Endoglin: An ‘Accessory’ Receptor Regulating Blood Cell Development and Inflammation



Abstract
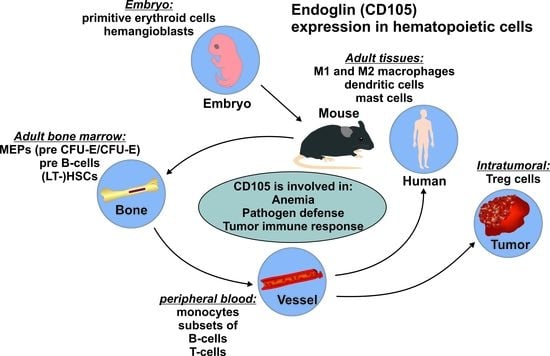
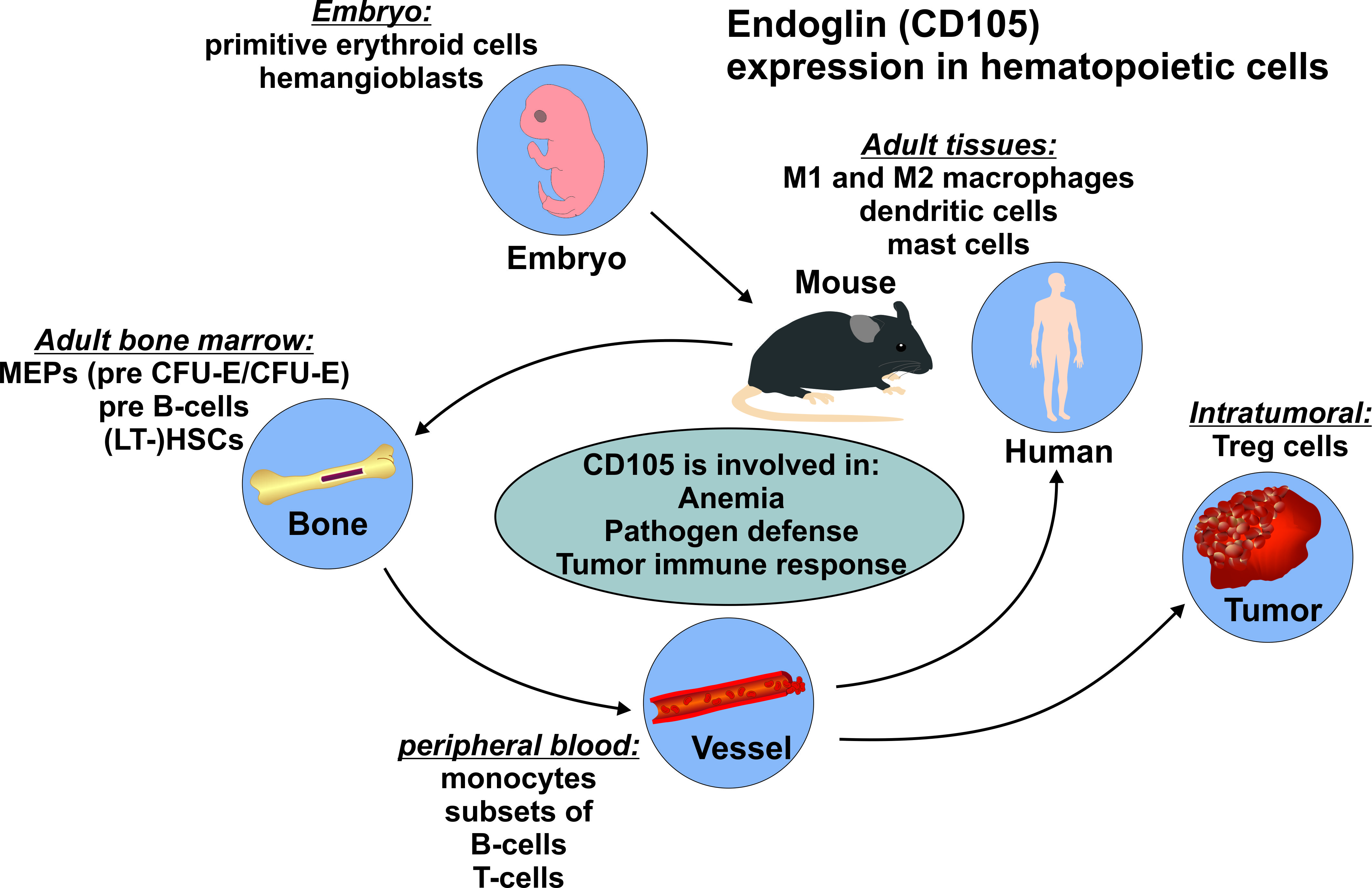{kind=link}
{kind=link}
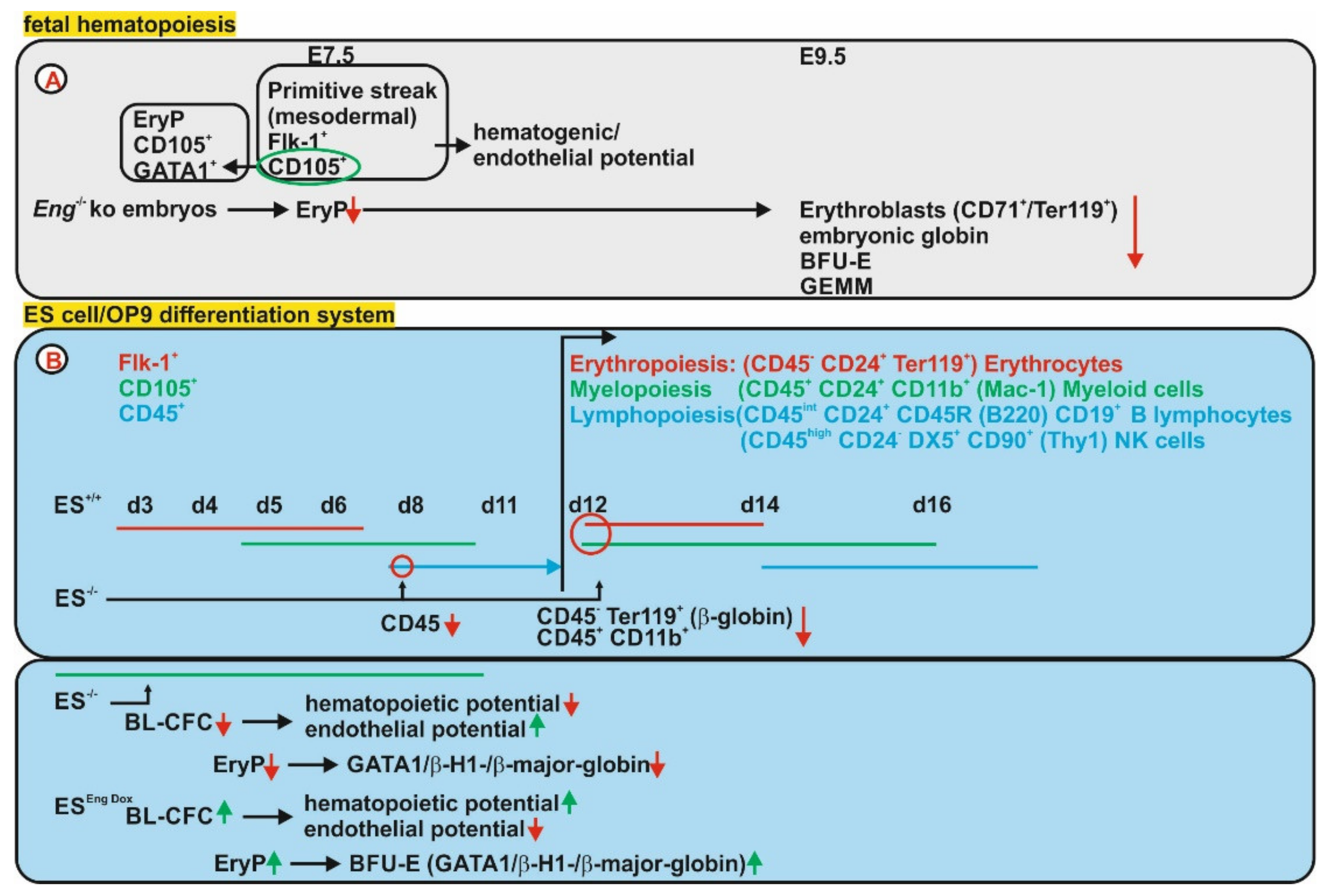{kind=link}
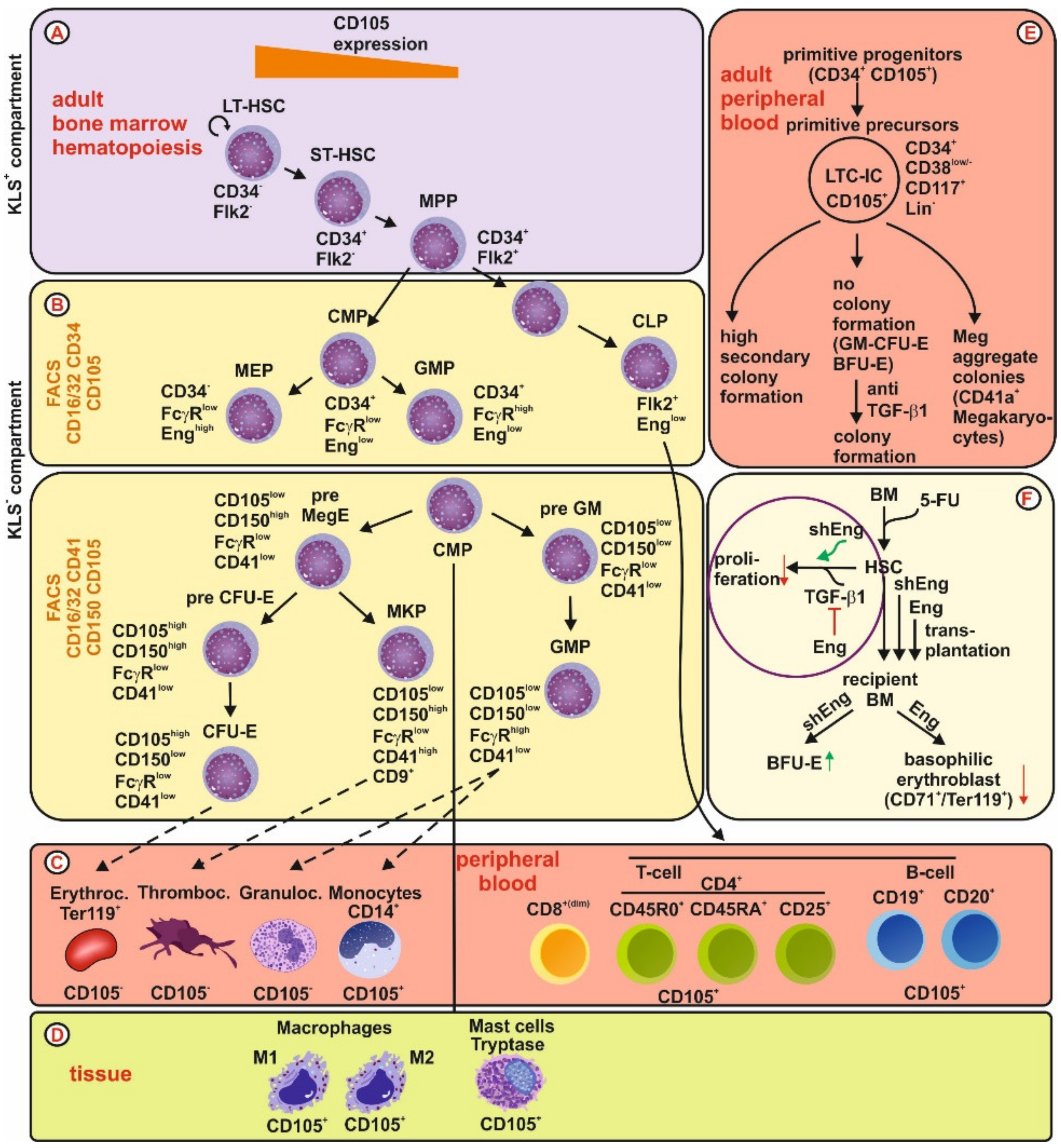{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Structural and Functional Aspects of Endoglin
2. Endoglin in Pathological Conditions
2.1. The Female Reproductive System
2.2. Vascular Homeostasis
3. Endoglin in the Hematopoietic System
3.1. Endoglin Expression in the Stem Cell/Niche
3.2. Endoglin Expression During Hematopoiesis
3.2.1. Fetal Hematopoiesis
3.2.2. Adult Hematopoiesis
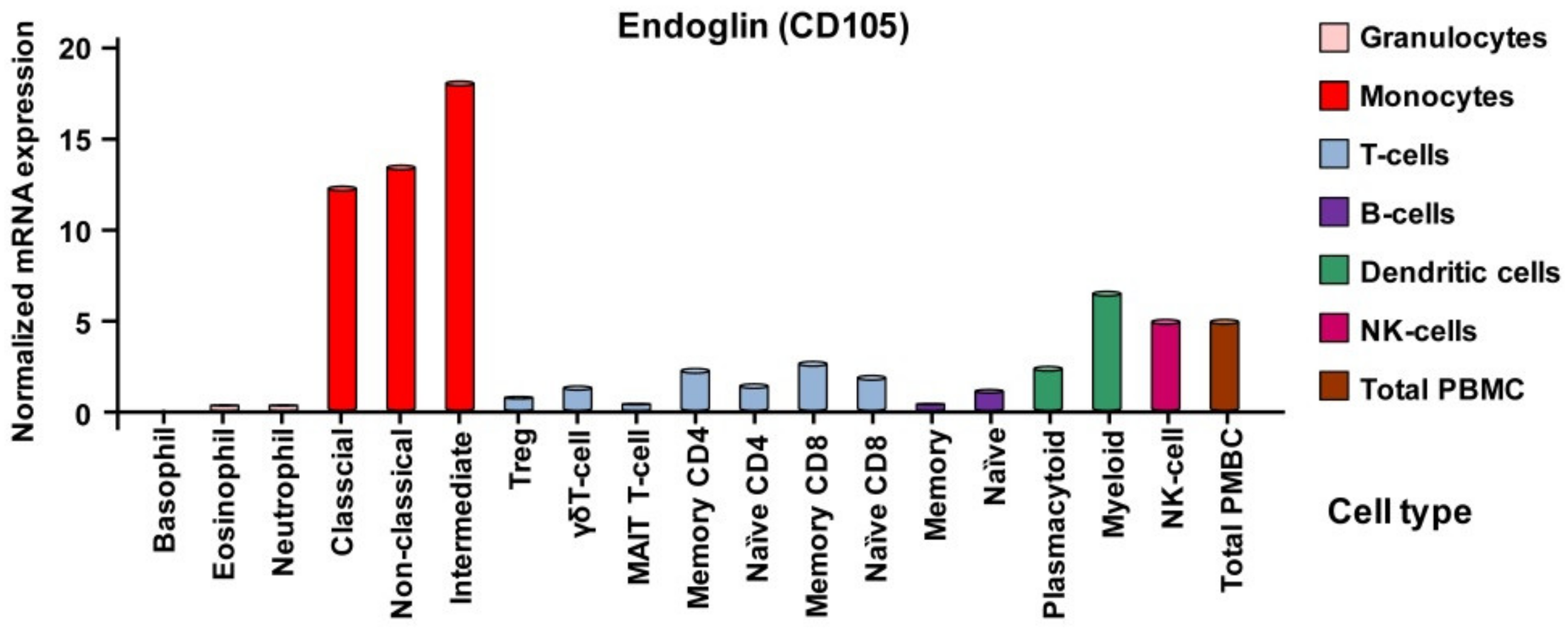
3.2.3. Expression of Endoglin in Individual Hematopoietic Lineages
Erythrocytes
Megakaryocytes/Thrombocytes
Granulocytes
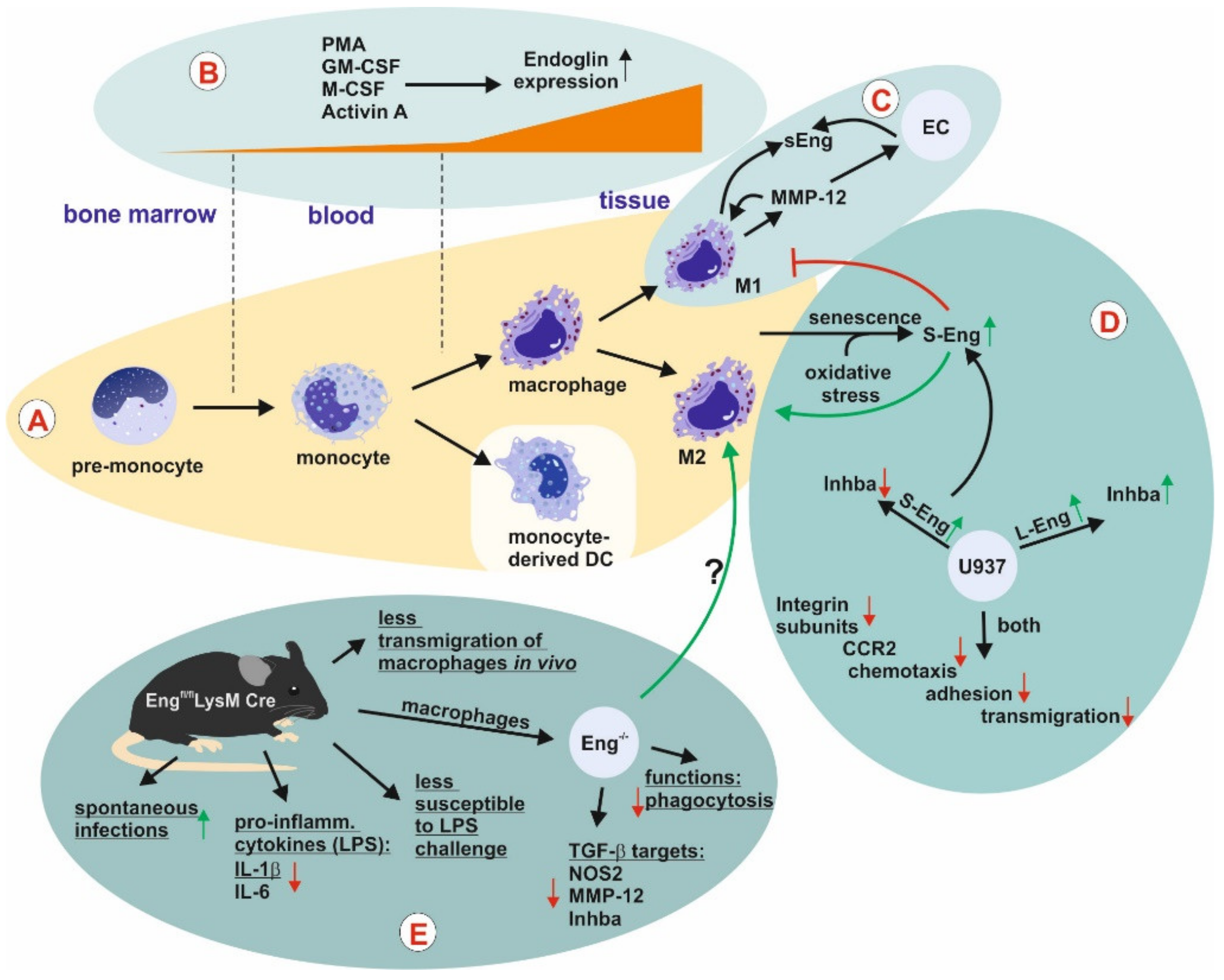
Monocytes and Macrophages
Mast Cells
- Mast Cells and TGF-β1
- Mast Cells and Endoglin
Lymphopoietic Cells
- Endoglin and Leukemia
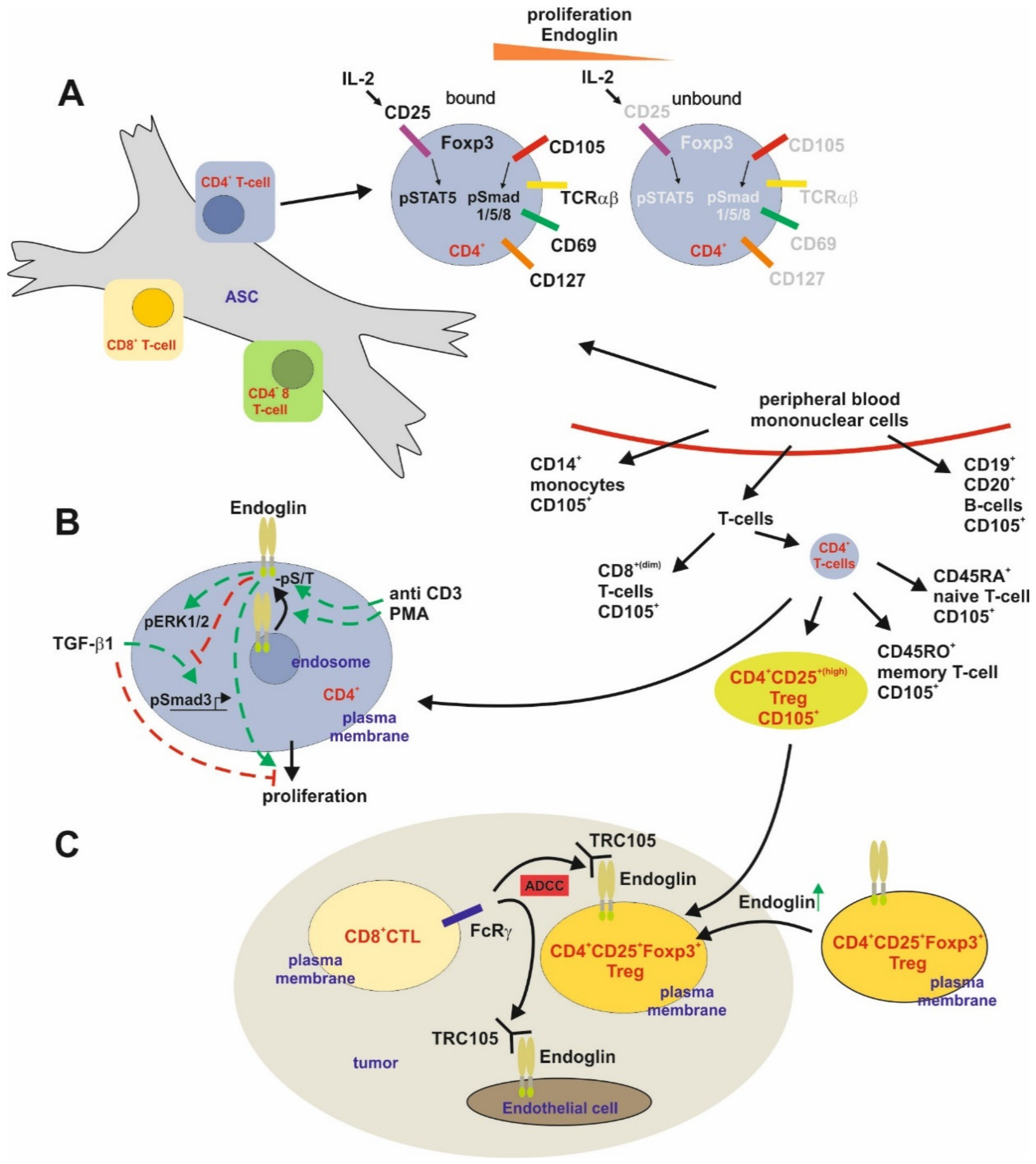
T-Cells
B-Cells
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADCC | Antibody-dependent cellular cytotoxicity |
| ALL | Acute lymphoblastic leukemia |
| AML | Acute myeloic leukemia |
| Ank | Ankylosis |
| BCSP | Bone cartilage, stromal progenitor |
| BFU-E | erythroid burst-forming unit(s) |
| BL-CFC | Blast-colony forming cell(s) |
| BMP | Bone morphogenetic protein |
| ccp | Clathrin-coated pits |
| CFU-E | Erythroid colony forming unit(s) |
| CFU-GM | Granulocyte-macrophage colony-forming unit(s) |
| E | Embryonic day |
| EC(s) | Endothelial cell(s) |
| EEA-1 | Early endosomal antigen-1 |
| Eng | Gene encoding Endoglin |
| EryP | Early erythroid precursor(s) |
| ES | Embryonic stem cell(s) |
| FACS | Fluorescence-activated cell sorting |
| Flk-1 | Fetal liver kinase-1 |
| GIPC | GAIP C-terminus-interacting protein |
| HHT-1 | Hereditary hemorrhagic telangiectasia-1 |
| HSC | Hematopoietic stem cell(s) |
| HUVECs | Human umbilical vein endothelial cells |
| iNOS | Inducible NO-synthase |
| IR | Intron retention |
| IUPR | Intrauterine growth restriction |
| L-Eng | Long form of Endoglin |
| mASC | Murine adipose tissue derived MSC |
| MC(s) | Mast cell(s) |
| MEP | Megakaryocyte-erythroid progenitor(s) |
| MPP | Multipotent progenitor(s) |
| MSC(s) | Mesenchymal stem cell(s) |
| pERK1/2 | Phosphorylated extracellular signal-regulated kinase 1/2 |
| PI3K/Akt | Phosphatidylinositol 3-kinase |
| PMA | Phorbol-12-myristate-13-acetate |
| PMN | Polymorphonuclear neutrophils |
| sEng | Soluble Endoglin |
| sFlt-1 | Soluble fms-like tyrosine kinase-1 (VEGF recptor) |
| S-Eng | Short variant of Endoglin |
| sVEGFR1 | Soluble VEGF receptor |
| TβRI/II | Transforming growth factor-β1 type I or II receptor |
| ZRP-1 | Zyxin-related protein (ZRP)-1 |
References
- Lux, A.; Gallione, C.J.; Marchuk, D.A. Expression analysis of endoglin missense and truncation mutations: Insights into protein structure and disease mechanisms. Hum. Mol. Genet. 2000, 9, 745–755. [Google Scholar] [CrossRef][Green Version]
- Guerrero-Esteo, M.; Sanchez-Elsner, T.; Letamendia, A.; Bernabeu, C. Extracellular and cytoplasmic domains of endoglin interact with the transforming growth factor-β receptors I and II. J. Biol Chem. 2002, 277, 29197–29209. [Google Scholar] [CrossRef]
- Kim, Y.W.; Park, J.; Lee, H.J.; Lee, S.Y.; Kim, S.J. TGF-β sensitivity is determined by N-linked glycosylation of the type II TGF-β receptor. Biochem. J. 2012, 445, 403–411. [Google Scholar] [CrossRef]
- Meurer, S.; Wimmer, A.E.; Leur, E.V.; Weiskirchen, R. Endoglin trafficking/exosomal targeting in liver cells depends on N-glycosylation. Cells 2019, 8, 997. [Google Scholar] [CrossRef] [PubMed]
- Letamendía, A.; Lastres, P.; Almendro, N.; Raab, U.; Bühring, H.J.; Kumar, S.; Bernabéu, C. Endoglin, a component of the TGF-β receptor system, is a differentiation marker of human choriocarcinoma cells. Int. J. Cancer 1998, 76, 541–546. [Google Scholar] [CrossRef]
- Pece-Barbara, N.; Cymerman, U.; Vera, S.; Marchuk, D.A.; Letarte, M. Expression analysis of four endoglin missense mutations suggests that haploinsufficiency is the predominant mechanism for hereditary hemorrhagic telangiectasia type 1. Hum. Mol. Genet. 1999, 8, 2171–2181. [Google Scholar] [CrossRef] [PubMed]
- Koleva, R.I.; Conley, B.A.; Romero, D.; Riley, K.S.; Marto, J.A.; Lux, A.; Vary, C.P. Endoglin structure and function: Determinants of endoglin phosphorylation by transforming growth factor-beta receptors. J. Biol. Chem. 2006, 281, 25110–25123. [Google Scholar] [CrossRef]
- Pan, C.C.; Kumar, S.; Shah, N.; Hoyt, D.G.; Hawinkels, L.J.; Mythreye, K.; Lee, N.Y. Src-mediated post-translational regulation of endoglin stability and function is critical for angiogenesis. J. Biol. Chem. 2014, 289, 25486–25496. [Google Scholar] [CrossRef]
- Kim, S.K.; Henen, M.A.; Hinck, A.P. Structural biology of betaglycan and endoglin, membrane-bound co-receptors of the TGF-β family. Exp. Biol. Med. 2019, 244, 1547–1558. [Google Scholar] [CrossRef]
- Conley, B.A.; Koleva, R.; Smith, J.D.; Kacer, D.; Zhang, D.; Bernabéu, C.; Vary, C.P. Endoglin controls cell migration and composition of focal adhesions: Function of the cytosolic domain. J. Biol. Chem. 2004, 279, 27440–27449. [Google Scholar] [CrossRef]
- Sanz-Rodriguez, F.; Guerrero-Esteo, M.; Botella, L.M.; Banville, D.; Vary, C.P.; Bernabéu, C. Endoglin regulates cytoskeletal organization through binding to ZRP-1, a member of the Lim family of proteins. J. Biol. Chem. 2004, 279, 32858–32868. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.Y.; Blobe, G.C.J. The interaction of endoglin with β-arrestin2 regulates transforming growth factor-β-mediated ERK activation and migration in endothelial cells. Biol. Chem. 2007, 282, 21507–21517. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.Y.; Ray, B.; How, T.; Blobe, G.C. Endoglin promotes transforming growth factor beta-mediated Smad 1/5/8 signaling and inhibits endothelial cell migration through its association with GIPC. J. Biol. Chem. 2008, 283, 32527–32533. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.Y.; Golzio, C.; Gatza, C.E.; Sharma, A.; Katsanis, N.; Blobe, G.C. Endoglin regulates PI3-kinase/Akt trafficking and signaling to alter endothelial capillary stability during angiogenesis. Mol. Biol. Cell. 2012, 23, 2412–2423. [Google Scholar] [CrossRef] [PubMed]
- Lastres, P.; Martín-Perez, J.; Langa, C.; Bernabéu, C. Phosphorylation of the human-transforming-growth-factor-beta-binding protein endoglin. Biochem. J. 1994, 301 Pt 3, 765–768. [Google Scholar] [CrossRef]
- Pérez-Gómez, E.; Eleno, N.; López-Novoa, J.M.; Ramirez, J.R.; Velasco, B.; Letarte, M.; Bernabéu, C.; Quintanilla, M. Characterization of murine S-endoglin isoform and its effects on tumor development. Oncogene 2005, 24, 4450–4461. [Google Scholar] [CrossRef]
- Meurer, S.K.; Tihaa, L.; Borkham-Kamphorst, E.; Weiskirchen, R. Expression and functional analysis of endoglin in isolated liver cells and its involvement in fibrogenic Smad signalling. Cell Signal. 2011, 23, 683–699. [Google Scholar] [CrossRef]
- Di Guglielmo, G.M.; Le Roy, C.; Goodfellow, A.F.; Wrana, J.L. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat. Cell Biol. 2003, 5, 410–421. [Google Scholar] [CrossRef]
- Budi, E.H.; Xu, J.; Derynck, R. Regulation of TGF-β Receptors. Methods Mol. Biol. 2016, 1344, 1–33. [Google Scholar] [CrossRef]
- Toporsian, M.; Gros, R.; Kabir, M.G.; Vera, S.; Govindaraju, K.; Eidelman, D.H.; Husain, M.; Letarte, M. A role for endoglin in coupling eNOS activity and regulating vascular tone revealed in hereditary hemorrhagic telangiectasia. Circ. Res. 2005, 96, 684–692. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, X.; Yan, Z.; Chen, Y.; Cui, Y.; Chen, B.; Huang, C.; Zhang, W.; Yin, X.; He, Q.Y.; et al. Detergent-insoluble proteome analysis revealed aberrantly aggregated proteins in human preeclampsia placentas. Proteome Res. 2017, 16, 4468–4480. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Massagué, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Lebrin, F.; Deckers, M.; Bertolino, P.; Ten Dijke, P. TGF-β receptor function in the endothelium. Cardiovasc. Res. 2005, 65, 599–608. [Google Scholar] [CrossRef]
- Schmidt-Weber, C.B.; Letarte, M.; Kunzmann, S.; Rückert, B.; Bernabéu, C.; Blaser, K. TGF-{beta} signaling of human T cells is modulated by the ancillary TGF-β receptor endoglin. Int. Immunol. 2005, 17, 921–930. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bernabeu, C.; Conley, B.A.; Vary, C.P. Novel biochemical pathways of endoglin in vascular cell physiology. J. Cell Biochem. 2007, 102, 1375–1388. [Google Scholar] [CrossRef] [PubMed]
- Santibanez, J.F.; Pérez-Gómez, E.; Fernandez-L, A.; Garrido-Martin, E.M.; Carnero, A.; Malumbres, M.; Vary, C.P.; Quintanilla, M.; Bernabéu, C. The TGF-beta co-receptor endoglin modulates the expression and transforming potential of H-Ras. Carcinogenesis 2010, 31, 2145–2154. [Google Scholar] [CrossRef]
- Park, S.; Dimaio, T.A.; Liu, W.; Wang, S.; Sorenson, C.M.; Sheibani, N. Endoglin regulates the activation and quiescence of endothelium by participating in canonical and non-canonical TGF-β signaling pathways. J. Cell Sci. 2013, 126 Pt 6, 1392–1405. [Google Scholar] [CrossRef]
- Sánchez-Elsner, T.; Botella, L.M.; Velasco, B.; Langa, C.; Bernabéu, C. Endoglin expression is regulated by transcriptional cooperation between the hypoxia and transforming growth factor-β pathways. J. Biol. Chem. 2002, 277, 43799–43808. [Google Scholar] [CrossRef]
- Botella, L.M.; Sanz-Rodriguez, F.; Komi, Y.; Fernandez-L, A.; Varela, E.; Garrido-Martin, E.M.; Narla, G.; Friedman, S.L.; Kojima, S. TGF-β regulates the expression of transcription factor KLF6 and its splice variants and promotes co-operative transactivation of common target genes through a Smad3-Sp1-KLF6 interaction. Biochem. J. 2009, 419, 485–495. [Google Scholar] [CrossRef]
- Sugden, W.W.; Siekmann, A.F. Endothelial cell biology of Endoglin in hereditary hemorrhagic telangiectasia. Curr. Opin. Hematol. 2018, 25, 237–244. [Google Scholar] [CrossRef]
- Seon, B.K.; Haba, A.; Matsuno, F.; Takahashi, N.; Tsujie, M.; She, X.; Harada, N.; Uneda, S.; Tsujie, T.; Toi, H.; et al. Endoglin-targeted cancer therapy. Curr. Drug Deliv. 2011, 8, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Ollauri-Ibáñez, C.; López-Novoa, J.M.; Pericacho, M. Endoglin-based biological therapy in the treatment of angiogenesis-dependent pathologies. Expert Opin. Biol. Ther. 2017, 17, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Cai, K.; Cheng, Y.; Wang, S.; Paun, B.; Hamilton, J.P.; Jin, Z.; Sato, F.; Berki, A.T.; Kan, T.; et al. A genome-wide search identifies epigenetic silencing of somatostatin, tachykinin-1, and 5 other genes in colon cancer. Gastroenterology 2006, 131, 797–808. [Google Scholar] [CrossRef]
- Wong, V.C.; Chan, P.L.; Bernabeu, C.; Law, S.; Wang, L.D.; Li, J.L.; Tsao, S.W.; Srivastava, G.; Lung, M.L. Identification of an invasion and tumor-suppressing gene, Endoglin (ENG), silenced by both epigenetic inactivation and allelic loss in esophageal squamous cell carcinoma. Int. J. Cancer 2008, 123, 2816–2823. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, K.; Shia, A.; Cavicchioli, F.; Haley, V.; Comino, A.; Merlano, M.; Mauri, F.; Walter, K.; Lackner, M.; Wischnewsky, M.B.; et al. Identification of Endoglin as an epigenetically regulated tumour-suppressor gene in lung cancer. Br. J. Cancer 2015, 113, 970–978. [Google Scholar] [CrossRef][Green Version]
- Rose, M.; Meurer, S.K.; Kloten, V.; Weiskirchen, R.; Denecke, B.; Antonopoulos, W.; Deckert, M.; Knüchel, R.; Dahl, E. ITIH5 induces a shift in TGF-β superfamily signaling involving Endoglin and reduces risk for breast cancer metastasis and tumor death. Mol. Carcinog. 2018, 57, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Paquet, M.E.; Pece-Barbara, N.; Vera, S.; Cymerman, U.; Karabegovic, A.; Shovlin, C.; Letarte, M. Analysis of several endoglin mutants reveals no endogenous mature or secreted protein capable of interfering with normal endoglin function. Hum. Mol. Genet. 2001, 10, 1347–1357. [Google Scholar] [CrossRef]
- Ali, B.R.; Ben-Rebeh, I.; John, A.; Akawi, N.A.; Milhem, R.M.; Al-Shehhi, N.A.; Al-Ameri, M.M.; Al-Shamisi, S.A.; Al-Gazali, L. Endoplasmic reticulum quality control is involved in the mechanism of endoglin-mediated hereditary haemorrhagic telangiectasia. PLoS ONE 2011, 6, e26206. [Google Scholar] [CrossRef]
- Förg, T.; Hafner, M.; Lux, A. Investigation of endoglin wild-type and missense mutant protein heterodimerisation using fluorescence microscopy based IF, BiFC and FRET analyses. PLoS ONE 2014, 9, e102998. [Google Scholar] [CrossRef][Green Version]
- Mallet, C.; Lamribet, K.; Giraud, S.; Dupuis-Girod, S.; Feige, J.J.; Bailly, S.; Tillet, E. Functional analysis of endoglin mutations from hereditary hemorrhagic telangiectasia type 1 patients reveals different mechanisms for endoglin loss of function. Hum. Mol. Genet. 2015, 24, 1142–1154. [Google Scholar] [CrossRef]
- Gariballa, N.; Ali, B.R. Endoplasmic reticulum associated protein degradation (ERAD) in the pathology of diseases related to TGFβ signaling pathway: Future therapeutic perspectives. Front. Mol. Biosci. 2020, 7, 575608. [Google Scholar] [CrossRef] [PubMed]
- Gallardo-Vara, E.; Ruiz-Llorente, L.; Casado-Vela, J.; Ruiz-Rodríguez, M.J.; López-Andrés, N.; Pattnaik, A.K.; Quintanilla, M.; Bernabeu, C. Endoglin protein interactome profiling identifies TRIM21 and Galectin-3 as new binding partners. Cells 2019, 8, 1082. [Google Scholar] [CrossRef] [PubMed]
- Bourdeau, A.; Faughnan, M.E.; Letarte, M. Endoglin-deficient mice, a unique model to study hereditary hemorrhagic telangiectasia. Trends Cardiovasc. Med. 2000, 10, 279–285. [Google Scholar] [CrossRef]
- Rossi, E.; Smadja, D.M.; Boscolo, E.; Langa, C.; Arevalo, M.A.; Pericacho, M.; Gamella-Pozuelo, L.; Kauskot, A.; Botella, L.M.; Gaussem, P.; et al. Endoglin regulates mural cell adhesion in the circulatory system. Cell Mol. Life Sci. 2016, 73, 1715–1739. [Google Scholar] [CrossRef] [PubMed]
- Hawinkels, L.J.; Kuiper, P.; Wiercinska, E.; Verspaget, H.W.; Liu, Z.; Pardali, E.; Sier, C.F.; ten Dijke, P. Matrix metalloproteinase-14 (MT1-MMP)-mediated endoglin shedding inhibits tumor angiogenesis. Cancer Res. 2010, 70, 4141–4150. [Google Scholar] [CrossRef] [PubMed]
- Venkatesha, S.; Toporsian, M.; Lam, C.; Hanai, J.; Mammoto, T.; Kim, Y.M.; Bdolah, Y.; Lim, K.H.; Yuan, H.T.; Libermann, T.A.; et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 2006, 12, 642–649. [Google Scholar] [CrossRef]
- St-Jacques, S.; Cymerman, U.; Pece, N.; Letarte, M. Molecular characterization and in situ localization of murine endoglin reveal that it is a transforming growth factor-beta binding protein of endothelial and stromal cells. Endocrinology 1994, 134, 2645–2657. [Google Scholar] [CrossRef]
- Cross, J.C.; Werb, Z.; Fisher, S.J. Implantation and the placenta: Key pieces of the development puzzle. Science 1994, 266, 1508–1518. [Google Scholar] [CrossRef]
- Zhou, Y.; Damsky, C.H.; Chiu, K.; Roberts, J.M.; Fisher, S.J. Preeclampsia is associated with abnormal expression of adhesion molecules by invasive cytotrophoblasts. J. Clin. Invest. 1993, 91, 950–960. [Google Scholar] [CrossRef]
- Sibai, B.; Dekker, G.; Kupferminc, M. Pre-eclampsia. Lancet 2005, 365, 785–799. [Google Scholar] [CrossRef]
- Weinstein, L. Syndrome of hemolysis, elevated liver enzymes, and low platelet count: A severe consequence of hypertension in pregnancy. 1982. Am. J. Obstet. Gynecol. 2005, 193, P859. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.E.; Min, J.Y.; Merchan, J.; Lim, K.H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Invest. 2003, 111, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Kingdom, J.; Huppertz, B.; Seaward, G.; Kaufmann, P. Development of the placental villous tree and its consequences for fetal growth. Eur. J. Obstet. Gynecol. Reprod. Biol. 2000, 92, 35–43. [Google Scholar] [CrossRef]
- Gougos, A.; St Jacques, S.; Greaves, A.; O’Connell, P.J.; d’Apice, A.J.; Bühring, H.J.; Bernabeu, C.; van Mourik, J.A.; Letarte, M. Identification of distinct epitopes of endoglin, an RGD-containing glycoprotein of endothelial cells, leukemic cells, and syncytiotrophoblasts. Int. Immunol. 1992, 4, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Dağdeviren, A.; Müftüoğlu, S.F.; Cakar, A.N.; Ors, U. Endoglin (CD 105) expression in human lymphoid organs and placenta. Ann. Anat. 1998, 180, 461–469. [Google Scholar] [CrossRef]
- Caniggia, I.; Taylor, C.V.; Ritchie, J.W.; Lye, S.J.; Letarte, M. Endoglin regulates trophoblast differentiation along the invasive pathway in human placental villous explants. Endocrinology 1997, 138, 4977–4988. [Google Scholar] [CrossRef]
- Yinon, Y.; Nevo, O.; Xu, J.; Many, A.; Rolfo, A.; Todros, T.; Post, M.; Caniggia, I. Severe intrauterine growth restriction pregnancies have increased placental endoglin levels: Hypoxic regulation via transforming growth factor-beta 3. Am. J. Pathol. 2008, 172, 77–85. [Google Scholar] [CrossRef][Green Version]
- Repnik, U.; Tilburgs, T.; Roelen, D.L.; van der Mast, B.J.; Kanhai, H.H.; Scherjon, S.; Claas, F.H. Comparison of macrophage phenotype between decidua basalis and decidua parietalis by flow cytometry. Placenta 2008, 29, 405–412. [Google Scholar] [CrossRef]
- Brusgaard, K.; Kjeldsen, A.D.; Poulsen, L.; Moss, H.; Vase, P.; Rasmussen, K.; Kruse, T.A.; Hørder, M. Mutations in endoglin and in activin receptor-like kinase 1 among Danish patients with hereditary haemorrhagic telangiectasia. Clin. Genet. 2004, 66, 556–561. [Google Scholar] [CrossRef]
- Lesca, G.; Plauchu, H.; Coulet, F.; Lefebvre, S.; Plessis, G.; Odent, S.; Rivière, S.; Leheup, B.; Goizet, C.; Carette, M.F.; et al. Molecular screening of ALK1/ACVRL1 and ENG genes in hereditary hemorrhagic telangiectasia in France. French Rendu-Osler Network. Hum. Mutat. 2004, 23, 289–299. [Google Scholar] [CrossRef]
- Schulte, C.; Geisthoff, U.; Lux, A.; Kupka, S.; Zenner, H.P.; Blin, N.; Pfister, M. High frequency of ENG and ALK1/ACVRL1 mutations in German HHT patients. Hum. Mutat. 2005, 25, 595. [Google Scholar] [CrossRef] [PubMed]
- Tørring, P.M.; Larsen, M.J.; Kjeldsen, A.D.; Ousager, L.B.; Tan, Q.; Brusgaard, K. Global gene expression profiling of telangiectasial tissue from patients with hereditary hemorrhagic telangiectasia. Microvasc. Res. 2015, 99, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.; Eyries, M.; Montagne, K.; Martin, S.; Agrapart, M.; Simerman-François, R.; Letarte, M.; Soubrier, F. Altered endothelial gene expression associated with hereditary haemorrhagic telangiectasia. Eur. J. Clin. Invest. 2007, 37, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Bataller, A.; Montalban-Bravo, G.; Soltysiak, K.A.; Garcia-Manero, G. The role of TGFβ in hematopoiesis and myeloid disorders. Leukemia 2019, 33, 1076–1089. [Google Scholar] [CrossRef]
- Braverman, I.M.; Keh, A.; Jacobson, B.S. Ultrastructure and three-dimensional organization of the telangiectases of hereditary hemorrhagic telangiectasia. J. Invest. Dermatol. 1990, 95, 422–427. [Google Scholar] [CrossRef]
- Jacobson, B.S. Hereditary hemorrhagic telangiectasia: A model for blood vessel growth and enlargement. Am. J. Pathol. 2000, 156, 737–742. [Google Scholar] [CrossRef]
- Cirulli, A.; Loria, M.P.; Dambra, P.; Di Serio, F.; Ventura, M.T.; Amati, L.; Jirillo, E.; Sabbà, C. Patients with Hereditary Hemorrhagic Telangectasia (HHT) exhibit a deficit of polymorphonuclear cell and monocyte oxidative burst and phagocytosis: A possible correlation with altered adaptive immune responsiveness in HHT. Curr. Pharm. Des. 2006, 12, 1209–1215. [Google Scholar] [CrossRef]
- Guilhem, A.; Malcus, C.; Clarivet, B.; Plauchu, H.; Dupuis-Girod, S. Immunological abnormalities associated with hereditary haemorrhagic telangiectasia. J. Intern. Med. 2013, 274, 351–362. [Google Scholar] [CrossRef]
- Amati, L.; Passeri, M.E.; Resta, F.; Triggiani, V.; Jirillo, E.; Sabbà, C. Ablation of T-helper 1 cell derived cytokines and of monocyte-derived tumor necrosis factor-alpha in hereditary hemorrhagic telangiectasia: Immunological consequences and clinical considerations. Curr. Pharm. Des. 2006, 12, 1201–1208. [Google Scholar] [CrossRef]
- Turley, J.M.; Falk, L.A.; Ruscetti, F.W.; Kasper, J.J.; Francomano, T.; Fu, T.; Bang, O.S.; Birchenall-Roberts, M.C. Transforming growth factor beta 1 functions in monocytic differentiation of hematopoietic cells through autocrine and paracrine mechanisms. Cell Growth Differ. 1996, 7, 1535–1544. [Google Scholar]
- Lagraoui, M.; Gagnon, L. Enhancement of human neutrophil survival and activation by TGF-beta 1. Cell Mol. Biol. 1997, 43, 313–318. [Google Scholar] [PubMed]
- Bourdeau, A.; Faughnan, M.E.; McDonald, M.L.; Paterson, A.D.; Wanless, I.R.; Letarte, M. Potential role of modifier genes influencing transforming growth factor-beta1 levels in the development of vascular defects in endoglin heterozygous mice with hereditary hemorrhagic telangiectasia. Am. J. Pathol. 2001, 158, 2011–2020. [Google Scholar] [CrossRef]
- Rossi, E.; Sanz-Rodriguez, F.; Eleno, N.; Düwell, A.; Blanco, F.J.; Langa, C.; Botella, L.M.; Cabañas, C.; Lopez-Novoa, J.M.; Bernabeu, C. Endothelial endoglin is involved in inflammation: Role in leukocyte adhesion and transmigration. Blood 2013, 121, 403–415. [Google Scholar] [CrossRef]
- Rossi, E.; Lopez-Novoa, J.M.; Bernabeu, C. Endoglin involvement in integrin-mediated cell adhesion as a putative pathogenic mechanism in hereditary hemorrhagic telangiectasia type 1 (HHT1). Front. Genet. 2015, 5, 457. [Google Scholar] [CrossRef] [PubMed]
- Marazuela, M.; Sánchez-Madrid, F.; Acevedo, A.; Larrañaga, E.; de Landázuri, M.O. Expression of vascular adhesion molecules on human endothelia in autoimmune thyroid disorders. Clin. Exp. Immunol. 1995, 102, 328–334. [Google Scholar] [CrossRef]
- Van de Kerkhof, P.C.; Rulo, H.F.; van Pelt, J.P.; van Vlijmen-Willems, I.M.; De Jong, E.M. Expression of endoglin in the transition between psoriatic uninvolved and involved skin. Acta Derm. Venereol. 1998, 78, 19–21. [Google Scholar] [CrossRef]
- Koç, O.N.; Gerson, S.L.; Cooper, B.W.; Dyhouse, S.M.; Haynesworth, S.E.; Caplan, A.I.; Lazarus, H.M. Rapid hematopoietic recovery after coinfusion of autologous-blood stem cells and culture-expanded marrow mesenchymal stem cells in advanced breast cancer patients receiving high-dose chemotherapy. J. Clin. Oncol. 2000, 18, 307–316. [Google Scholar] [CrossRef]
- Da Silva Meirelles, L.; Chagastelles, P.C.; Nardi, N.B. Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J. Cell Sci. 2006, 119 Pt 11, 2204–2213. [Google Scholar] [CrossRef]
- Barry, F.P.; Boynton, R.E.; Haynesworth, S.; Murphy, J.M.; Zaia, J. The monoclonal antibody SH-2, raised against human mesenchymal stem cells, recognizes an epitope on endoglin (CD105). Biochem. Biophys. Res. Commun. 1999, 265, 134–139. [Google Scholar] [CrossRef]
- Bühring, H.J.; Battula, V.L.; Treml, S.; Schewe, B.; Kanz, L.; Vogel, W. Novel markers for the prospective isolation of human MSC. Ann. NY Acad. Sci. 2007, 1106, 262–271. [Google Scholar] [CrossRef]
- Bochev, I.; Elmadjian, G.; Kyurkchiev, D.; Tzvetanov, L.; Altankova, I.; Tivchev, P.; Kyurkchiev, S. Mesenchymal stem cells from human bone marrow or adipose tissue differently modulate mitogen-stimulated B-cell immunoglobulin production in vitro. Cell Biol. Int. 2008, 32, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xu, H.; Wan, C.; McCaigue, M.; Li, G. Bioreactor expansion of human adult bone marrow-derived mesenchymal stem cells. Stem Cells 2006, 24, 2052–2059. [Google Scholar] [CrossRef][Green Version]
- Anderson, P.; Carrillo-Gálvez, A.B.; García-Pérez, A.; Cobo, M.; Martín, F. CD105 (endoglin)-negative murine mesenchymal stromal cells define a new multipotent subpopulation with distinct differentiation and immunomodulatory capacities. PLoS ONE 2013, 8, e76979. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.A.; Lemischka, I.R. Stem cells and their niches. Science 2006, 311, 1880–1885. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.K.; Lindau, P.; Jiang, W.; Chen, J.Y.; Zhang, L.F.; Chen, C.C.; Seita, J.; Sahoo, D.; Kim, J.B.; Lee, A.; et al. Clonal precursor of bone, cartilage, and hematopoietic niche stromal cells. Proc. Natl. Acad. Sci. USA 2013, 110, 12643–12648. [Google Scholar] [CrossRef] [PubMed]
- Borges, L.; Iacovino, M.; Mayerhofer, T.; Koyano-Nakagawa, N.; Baik, J.; Garry, D.J.; Kyba, M.; Letarte, M.; Perlingeiro, R.C. A critical role for endoglin in the emergence of blood during embryonic development. Blood 2012, 119, 5417–5428. [Google Scholar] [CrossRef]
- Arthur, H.M.; Ure, J.; Smith, A.J.; Renforth, G.; Wilson, D.I.; Torsney, E.; Charlton, R.; Parums, D.V.; Jowett, T.; Marchuk, D.A.; et al. Endoglin, an ancillary TGFbeta receptor, is required for extraembryonic angiogenesis and plays a key role in heart development. Dev. Biol. 2000, 217, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.K.; Bourdeau, A.; Letarte, M.; Zúñiga-Pflücker, J.C. Expression and function of CD105 during the onset of hematopoiesis from Flk1(+) precursors. Blood 2001, 98, 3635–3642. [Google Scholar] [CrossRef]
- Perlingeiro, R.C. Endoglin is required for hemangioblast and early hematopoietic development. Development 2007, 134, 3041–3048. [Google Scholar] [CrossRef]
- Li, D.Y.; Sorensen, L.K.; Brooke, B.S.; Urness, L.D.; Davis, E.C.; Taylor, D.G.; Boak, B.B.; Wendel, D.P. Defective angiogenesis in mice lacking endoglin. Science 1999, 284, 1534–1537. [Google Scholar] [CrossRef]
- Baik, J.; Borges, L.; Magli, A.; Thatava, T.; Perlingeiro, R.C. Effect of endoglin overexpression during embryoid body development. Exp. Hematol. 2012, 40, 837–846. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Baik, J.; Magli, A.; Tahara, N.; Swanson, S.A.; Koyano-Nakagawa, N.; Borges, L.; Stewart, R.; Garry, D.J.; Kawakami, Y.; Thomson, J.A.; et al. Endoglin integrates BMP and Wnt signalling to induce haematopoiesis through JDP2. Nat. Commun. 2016, 7, 13101. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Z.; Li, M.; de Graaf, D.; Monti, S.; Göttgens, B.; Sanchez, M.J.; Lander, E.S.; Golub, T.R.; Green, A.R.; Lodish, H.F. Identification of endoglin as a functional marker that defines long-term repopulating hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 2002, 99, 15468–15473. [Google Scholar] [CrossRef] [PubMed]
- Pronk, C.J.; Rossi, D.J.; Månsson, R.; Attema, J.L.; Norddahl, G.L.; Chan, C.K.; Sigvardsson, M.; Weissman, I.L.; Bryder, D. Elucidation of the phenotypic, functional, and molecular topography of a myeloerythroid progenitor cell hierarchy. Cell Stem Cell 2007, 1, 428–442. [Google Scholar] [CrossRef]
- Chen, C.Z.; Li, L.; Li, M.; Lodish, H.F. The endoglin(positive) sca-1(positive) rhodamine(low) phenotype defines a near-homogeneous population of long-term repopulating hematopoietic stem cells. Immunity 2003, 19, 525–533. [Google Scholar] [CrossRef]
- Pronk, C.J.; Attema, J.; Rossi, D.J.; Sigvardsson, M.; Bryder, D. Deciphering developmental stages of adult myelopoiesis. Cell Cycle 2008, 7, 706–713. [Google Scholar] [CrossRef]
- Ng, A.P.; Kauppi, M.; Metcalf, D.; Di Rago, L.; Hyland, C.D.; Alexander, W.S. Characterization of thrombopoietin (TPO)-responsive progenitor cells in adult mouse bone marrow with in vivo megakaryocyte and erythroid potential. Proc. Natl. Acad. Sci. USA 2012, 109, 2364–2369. [Google Scholar] [CrossRef]
- Pierelli, L.; Scambia, G.; Fattorossi, A.; Bonanno, G.; Battaglia, A.; Rumi, C.; Marone, M.; Mozzetti, S.; Rutella, S.; Menichella, G.; et al. Functional, phenotypic and molecular characterization of cytokine low-responding circulating CD34+ haemopoietic progenitors. Br. J. Haematol. 1998, 102, 1139–1150. [Google Scholar] [CrossRef]
- Pierelli, L.; Scambia, G.; Bonanno, G.; Rutella, S.; Puggioni, P.; Battaglia, A.; Mozzetti, S.; Marone, M.; Menichella, G.; Rumi, C.; et al. CD34+/CD105+ cells are enriched in primitive circulating progenitors residing in the G0 phase of the cell cycle and contain all bone marrow and cord blood CD34+/CD38low/- precursors. Br. J. Haematol. 2000, 108, 610–620. [Google Scholar] [CrossRef]
- Moody, J.L.; Singbrant, S.; Karlsson, G.; Blank, U.; Aspling, M.; Flygare, J.; Bryder, D.; Karlsson, S. Endoglin is not critical for hematopoietic stem cell engraftment and reconstitution but regulates adult erythroid development. Stem Cells 2007, 25, 2809–2819. [Google Scholar] [CrossRef]
- The Human Protein Atlas. ENG. Available online: https://www.proteinatlas.org/ENSG00000106991-ENG/blood (accessed on 7 November 2020).
- Bühring, H.J.; Müller, C.A.; Letarte, M.; Gougos, A.; Saalmüller, A.; van Agthoven, A.J.; Busch, F.W. Endoglin is expressed on a subpopulation of immature erythroid cells of normal human bone marrow. Leukemia 1991, 5, 841–847. [Google Scholar] [PubMed]
- Della Porta, M.G.; Malcovati, L.; Invernizzi, R.; Travaglino, E.; Pascutto, C.; Maffioli, M.; Gallì, A.; Boggi, S.; Pietra, D.; Vanelli, L.; et al. Flow cytometry evaluation of erythroid dysplasia in patients with myelodysplastic syndrome. Leukemia 2006, 20, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Wu, L.; He, Q.; Zhang, Z.; Chang, C.; Li, X. Immunophenotypic analysis of erythroid dysplasia and its diagnostic application in myelodysplastic syndromes. Intern. Med. J. 2012, 42, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, H.S.; Shen, H.M.; Yan, H.C.; DeLisser, H.M.; Chung, A.; Mickanin, C.; Trask, T.; Kirschbaum, N.E.; Newman, P.J.; Albelda, S.M.; et al. Platelet endothelial cell adhesion molecule-1 (PECAM-1/CD31): Alternatively spliced, functionally distinct isoforms expressed during mammalian cardiovascular development. Development 1994, 120, 2539–2553. [Google Scholar]
- Young, P.E.; Baumhueter, S.; Lasky, L.A. The sialomucin CD34 is expressed on hematopoietic cells and blood vessels during murine development. Blood 1995, 85, 96–105. [Google Scholar] [CrossRef]
- Breier, G.; Breviario, F.; Caveda, L.; Berthier, R.; Schnürch, H.; Gotsch, U.; Vestweber, D.; Risau, W.; Dejana, E. Molecular cloning and expression of murine vascular endothelial-cadherin in early stage development of cardiovascular system. Blood 1996, 87, 630–641. [Google Scholar] [CrossRef]
- Rakocevic, J.; Orlic, D.; Mitrovic-Ajtic, O.; Tomasevic, M.; Dobric, M.; Zlatic, N.; Milasinovic, D.; Stankovic, G.; Ostojić, M.; Labudovic-Borovic, M. Endothelial cell markers from clinician’s perspective. Exp. Mol. Pathol. 2017, 102, 303–313. [Google Scholar] [CrossRef]
- Ema, M.; Yokomizo, T.; Wakamatsu, A.; Terunuma, T.; Yamamoto, M.; Takahashi, S. Primitive erythropoiesis from mesodermal precursors expressing VE-cadherin, PECAM-1, Tie2, endoglin, and CD34 in the mouse embryo. Blood 2006, 108, 4018–4024. [Google Scholar] [CrossRef]
- Dzierzak, E. The emergence of definitive hematopoietic stem cells in the mammal. Curr. Opin. Hematol. 2005, 12, 197–202. [Google Scholar] [CrossRef]
- Iskander, D.; Psaila, B.; Gerrard, G.; Chaidos, A.; En Foong, H.; Harrington, Y.; Karnik, L.C.; Roberts, I.; de la Fuente, J.; Karadimitris, A. Elucidation of the EP defect in Diamond-Blackfan anemia by characterization and prospective isolation of human EPs. Blood 2015, 125, 2553–2557. [Google Scholar] [CrossRef]
- Mori, Y.; Chen, J.Y.; Pluvinage, J.V.; Seita, J.; Weissman, I.L. Prospective isolation of human erythroid lineage-committed progenitors. Proc. Natl. Acad. Sci. USA 2015, 112, 9638–9643. [Google Scholar] [CrossRef] [PubMed]
- Shovlin, C.L. Hereditary haemorrhagic telangiectasia: Pathophysiology, diagnosis and treatment. Blood Rev. 2010, 24, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Shovlin, C.L.; Sulaiman, N.L.; Govani, F.S.; Jackson, J.E.; Begbie, M.E. Elevated factor VIII in hereditary haemorrhagic telangiectasia (HHT): Association with venous thromboembolism. Thromb. Haemost. 2007, 98, 1031–1039. [Google Scholar] [PubMed]
- Rossi, E.; Pericacho, M.; Bachelot-Loza, C.; Pidard, D.; Gaussem, P.; Poirault-Chassac, S.; Blanco, F.J.; Langa, C.; González-Manchón, C.; Novoa, J.M.L.; et al. Human endoglin as a potential new partner involved in platelet-endothelium interactions. Cell Mol. Life Sci. 2018, 75, 1269–1284. [Google Scholar] [CrossRef]
- Gougos, A.; Letarte, M. Identification of a human endothelial cell antigen with monoclonal antibody 44G4 produced against a pre-B leukemic cell line. J. Immunol. 1988, 141, 1925–1933. [Google Scholar]
- Ojeda-Fernández, L.; Recio-Poveda, L.; Aristorena, M.; Lastres, P.; Blanco, F.J.; Sanz-Rodríguez, F.; Gallardo-Vara, E.; de las Casas-Engel, M.; Corbí, Á.; Arthur, H.M.; et al. Mice Lacking Endoglin in Macrophages Show an Impaired Immune Response. PLoS Genet. 2016, 12, e1005935. [Google Scholar] [CrossRef]
- Torsney, E.; Charlton, R.; Parums, D.; Collis, M.; Arthur, H.M. Inducible expression of human endoglin during inflammation and wound healing in vivo. Inflamm. Res. 2002, 51, 464–470. [Google Scholar] [CrossRef]
- Quackenbush, E.J.; Letarte, M. Identification of several cell surface proteins of non-T, non-B acute lymphoblastic leukemia by using monoclonal antibodies. J. Immunol. 1985, 134, 1276–1285. [Google Scholar]
- Gougos, A.; Letarte, M. Biochemical characterization of the 44G4 antigen from the HOON pre-B leukemic cell line. J. Immunol. 1988, 141, 1934–1940. [Google Scholar]
- Lastres, P.; Bellon, T.; Cabañas, C.; Sanchez-Madrid, F.; Acevedo, A.; Gougos, A.; Letarte, M.; Bernabeu, C. Regulated expression on human macrophages of endoglin, an Arg-Gly-Asp-containing surface antigen. Eur. J. Immunol. 1992, 22, 393–397. [Google Scholar] [CrossRef]
- O’Connell, P.J.; McKenzie, A.; Fisicaro, N.; Rockman, S.P.; Pearse, M.J.; d’Apice, A.J. Endoglin: A 180-kD endothelial cell and macrophage restricted differentiation molecule. Clin. Exp. Immunol. 1992, 90, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Docherty, N.G.; López-Novoa, J.M.; Arevalo, M.; Düwel, A.; Rodriguez-Peña, A.; Pérez-Barriocanal, F.; Bernabeu, C.; Eleno, N. Endoglin regulates renal ischaemia-reperfusion injury. Nephrol. Dial. Transplant. 2006, 21, 2106–2119. [Google Scholar] [CrossRef] [PubMed]
- Scharpfenecker, M.; Floot, B.; Russell, N.S.; Stewart, F.A. The TGF-β co-receptor endoglin regulates macrophage infiltration and cytokine production in the irradiated mouse kidney. Radiother. Oncol. 2012, 105, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Szekanecz, Z.; Haines, G.K.; Harlow, L.A.; Shah, M.R.; Fong, T.W.; Fu, R.; Lin, S.J.; Rayan, G.; Koch, A.E. Increased synovial expression of transforming growth factor (TGF)-beta receptor endoglin and TGF-beta 1 in rheumatoid arthritis: Possible interactions in the pathogenesis of the disease. Clin. Immunol. Immunopathol. 1995, 76, 187–194. [Google Scholar] [CrossRef]
- Wang, J.; Wang, C.; Tsui, H.W.; Las Heras, F.; Cheng, E.Y.; Iscove, N.N.; Chiu, B.; Inman, R.D.; Pritzker, K.P.; Tsui, F.W. Microcytosis in ank/ank mice and the role of ANKH in promoting erythroid differentiation. Exp. Cell Res. 2007, 313, 4120–4129. [Google Scholar] [CrossRef]
- Aristorena, M.; Gallardo-Vara, E.; Vicen, M.; de Las Casas-Engel, M.; Ojeda-Fernandez, L.; Nieto, C.; Blanco, F.J.; Valbuena-Diez, A.C.; Botella, L.M.; Nachtigal, P.; et al. MMP-12, secreted by pro-inflammatory macrophages, targets endoglin in human macrophages and endothelial cells. Int. J. Mol. Sci. 2019, 20, 3107. [Google Scholar] [CrossRef]
- Varejckova, M.; Gallardo-Vara, E.; Vicen, M.; Vitverova, B.; Fikrova, P.; Dolezelova, E.; Rathouska, J.; Prasnicka, A.; Blazickova, K.; Micuda, S.; et al. Soluble endoglin modulates the pro-inflammatory mediators NF-κB and IL-6 in cultured human endothelial cells. Life Sci. 2017, 175, 52–60. [Google Scholar] [CrossRef]
- Bellón, T.; Corbí, A.; Lastres, P.; Calés, C.; Cebrián, M.; Vera, S.; Cheifetz, S.; Massague, J.; Letarte, M.; Bernabéu, C. Identification and expression of two forms of the human transforming growth factor-beta-binding protein endoglin with distinct cytoplasmic regions. Eur. J. Immunol. 1993, 23, 2340–2345. [Google Scholar] [CrossRef]
- Lastres, P.; Letamendía, A.; Zhang, H.; Rius, C.; Almendro, N.; Raab, U.; López, L.A.; Langa, C.; Fabra, A.; Letarte, M.; et al. Endoglin modulates cellular responses to TGF-beta 1. J. Cell Biol. 1996, 133, 1109–1121. [Google Scholar] [CrossRef]
- Blanco, F.J.; Grande, M.T.; Langa, C.; Oujo, B.; Velasco, S.; Rodriguez-Barbero, A.; Perez-Gomez, E.; Quintanilla, M.; López-Novoa, J.M.; Bernabeu, C. S-endoglin expression is induced in senescent endothelial cells and contributes to vascular pathology. Circ. Res. 2008, 103, 1383–1392. [Google Scholar] [CrossRef]
- Velasco, S.; Alvarez-Muñoz, P.; Pericacho, M.; Dijke, P.T.; Bernabéu, C.; López-Novoa, J.M.; Rodríguez-Barbero, A. L- and S-endoglin differentially modulate TGFbeta1 signaling mediated by ALK1 and ALK5 in L6E9 myoblasts. J. Cell Sci. 2008, 121 Pt 6, 913–919. [Google Scholar] [CrossRef]
- Blanco, F.J.; Ojeda-Fernandez, L.; Aristorena, M.; Gallardo-Vara, E.; Benguria, A.; Dopazo, A.; Langa, C.; Botella, L.M.; Bernabeu, C. Genome-wide transcriptional and functional analysis of endoglin isoforms in the human promonocytic cell line U937. J. Cell Physiol. 2015, 230, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Post, S.; Smits, A.M.; van den Broek, A.J.; Sluijter, J.P.; Hoefer, I.E.; Janssen, B.J.; Snijder, R.J.; Mager, J.J.; Pasterkamp, G.; Mummery, C.L.; et al. Impaired recruitment of HHT-1 mononuclear cells to the ischaemic heart is due to an altered CXCR4/CD26 balance. Cardiovasc. Res. 2010, 85, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Aristorena, M.; Blanco, F.J.; de Las Casas-Engel, M.; Ojeda-Fernandez, L.; Gallardo-Vara, E.; Corbi, A.; Botella, L.M.; Bernabeu, C. Expression of endoglin isoforms in the myeloid lineage and their role during aging and macrophage polarization. J. Cell Sci. 2014, 127 Pt 12, 2723–2735. [Google Scholar] [CrossRef]
- Green, I.D.; Pinello, N.; Song, R.; Lee, Q.; Halstead, J.M.; Kwok, C.T.; Wong, A.C.H.; Nair, S.S.; Clark, S.J.; Roediger, B.; et al. Macrophage development and activation involve coordinated intron retention in key inflammatory regulators. Nucleic Acids Res. 2020, 48, 6513–6529. [Google Scholar] [CrossRef] [PubMed]
- Braunschweig, U.; Barbosa-Morais, N.L.; Pan, Q.; Nachman, E.N.; Alipanahi, B.; Gonatopoulos-Pournatzis, T.; Frey, B.; Irimia, M.; Blencowe, B.J. Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res. 2014, 24, 1774–1786. [Google Scholar] [CrossRef]
- Schmitz, U.; Pinello, N.; Jia, F.; Alasmari, S.; Ritchie, W.; Keightley, M.C.; Shini, S.; Lieschke, G.J.; Wong, J.J.; Rasko, J.E.J. Intron retention enhances gene regulatory complexity in vertebrates. Genome Biol. 2017, 18, 216. [Google Scholar] [CrossRef]
- Edwards, C.R.; Ritchie, W.; Wong, J.J.; Schmitz, U.; Middleton, R.; An, X.; Mohandas, N.; Rasko, J.E.; Blobel, G.A. A dynamic intron retention program in the mammalian megakaryocyte and erythrocyte lineages. Blood 2016, 127, e24–e34. [Google Scholar] [CrossRef]
- Ni, T.; Yang, W.; Han, M.; Zhang, Y.; Shen, T.; Nie, H.; Zhou, Z.; Dai, Y.; Yang, Y.; Liu, P.; et al. Global intron retention mediated gene regulation during CD4+ T cell activation. Nucleic Acids Res. 2016, 44, 6817–6829. [Google Scholar] [CrossRef]
- Blanco, F.J.; Bernabeu, C. Alternative splicing factor or splicing factor-2 plays a key role in intron retention of the endoglin gene during endothelial senescence. Aging Cell 2011, 10, 896–907. [Google Scholar] [CrossRef]
- Boutz, P.L.; Bhutkar, A.; Sharp, P.A. Detained introns are a novel, widespread class of post-transcriptionally spliced introns. Genes Dev. 2015, 29, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Lykke-Andersen, S.; Jensen, T.H. Nonsense-mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015, 16, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Mauger, O.; Lemoine, F.; Scheiffele, P. Targeted intron retention and excision for rapid gene regulation in response to neuronal activity. Neuron 2016, 92, 1266–1278. [Google Scholar] [CrossRef] [PubMed]
- Norozian, F.; Kashyap, M.; Ramirez, C.D.; Patel, N.; Kepley, C.L.; Barnstein, B.O.; Ryan, J.J. TGFβ1 induces mast cell apoptosis. Exp. Hematol. 2006, 34, 579–587. [Google Scholar] [CrossRef]
- Miller, H.R.; Wright, S.H.; Knight, P.A.; Thornton, E.M. A novel function for transforming growth factor-β1, upregulation of the expression and the IgE-independent extracellular release of a mucosal mast cell granule-specific β-chymase, mouse mast cell protease-1. Blood 1999, 93, 3473–3486. [Google Scholar] [CrossRef]
- Yamazaki, S.; Nakano, N.; Honjo, A.; Hara, M.; Maeda, K.; Nishiyama, C.; Kitaura, J.; Ohtsuka, Y.; Okumura, K.; Ogawa, H.; et al. The transcription factor Ehf is involved in TGF-β-induced suppression of FcεRI and c-Kit expression and FcεRI-mediated activation in mast cells. J. Immunol. 2015, 195, 3427–3435. [Google Scholar] [CrossRef]
- Ndaw, V.S.; Abebayehu, D.; Spence, A.J.; Paez, P.A.; Kolawole, E.M.; Taruselli, M.T.; Caslin, H.L.; Chumanevich, A.P.; Paranjape, A.; Baker, B.; et al. TGF-β1 suppresses IL-33-induced mast cell function. J. Immunol. 2017, 199, 866–873. [Google Scholar] [CrossRef]
- Olsson, N.; Piek, E.; Sundström, M.; ten Dijke, P.; Nilsson, G. Transforming growth factor-β-mediated mast cell migration depends on mitogen-activated protein kinase activity. Cell Signal. 2001, 13, 483–490. [Google Scholar] [CrossRef]
- Ramírez-Valadez, K.A.; Vázquez-Victorio, G.; Macías-Silva, M.; González-Espinosa, C. Fyn kinase mediates cortical actin ring depolymerization required for mast cell migration in response to TGF-β in mice. Eur. J. Immunol. 2017, 47, 1305–1316. [Google Scholar] [CrossRef]
- Olsson, N.; Piek, E.; ten Dijke, P.; Nilsson, G. Human mast cell migration in response to members of the transforming growth factor-beta family. J. Leukoc. Biol. 2000, 67, 350–356. [Google Scholar] [CrossRef]
- Gebhardt, T.; Lorentz, A.; Detmer, F.; Trautwein, C.; Bektas, H.; Manns, M.P.; Bischoff, S.C. Growth, phenotype, and function of human intestinal mast cells are tightly regulated by transforming growth factor β1. Gut 2005, 54, 928–934. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kashyap, M.; Bailey, D.P.; Gomez, G.; Rivera, J.; Huff, T.F.; Ryan, J.J. TGF-β1 inhibits late-stage mast cell maturation. Exp. Hematol. 2005, 33, 1281–1291. [Google Scholar] [CrossRef] [PubMed]
- Gomez, G.; Ramirez, C.D.; Rivera, J.; Patel, M.; Norozian, F.; Wright, H.V.; Kashyap, M.V.; Barnstein, B.O.; Fischer-Stenger, K.; Schwartz, L.B.; et al. TGF-β1 inhibits mast cell FcεRI expression. J. Immunol. 2005, 174, 5987–5993. [Google Scholar] [CrossRef] [PubMed]
- Groneberg, D.A.; Bester, C.; Grützkau, A.; Serowka, F.; Fischer, A.; Henz, B.M.; Welker, P. Mast cells and vasculature in atopic dermatitis--potential stimulus of neoangiogenesis. Allergy 2005, 60, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Lima, S.C.; Rizo, V.H.; Silva-Sousa, Y.T.; Almeida, L.Y.; Almeida, O.P.; León, J.E. Immunohistochemical evaluation of angiogenesis and tryptase-positive mast cell infiltration in periapical lesions. J. Endod. 2011, 37, 1642–1646. [Google Scholar] [CrossRef]
- Białas, M.; Dyduch, G.; Szpor, J.; Demczuk, S.; Okoń, K. Microvascular density and mast cells in benign and malignant pheochromocytomas. Pol. J. Pathol. 2012, 63, 235–242. [Google Scholar] [CrossRef]
- Micu, G.V.; Stăniceanu, F.; Sticlaru, L.C.; Popp, C.G.; Bastian, A.E.; Gramada, E.; Pop, G.; Mateescu, R.B.; Rimbaş, M.; Archip, B.; et al. Correlations between the density of tryptase positive mast cells (DMCT) and that of new blood vessels (CD105+) in patients with gastric cancer. Rom. J. Intern. Med. 2016, 54, 113–120. [Google Scholar] [CrossRef][Green Version]
- Munteanu, A.I.; Raica, M.; Zota, E.G. Immunohistochemical study of the role of mast cells and macrophages in the process of angiogenesis in the atherosclerotic plaques in patients with metabolic syndrome. Arkhiv Patologii 2016, 78, 19–28. [Google Scholar] [CrossRef]
- McKittrick, C.M.; Lawrence, C.E.; Carswell, H.V. Mast cells promote blood brain barrier breakdown and neutrophil infiltration in a mouse model of focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2015, 35, 638–647. [Google Scholar] [CrossRef]
- Gruber, B.L.; Marchese, M.J.; Kew, R.R. Transforming growth factor-beta 1 mediates mast cell chemotaxis. J. Immunol. 1994, 152, 5860–5867. [Google Scholar]
- Zhao, W.; Gomez, G.; Yu, S.H.; Ryan, J.J.; Schwartz, L.B. TGF-beta1 attenuates mediator release and de novo Kit expression by human skin mast cells through a Smad-dependent pathway. J. Immunol. 2008, 181, 7263–7272. [Google Scholar] [CrossRef] [PubMed]
- Trentin Brum, S.; Demasi, A.P.; Fantelli Stelini, R.; Cintra, M.L.; Cavalcanti de Araujo, V.; Borges Soares, A. Endoglin is highly expressed in human mast cells. Appl. Immunohistochem. Mol. Morphol. 2019, 27, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Dourado, K.M.C.; Baik, J.; Oliveira, V.K.P.; Beltrame, M.; Yamamoto, A.; Theuer, C.P.; Figueiredo, C.A.V.; Verneris, M.R.; Perlingeiro, R.C.R. Endoglin: A novel target for therapeutic intervention in acute leukemias revealed in xenograft mouse models. Blood 2017, 129, 2526–2536. [Google Scholar] [CrossRef] [PubMed]
- Al-Mowallad, A.; Carr, T.; Al-Qouzi, A.; Li, C.; Byers, R.; Kumar, S. Plasma CD105, TGFbeta-1, TGFbeta-3 and the ligand/receptor complexes in children with acute lymphoblastic leukaemia. Anticancer Res. 2006, 26 (1B), 543–547. [Google Scholar]
- Cosimato, V.; Scalia, G.; Raia, M.; Gentile, L.; Cerbone, V.; Visconte, F.; Statuto, T.; Valvano, L.; D’Auria, F.; Calice, G.; et al. Surface endoglin (CD105) expression on acute leukemia blast cells: An extensive flow cytometry study of 1002 patients. Leuk. Lymphoma 2018, 59, 2242–2245. [Google Scholar] [CrossRef]
- Bartholomew, A.; Sturgeon, C.; Siatskas, M.; Ferrer, K.; McIntosh, K.; Patil, S.; Hardy, W.; Devine, S.; Ucker, D.; Deans, R.; et al. Mesenchymal stem cells suppress lymphocyte proliferation in vitro and prolong skin graft survival in vivo. Exp. Hematol. 2002, 30, 42–48. [Google Scholar] [CrossRef]
- Di Nicola, M.; Carlo-Stella, C.; Magni, M.; Milanesi, M.; Longoni, P.D.; Matteucci, P.; Grisanti, S.; Gianni, A.M. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood 2002, 99, 3838–3843. [Google Scholar] [CrossRef]
- Krampera, M.; Glennie, S.; Dyson, J.; Scott, D.; Laylor, R.; Simpson, E.; Dazzi, F. Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen-specific T cells to their cognate peptide. Blood 2003, 101, 3722–3729. [Google Scholar] [CrossRef]
- Rasmusson, I.; Le Blanc, K.; Sundberg, B.; Ringdén, O. Mesenchymal stem cells stimulate antibody secretion in human B cells. Scand. J. Immunol. 2007, 65, 336–343. [Google Scholar] [CrossRef]
- Quaedackers, M.E.; Baan, C.C.; Weimar, W.; Hoogduijn, M.J. Cell contact interaction between adipose-derived stromal cells and allo-activated T lymphocytes. Eur. J. Immunol. 2009, 39, 3436–3446. [Google Scholar] [CrossRef]
- Dennler, S.; Itoh, S.; Vivien, D.; ten Dijke, P.; Huet, S.; Gauthier, J.M. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998, 17, 3091–3100. [Google Scholar] [CrossRef] [PubMed]
- Schoonderwoerd, M.J.A.; Koops, M.F.M.; Angela, R.A.; Koolmoes, B.; Toitou, M.; Paauwe, M.; Barnhoorn, M.C.; Liu, Y.; Sier, C.F.M.; Hardwick, J.C.H.; et al. Targeting endoglin-expressing regulatory T cells in the tumor microenvironment enhances the effect of PD1 checkpoint inhibitor immunotherapy. Clin. Cancer Res. 2020, 26, 3831–3842. [Google Scholar] [CrossRef] [PubMed]
- Rokhlin, O.W.; Cohen, M.B.; Kubagawa, H.; Letarte, M.; Cooper, M.D. Differential expression of endoglin on fetal and adult hematopoietic cells in human bone marrow. J. Immunol. 1995, 154, 4456–4465. [Google Scholar] [PubMed]
- Gougos, A.; Letarte, M.J. Primary structure of endoglin, an RGD-containing glycoprotein of human endothelial cells. Biol. Chem. 1990, 265, 8361–8364. [Google Scholar]
- Moos, P.J.; Raetz, E.A.; Carlson, M.A.; Szabo, A.; Smith, F.E.; Willman, C.; Wei, Q.; Hunger, S.P.; Carroll, W.L. Identification of gene expression profiles that segregate patients with childhood leukemia. Clin. Cancer Res. 2002, 8, 3118–3130. [Google Scholar]
- Zhang, H.; Shaw, A.R.; Mak, A.; Letarte, M. Endoglin is a component of the transforming growth factor (TGF)-β receptor complex of human pre-B leukemic cells. J. Immunol. 1996, 156, 564–573. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meurer, S.K.; Weiskirchen, R. Endoglin: An ‘Accessory’ Receptor Regulating Blood Cell Development and Inflammation. Int. J. Mol. Sci. 2020, 21, 9247. https://doi.org/10.3390/ijms21239247
Meurer SK, Weiskirchen R. Endoglin: An ‘Accessory’ Receptor Regulating Blood Cell Development and Inflammation. International Journal of Molecular Sciences. 2020; 21(23):9247. https://doi.org/10.3390/ijms21239247
Chicago/Turabian StyleMeurer, Steffen K., and Ralf Weiskirchen. 2020. "Endoglin: An ‘Accessory’ Receptor Regulating Blood Cell Development and Inflammation" International Journal of Molecular Sciences 21, no. 23: 9247. https://doi.org/10.3390/ijms21239247
APA StyleMeurer, S. K., & Weiskirchen, R. (2020). Endoglin: An ‘Accessory’ Receptor Regulating Blood Cell Development and Inflammation. International Journal of Molecular Sciences, 21(23), 9247. https://doi.org/10.3390/ijms21239247