Inhibition of the FGF/FGFR System Induces Apoptosis in Lung Cancer Cells via c-Myc Downregulation and Oxidative Stress


 , ,
, ,  ,
,  and
and 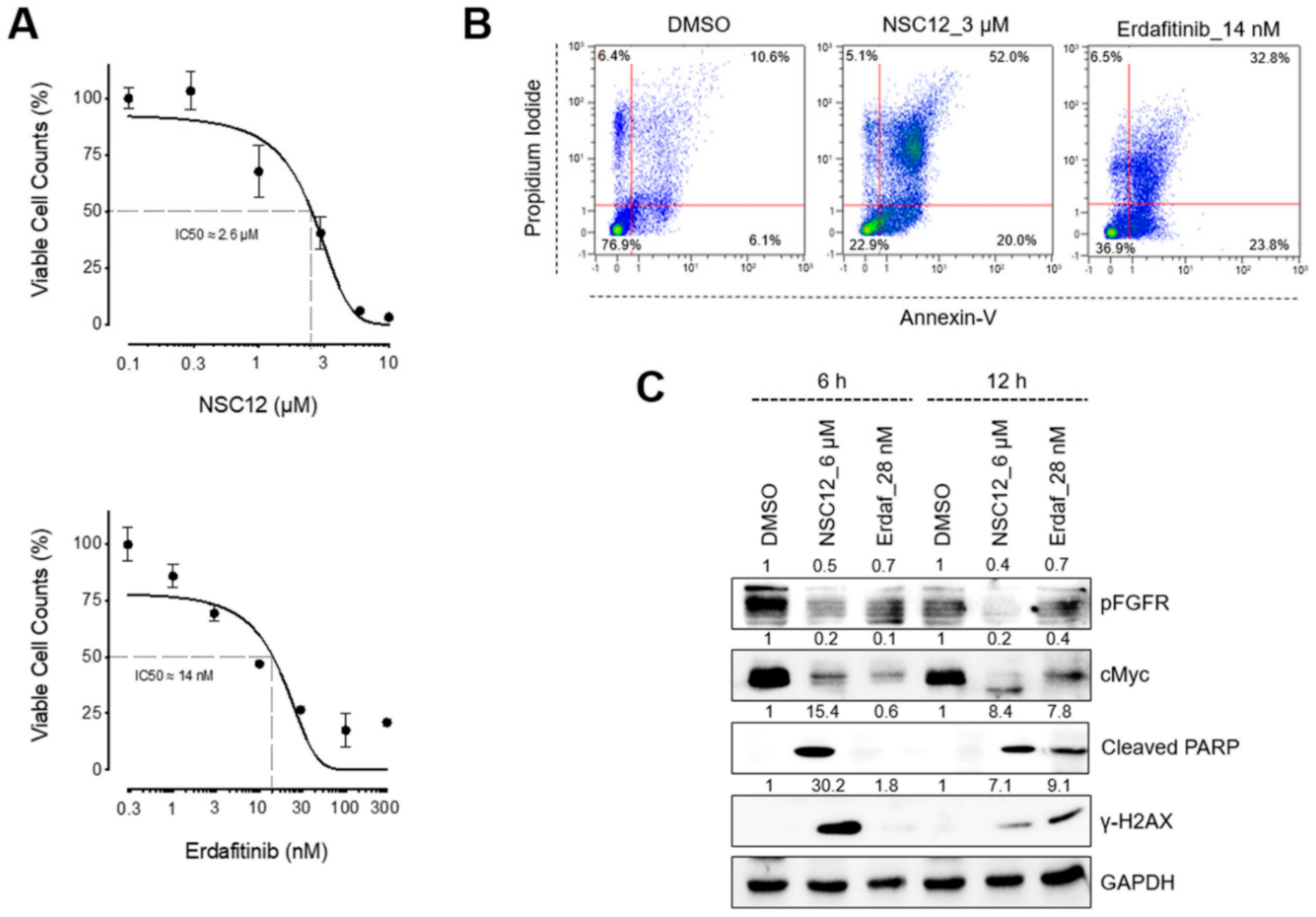{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. FGF/FGFR Inhibition Triggers Apoptosis in FGF/FGFR-Dependent Lung Cancer Cells
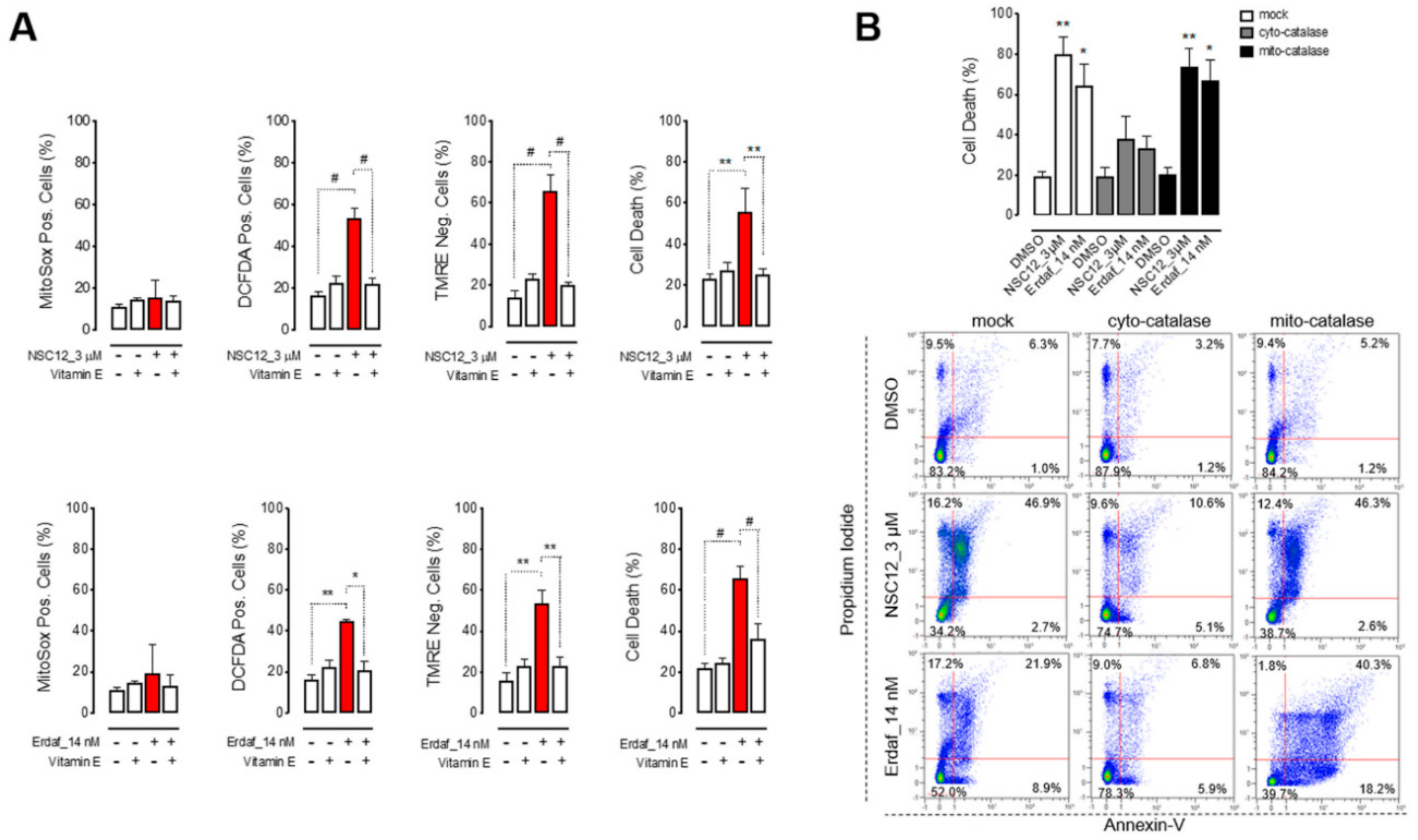
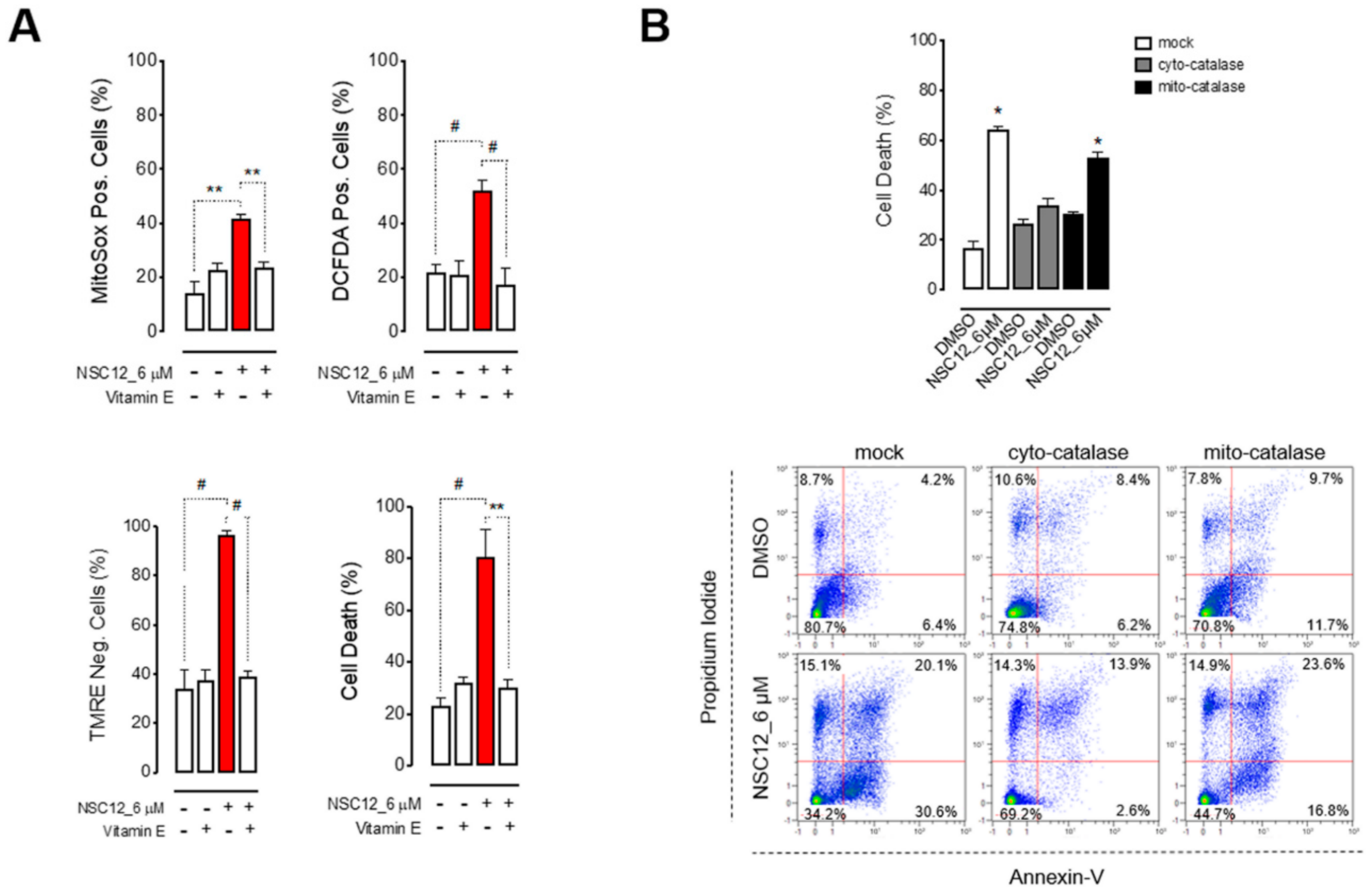
2.2. Apoptosis Upon FGF/FGFR Inhibition is Induced by Oxidative Stress
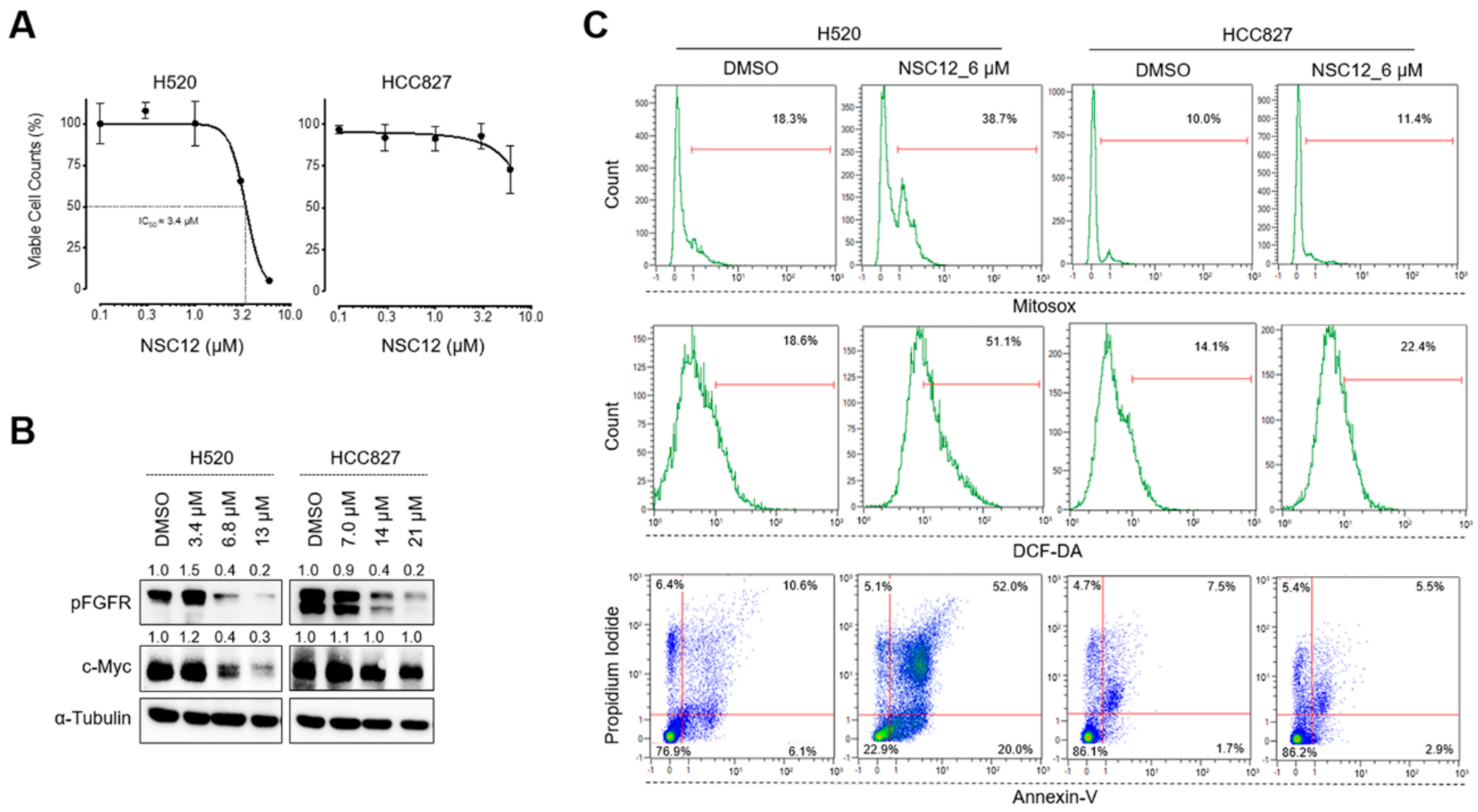
2.3. Fgf Trapping-Mediated C-Myc Modulation and Consequent Oxidative Stress Are Specific for Fgf-Dependent Lung Cancer Cells
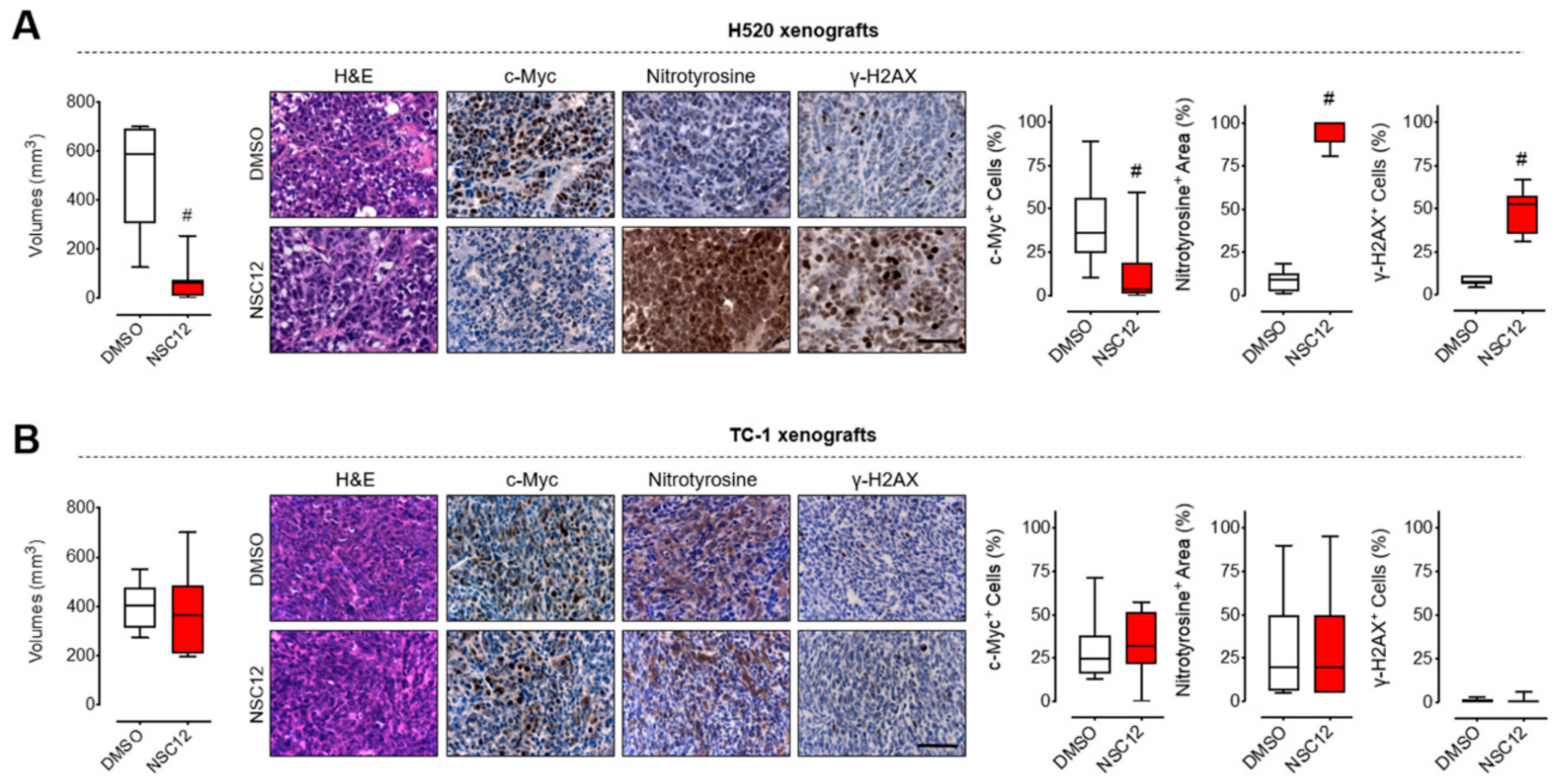
2.4. FGF Trapping Inhibits Tumor Growth and Induces Oxidative Stress and DNA Damage in Vivo in FGF-Dependent Lung Cancer
3. Discussion
4. Materials and Methods
4.1. Cell Cultures and Reagents
4.2. Cytofluorimetric Analyses
4.3. Western Blot Analysis
4.4. RT-qPCR
4.5. Subcutaneous Human Xenografts
4.6. Histological Analyses
4.7. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| NSCLC | Non-small-cell lung cancer |
| TKi | Tyrosine kinase inhibitors |
| TK | Tyrosine kinase |
| FGFR | Fibroblast growth factor receptor |
| FGF | Fibroblast growth factor |
| SCC | Squamous cell carcinomas |
| SCLC | Small cell lung cancer |
| ROS | Reactive oxygen species |
References
- Hirsch, F.R.; Scagliotti, G.V.; Mulshine, J.L.; Kwon, R.; Curran, W.J.; Wu, Y.-L.; Paz-Ares, L. Lung cancer: Current therapies and new targeted treatments. Lancet 2017, 389, 299–311. [Google Scholar] [CrossRef]
- Kwabi-Addo, B.; Ozen, M.; Ittmann, M. The role of fibroblast growth factors and their receptors in prostate cancer. Endocr. Relat. Cancer 2004, 11, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Williams, L.T. Structural and Functional Diversity in the FGf Receptor Multigene Family. Adv. Cancer Res. 1992, 60, 1–41. [Google Scholar] [CrossRef]
- Presta, M.; Chiodelli, P.; Giacomini, A.; Rusnati, M.; Ronca, R. Fibroblast growth factors (FGFs) in cancer: FGF traps as a new therapeutic approach. Pharmacol. Ther. 2017, 179, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Dieci, M.V.; Arnedos, M.; Andre, F.; Soria, J.-C. Fibroblast Growth Factor Receptor Inhibitors as a Cancer Treatment: From a Biologic Rationale to Medical Perspectives. Cancer Discov. 2013, 3, 264–279. [Google Scholar] [CrossRef]
- Katoh, M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat. Rev. Clin. Oncol. 2019, 16, 105–122. [Google Scholar] [CrossRef]
- Ghedini, G.C.; Ronca, R.; Presta, M.; Giacomini, A. Future applications of FGF/FGFR inhibitors in cancer. Expert Rev. Anticancer. Ther. 2018, 18, 861–872. [Google Scholar] [CrossRef]
- Harding, T.C.; Long, L.; Palencia, S.; Zhang, H.; Sadra, A.; Hestir, K.; Patil, N.; Levin, A.; Hsu, A.W.; Charych, D.H.; et al. Blockade of Nonhormonal Fibroblast Growth Factors by FP-1039 Inhibits Growth of Multiple Types of Cancer. Sci. Transl. Med. 2013, 5, 178ra39. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Salvador, M.K.; Prencipe, F.; Lopez-Cara, L.C.; Ortega, S.S.; Brancale, A.; Hamel, E.; Castagliuolo, I.; Mitola, S.; et al. Design, Synthesis, in Vitro, and in Vivo Anticancer and Antiangiogenic Activity of Novel 3-Arylaminobenzofuran Derivatives Targeting the Colchicine Site on Tubulin. J. Med. Chem. 2015, 58, 3209–3222. [Google Scholar] [CrossRef]
- Castelli, R.; Giacomini, A.; Anselmi, M.; Bozza, N.; Vacondio, F.; Rivara, S.; Matarazzo, S.; Presta, M.; Mor, M.; Ronca, R. Synthesis, Structural Elucidation, and Biological Evaluation of NSC12, an Orally Available Fibroblast Growth Factor (FGF) Ligand Trap for the Treatment of FGF-Dependent Lung Tumors. J. Med. Chem. 2016, 59, 4651–4663. [Google Scholar] [CrossRef]
- Peifer, M.; Fernández-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, F.R.; Suda, K.; Wiens, J.; Bunn, P.A. New and emerging targeted treatments in advanced non-small-cell lung cancer. Lancet 2016, 388, 1012–1024. [Google Scholar] [CrossRef]
- Campbell, J.D.; Cancer Genome Atlas Research Network; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.; Sos, M.L.; Seidel, D.; Peifer, M.; Zander, T.; Heuckmann, J.M.; Ullrich, R.T.; Menon, R.; Maier, S.; Soltermann, A.; et al. Frequent and Focal FGFR1 Amplification Associates with Therapeutically Tractable FGFR1 Dependency in Squamous Cell Lung Cancer. Sci. Transl. Med. 2010, 2, 62ra93. [Google Scholar] [CrossRef]
- Dutt, A.; Ramos, A.H.; Hammerman, P.S.; Mermel, C.; Cho, J.; Sharifnia, T.; Chande, A.; Tanaka, K.E.; Stransky, N.; Greulich, H.; et al. Inhibitor-Sensitive FGFR1 Amplification in Human Non-Small Cell Lung Cancer. PLoS ONE 2011, 6, e20351. [Google Scholar] [CrossRef]
- Marek, L.; Ware, K.E.; Fritzsche, A.; Hercule, P.; Helton, W.R.; Smith, J.E.; McDermott, L.A.; Coldren, C.D.; Nemenoff, R.A.; Merrick, D.T.; et al. Fibroblast Growth Factor (FGF) and FGF Receptor-Mediated Autocrine Signaling in Non-Small-Cell Lung Cancer Cells. Mol. Pharmacol. 2008, 75, 196–207. [Google Scholar] [CrossRef]
- Wesche, J.; Haglund, K.; Haugsten, E.M. Fibroblast growth factors and their receptors in cancer. Biochem. J. 2011, 437, 199–213. [Google Scholar] [CrossRef]
- Wynes, M.W.; Hinz, T.K.; Gao, D.; Martini, M.; Marek, L.A.; Ware, K.E.; Edwards, M.G.; Böhm, D.; Perner, S.; Helfrich, B.A.; et al. FGFR1 mRNA and protein expression, not gene copy number, predict FGFR TKI sensitivity across all lung cancer histologies. Clin. Cancer Res. 2014, 20, 3299–3309. [Google Scholar] [CrossRef]
- Rooney, C.; Geh, C.; Williams, V.; Heuckmann, J.M.; Menon, R.; Schneider, P.; Al-Kadhimi, K.; Dymond, M.; Smith, N.R.; Baker, D.; et al. Characterization of FGFR1 Locus in sqNSCLC Reveals a Broad and Heterogeneous Amplicon. PLoS ONE 2016, 11, e0149628. [Google Scholar] [CrossRef]
- Bahleda, R.; Italiano, A.; Hierro, C.; Mita, A.; Cervantes, A.; Chan, N.; Awad, M.; Calvo, E.; Moreno, V.; Govindan, R.; et al. Multicenter Phase I Study of Erdafitinib (JNJ-42756493), Oral Pan-Fibroblast Growth Factor Receptor Inhibitor, in Patients with Advanced or Refractory Solid Tumors. Clin. Cancer Res. 2019, 25, 4888–4897. [Google Scholar] [CrossRef]
- Ronca, R.; Giacomini, A.; Di Salle, E.; Coltrini, D.; Pagano, K.; Ragona, L.; Matarazzo, S.; Rezzola, S.; Maiolo, D.; Torrella, R.; et al. Long-Pentraxin 3 Derivative as a Small-Molecule FGF Trap for Cancer Therapy. Cancer Cell 2015, 28, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Ronca, R.; Ghedini, G.C.; Maccarinelli, F.; Sacco, A.; Locatelli, S.L.; Foglio, E.; Taranto, S.; Grillo, E.; Matarazzo, S.; Castelli, R.; et al. FGF Trapping Inhibits Multiple Myeloma Growth through c-Myc Degradation–Induced Mitochondrial Oxidative Stress. Cancer Res. 2020, 80, 2340–2354. [Google Scholar] [CrossRef] [PubMed]
- Ladas, E.J.; Jacobson, J.S.; Kennedy, D.D.; Teel, K.; Fleischauer, A.; Kelly, K.M. Antioxidants and Cancer Therapy: A Systematic Review. J. Clin. Oncol. 2004, 22, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ai, J.; Shen, A.; Chen, Y.; Wang, X.; Peng, X.; Chen, H.; Shen, Y.; Huang, M.; Ding, J.; et al. c-Myc Alteration Determines the Therapeutic Response to FGFR Inhibitors. Clin. Cancer Res. 2016, 23, 974–984. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.-M.; Zhao, X.; Christensen, J.; et al. MET Amplification Leads to Gefitinib Resistance in Lung Cancer by Activating ERBB3 Signaling. Sci. 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Giaccone, G. The Role of Gefitinib in Lung Cancer Treatment. Clin. Cancer Res. 2004, 10, 4233–4237. [Google Scholar] [CrossRef]
- Drilon, A.; Clark, J.W.; Weiss, J.; Ou, S.-H.I.; Camidge, D.R.; Solomon, B.J.; Otterson, G.A.; Villaruz, L.C.; Riely, G.J.; Heist, R.S.; et al. Antitumor activity of crizotinib in lung cancers harboring a MET exon 14 alteration. Nat. Med. 2020, 26, 47–51. [Google Scholar] [CrossRef]
- Solomon, B.; Mok, T.; Kim, D.-W.; Wu, Y.-L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-Line Crizotinib versus Chemotherapy in ALK-Positive Lung Cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef]
- Fischer, H.; Taylor, N.; Allerstorfer, S.; Grusch, M.; Sonvilla, G.; Holzmann, K.; Setinek, U.; Elbling, L.; Cantonati, H.; Grasl-Kraupp, B.; et al. Fibroblast growth factor receptor-mediated signals contribute to the malignant phenotype of non-small cell lung cancer cells: Therapeutic implications and synergism with epidermal growth factor receptor inhibition. Mol. Cancer Ther. 2008, 7, 3408–3419. [Google Scholar] [CrossRef]
- Giacomini, A.; Chiodelli, P.; Matarazzo, S.; Rusnati, M.; Presta, M.; Ronca, R. Blocking the FGF/FGFR system as a two-compartment antiangiogenic/antitumor approach in cancer therapy. Pharmacol. Res. 2016, 107, 172–185. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Malchers, F.; Dietlein, F.; Schöttle, J.; Lu, X.; Nogova, L.; Albus, K.; Fernandez-Cuesta, L.; Heuckmann, J.M.; Gautschi, O.; Diebold, J.; et al. Cell-Autonomous and Non-Cell-Autonomous Mechanisms of Transformation by Amplified FGFR1 in Lung Cancer. Cancer Discov. 2013, 4, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Mut-Salud, N.; Álvarez, P.J.; Garrido, J.M.; Carrasco, E.; Aránega, A.; Rodríguez-Serrano, F. Antioxidant Intake and Antitumor Therapy: Toward Nutritional Recommendations for Optimal Results. Oxidative Med. Cell. Longev. 2015, 2016, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Accardi, L.; Paolini, F.; Mandarino, A.; Percario, Z.; Di Bonito, P.; Di Carlo, V.; Affabris, E.; Giorgi, C.; Amici, C.; Venuti, A. In vivoantitumor effect of an intracellular single-chain antibody fragment against the E7 oncoprotein of human papillomavirus 16. Int. J. Cancer 2013, 134, 2742–2747. [Google Scholar] [CrossRef] [PubMed]
- Gurgul-Convey, E.; Lortz, S.; Tiedge, M.; Jörns, A.; Lenzen, S. Mitochondrial catalase overexpression protects insulin-producing cells against toxicity of reactive oxygen species and proinflammatory cytokines. Diabetes 2004, 53, 2271–2280. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giacomini, A.; Taranto, S.; Rezzola, S.; Matarazzo, S.; Grillo, E.; Bugatti, M.; Scotuzzi, A.; Guerra, J.; Di Trani, M.; Presta, M.; et al. Inhibition of the FGF/FGFR System Induces Apoptosis in Lung Cancer Cells via c-Myc Downregulation and Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 9376. https://doi.org/10.3390/ijms21249376
Giacomini A, Taranto S, Rezzola S, Matarazzo S, Grillo E, Bugatti M, Scotuzzi A, Guerra J, Di Trani M, Presta M, et al. Inhibition of the FGF/FGFR System Induces Apoptosis in Lung Cancer Cells via c-Myc Downregulation and Oxidative Stress. International Journal of Molecular Sciences. 2020; 21(24):9376. https://doi.org/10.3390/ijms21249376
Chicago/Turabian StyleGiacomini, Arianna, Sara Taranto, Sara Rezzola, Sara Matarazzo, Elisabetta Grillo, Mattia Bugatti, Alessia Scotuzzi, Jessica Guerra, Martina Di Trani, Marco Presta, and et al. 2020. "Inhibition of the FGF/FGFR System Induces Apoptosis in Lung Cancer Cells via c-Myc Downregulation and Oxidative Stress" International Journal of Molecular Sciences 21, no. 24: 9376. https://doi.org/10.3390/ijms21249376
APA StyleGiacomini, A., Taranto, S., Rezzola, S., Matarazzo, S., Grillo, E., Bugatti, M., Scotuzzi, A., Guerra, J., Di Trani, M., Presta, M., & Ronca, R. (2020). Inhibition of the FGF/FGFR System Induces Apoptosis in Lung Cancer Cells via c-Myc Downregulation and Oxidative Stress. International Journal of Molecular Sciences, 21(24), 9376. https://doi.org/10.3390/ijms21249376