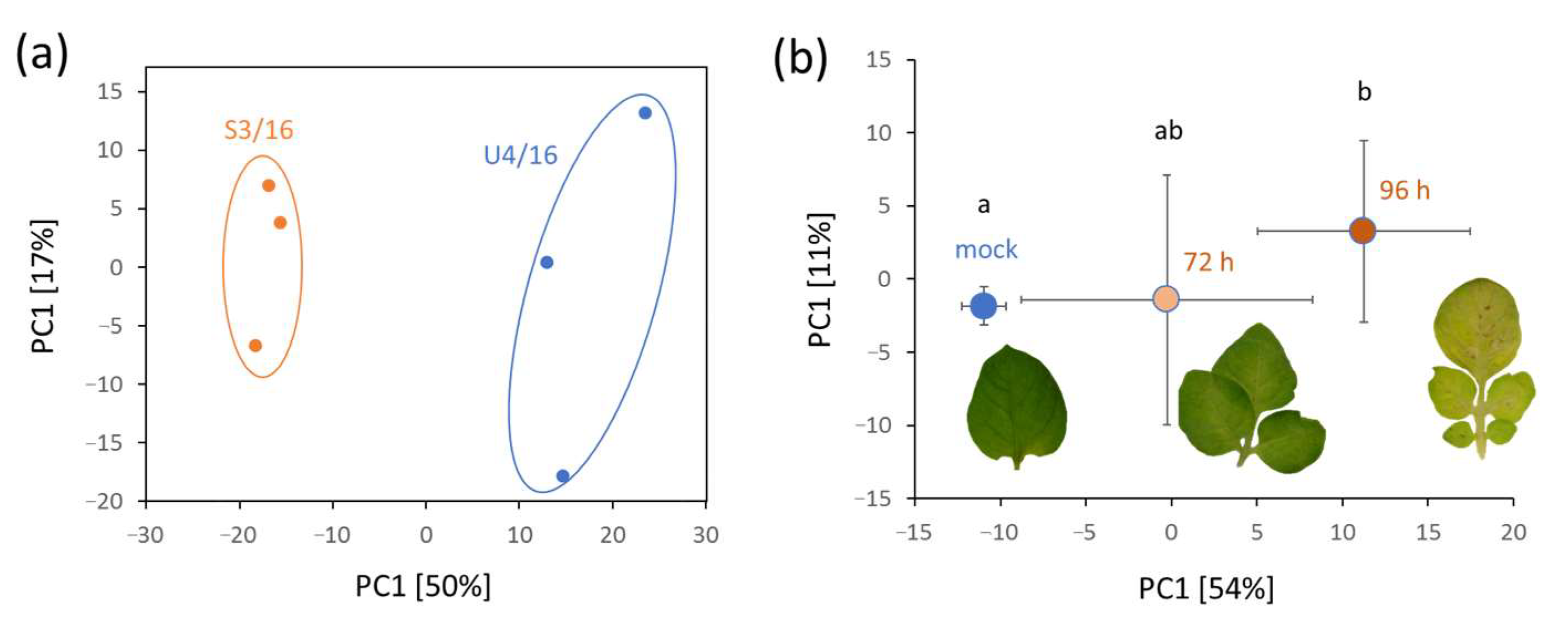
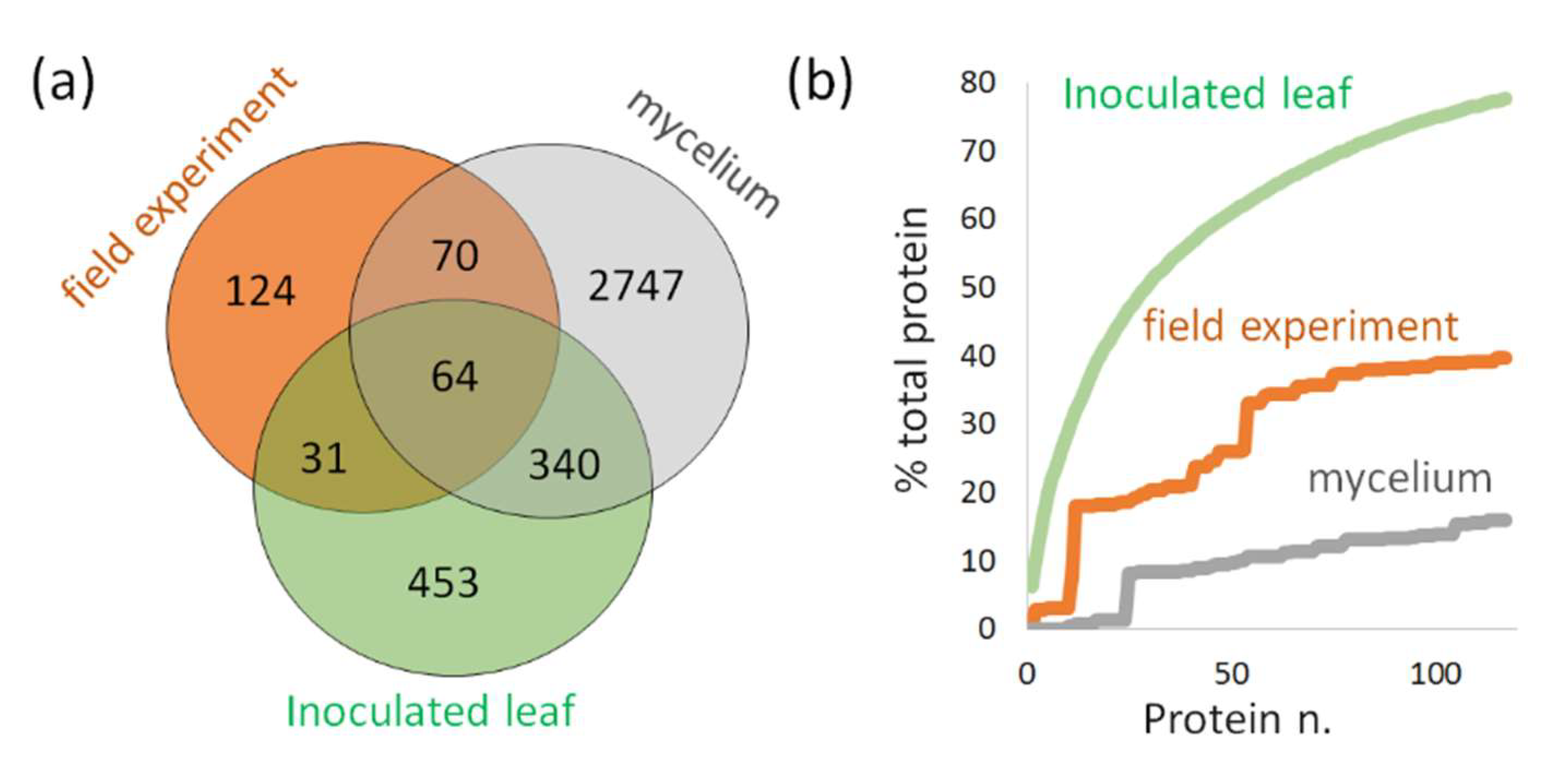
2.3.3. Detection of P. infestans in Inoculated Detached Leaves of Solanum tuberosum
2.3.4. Detection of P. infestans in Field-Grown Solanum tuberosum
2.3.5. Validation of P. infestans Presence in Planta
3. Discussion
3.1. Peptide-Based Analysis May Be the Future of Phytophthora Detection and Classification
3.2. Highly Abundant Mycelium Proteins Are Not the Best Targets for Phytophthora Detection in Planta
3.3. Evolutionary Conserved Protein Sequences Present an Obstacle for Unbiased Phytophthora Detection
3.4. P. palmivora-Induced Alteration of Polyamine and Melatonin Biosynthetic Pathways May Be a Part of Host Defense Suppression Mechanism
3.5. Protein-Based Early Detection of Phytopthora in Planta
4. Materials and Methods
4.1. Isolates and Phytophthora Cultivation
4.2. P. palmivora Response in Barley
4.3. S. tuberosum Leaf Inoculation
4.4. S. tuberosum Field Experiment
4.5. Protein Extraction and LC-MS Analysis
4.6. Proteomics Data Processing
- qTOF data (Supplementary Tables S1 and S2)—mass tolerance MS1 35 ppm, MS2 0.05 Da; enzyme—trypsin, maximum two missed cleavage sites; and modifications—up to three dynamic modifications including Met oxidation and Asn/Gln deamidation.
- Lumos data (in planta experiments, P. infestans)—mass tolerance MS1 5 ppm, MS2 0.02 Da; enzyme—trypsin, max two missed cleavage sites; modifications—up to three dynamic modifications including Met oxidation and Asn/Gln deamidation; Met-loss (protein N-terminus); Cys carbamidomethylation; and the MS Fragger algorithm was employed exclusively with Lumos data and the default settings for mass-tolerant search (MS1 tolerance 500 Da). The quantitative differences were determined by employing precursor ion quantification by Profile Analysis 2.0 and the spectral counting method (qTOF), and by Minora, followed by normalization and a background-based t-test for peptide- and protein-based quantitation. For selected candidate proteins, the corresponding peptide peak areas were manually evaluated in Skyline [42]. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [43] partner repository with the dataset identifier PXD022569.
4.7. Detection of P. infestans by qPCR
4.8. GC–MS Metabolomics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Burgess, T.I.; McDougall, K.L.; Scott, P.M.; Hardy, G.E.S.S.; Garnas, J. Predictors of Phytophthora diversity and community composition in natural areas across diverse Australian ecoregions. Ecography 2019, 42, 565–577. [Google Scholar] [CrossRef]
- Erwin, D.C.; Ribeiro, O.K. Phytophthora Diseases Worldwide; APS Press: St. Paul, MN, USA, 1996; ISBN 9780890542125. [Google Scholar]
- Judelson, H.S.; Ah-Fong, A.M.V. Exchanges at the plant-Oomycete interface that influence disease. Plant Physiol. 2019, 179, 1198–1211. [Google Scholar] [CrossRef] [PubMed]
- Fawke, S.; Doumane, M.; Schornack, S. Oomycete interactions with plants: Infection strategies and resistance principles. Microbiol. Mol. Biol. Rev. 2015, 79, 263–280. [Google Scholar] [CrossRef] [PubMed]
- Werres, S.; Marwitz, R.; Man In’t veld, W.A.; De Cock, A.W.A.M.; Bonants, P.J.M.; De Weerdt, M.; Themann, K.; Ilieva, E.; Baayen, R.P. Phytophthora ramorum sp. nov., a new pathogen on Rhododendron and Viburnum. Mycol. Res. 2001, 105, 1155–1165. [Google Scholar] [CrossRef]
- Vettraino, A.M.; Morel, O.; Perlerou, C.; Robin, C.; Diamandis, S.; Vannini, A. Occurrence and distribution of Phytophthora species in European chestnut stands, and their association with ink disease and crown decline. Eur. J. Plant Pathol. 2005, 111, 169–180. [Google Scholar] [CrossRef]
- Hansen, E.M. Phytophthora species emerging as pathogens of forest trees. Curr. For. Reports 2015, 1, 16–24. [Google Scholar] [CrossRef]
- Jung, T.; Orlikowski, L.; Henricot, B.; Abad-Campos, P.; Aday, A.G.; Aguín Casal, O.; Bakonyi, J.; Cacciola, S.O.; Cech, T.; Chavarriaga, D.; et al. Widespread Phytophthora infestations in European nurseries put forest, semi-natural and horticultural ecosystems at high risk of Phytophthora diseases. For. Pathol. 2016, 46, 134–163. [Google Scholar] [CrossRef]
- Jung, T.; Stukely, M.J.C.; Hardy, G.E.S.J.; White, D.; Paap, T.; Dunstan, W.A.; Burgess, T.I. Multiple new Phytophthora species from ITS Clade 6 associated with natural ecosystems in Australia: Evolutionary and ecological implications. Pers. Mol. Phylogeny Evol. Fungi 2011, 26, 13–39. [Google Scholar] [CrossRef]
- Kroon, L.P.N.M.; Brouwer, H.; de Cock, A.W.A.M.; Govers, F. The genus Phytophthora anno 2012. Phytopathology 2012, 102, 348–364. [Google Scholar] [CrossRef]
- O’Brien, P.A.; Williams, N.; Hardy, G.E.S. Detecting Phytophthora. Crit. Rev. Microbiol. 2009, 35, 169–181. [Google Scholar] [CrossRef]
- Yang, X.; Tyler, B.M.; Hong, C. An expanded phylogeny for the genus Phytophthora. IMA Fungus 2017, 8, 355–384. [Google Scholar] [CrossRef] [PubMed]
- Cooke, D.E.L.; Schena, L.; Cacciola, S.O. Tools to detect, identify and monitor Phytophthora species in natural ecosystems. J. Plant Pathol. 2007, 89, 13–28. [Google Scholar]
- Yang, X.; Hong, C. Differential usefulness of nine commonly used genetic markers for identifying Phytophthora species. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Strejcek, M.; Smrhova, T.; Junkova, P.; Uhlik, O. Whole-Cell MALDI-TOF MS Versus 16S rRNA Gene analysis for identification and dereplication of recurrent bacterial isolates. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Hayoun, K.; Gaillard, J.-C.; Pible, O.; Alpha-Bazin, B.; Armengaud, J. High-throughput proteotyping of bacterial isolates by double barrel chromatography-tandem mass spectrometry based on microplate paramagnetic beads and phylopeptidomics. J. Proteomics 2020, 226, 103887. [Google Scholar] [CrossRef]
- Hayoun, K.; Pible, O.; Petit, P.; Allain, F.; Jouffret, V.; Culotta, K.; Rivasseau, C.; Armengaud, J.; Alpha-Bazin, B. Proteotyping environmental microorganisms by Phylopeptidomics: Case study screening water from a radioactive material storage pool. Microorganisms 2020, 8, 1525. [Google Scholar] [CrossRef]
- Le Fevre, R.; O’Boyle, B.; Moscou, M.J.; Schornack, S. Colonization of barley by the broad-host hemibiotrophic pathogen Phytophthora palmivora uncovers a leaf development–Dependent involvement of Mlo. Mol. Plant Microbe Interact. 2016, 29, 385–395. [Google Scholar] [CrossRef]
- Shang, Y.; Wang, K.; Sun, S.; Zhou, J.; Yu, J.Q. COP9 Signalosome CSN4 and CSN5 subunits are involved in Jasmonate-dependent defense against root-knot nematode in tomato. Front. Plant Sci. 2019, 10. [Google Scholar] [CrossRef]
- Barreto, P.; Okura, V.; Pena, I.A.; Maia, R.; Maia, I.G.; Arruda, P. Overexpression of mitochondrial uncoupling protein 1 (UCP1) induces a hypoxic response in Nicotiana tabacum leaves. J. Exp. Bot. 2016, 67, 301–313. [Google Scholar] [CrossRef]
- Ichikawa, M.; Hirano, T.; Enami, K.; Fuselier, T.; Kato, N.; Kwon, C.; Voigt, B.; Schulze-Lefert, P.; Baluška, F.; Sato, M.H. Syntaxin of plant proteins SYP123 and SYP132 mediate root hair tip growth in Arabidopsis thaliana. Plant Cell Physiol. 2014, 55, 790–800. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.; Scanu, B.; Brasier, C.; Webber, J.; Milenković, I.; Corcobado, T.; Tomšovský, M.; Pánek, M.; Bakonyi, J.; Maia, C.; et al. Survey in natural forest ecosystems of Vietnam reveals high diversity of both new and described Phytophthora taxa including P. ramorum. Forests 2020, 11, 93. [Google Scholar] [CrossRef]
- Park, B.; Martin, F.; Geiser, D.M.; Kim, H.-S.; Mansfield, M.A.; Nikolaeva, E.; Park, S.-Y.; Coffey, M.D.; Russo, J.; Kim, S.H.; et al. Phytophthora database 2.0: Update and future direction. Phytopathology 2013, 103, 1204–1208. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.; Durán, A.; Sanfuentes von Stowasser, E.; Schena, L.; Mosca, S.; Fajardo, S.; González, M.; Navarro Ortega, A.D.; Bakonyi, J.; Seress, D.; et al. Diversity of Phytophthora species in Valdivian rainforests and association with severe dieback symptoms. For. Pathol. 2018, 48, e12443. [Google Scholar] [CrossRef]
- Asandei, A.; Di Muccio, G.; Schiopu, I.; Mereuta, L.; Dragomir, I.S.; Chinappi, M.; Luchian, T. Nanopore-based protein sequencing using biopores: Current achievements and open challenges. Small Methods 2020, 4, 1900595. [Google Scholar] [CrossRef]
- Ouldali, H.; Sarthak, K.; Ensslen, T.; Piguet, F.; Manivet, P.; Pelta, J.; Behrends, J.C.; Aksimentiev, A.; Oukhaled, A. Electrical recognition of the twenty proteinogenic amino acids using an aerolysin nanopore. Nat. Biotechnol. 2020, 38, 176–181. [Google Scholar] [CrossRef]
- Khan, Z.; Ford, M.J.; Cusanovich, D.A.; Mitrano, A.; Pritchard, J.K.; Gilad, Y. Primate transcript and protein expression levels evolve under compensatory selection pressures. Science 2013, 342, 1100–1104. [Google Scholar] [CrossRef]
- Savidor, A.; Donahoo, R.S.; Hurtado-Gonzales, O.; Land, M.L.; Shah, M.B.; Lamour, K.H.; McDonald, W.H. Cross-species global proteomics reveals conserved and unique processes in Phytophthora sojae and Phytophthora ramorum. Mol. Cell. Proteom. 2008, 7, 1501–1516. [Google Scholar] [CrossRef]
- Saiz-Fernández, I.; Milenković, I.; Berka, M.; Černý, M.; Tomšovský, M.; Brzobohatý, B.; Kerchev, P. Integrated proteomic and metabolomic profiling of Phytophthora cinnamomi attack on sweet chestnut (Castanea sativa) reveals distinct molecular reprogramming proximal to the infection site and away from it. Int. J. Mol. Sci. 2020, 21, 8525. [Google Scholar] [CrossRef]
- Wang, Y.; Kim, S.G.; Kim, S.T.; Agrawal, G.K.; Rakwal, R.; Kang, K.Y. Biotic stress-responsive rice proteome: An overview. J. Plant Biol. 2011, 54, 219–226. [Google Scholar] [CrossRef]
- Henty-Ridilla, J.L.; Shimono, M.; Li, J.; Chang, J.H.; Day, B.; Staiger, C.J. The plant actin Cytoskeleton responds to signals from microbe-associated molecular patterns. PLoS Pathog. 2013, 9, e1003290. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Pang, X.; Cheng, Y.; Yin, Y.; Zhang, Q.; Su, W.; Hu, B.; Guo, Q.; Ha, S.; Zhang, J.; et al. The Hsp70 gene family in Solanum tuberosum: Genome-wide identification, phylogeny, and expression patterns. Sci. Rep. 2018, 8, 16628. [Google Scholar] [CrossRef] [PubMed]
- Zuluaga, A.P.; Vega-Arreguín, J.C.; Fei, Z.; Ponnala, L.; Lee, S.J.; Matas, A.J.; Patev, S.; Fry, W.E.; Rose, J.K.C. Transcriptional dynamics of Phytophthora infestans during sequential stages of hemibiotrophic infection of tomato. Mol. Plant Pathol. 2016, 17, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, E.; Gogleva, A.; Hainaux, T.; Doumane, M.; Tulin, F.; Quan, C.; Yunusov, T.; Floch, K.; Schornack, S. Time-resolved dual transcriptomics reveal early induced Nicotiana benthamiana root genes and conserved infection-promoting Phytophthora palmivora effectors. BMC Biol. 2017, 15, 39. [Google Scholar] [CrossRef]
- Cerna, H.; Černý, M.; Habánová, H.; Šafářová, D.; Abushamsiya, K.; Navrátil, M.; Brzobohatý, B. Proteomics offers insight to the mechanism behind Pisum sativum L. response to pea seed-borne mosaic virus (PSbMV). J. Proteomics 2017, 153, 78–88. [Google Scholar] [CrossRef]
- Dufková, H.; Berka, M.; Luklová, M.; Rashotte, A.M.; Brzobohatý, B.; Černý, M. Eggplant germination is promoted by hydrogen peroxide and temperature in an independent but overlapping manner. Molecules 2019, 24, 4270. [Google Scholar] [CrossRef]
- Berková, V.; Kameniarová, M.; Ondrisková, V.; Berka, M.; Menšíková, S.; Kopecká, R.; Luklová, M.; Novák, J.; Spíchal, L.; Rashotte, A.M.; et al. Arabidopsis response to inhibitor of Cytokinin degradation INCYDE: Modulations of Cytokinin signaling and plant proteome. Plants 2020, 9, 1563. [Google Scholar] [CrossRef]
- Mierswa, I.; Wurst, M.; Klinkenberg, R.; Scholz, M.; Euler, T. YALE: Rapid prototyping for complex data mining tasks. In Proceedings of the 12th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining—KDD ’06; Association for Computing Machinery (ACM), Philadelphia, PA, USA, 20–23 August 2006; p. 935. [Google Scholar]
- Dorfer, V.; Pichler, P.; Stranzl, T.; Stadlmann, J.; Taus, T.; Winkler, S.; Mechtler, K. MS Amanda, a universal identification algorithm optimized for high accuracy tandem mass spectra. J. Proteome Res. 2014, 13, 3679–3684. [Google Scholar] [CrossRef]
- Kong, A.T.; Leprevost, F.V.; Avtonomov, D.M.; Mellacheruvu, D.; Nesvizhskii, A.I. MSFragger: Ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods 2017, 14, 513–520. [Google Scholar] [CrossRef]
- Pino, L.K.; Searle, B.C.; Bollinger, J.G.; Nunn, B.; MacLean, B.; MacCoss, M.J. The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass Spectrom. Rev. 2020, 39, 229–244. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Llorente, B.; Bravo-Almonacid, F.; Cvitanich, C.; Orlowska, E.; Torres, H.N.; Flawiá, M.M.; Alonso, G.D. A quantitative real-time PCR method for in planta monitoring of Phytophthora infestans growth. Lett. Appl. Microbiol. 2010, 51, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Berka, M.; Luklová, M.; Dufková, H.; Malých, V.; Novák, J.; Saiz-Fernández, I.; Rashotte, A.M.; Brzobohaty, B.; Cerny, M. Barley root proteome and metabolome in response to cytokinin and abiotic stimuli. Front. Plant Sci. 2020, 11, 1647. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berka, M.; Greplová, M.; Saiz-Fernández, I.; Novák, J.; Luklová, M.; Zelená, P.; Tomšovský, M.; Brzobohatý, B.; Černý, M. Peptide-Based Identification of Phytophthora Isolates and Phytophthora Detection in Planta. Int. J. Mol. Sci. 2020, 21, 9463. https://doi.org/10.3390/ijms21249463
Berka M, Greplová M, Saiz-Fernández I, Novák J, Luklová M, Zelená P, Tomšovský M, Brzobohatý B, Černý M. Peptide-Based Identification of Phytophthora Isolates and Phytophthora Detection in Planta. International Journal of Molecular Sciences. 2020; 21(24):9463. https://doi.org/10.3390/ijms21249463
Chicago/Turabian StyleBerka, Miroslav, Marie Greplová, Iñigo Saiz-Fernández, Jan Novák, Markéta Luklová, Pavla Zelená, Michal Tomšovský, Břetislav Brzobohatý, and Martin Černý. 2020. "Peptide-Based Identification of Phytophthora Isolates and Phytophthora Detection in Planta" International Journal of Molecular Sciences 21, no. 24: 9463. https://doi.org/10.3390/ijms21249463
APA StyleBerka, M., Greplová, M., Saiz-Fernández, I., Novák, J., Luklová, M., Zelená, P., Tomšovský, M., Brzobohatý, B., & Černý, M. (2020). Peptide-Based Identification of Phytophthora Isolates and Phytophthora Detection in Planta. International Journal of Molecular Sciences, 21(24), 9463. https://doi.org/10.3390/ijms21249463