Endocrine-Disrupting Chemicals’ (EDCs) Effects on Tumour Microenvironment and Cancer Progression: Emerging Contribution of RACK1




{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Concepts of Endocrine Disruption
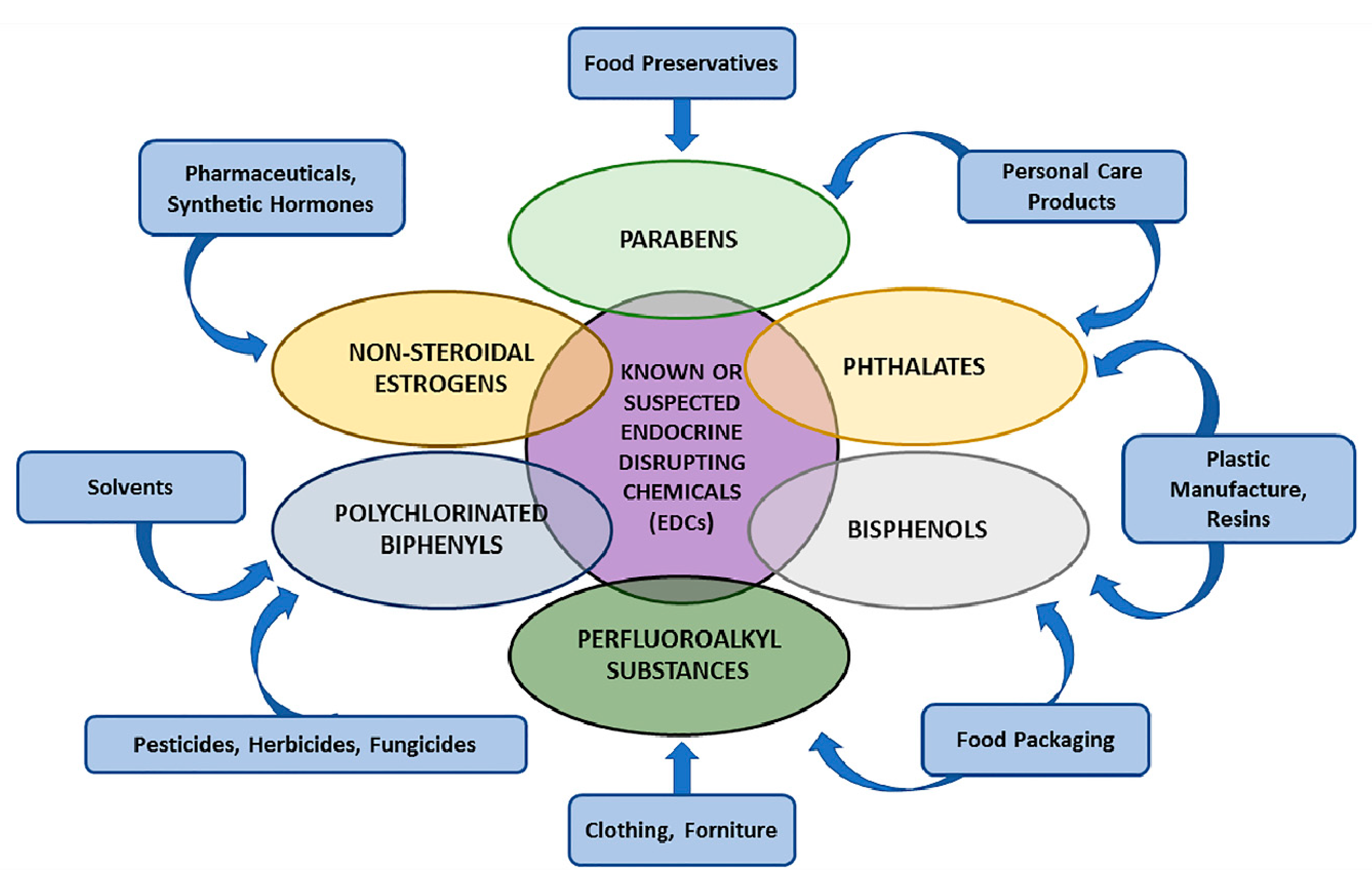
2.1. EDCs: Definition and Characterisation
2.2. EDCs’ Molecular Mechanisms: From Dichlorodiphenyltrichloroethane (DTT) and Diethylstilbestrol (DES) as Proof of Concept to Other Sustances
3. Cancer Risk Linked to EDC Exposure
3.1. EDCs Associated with Hormone-Sensitive Cancers in Females and Males: Focus on BC and PC
3.1.1. Non-Steroidal Oestrogens (DES and Zearalenone)
3.1.2. DDT
3.1.3. Bisphenols
3.1.4. Phthalates
3.1.5. PFASs
4. Tumour Microenvironment (TME) and EDCs
4.1. Tumour Microenvironment as Promoter of Cancer Progression
4.2. Immune System in TME and Its Tumour-Associated Macrophages
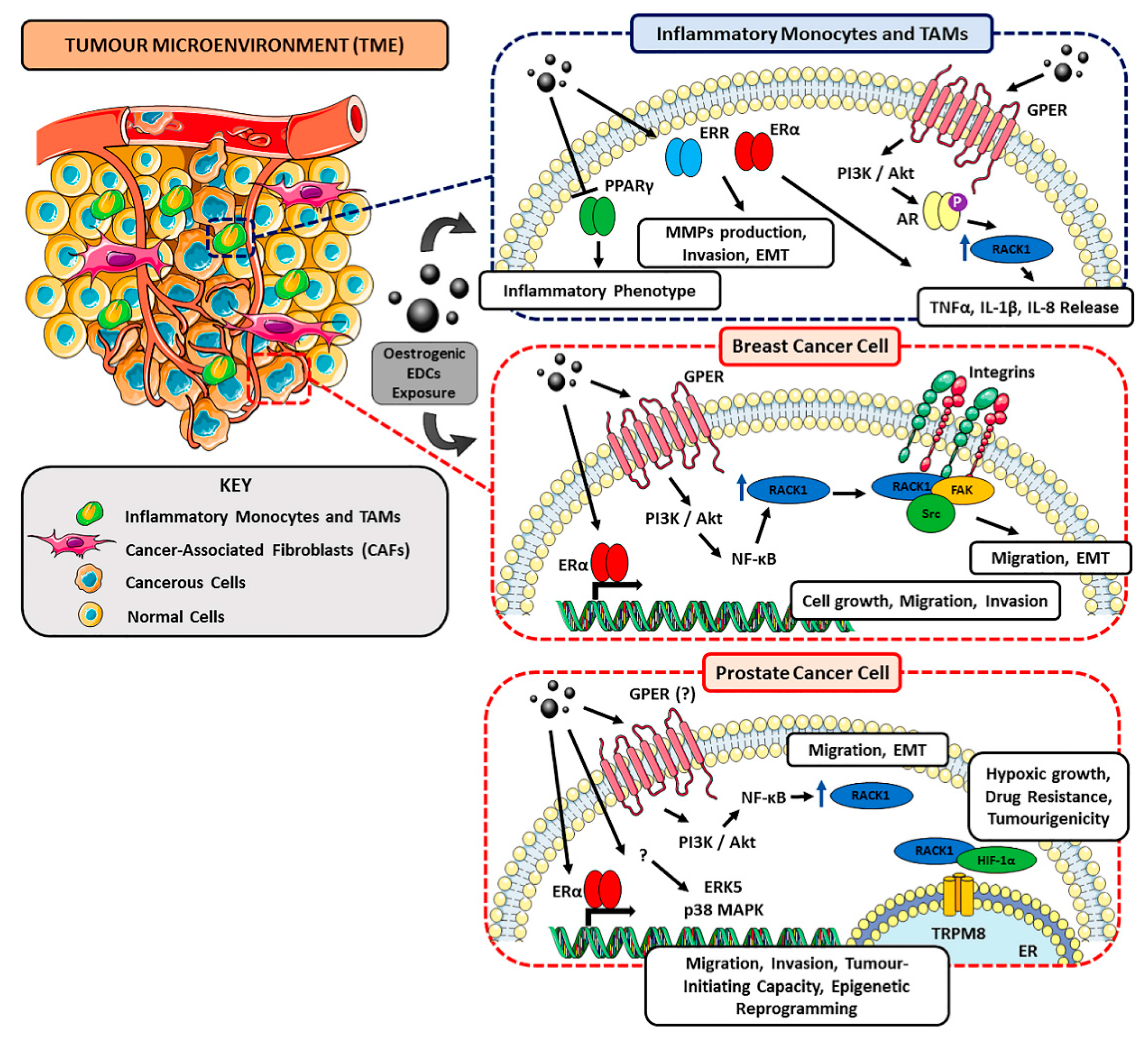
4.3. EDCs as Landscape Shapers in BC- and PC-Associated TME
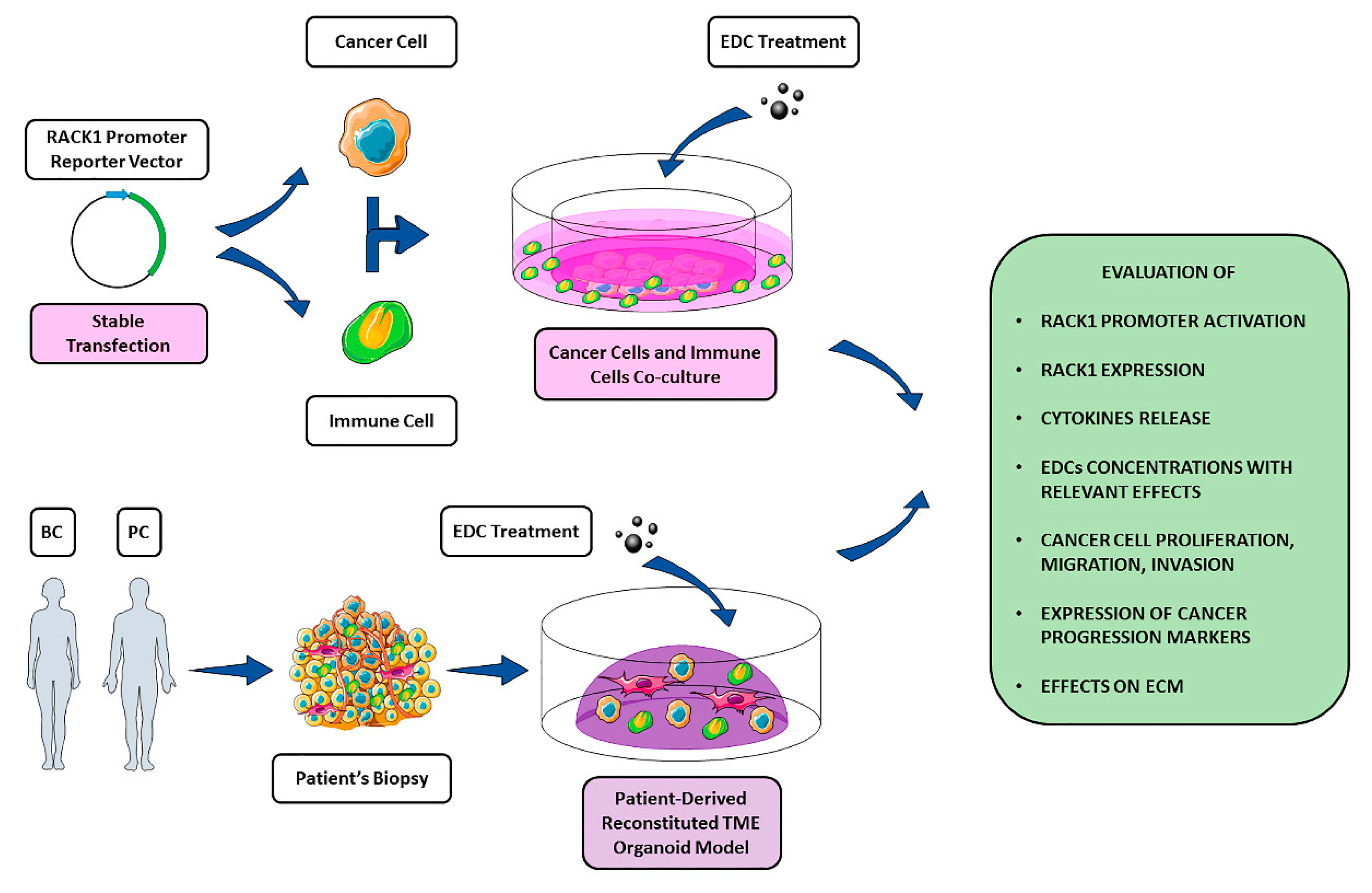
5. RACK1 as a Possible Target of EDCs
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Whirledge, S.; Cidlowski, J.A. Chapter 5—Steroid Hormone Action. Yen and Jaffe’s Reproductive Endocrinology (Eighth Edition), Physiology, Pathophysiology, and Clinical Management; Strauss, J., Barbieri, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 115–131. [Google Scholar]
- La Merrill, M.A.; Vandenberg, L.N.; Smith, J.R.; Goodson, W.; Browne, P.; Patisaul, H.B.; Guyton, K.Z.; Kortenkamp, A.; Cogliano, V.J.; Woodruff, T.; et al. Consensus on the key characteristics of endocrine-disrupting chemicals as a basis for hazard identification. Nat. Rev. Endocrinol. 2020, 16, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Axelstad, M.; Hass, U.; Scholze, M.; Christiansen, S.; Kortenkamp, A.; Boberg, J. EDC IMPACT: Reduced sperm counts in rats exposed to human relevant mixtures of endocrine disrupters. Endocr. Connect. 2018, 7, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Johansson, H.K.L.; Svingen, T.; Fowler, P.A.; Vinggaard, A.M.; Boberg, J. Environmental influences on ovarian dysgenesis—Developmental windows sensitive to chemical exposures. Nat. Rev. Endocrinol. 2017, 13, 400–414. [Google Scholar] [CrossRef]
- Skakkebaek, N.E. A Brief Review of the Link between Environment and Male Reproductive Health: Lessons from Studies of Testicular Germ Cell Cancer. Horm. Res. Paediatr. 2016, 86, 240–246. [Google Scholar] [CrossRef]
- Amano, I.; Takatsuru, Y.; Khairinisa, M.A.; Kokubo, M.; Haijima, A.; Koibuchi, N. Effects of Mild Perinatal Hypothyroidism on Cognitive Function of Adult Male Offspring. Endocrinology 2018, 159, 1910–1921. [Google Scholar] [CrossRef]
- Ghassabian, A.; Trasande, L. Disruption in Thyroid Signaling Pathway: A Mechanism for the Effect of Endocrine-Disrupting Chemicals on Child Neurodevelopment. Front. Endocrinol. 2018, 9, 204. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, W.N.; Kinyamu, H.K.; Wang, T.; Miranda, A.; Padilla-Banks, E.; Suen, A.A.; Williams, C.J. Widespread enhancer activation via ERα mediates estrogen response in vivo during uterine development. Nucleic Acids Res. 2018, 46, 5487–5503. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Magdalena, P.; Vieira, E.; Soriano, S.; Menes, L.; Burks, D.; Quesada, I.; Nadal, A. Bisphenol A Exposure during Pregnancy Disrupts Glucose Homeostasis in Mothers and Adult Male Offspring. Environ. Health Perspect. 2010, 118, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Cano-Sancho, G.; Salmon, A.G.; La Merrill, M.A. Association between Exposure to p,p ′-DDT and Its Metabolite p,p ′-DDE with Obesity: Integrated Systematic Review and Meta-Analysis. Environ. Health Perspect. 2017, 125, 096002. [Google Scholar] [CrossRef] [PubMed]
- Heindel, J.J.; Skalla, L.A.; Joubert, B.R.; Dilworth, C.H.; Gray, K.A. Review of developmental origins of health and disease publications in environmental epidemiology. Reprod. Toxicol. 2017, 68, 34–48. [Google Scholar] [CrossRef]
- Sifakis, S.; Androutsopoulos, V.P.; Tsatsakis, A.M.; Spandidos, D.A. Human exposure to endocrine disrupting chemicals: Effects on the male and female reproductive systems. Environ. Toxicol. Pharmacol. 2017, 51, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Giulivo, M.; De Alda, M.L.; Capri, E.; Barcelo, D. Human exposure to endocrine disrupting compounds: Their role in reproductive systems, metabolic syndrome and breast cancer. A review. Environ. Res. 2016, 151, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Scsukova, S.; Rollerova, E.; Mlynarcikova, A.B. Impact of endocrine disrupting chemicals on onset and development of female reproductive disorders and hormone-related cancer. Reprod. Biol. 2016, 16, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Lucaccioni, L.; Trevisani, V.; Marrozzini, L.; Bertoncelli, N.; Predieri, B.; Lugli, L.; Berardi, A.; Iughetti, L. Endocrine-Disrupting Chemicals and Their Effects during Female Puberty: A Review of Current Evidence. Int. J. Mol. Sci. 2020, 21, 2078. [Google Scholar] [CrossRef]
- Corsini, E.; Ruffo, F.; Racchi, M. Steroid hormones, endocrine disrupting compounds and immunotoxicology. Curr. Opin. Toxicol. 2018, 10, 69–73. [Google Scholar] [CrossRef]
- Buoso, E.; Galasso, M.; Serafini, M.M.; Ronfani, M.; Lanni, C.; Corsini, E.; Racchi, M. Transcriptional regulation of RACK1 and modulation of its expression: Role of steroid hormones and significance in health and aging. Cell. Signal. 2017, 35, 264–271. [Google Scholar] [CrossRef]
- Buoso, E.; Galasso, M.; Ronfani, M.; Papale, A.; Galbiati, V.; Eberini, I.; Marinovich, M.; Racchi, M.; Corsini, E. The scaffold protein RACK1 is a target of endocrine disrupting chemicals (EDCs) with important implication in immunity. Toxicol. Appl. Pharmacol. 2017, 325, 37–47. [Google Scholar] [CrossRef]
- Buoso, E.; Masi, M.; Galbiati, V.; Maddalon, A.; Iulini, M.; Kenda, M.; Dolenc, M.S.; Marinovich, M.; Racchi, M.; Corsini, E. Effect of estrogen-active compounds on the expression of RACK1 and immunological implications. Arch. Toxicol. 2020, 94, 2081–2095. [Google Scholar] [CrossRef]
- Racchi, M.; Buoso, E.; Ronfani, M.; Serafini, M.M.; Galasso, M.; Lanni, C.; Corsini, E. Role of Hormones in the Regulation of RACK1 Expression as a Signaling Checkpoint in Immunosenescence. Int. J. Mol. Sci. 2017, 18, 1453. [Google Scholar] [CrossRef]
- Buoso, E.; Masi, M.; Long, A.; Chiappini, C.; Travelli, C.; Govoni, S.; Racchi, M. Ribosomes as a nexus between translation and cancer progression: Focus on ribosomal Receptor for Activated C Kinase 1 (RACK1) in breast cancer. Br. J. Pharmacol. 2020. [Google Scholar] [CrossRef]
- WHO/IPCS. Global Assessment of the State-of-the-Science of Endocrine Disruptors. 2002. Available online: http://www.who.int/ipcs/publications/new_issues/endocrine_disruptors/en/ (accessed on 26 September 2020).
- Schug, T.T.; Janesick, A.; Blumberg, B.; Heindel, J.J. Endocrine disrupting chemicals and disease susceptibility. J. Steroid Biochem. Mol. Biol. 2011, 127, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, L.N.; Colborn, T.; Hayes, T.B.; Heindel, J.J.; Jacobs, D.R., Jr.; Lee, D.H.; Shioda, T.; Soto, A.M.; vom Saal, F.S.; Welshons, W.V.; et al. Hormones and Endocrine-Disrupting Chemicals: Low-Dose Effects and Nonmonotonic Dose Responses. Endocr. Rev. 2012, 33, 378–455. [Google Scholar] [CrossRef] [PubMed]
- Ropero, A.B.; Alonso-Magdalena, P.; García-García, E.; Ripoll, C.; Fuentes, E.; Nadal, A. Bisphenol-A disruption of the endocrine pancreas and blood glucose homeostasis. Int. J. Androl. 2007, 31, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Cohn, B.A.; La Merrill, M.; Krigbaum, N.Y.; Yeh, G.; Park, J.-S.; Zimmermann, L.; Cirillo, P.M. DDT Exposure in Utero and Breast Cancer. J. Clin. Endocrinol. Metab. 2015, 100, 2865–2872. [Google Scholar] [CrossRef]
- Cooper, G.S.; Martin, S.A.; Longnecker, M.P.; Sandler, D.P.; Germolec, D.R. Associations between Plasma DDE Levels and Immunologic Measures in African-American Farmers in North Carolina. Environ. Health Perspect. 2004, 112, 1080–1084. [Google Scholar] [CrossRef][Green Version]
- Hermanowicz, A.; Nawarska, Z.; Borys, D.; Maślankiewicz, A. The neutrophil function and infectious diseases in workers occupationally exposed to organochloride insecticides. Int. Arch. Occup. Environ. Health 1982, 50, 329–340. [Google Scholar] [CrossRef]
- Newbold, R.R. Prenatal exposure to diethylstilbestrol (DES). Fertil. Steril. 2008, 89, e55–e56. [Google Scholar] [CrossRef]
- WHO. Endocrine Disrupters and Child Health—Possible Developmental Early Effects of Endocrine Disrupters on Child Health; Toppari, J., Adamsson, A., Boas, M., Juul, A., Main, K.M., Skakkebaek, N.E., Virtanen, H.E., Eds.; World Health Organization: Geneva, Switzerland, 2002; Available online: https://www.who.int/iris/bitstream/10665/75342/1/9789241503761_eng.pdf (accessed on 30 September 2020).
- Schrager, S.; Potter, B.E. Diethylstilbestrol Exposure. Am. Fam. Physician 2004, 15, 2395–2400. [Google Scholar] [CrossRef]
- Monneret, C. What is an endocrine disruptor? Comptes Rendus Biol. 2017, 340, 403–405. [Google Scholar] [CrossRef]
- Geens, T.T.; Aerts, D.D.; Berthot, C.C.; Bourguignon, J.-P.; Goeyens, L.; LeComte, P.P.; Maghuin-Rogister, G.G.; Pironnet, A.M.A.; Pussemier, L.; Scippo, M.L.M.; et al. A review of dietary and non-dietary exposure to bisphenol-A. Food Chem. Toxicol. 2012, 50, 3725–3740. [Google Scholar] [CrossRef]
- Murata, M.; Kang, J.-H. Bisphenol A (BPA) and cell signaling pathways. Biotechnol. Adv. 2018, 36, 311–327. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, L.N.; Ehrlich, S.; Belcher, S.M.; Ben-Jonathan, N.; Dolinoy, D.; Hugo, E.R.; Hunt, P.A.; Newbold, R.R.; Rubin, B.S.; Saili, K.S.; et al. Low dose effects of bisphenol A. Endocr. Disruptors 2013, 1, e26490. [Google Scholar] [CrossRef]
- Le Magueresse-Battistoni, B.; Multigner, L.; Beausoleil, C.; Rousselle, C. Effects of bisphenol A on metabolism and evidences of a mode of action mediated through endocrine disruption. Mol. Cell. Endocrinol. 2018, 475, 74–91. [Google Scholar] [CrossRef] [PubMed]
- Wassenaar, P.N.H.; Trasande, L.; Legler, J. Systematic Review and Meta-Analysis of Early-Life Exposure to Bisphenol A and Obesity-Related Outcomes in Rodents. Environ. Health Perspect. 2017, 125, 106001. [Google Scholar] [CrossRef] [PubMed]
- Mhaouty-Kodja, S.; Belzunces, L.P.; Canivenc, M.-C.; Schroeder, H.; Chevrier, C.; Pasquier, E. Impairment of learning and memory performances induced by BPA: Evidences from the literature of a MoA mediated through an ED. Mol. Cell. Endocrinol. 2018, 475, 54–73. [Google Scholar] [CrossRef] [PubMed]
- Rebuli, M.E.; Camacho, L.; Adonay, M.E.; Reif, D.M.; Aylor, D.L.; Patisaul, H.B. Impact of Low-Dose Oral Exposure to Bisphenol A (BPA) on Juvenile and Adult Rat Exploratory and Anxiety Behavior: A CLARITY-BPA Consortium Study. Toxicol. Sci. 2015, 148, 341–354. [Google Scholar] [CrossRef]
- Tomza-Marciniak, A.; Stepkowska, P.; Kuba, J.; Pilarczyk, B. Effect of bisphenol A on reproductive processes: A review of in vitro, in vivo and epidemiological studies. J. Appl. Toxicol. 2018, 38, 51–80. [Google Scholar] [CrossRef]
- Peretz, J.; Vrooman, L.; Ricke, W.A.; Hunt, P.A.; Ehrlich, S.; Hauser, R.; Padmanabhan, V.; Taylor, H.S.; Swan, S.H.; Vandevoort, C.A.; et al. Bisphenol A and Reproductive Health: Update of Experimental and Human Evidence, 2007–2013. Environ. Health Perspect. 2014, 122, 775–786. [Google Scholar] [CrossRef]
- Bae, S.; Hong, Y.C. Exposure to bisphenol A from drinking canned beverages increases blood pressure: Randomized crossover trial. Hypertension 2015, 65, 313–319. [Google Scholar] [CrossRef]
- Melzer, D.; Osborne, N.J.; Henley, W.E.; Cipelli, R.; Young, A.; Money, C.; McCormack, P.; Luben, R.; Khaw, K.-T.; Wareham, N.J.; et al. Urinary Bisphenol A Concentration and Risk of Future Coronary Artery Disease in Apparently Healthy Men and Women. Circulation 2012, 125, 1482–1490. [Google Scholar] [CrossRef]
- Rochester, J.R. Bisphenol A and human health: A review of the literature. Reprod. Toxicol. 2013, 42, 132–155. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.H.E.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.-L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Heemers, H.; Sharifi, N. Androgen Signaling in Prostate Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a030452. [Google Scholar] [CrossRef] [PubMed]
- Perera, L.; Lalith, P.; Coons, L.A.; Houtman, R.; Van Beuningen, R.; Goodwin, B.; Auerbach, S.S.; Teng, C.T. Binding of bisphenol A, bisphenol AF, and bisphenol S on the androgen receptor: Coregulator recruitment and stimulation of potential interaction sites. Toxicol. Vitr. 2017, 44, 287–302. [Google Scholar] [CrossRef]
- Maske, P.; Dighe, V.; Vanage, G. n-butylparaben exposure during perinatal period impairs fertility of the F1 generation female rats. Chemosphere 2018, 213, 114–123. [Google Scholar] [CrossRef]
- Pollock, T.; Weaver, R.E.; Ghasemi, R.; Decatanzaro, D. Butyl paraben and propyl paraben modulate bisphenol A and estradiol concentrations in female and male mice. Toxicol. Appl. Pharmacol. 2017, 325, 18–24. [Google Scholar] [CrossRef]
- European Medicines Agency, Committee For Medicinal Products For Human Use (CHMP). Guideline on the Environmental Risk Assessment of Medicinal Products for Human Use; EMEA: London, UK, 2006. [Google Scholar]
- Guo, J.; Wu, C.; Lu, D.; Jiang, S.; Liang, W.; Chang, X.; Xu, H.; Wang, G.; Zhou, Z. Urinary paraben concentrations and their associations with anthropometric measures of children aged 3 years. Environ. Pollut. 2017, 222, 307–314. [Google Scholar] [CrossRef]
- Philippat, C.; Botton, J.; Calafat, A.M.; Ye, X.; Charles, M.-A.; Slama, R. Prenatal Exposure to Phenols and Growth in Boys. Epidemiology 2014, 25, 625–635. [Google Scholar] [CrossRef]
- Aker, A.M.; Watkins, D.J.; Johns, L.E.; Ferguson, K.K.; Soldin, O.P.; Del Toro, L.V.A.; Alshawabkeh, A.N.; Cordero, J.F.; Meeker, J.D. Phenols and parabens in relation to reproductive and thyroid hormones in pregnant women. Environ. Res. 2016, 151, 30–37. [Google Scholar] [CrossRef]
- Philippat, C.; Wolff, M.S.; Calafat, A.M.; Ye, X.; Bausell, R.; Meadows, M.; Stone, J.; Slama, R.; Engel, S.M. Prenatal Exposure to Environmental Phenols: Concentrations in Amniotic Fluid and Variability in Urinary Concentrations during Pregnancy. Environ. Health Perspect. 2013, 121, 1225–1231. [Google Scholar] [CrossRef]
- Viñas, P.; Campillo, N.; Pastor-Belda, M.; Oller, A.; Hernández-Córdoba, M. Determination of phthalate esters in cleaning and personal care products by dispersive liquid–liquid microextraction and liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2015, 1376, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Varshavsky, J.R.; Morello-Frosch, R.; Woodruff, T.J.; Zota, A.R. Dietary sources of cumulative phthalates exposure among the U.S. general population in NHANES 2005–2014. Environ. Int. 2018, 115, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Faessler, D.; McCombie, G.; Biedermann, M.; Felder, F.; Subotic, U. Leaching of plasticizers from polyvinylchloride perfusion lines by different lipid emulsions for premature infants under clinical conditions. Int. J. Pharm. 2017, 520, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Casas, M.; Chevrier, C.; Hond, E.D.; Fernandez, M.F.; Pierik, F.; Philippat, C.; Slama, R.; Toft, G.; Vandentorren, S.; Wilhelm, M.; et al. Exposure to brominated flame retardants, perfluorinated compounds, phthalates and phenols in European birth cohorts: ENRIECO evaluation, first human biomonitoring results, and recommendations. Int. J. Hyg. Environ. Health 2013, 216, 230–242. [Google Scholar] [CrossRef]
- Haug, L.; Sakhi, A.K.; Cequier, E.; Casas, M.; Maitre, L.; Basagana, X.; Andrusaityte, S.; Chalkiadaki, G.; Chatzi, L.; Coen, M.; et al. In-utero and childhood chemical exposome in six European mother-child cohorts. Environ. Int. 2018, 121, 751–763. [Google Scholar] [CrossRef]
- Koch, H.M.; Rüther, M.; Schütze, A.; Conrad, A.; Pälmke, C.; Apel, P.; Brüning, T.; Kolossa-Gehring, M. Phthalate metabolites in 24-h urine samples of the German Environmental Specimen Bank (ESB) from 1988 to 2015 and a comparison with US NHANES data from 1999 to 2012. Int. J. Hyg. Environ. Health 2017, 220, 130–141. [Google Scholar] [CrossRef]
- Wójtowicz, A.K.; Sitarz-Głownia, A.M.; Szczęsna, M.; Szychowski, K.A. The Action of Di-(2-Ethylhexyl) Phthalate (DEHP) in Mouse Cerebral Cells Involves an Impairment in Aryl Hydrocarbon Receptor (AhR) Signaling. Neurotox. Res. 2019, 35, 183–195. [Google Scholar] [CrossRef]
- Nassan, F.L.; Korevaar, T.I.; Coull, B.A.; Skakkebæk, N.E.; Krawetz, S.A.; Estill, M.; Hait, E.J.; Korzenik, J.R.; Ford, J.B.; De Poortere, R.A.; et al. Dibutyl-phthalate exposure from mesalamine medications and serum thyroid hormones in men. Int. J. Hyg. Environ. Health 2019, 222, 101–110. [Google Scholar] [CrossRef]
- Boas, M.; Frederiksen, H.; Feldt-Rasmussen, U.; Skakkebæk, N.E.; Hegedüs, L.; Hilsted, L.; Juul, A.; Main, K.M. Childhood Exposure to Phthalates: Associations with Thyroid Function, Insulin-like Growth Factor I, and Growth. Environ. Health Perspect. 2010, 118, 1458–1464. [Google Scholar] [CrossRef]
- Wang, C.; Yang, L.; Wang, S.; Zhang, Z.; Yu, Y.; Wang, M.; Cromie, M.; Gao, W.; Wang, S.-L. The classic EDCs, phthalate esters and organochlorines, in relation to abnormal sperm quality: A systematic review with meta-analysis. Sci. Rep. 2016, 6, 19982. [Google Scholar] [CrossRef]
- Radke, E.G.; Braun, J.M.; Meeker, J.D.; Cooper, G.S. Phthalate exposure and male reproductive outcomes: A systematic review of the human epidemiological evidence. Environ. Int. 2018, 121, 764–793. [Google Scholar] [CrossRef] [PubMed]
- Philippat, C.; Bennett, D.H.; Krakowiak, P.; Rose, M.; Hwang, H.-M.; Hertz-Picciotto, I. Phthalate concentrations in house dust in relation to autism spectrum disorder and developmental delay in the CHildhood Autism Risks from Genetics and the Environment (CHARGE) study. Environ. Health 2015, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Miodovnik, A.; Edwards, A.; Bellinger, D.C.; Hauser, R. Developmental neurotoxicity of ortho-phthalate diesters: Review of human and experimental evidence. NeuroToxicology 2014, 41, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Olesen, T.S.; Bleses, D.; Andersen, H.R.; Grandjean, P.; Frederiksen, H.; Trecca, F.; Bilenberg, N.; Kyhl, H.B.; Dalsager, L.; Jensen, I.K.; et al. Prenatal phthalate exposure and language development in toddlers from the Odense Child Cohort. Neurotoxicol. Teratol. 2018, 65, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Sathyanarayana, S.; Butts, S.; Wang, C.; Barrett, E.; Nguyen, R.; Schwartz, S.M.; Haaland, W.; Swan, S.H. Tides the TIDES Team Early Prenatal Phthalate Exposure, Sex Steroid Hormones, and Birth Outcomes. J. Clin. Endocrinol. Metab. 2017, 102, 1870–1878. [Google Scholar] [CrossRef]
- Ferguson, K.K.; McElrath, T.F.; Meeker, J.D. Environmental Phthalate Exposure and Preterm Birth. JAMA Pediatr. 2014, 168, 61–67. [Google Scholar] [CrossRef]
- Huang, P.-C.; Tsai, C.-H.; Liang, W.-Y.; Li, S.-S.; Huang, H.-B.; Kuo, P.-L. Early Phthalates Exposure in Pregnant Women Is Associated with Alteration of Thyroid Hormones. PLoS ONE 2016, 11, e0159398. [Google Scholar] [CrossRef]
- EFSA Panel on Contaminants in the Food Chain (EFSA CONTAM Pane); Schrenk, D.; Bignami, M.; Bodin, L.; Chipman, J.K.; Del Mazo, J.; Grasl-Kraupp, B.; Hogstrand, C.; Hoogenboom, L. (Ron); Leblanc, J.; et al. Risk to human health related to the presence of perfluoroalkyl substances in food. EFSA J. 2020, 18. [Google Scholar] [CrossRef]
- Averina, M.; Brox, J.; Huber, S.; Furberg, A.-S. Perfluoroalkyl substances in adolescents in northern Norway: Lifestyle and dietary predictors. The Tromsø study, Fit Futures 1. Environ. Int. 2018, 114, 123–130. [Google Scholar] [CrossRef]
- Trier, X.; Granby, K.; Christensen, J.H. Polyfluorinated surfactants (PFS) in paper and board coatings for food packaging. Environ. Sci. Pollut. Res. 2011, 18, 1108–1120. [Google Scholar] [CrossRef]
- Ji, K.; Kim, S.; Kho, Y.; Paek, D.; Sakong, J.; Ha, J.; Kim, S.; Choi, K. Serum concentrations of major perfluorinated compounds among the general population in Korea: Dietary sources and potential impact on thyroid hormones. Environ. Int. 2012, 45, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Shah-Kulkarni, S.; Kim, B.-M.; Hong, Y.-C.; Kim, H.S.; Kwon, E.J.; Park, H.; Kim, Y.J.; Ha, E. Prenatal exposure to perfluorinated compounds affects thyroid hormone levels in newborn girls. Environ. Int. 2016, 94, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Dufour, P.; Pirard, C.; Seghaye, M.-C.; Charlier, C. Association between organohalogenated pollutants in cord blood and thyroid function in newborns and mothers from Belgian population. Environ. Pollut. 2018, 238, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Sepúlveda, M.S.; Roy, K.; Leszczynski, J. Endocrine-disrupting activity of per- and polyfluoroalkyl substances: Exploring combined approaches of ligand and structure based modeling. Chemosphere 2017, 184, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Long, M.; Ghisari, M.; Bonefeld-Jørgensen, E.C. Effects of perfluoroalkyl acids on the function of the thyroid hormone and the aryl hydrocarbon receptor. Environ. Sci. Pollut. Res. 2013, 20, 8045–8056. [Google Scholar] [CrossRef]
- Halldorsson, T.I.; Rytter, D.; Haug, L.S.; Bech, B.H.; Danielsen, I.; Becher, G.; Henriksen, T.B.; Olsen, S.F. Prenatal Exposure to Perfluorooctanoate and Risk of Overweight at 20 Years of Age: A Prospective Cohort Study. Environ. Health Perspect. 2012, 120, 668–673. [Google Scholar] [CrossRef]
- Steenland, K.; Zhao, L.; Winquist, A.; Parks, C. Ulcerative Colitis and Perfluorooctanoic Acid (PFOA) in a Highly Exposed Population of Community Residents and Workers in the Mid-Ohio Valley. Environ. Health Perspect. 2013, 121, 900–905. [Google Scholar] [CrossRef]
- Darrow, L.A.; Groth, A.C.; Winquist, A.; Shin, H.-M.; Bartell, S.M.; Steenland, K. Modeled Perfluorooctanoic Acid (PFOA) Exposure and Liver Function in a Mid-Ohio Valley Community. Environ. Health Perspect. 2016, 124, 1227–1233. [Google Scholar] [CrossRef]
- Oulhote, Y.; Steuerwald, U.; Debes, F.; Weihe, P.; Grandjean, P. Behavioral difficulties in 7-year old children in relation to developmental exposure to perfluorinated alkyl substances. Environ. Int. 2016, 97, 237–245. [Google Scholar] [CrossRef]
- Timmermann, C.A.G.; Budtz-Jørgensen, E.; Jensen, T.K.; Osuna, C.E.; Petersen, M.S.; Steuerwald, U.; Nielsen, F.; Poulsen, L.K.; Weihe, P.; Grandjean, P. Association between perfluoroalkyl substance exposure and asthma and allergic disease in children as modified by MMR vaccination. J. Immunotoxicol. 2017, 14, 39–49. [Google Scholar] [CrossRef]
- Dalsager, L.; Christensen, N.; Husby, S.; Kyhl, H.; Nielsen, F.; Høst, A.; Grandjean, P.; Jensen, T.K. Association between prenatal exposure to perfluorinated compounds and symptoms of infections at age 1–4years among 359 children in the Odense Child Cohort. Environ. Int. 2016, 96, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Zota, A.R.; Geller, R.J.; Romano, L.E.; Coleman-Phox, K.; Adler, N.E.; Parry, E.; Wang, M.; Park, J.-S.; Elmi, A.F.; Laraia, B.A.; et al. Association between persistent endocrine-disrupting chemicals (PBDEs, OH-PBDEs, PCBs, and PFASs) and biomarkers of inflammation and cellular aging during pregnancy and postpartum. Environ. Int. 2018, 115, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Howie, D.; Bokum, A.T.; Necula, A.S.; Cobbold, S.P.; Waldmann, H.; Howie, D.; Bokum, A.T.; Necula, A.S.; Cobbold, S.P.; Waldmann, H. The Role of Lipid Metabolism in T Lymphocyte Differentiation and Survival. Front. Immunol. 2018, 8, 1949. [Google Scholar] [CrossRef] [PubMed]
- Herbst, A.L.; Ulfelder, H.; Poskanzer, D.C. Adenocarcinoma of the vagina. Association of maternal stilbestrol therapy with tumor appearance in young women. Am. J. Obstet. Gynecol. 1999, 181, 1574–1575. [Google Scholar] [CrossRef]
- Multigner, L.; Ndong, J.R.; Giusti, A.; Romana, M.; Delacroix-Maillard, H.; Cordier, S.; Jégou, B.; Thomé, J.-P.; Blanchet, P. Chlordecone Exposure and Risk of Prostate Cancer. J. Clin. Oncol. 2010, 28, 3457–3462. [Google Scholar] [CrossRef]
- Seachrist, D.D.; Bonk, K.W.; Ho, S.-M.; Prins, G.S.; Soto, A.M.; Keri, R.A. A review of the carcinogenic potential of bisphenol A. Reprod. Toxicol. 2016, 59, 167–182. [Google Scholar] [CrossRef]
- Köhrle, J. Environment and endocrinology: The case of thyroidology. Ann. Endocrinol. 2008, 69, 116–122. [Google Scholar] [CrossRef]
- Perdichizzi, S.; Mascolo, M.G.; Silingardi, P.; Morandi, E.; Rotondo, F.; Guerrini, A.; Prete, L.; Vaccari, M.; Colacci, A. Cancer-related genes transcriptionally induced by the fungicide penconazole. Toxicol. Vitr. 2014, 28, 125–130. [Google Scholar] [CrossRef]
- Hoffman, K.; Sosa, J.A.; Stapleton, H.M. Do flame retardant chemicals increase the risk for thyroid dysregulation and cancer? Curr. Opin. Oncol. 2017, 29, 7–13. [Google Scholar] [CrossRef]
- Barry, V.; Winquist, A.; Steenland, K. Perfluorooctanoic Acid (PFOA) Exposures and Incident Cancers among Adults Living Near a Chemical Plant. Environ. Health Perspect. 2013, 121, 1313–1318. [Google Scholar] [CrossRef]
- Judson, R.; Houck, K.A.; Watt, E.D.; Thomas, R.S. On selecting a minimal set of in vitro assays to reliably determine estrogen agonist activity. Regul. Toxicol. Pharmacol. 2017, 91, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, G.B.; Khurana, S.M.P. In VitroReporter Assays for Screening of Chemicals That Disrupt Androgen Signaling. J. Toxicol. 2014, 2014, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Patel, S. Disruption of aromatase homeostasis as the cause of a multiplicity of ailments: A comprehensive review. J. Steroid Biochem. Mol. Biol. 2017, 168, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Gore, A.C.; Chappell, V.A.; Fenton, S.E.; Flaws, J.A.; Nadal, A.; Prins, G.S.; Toppari, J.; Zoeller, R.T. EDC-2: The Endocrine Society’s Second Scientific Statement on Endocrine-Disrupting Chemicals. Endocr. Rev. 2015, 36, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Islami, F.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer in Women: Burden and Trends. Cancer Epidemiology Biomarkers Prev. 2017, 26, 444–457. [Google Scholar] [CrossRef] [PubMed]
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. Rev. 2017, 7, 1016–1036. [Google Scholar] [CrossRef]
- Grönberg, H. Prostate cancer epidemiology. Lancet 2003, 361, 859–864. [Google Scholar] [CrossRef]
- Harbeck, N.; Dettmar, P.; Thomssen, C.; Berger, U.; Ulm, K.; Kates, R.; Höfler, H.; Jänicke, F.; Graeff, H.; Schmitt, M. Risk-group discrimination in node-negative breast cancer using invasion and proliferation markers: 6-year median follow-up. Br. J. Cancer 1999, 80, 419–426. [Google Scholar] [CrossRef]
- Leevers, S.J.; Vanhaesebroeck, B.; Waterfield, M.D. Signalling through phosphoinositide 3-kinases: The lipids take centre stage. Curr. Opin. Cell Biol. 1999, 11, 219–225. [Google Scholar] [CrossRef]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase–AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef]
- Fang, J.; Ding, M.; Yang, L.; Liu, L.-Z.; Jiang, B.-H. PI3K/PTEN/AKT signaling regulates prostate tumor angiogenesis. Cell. Signal. 2007, 19, 2487–2497. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.R.; Wise, L.A.; Hatch, E.E.; Troisi, R.; Titus-Ernstoff, L.; Strohsnitter, W.; Kaufman, R.; Herbst, A.L.; Noller, K.L.; Hyer, M.; et al. Prenatal Diethylstilbestrol Exposure and Risk of Breast Cancer. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1509–1514. [Google Scholar] [CrossRef] [PubMed]
- Conlon, J.L. Diethylstilbestrol: Potential health risks for women exposed in utero and their offspring. J. Am. Acad. Physician Assist. 2017, 30, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Hilakivi-Clarke, L. Maternal exposure to diethylstilbestrol during pregnancy and increased breast cancer risk in daughters. Breast Cancer Res. 2014, 16, 208. [Google Scholar] [CrossRef]
- Rothman, K.J. Is Breast Cancer Initiated In Utero? Epidemiology 1990, 1, 95–96. [Google Scholar] [CrossRef]
- Braun, M.M.; Ahlbom, A.; Floderus, B.; Brinton, L.A.; Hoover, R.N. Effect of twinship on incidence of cancer of the testis, breast, and other sites (Sweden). Cancer Causes Control. 1995, 6, 519–524. [Google Scholar] [CrossRef]
- Potischman, N.; Troisi, R. In-utero and early life exposures in relation to risk of breast cancer. Cancer Causes Control. 1999, 10, 561–573. [Google Scholar] [CrossRef]
- Wormsbaecher, C.; Hindman, A.R.; Avendano, A.; Cortes-Medina, M.; Jones, C.E.; Bushman, A.; Onua, L.; Kovalchin, C.E.; Murphy, A.R.; Helber, H.L.; et al. In utero estrogenic endocrine disruption alters the stroma to increase extracellular matrix density and mammary gland stiffness. Breast Cancer Res. 2020, 22, 1–12. [Google Scholar] [CrossRef]
- Loudig, O.; Babichuk, C.; White, J.; Abu-Abed, S.; Mueller, C.; Petkovich, M. Cytochrome P450RAI(CYP26) promoter: A distinct composite retinoic acid response element underlies the complex regulation of retinoic acid metabolism. Mol. Endocrinol. 2000, 14, 1483–1497. [Google Scholar] [CrossRef]
- Thatcher, J.E.; Isoherranen, N. The role of CYP26 enzymes in retinoic acid clearance. Expert Opin. Drug Metab. Toxicol. 2009, 5, 875–886. [Google Scholar] [CrossRef]
- Doherty, L.F.; Bromer, J.G.; Zhou, Y.; Aldad, T.S.; Taylor, H.S. In Utero Exposure to Diethylstilbestrol (DES) or Bisphenol-A (BPA) Increases EZH2 Expression in the Mammary Gland: An Epigenetic Mechanism Linking Endocrine Disruptors to Breast Cancer. Horm. Cancer 2010, 1, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Bhan, A.; Hussain, I.; Ansari, K.I.; Bobzean, S.A.; Perrotti, L.I.; Mandal, S.S. Histone Methyltransferase EZH2 Is Transcriptionally Induced by Estradiol as Well as Estrogenic Endocrine Disruptors Bisphenol-A and Diethylstilbestrol. J. Mol. Biol. 2014, 426, 3426–3441. [Google Scholar] [CrossRef] [PubMed]
- Adam, A.H.B.; De Haan, L.H.J.; Estruch, I.M.; Hooiveld, G.J.E.J.; Louisse, J.; Rietjens, I.M.C.M. Estrogen receptor alpha (ERα)–mediated coregulator binding and gene expression discriminates the toxic ERα agonist diethylstilbestrol (DES) from the endogenous ERα agonist 17β-estradiol (E2). Cell Biol. Toxicol. 2020, 36, 417–435. [Google Scholar] [CrossRef] [PubMed]
- Prossnitz, E.R.; Barton, M. Estrogen biology: New insights into GPER function and clinical opportunities. Mol. Cell. Endocrinol. 2014, 389, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, K.; Habrowska-Górczyńska, D.E.; Domińska, K.; Piastowska-Ciesielska, A.W. The dose-dependent effect of zearalenone on mitochondrial metabolism, plasma membrane permeabilization and cell cycle in human prostate cancer cell lines. Chemosphere 2017, 180, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Belhassen, H.; Jiménez-Díaz, I.; Arrebola, J.; Ghali, R.; Ghorbel, H.; Olea, N.; Hedili, A. Zearalenone and its metabolites in urine and breast cancer risk: A case-control study in Tunisia. Chemosphere 2015, 128, 1–6. [Google Scholar] [CrossRef]
- Nittoli, A.C.; Costantini, S.; Sorice, A.; Capone, F.; Ciarcia, R.; Marzocco, S.; Budillon, A.; Severino, L. Effects of α-zearalenol on the metabolome of two breast cancer cell lines by 1H-NMR approach. Metabolomics 2018, 14, 33. [Google Scholar] [CrossRef]
- Kowalska, K.; Habrowska-Górczyńska, D.E.; Urbanek, K.A.; Domińska, K.; Piastowska-Ciesielska, A.W. Estrogen Receptor α Is Crucial in Zearalenone-Induced Invasion and Migration of Prostate Cancer Cells. Toxins 2018, 10, 98. [Google Scholar] [CrossRef]
- Kowalska, K.; Habrowska-Górczyńska, D.E.; Domińska, K.; Urbanek, K.A.; Piastowska-Ciesielska, A.W. ERβ and NFκB—Modulators of Zearalenone-Induced Oxidative Stress in Human Prostate Cancer Cells. Toxins 2020, 12, 199. [Google Scholar] [CrossRef]
- Rodgers, K.M.; Udesky, J.O.; Rudel, R.A.; Brody, J.G. Environmental chemicals and breast cancer: An updated review of epidemiological literature informed by biological mechanisms. Environ. Res. 2018, 160, 152–182. [Google Scholar] [CrossRef]
- Zeinomar, N.; Oskar, S.; Kehm, R.D.; Sahebzeda, S.; Terry, M.B. Environmental exposures and breast cancer risk in the context of underlying susceptibility: A systematic review of the epidemiological literature. Environ. Res. 2020, 187, 109346. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.A.; Cirillo, P.M.; Tehranifar, P.; Krigbaum, N.Y.; Engmann, N.J.; Cohn, B.A.; Terry, M.B. In utero DDT exposure and breast density in early menopause by maternal history of breast cancer. Reprod. Toxicol. 2020, 92, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Krigbaum, N.Y.; Cirillo, P.M.; Flom, J.D.; McDonald, J.A.; Terry, M.B.; Cohn, B.A. In utero DDT exposure and breast density before age 50. Reprod. Toxicol. 2020, 92, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; El-Zaemey, S.; Heyworth, J.; Tang, M.-C. DDT exposure in early childhood and female breast cancer: Evidence from an ecological study in Taiwan. Environ. Int. 2018, 121, 1106–1112. [Google Scholar] [CrossRef] [PubMed]
- Cohn, B.A.; Wolff, M.S.; Cirillo, P.M.; Sholtz, R.I. DDT and breast cancer in young women: New data on the significance of age at exposure. Environ. Health Perspect. 2007, 115, 1406–1414. [Google Scholar] [CrossRef] [PubMed]
- Cohn, B.A.; Cirillo, P.M.; Terry, M.B. DDT and Breast Cancer: Prospective Study of Induction Time and Susceptibility Windows. J. Natl. Cancer Inst. 2019, 111, 803–810. [Google Scholar] [CrossRef]
- Pavuk, M.; Cerhan, J.R.; Lynch, C.F.; Kocan, A.; Petrik, J.; Chovancova, J. Case-control study of PCBs, other organochlorines and breast cancer in Eastern Slovakia. J. Expo. Sci. Environ. Epidemiol. 2003, 13, 267–275. [Google Scholar] [CrossRef]
- Williams, G.P.; Darbre, P.D. Low-dose environmental endocrine disruptors, increase aromatase activity, estradiol biosynthesis and cell proliferation in human breast cells. Mol. Cell. Endocrinol. 2019, 486, 55–64. [Google Scholar] [CrossRef]
- Eldakroory, S.A.; Morsi, D.E.; Abdel-Rahman, R.H.; Roshdy, S.; Gouida, M.S.; Khashaba, E.O. Correlation between toxic organochlorine pesticides and breast cancer. Hum. Exp. Toxicol. 2017, 36, 1326–1334. [Google Scholar] [CrossRef]
- Wu, H.-C.; Cohn, B.A.; Cirillo, P.M.; Santella, R.M.; Terry, M.B. DDT exposure during pregnancy and DNA methylation alterations in female offspring in the Child Health and Development Study. Reprod. Toxicol. 2020, 92, 138–147. [Google Scholar] [CrossRef]
- Hengstler, J.G.; Foth, H.; Gebel, T.; Kramer, P.-J.; Lilienblum, W.; Schweinfurth, H.; Völkel, W.; Wollin, K.-M.; Gundert-Remy, U. Critical evaluation of key evidence on the human health hazards of exposure to bisphenol A. Crit. Rev. Toxicol. 2011, 41, 263–291. [Google Scholar] [CrossRef] [PubMed]
- Soto, A.M.; Brisken, C.; Schaeberle, C.; Sonnenschein, C. Does cancer start in the womb? altered mammary gland development and predisposition to breast cancer due to in utero exposure to endocrine disruptors. J. Mammary Gland. Biol. Neoplasia 2013, 18, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Dairkee, S.H.; Seok, J.; Champion, S.; Sayeed, A.; Mindrinos, M.; Xiao, W.; Davis, R.W.; Goodson, W.H. Bisphenol A induces a profile of tumor aggressiveness in high-risk cells from breast cancer patients. Cancer Res. 2008, 68, 2076–2080. [Google Scholar] [CrossRef] [PubMed]
- Montes-Grajales, D.; Bernardes, G.J.L.; Olivero-Verbel, J.T. Urban Endocrine Disruptors Targeting Breast Cancer Proteins. Chem. Res. Toxicol. 2016, 29, 150–161. [Google Scholar] [CrossRef]
- Böckers, M.; Paul, N.W.; Efferth, T. Bisphenolic compounds alter gene expression in MCF-7 cells through interaction with estrogen receptor α. Toxicol. Appl. Pharmacol. 2020, 399, 115030. [Google Scholar] [CrossRef]
- Merzoug-Larabi, M.; Youssef, I.; Bui, A.T.; Legay, C.; Loiodice, S.; Lognon, S.; Babajko, S.; Ricort, J.-M. Protein Kinase D1 (PKD1) Is a New Functional Non-Genomic Target of Bisphenol A in Breast Cancer Cells. Front. Pharmacol. 2020, 10. [Google Scholar] [CrossRef]
- Donini, C.F.; El Helou, M.; Wierinckx, A.; Győrffy, B.; Aires, S.; Escande, A.; Croze, S.; Clezardin, P.; Lachuer, J.; Diab-Assaf, M.; et al. Long-Term Exposure of Early-Transformed Human Mammary Cells to Low Doses of Benzo[a]pyrene and/or Bisphenol A Enhances Their Cancerous Phenotype via an AhR/GPR30 Interplay. Front. Oncol. 2020, 10, 712. [Google Scholar] [CrossRef]
- Castillo-Sanchez, R.; Ramirez-Ricardo, J.; Martinez-Baeza, E.; Cortes-Reynosa, P.; Candanedo-Gonzales, F.; Gomez, R.; Salazar, E.P. Bisphenol A induces focal adhesions assembly and activation of FAK, Src and ERK2 via GPER in MDA-MB-231 breast cancer cells. Toxicol. Vitr. 2020, 66, 104871. [Google Scholar] [CrossRef]
- Bosland, M.C.; Der Meulen, H.C.D.-V.; Sukumar, S.; Ofner, P.; Leav, I.; Han, X.; Liehr, J.G. Multistage prostate carcinogenesis: The role of hormones. Princess Takamatsu Symp. 1991, 22, 109–123. [Google Scholar]
- Ho, S.-M.; Tang, W.-Y.; De Frausto, J.B.; Prins, G.S. Developmental Exposure to Estradiol and Bisphenol A Increases Susceptibility to Prostate Carcinogenesis and Epigenetically Regulates Phosphodiesterase Type 4 Variant 4. Cancer Res. 2006, 66, 5624–5632. [Google Scholar] [CrossRef]
- Zhao, Q.; Howard, E.W.; Parris, A.B.; Ma, Z.; Xing, Y.; Yang, X. Bisphenol AF promotes estrogen receptor-positive breast cancer cell proliferation through amphiregulin-mediated crosstalk with receptor tyrosine kinase signaling. PLoS ONE 2019, 14, e0216469. [Google Scholar] [CrossRef] [PubMed]
- Lei, B.; Sun, S.; Zhang, X.; Feng, C.; Xu, J.; Wen, Y.; Huang, Y.; Wu, M.; Yu, Y. Bisphenol AF exerts estrogenic activity in MCF-7 cells through activation of Erk and PI3K/Akt signals via GPER signaling pathway. Chemosphere 2019, 220, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Zhao, C.; Zhong, H.; Zhang, S.; Xia, Y.; Cai, Z. Bisphenol S induced epigenetic and transcriptional changes in human breast cancer cell line MCF-7. Environ. Pollut. 2019, 246, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Lovekamp-Swan, T.; Davis, B.J. Mechanisms of phthalate ester toxicity in the female reproductive system. Environ. Health Perspect. 2003, 111, 139–145. [Google Scholar] [CrossRef]
- Chen, F.-P.; Chien, M.-H. Lower concentrations of phthalates induce proliferation in human breast cancer cells. Climacteric 2013, 17, 377–384. [Google Scholar] [CrossRef]
- Zuccarello, P.; Conti, G.O.; Cavallaro, F.; Copat, C.; Cristaldi, A.; Fiore, M.; Ferrante, M. Implication of dietary phthalates in breast cancer. A systematic review. Food Chem. Toxicol. 2018, 118, 667–674. [Google Scholar] [CrossRef]
- Zhu, M.; Huang, C.; Ma, X.; Wu, R.; Zhu, W.; Li, X.; Liang, Z.; Deng, F.; Wu, J.; Geng, S.; et al. Phthalates promote prostate cancer cell proliferation through activation of ERK5 and p38. Environ. Toxicol. Pharmacol. 2018, 63, 29–33. [Google Scholar] [CrossRef]
- Chuang, S.-C.; Chen, H.-C.; Sun, C.-W.; Chen, Y.-A.; Wang, Y.-H.; Chiang, C.-J.; Chen, C.-C.; Wang, S.-L.; Chen, C.-J.; Hsiung, C.A. Phthalate exposure and prostate cancer in a population-based nested case-control study. Environ. Res. 2020, 181, 108902. [Google Scholar] [CrossRef]
- Sonthithai, P.; Suriyo, T.; Thiantanawat, A.; Watcharasit, P.; Ruchirawat, M.; Satayavivad, J. Perfluorinated chemicals, PFOS and PFOA, enhance the estrogenic effects of 17β-estradiol in T47D human breast cancer cells. J. Appl. Toxicol. 2015, 36, 790–801. [Google Scholar] [CrossRef]
- Pierozan, P.; Jernerén, F.; Karlsson, O. Perfluorooctanoic acid (PFOA) exposure promotes proliferation, migration and invasion potential in human breast epithelial cells. Arch. Toxicol. 2018, 92, 1729–1739. [Google Scholar] [CrossRef]
- Pierozan, P.; Karlsson, O. PFOS induces proliferation, cell-cycle progression, and malignant phenotype in human breast epithelial cells. Arch. Toxicol. 2018, 92, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Hurley, S.; Goldberg, D.; Wang, M.; Park, J.-S.; Petreas, M.; Bernstein, L.; Anton-Culver, H.; Nelson, D.O.; Reynolds, P. Breast cancer risk and serum levels of per- and poly-fluoroalkyl substances: A case-control study nested in the California Teachers Study. Environ. Health 2018, 17, 83. [Google Scholar] [CrossRef] [PubMed]
- Mancini, F.R.; Cano-Sancho, G.; Gambaretti, J.; Marchand, P.; Boutron-Ruault, M.; Severi, G.; Arveux, P.; Antignac, J.; Kvaskoff, M.; Cano-Sancho, G. Perfluorinated alkylated substances serum concentration and breast cancer risk: Evidence from a nested case-control study in the French E3N cohort. Int. J. Cancer 2020, 146, 917–928. [Google Scholar] [CrossRef]
- Chang, E.T.; Adami, H.-O.; Boffetta, P.; Cole, P.; Starr, T.B.; Mandel, J.S. A critical review of perfluorooctanoate and perfluorooctanesulfonate exposure and cancer risk in humans. Crit. Rev. Toxicol. 2014, 44, 1–81. [Google Scholar] [CrossRef] [PubMed]
- Arneth, B. Tumor Microenvironment. Medicina 2019, 56, 15. [Google Scholar] [CrossRef] [PubMed]
- Jahanban-Esfahlan, R.; Seidi, K.; Banimohamad-Shotorbani, B.; Jahanban-Esfahlan, A.; Yousefi, B. Combination of nanotechnology with vascular targeting agents for effective cancer therapy. J. Cell. Physiol. 2018, 233, 2982–2992. [Google Scholar] [CrossRef] [PubMed]
- Jahanban-Esfahlan, R.; Seidi, K.; Zarghami, N. Tumor vascular infarction: Prospects and challenges. Int. J. Hematol. 2017, 105, 244–256. [Google Scholar] [CrossRef]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 1–19. [Google Scholar] [CrossRef]
- Denisenko, T.V.; Budkevich, I.N.; Zhivotovsky, B. Cell death-based treatment of lung adenocarcinoma. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef]
- Jahanban-Esfahlan, R.; Seidi, K.; Manjili, M.H.; Jahanban-Esfahlan, A.; Javaheri, T.; Zare, P. Tumor Cell Dormancy: Threat or Opportunity in the Fight against Cancer. Cancers 2019, 11, 1207. [Google Scholar] [CrossRef] [PubMed]
- Seidi, K.; Neubauer, H.A.; Moriggl, R.; Jahanban-Esfahlan, R.; Javaheri, T. Tumor target amplification: Implications for nano drug delivery systems. J. Control. Release 2018, 275, 142–161. [Google Scholar] [CrossRef] [PubMed]
- Kloc, M.; Kubiak, J.Z.; Li, X.C.; Ghobrial, R.M. Pericytes, Microvasular Dysfunction, and Chronic Rejection. Transplantation 2015, 99, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Baluk, P.; Morikawa, S.; Haskell, A.; Mancuso, M.; McDonald, D.M. Abnormalities of Basement Membrane on Blood Vessels and Endothelial Sprouts in Tumors. Am. J. Pathol. 2003, 163, 1801–1815. [Google Scholar] [CrossRef]
- Dudley, A.C. Tumor Endothelial Cells. Cold Spring Harb. Perspect. Med. 2011, 2, a006536. [Google Scholar] [CrossRef]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 1–15. [Google Scholar] [CrossRef]
- Zhou, W.; Xu, G.; Wang, Y.; Xu, Z.; Liu, X.; Xu, X.; Ren, G.; Tian, K. Oxidative stress induced autophagy in cancer associated fibroblast enhances proliferation and metabolism of colorectal cancer cells. Cell Cycle 2017, 16, 73–81. [Google Scholar] [CrossRef]
- Zhang, R.; Qi, F.; Zhao, F.; Li, G.; Shao, S.; Zhang, X.; Yuan, L.; Feng, Y. Cancer-associated fibroblasts enhance tumor-associated macrophages enrichment and suppress NK cells function in colorectal cancer. Cell Death Dis. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Solinas, G.; Schiarea, S.; Liguori, M.; Fabbri, M.; Pesce, S.; Zammataro, L.; Pasqualini, F.; Nebuloni, M.; Chiabrando, C.; Mantovani, A.; et al. Tumor-Conditioned Macrophages Secrete Migration-Stimulating Factor: A New Marker for M2-Polarization, Influencing Tumor Cell Motility. J. Immunol. 2010, 185, 642–652. [Google Scholar] [CrossRef]
- Wei, C.; Yang, C.; Wang, S.; Shi, D.; Zhang, C.; Lin, X.; Liu, Q.; Dou, R.; Xiong, B. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol. Cancer 2019, 18, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tan, W.; Wang, C. Tumor-associated macrophage-derived cytokines enhance cancer stem-like characteristics through epithelial–mesenchymal transition. OncoTargets Ther. 2018, 11, 3817–3826. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhou, L.; Li, D.; Andl, T.; Zhang, Y. Cancer-Associated Fibroblasts Build and Secure the Tumor Microenvironment. Front. Cell Dev. Biol. 2019, 7, 60. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.; Mojares, E.; Hernández, A.E.D.R. Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef]
- Watnick, R.S. The Role of the Tumor Microenvironment in Regulating Angiogenesis. Cold Spring Harb. Perspect. Med. 2012, 2, a006676. [Google Scholar] [CrossRef]
- Angell, H.; Galon, J. From the immune contexture to the Immunoscore: The role of prognostic and predictive immune markers in cancer. Curr. Opin. Immunol. 2013, 25, 261–267. [Google Scholar] [CrossRef]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef]
- Wang, T.; Niu, G.; Kortylewski, M.; Burdelya, L.; Shain, K.H.; Zhang, S.; Bhattacharya, R.; Gabrilovich, D.I.; Heller, R.; Coppola, D.; et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. 2004, 10, 48–54. [Google Scholar] [CrossRef]
- Hivroz, C.; Chemin, K.; Tourret, M.; Bohineust, A. Crosstalk between T Lymphocytes and Dendritic Cells. Crit. Rev. Immunol. 2012, 32, 139–155. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Gattinoni, L.; Restifo, N.P. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol. Rev. 2006, 211, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.; Pan, K.; Li, X.-D.; She, K.-L.; Zhao, J.-J.; Wang, W.; Chen, J.-G.; Chen, Y.-B.; Yun, J.-P.; Xia, J.-C. The Accumulation and Prognosis Value of Tumor Infiltrating IL-17 Producing Cells in Esophageal Squamous Cell Carcinoma. PLoS ONE 2011, 6, e18219. [Google Scholar] [CrossRef] [PubMed]
- Schioppa, T.; Moore, R.; Thompson, R.G.; Rosser, E.C.; Kulbe, H.; Nedospasov, S.; Mauri, C.; Coussens, L.M.; Balkwill, F.R. B regulatory cells and the tumor-promoting actions of TNF- during squamous carcinogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 10662–10667. [Google Scholar] [CrossRef] [PubMed]
- Olkhanud, P.B.; Damdinsuren, B.; Bodogai, M.; Gress, R.E.; Sen, R.; Wejksza, K.; Malchinkhuu, E.; Wersto, R.P.; Biragyn, A. Tumor-Evoked Regulatory B Cells Promote Breast Cancer Metastasis by Converting Resting CD4+ T Cells to T-Regulatory Cells. Cancer Res. 2011, 71, 3505–3515. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wang, J.H.-C. Fibroblasts and myofibroblasts in wound healing: Force generation and measurement. J. Tissue Viability 2011, 20, 108–120. [Google Scholar] [CrossRef]
- Xing, F. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front. Biosci. 2010, 15, 166–179. [Google Scholar] [CrossRef]
- Spaeth, E.L.; Dembinski, J.L.; Sasser, A.K.; Watson, K.; Klopp, A.; Hall, B.; Andreeff, M.; Marini, F. Mesenchymal Stem Cell Transition to Tumor-Associated Fibroblasts Contributes to Fibrovascular Network Expansion and Tumor Progression. PLoS ONE 2009, 4, e4992. [Google Scholar] [CrossRef]
- Birbrair, A.; Zhang, T.; Wang, Z.-M.; Messi, M.L.; Olson, J.D.; Mintz, A.; Delbono, O. Type-2 pericytes participate in normal and tumoral angiogenesis. Am. J. Physiol. Physiol. 2014, 307, C25–C38. [Google Scholar] [CrossRef]
- Wang, S.-C.; Hong, J.-H.; Hsueh, C.; Chiang, C.-S. Tumor-secreted SDF-1 promotes glioma invasiveness and TAM tropism toward hypoxia in a murine astrocytoma model. Lab. Investig. 2012, 92, 151–162. [Google Scholar] [CrossRef]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M.O. The cellular and molecular origin of tumor-associated macrophages. Sci. 2014, 344, 921–925. [Google Scholar] [CrossRef]
- Fisher, D.T.; Appenheimer, M.M.; Evans, S.S. The two faces of IL-6 in the tumor microenvironment. Semin. Immunol. 2014, 26, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Strong, A.L.; Burow, M.E.; Gimble, J.M.; Bunnell, B.A. Concise Review: The Obesity Cancer Paradigm: Exploration of the Interactions and Crosstalk with Adipose Stem Cells. STEM CELLS 2015, 33, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Ohlstein, J.F.; Strong, A.L.; McLachlan, J.A.; Gimble, J.M.; Burow, M.E.; Bunnell, B.A. Bisphenol A enhances adipogenic differentiation of human adipose stromal/stem cells. J. Mol. Endocrinol. 2014, 53, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Gassman, N.R.; Coşkun, E.; Stefanick, N.F.; Horton, J.K.; Jaruga, P.; Dizdaroglu, M.; Wilson, S.H. Bisphenol A Promotes Cell Survival Following Oxidative DNA Damage in Mouse Fibroblasts. PLoS ONE 2015, 10, e0118819. [Google Scholar] [CrossRef]
- Rochester, J.R.; Bolden, A.L. Bisphenol S and F: A Systematic Review and Comparison of the Hormonal Activity of Bisphenol A Substitutes. Environ. Health Perspect. 2015, 123, 643–650. [Google Scholar] [CrossRef]
- Kuruto-Niwa, R.; Nozawa, R.; Miyakoshi, T.; Shiozawa, T.; Terao, Y. Estrogenic activity of alkylphenols, bisphenol S, and their chlorinated derivatives using a GFP expression system. Environ. Toxicol. Pharmacol. 2005, 19, 121–130. [Google Scholar] [CrossRef]
- Liu, X.; Matsushima, A.; Okada, H.; Tokunaga, T.; Isozaki, K.; Shimohigashi, Y. Receptor binding characteristics of the endocrine disruptor bisphenol A for the human nuclear estrogen-related receptor γ. Chief and corroborative hydrogen bonds of the bisphenol A phenol-hydroxyl group with Arg316 and Glu275 residues. FEBS J. 2007, 274, 6340–6351. [Google Scholar] [CrossRef]
- Kubo, M.; Ijichi, N.; Ikeda, K.; Horie-Inoue, K.; Takeda, S.; Inoue, S. Modulation of adipogenesis-related gene expression by estrogen-related receptor γ during adipocytic differentiation. Biochim. Biophys. Acta 2009, 1789, 71–77. [Google Scholar] [CrossRef]
- Zoubouliss, C.C.; Chen, W.-C.; Thornton, M.J.; Qin, K.; Rosenfield, R. Sexual Hormones in Human Skin. Horm. Metab. Res. 2007, 39, 85–95. [Google Scholar] [CrossRef]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix Crosslinking Forces Tumor Progression by Enhancing Integrin Signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.A.; Elbakry, R.H.; Bayomy, N.A. Effect of bisphenol A on morphology, apoptosis and proliferation in the resting mammary gland of the adult albino rat. Int. J. Exp. Pathol. 2016, 97, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Leśniewska, M.; Miltyk, W.; Światecka, J.; Tomaszewska, M.; Kuzmicki, M.; Palka, J.; Wolczyński, S. Estrogen receptor beta participate in the regulation of metabolizm of extracellular matrix in estrogen alpha negative breast cancer. Folia Histochem. et Cytobiol. 2010, 47, 107–112. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kousidou, O.C.; Berdiaki, A.; Kletsas, D.; Zafiropoulos, A.; Theocharis, A.D.; Tzanakakis, G.N.; Karamanos, N.K. Estradiol-estrogen receptor: A key interplay of the expression of syndecan-2 and metalloproteinase-9 in breast cancer cells. Mol. Oncol. 2008, 2, 223–232. [Google Scholar] [CrossRef]
- Nilsson, U.W.; Garvin, S.; Dabrosin, C. MMP-2 and MMP-9 activity is regulated by estradiol and tamoxifen in cultured human breast cancer cells. Breast Cancer Res. Treat. 2006, 102, 253–261. [Google Scholar] [CrossRef]
- Burks, H.; Pashos, N.; Martin, E.; McLachlan, J.; Bunnell, B.; Burow, M. Endocrine disruptors and the tumor microenvironment: A new paradigm in breast cancer biology. Mol. Cell. Endocrinol. 2017, 457, 13–19. [Google Scholar] [CrossRef]
- Hu, W.-Y.; Shi, G.-B.; Hu, D.-P.; Nelles, J.L.; Prins, G.S. Actions of estrogens and endocrine disrupting chemicals on human prostate stem/progenitor cells and prostate cancer risk. Mol. Cell. Endocrinol. 2012, 354, 63–73. [Google Scholar] [CrossRef]
- Aghajanova, L.; Giudice, L.C. Effect of bisphenol A on human endometrial stromal fibroblasts in vitro. Reprod. Biomed. Online 2010, 22, 249–256. [Google Scholar] [CrossRef]
- Csaba, G. Effect of endocrine disruptor phytoestrogens on the immune system: Present and future. Acta Microbiol. Immunol. Hung. 2018, 65, 1–14. [Google Scholar] [CrossRef]
- Malaisé, Y.; Ménard, S.; Cartier, C.; Lencina, C.; Sommer, C.; Gaultier, E.; Houdeau, E.; Guzylack-Piriou, L. Consequences of bisphenol a perinatal exposure on immune responses and gut barrier function in mice. Arch. Toxicol. 2017, 92, 347–358. [Google Scholar] [CrossRef]
- Anwer, F.; Chaurasia, S.; Khan, A.A. Hormonally active agents in the environment: A state-of-the-art review. Rev. Environ. Health 2016, 31, 415–433. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Schafer, R.; Barnett, J. The Immunomodulatory Effects of the Herbicide Propanil on Murine Macrophage Interleukin-6 and Tumor Necrosis Factor-α Production. Toxicol. Appl. Pharmacol. 1997, 145, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, J.; Iwahara, C.; Kawasaki, M.; Yoshizaki, F.; Nakayama, H.; Takamori, K.; Ogawa, H.; Iwabuchi, K. Di-(2-ethylhexyl) phthalate induces production of inflammatory molecules in human macrophages. Inflamm. Res. 2012, 61, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Sugita-Konishi, Y.; Shimura, S.; Nishikawa, T.; Sunaga, F.; Naito, H.; Suzuki, Y. Effect of Bisphenol A on non-specific immunodefenses against non-pathogenic Escherichia coli. Toxicol. Lett. 2003, 136, 217–227. [Google Scholar] [CrossRef]
- Švajger, U.; Dolenc, M.S.; Jeras, M. In vitro impact of bisphenols BPA, BPF, BPAF and 17β-estradiol (E2) on human monocyte-derived dendritic cell generation, maturation and function. Int. Immunopharmacol. 2016, 34, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Liu, T.; Uemura, Y.; Jiao, S.; Wang, D.; Lin, Z.; Narita, Y.; Suzuki, M.; Hirosawa, N.; Ichihara, Y.; et al. Bisphenol A in combination with TNF-α selectively induces Th2 cell-promoting dendritic cells in vitro with an estrogen-like activity. Cell. Mol. Immunol. 2010, 7, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Ndebele, K.; Tchounwou, P.B.; McMurray, R.W. Coumestrol, Bisphenol-A, DDT, and TCDD Modulation of Interleukin-2 Expression in Activated CD+4 Jurkat T Cells. Int. J. Environ. Res. Public Health 2004, 1, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.; Jabłońska, E.; Ratajczak-Wrona, W. Immunomodulatory effects of synthetic endocrine disrupting chemicals on the development and functions of human immune cells. Environ. Int. 2019, 125, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Bansal, A.; Henao-Mejia, J.; Simmons, R.A. Immune System: An Emerging Player in Mediating Effects of Endocrine Disruptors on Metabolic Health. Endocrinology 2017, 159, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Blauer, K.L.; Poth, M.; Rogers, W.M.; Bernton, E.W. Dehydroepiandrosterone Antagonizes the Suppressive Effects of Dexamethasone on Lymphocyte Proliferation. Endocrinology 1991, 129, 3174–3179. [Google Scholar] [CrossRef] [PubMed]
- Daynes, R.A.; Araneo, B.A.; Bogan, R.L.; Enioutina, E.; Mu, H.H. Steroids as Regulators of the Mammalian Immune Response. J. Investig. Dermatol. 1995, 105, 14S–19S. [Google Scholar] [CrossRef] [PubMed]
- Butcher, S.K.; Killampalli, V.; Lascelles, D.; Wang, K.; Alpar, E.K.; Lord, J.M. Raised cortisol:DHEAS ratios in the elderly after injury: Potential impact upon neutrophil function and immunity. Aging Cell 2005, 4, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Buoso, E.; Lanni, C.; Molteni, E.; Rousset, F.; Corsini, E.; Racchi, M. Opposing effects of cortisol and dehydroepiandrosterone on the expression of the receptor for Activated C Kinase 1: Implications in immunosenescence. Exp. Gerontol. 2011, 46, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Mochly-Rosen, D.; Khaner, H.; Lopez, J. Identification of intracellular receptor proteins for activated protein kinase C. Proc. Natl. Acad. Sci. USA 1991, 88, 3997–4000. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T. Signal transduction changes in granulocytes and lymphocytes with ageing. Immunol. Lett. 1994, 40, 259–268. [Google Scholar] [CrossRef]
- Delpedro, A.D.; Barjavel, M.J.; Mamdouh, Z.; Faure, S.; Bakouche, O. Signal Transduction in LPS-Activated Aged and Young Monocytes. J. Interf. Cytokine Res. 1998, 18, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Corsini, E.; Vismara, L.; Lucchi, L.; Viviani, B.; Govoni, S.; Galli, C.L.; Marinovich, M.; Racchi, M. High interleukin-10 production is associated with low antibody response to influenza vaccination in the elderly. J. Leukoc. Biol. 2006, 80, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Corsini, E.; Battaini, F.; Lucchi, L.; Marinovich, M.; Racchi, M.; Govoni, S.; Galli, C.L. A defective protein kinase C anchoring system underlying age-associated impairment in TNF-alpha production in rat macrophages. J. Immunol. 1999, 163, 3468–3473. [Google Scholar]
- Corsini, E.; Lucchi, L.; Meroni, M.; Racchi, M.; Solerte, B.; Fioravanti, M.; Viviani, B.; Marinovich, M.; Govoni, S.; Galli, C.L. In vivo dehydroepiandrosterone restores age-associated defects in the protein kinase C signal transduction pathway and related functional responses. J. Immunol. 2002, 168, 1753–1758. [Google Scholar] [CrossRef] [PubMed]
- Corsini, E.; Racchi, M.; Sinforiani, E.; Lucchi, L.; Viviani, B.; Rovati, G.E.; Govoni, S.; Galli, C.L.; Marinovich, M. Age-related decline in RACK-1 expression in human leukocytes is correlated to plasma levels of dehydroepiandrosterone. J. Leukoc. Biol. 2004, 77, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Battaini, F.; Pascale, A.; Paoletti, R.; Govoni, S. The role of anchoring protein rack1 in pkc activation in the ageing rat brain. Trends Neurosci. 1997, 20, 410–415. [Google Scholar] [CrossRef]
- Corsini, E.; Pinto, A.; Galbiati, V.; Viviani, B.; Galli, C.L.; Marinovich, M.; Racchi, M. Corticosteroids modulate the expression of the PKC-anchoring protein RACK-1 and cytokine release in THP-1 cells. Pharmacol. Res. 2014, 81, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Buoso, E.; Galasso, M.; Ronfani, M.; Serafini, M.M.; Lanni, C.; Corsini, E.; Racchi, M. Role of spliceosome proteins in the regulation of glucocorticoid receptor isoforms by cortisol and dehydroepiandrosterone. Pharmacol. Res. 2017, 120, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Kino, T.; Su, Y.A.; Chrousos, G.P. Human glucocorticoid receptor isoform β: Recent understanding of its potential implications in physiology and pathophysiology. Cell. Mol. Life Sci. 2009, 66, 3435–3448. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, B.D. Sub-chronic effect of DDT on humoral immune response to a thymus-independent antigen (bacterial lipopolysaccharide) in mice. Bull. Environ. Contam. Toxicol. 1987, 39, 822–826. [Google Scholar] [CrossRef]
- Banerjee, B.D. Effects of sub-chronic DDT exposure on humoral and cell-mediated immune responses in albino rats. Bull. Environ. Contam. Toxicol. 1987, 39, 827–834. [Google Scholar] [CrossRef]
- Corsini, E.; Galbiati, V.; Esser, P.R.; Pinto, A.; Racchi, M.; Marinovich, M.; Martin, S.F.; Galli, C.L. Role of PKC-β in chemical allergen-induced CD86 expression and IL-8 release in THP-1 cells. Arch. Toxicol. 2013, 88, 415–424. [Google Scholar] [CrossRef]
- Adams, D.R.; Ron, D.; A Kiely, P. RACK1, A multifaceted scaffolding protein: Structure and function. Cell Commun. Signal. 2011, 9, 22. [Google Scholar] [CrossRef]
- Buoso, E.; Biundo, F.; Lanni, C.; Aiello, S.; Beer, H.; Schettini, G.; Govoni, S.; Racchi, M. Modulation of Rack-1/PKCβII Signalling By Soluble AβPPα in SH-SY5Y Cells. Curr. Alzheimer Res. 2013, 10, 697–705. [Google Scholar] [CrossRef]
- Li, J.-J.; Xie, D. RACK1, a versatile hub in cancer. Oncogene 2014, 34, 1890–1898. [Google Scholar] [CrossRef]
- Buoso, E.; Ronfani, M.; Galasso, M.; Ventura, D.; Corsini, E.; Racchi, M. Cortisol-induced SRSF3 expression promotes GR splicing, RACK1 expression and breast cancer cells migration. Pharmacol. Res. 2019, 143, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, J.; Mangé, A.; Jarlier, M.; Bascoul-Mollevi, C.; Rouanet, P.; Lamy, P.-J.; Maudelonde, T.; Solassol, J. Identification and validation of new autoantibodies for the diagnosis of DCIS and node negative early-stage breast cancers. Int. J. Cancer 2012, 132, 1105–1113. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, J.; Mangé, A.; Bougnoux, A.-C.; Prassas, I.; Solassol, J. A Multiparametric Serum Marker Panel as a Complementary Test to Mammography for the Diagnosis of Node-Negative Early-Stage Breast Cancer and DCIS in Young Women. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 1834–1842. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.-X.; Xu, J.-D.; Liu, X.-L.; Xu, J.-W.; Wang, W.-J.; Li, Q.-Q.; Chen, Q.; Xu, Z.-D.; Liu, X.-P. RACK1: A superior independent predictor for poor clinical outcome in breast cancer. Int. J. Cancer 2009, 127, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.-X.; Xu, J.-D.; Xu, J.-W.; Liu, X.-L.; Cheng, Y.-Y.; Wang, W.-J.; Li, Q.-Q.; Chen, Q.; Xu, Z.-D.; Liu, X.-P. RACK1 promotes breast carcinoma proliferation and invasion/metastasis in vitro and in vivo. Breast Cancer Res. Treat. 2009, 123, 375–386. [Google Scholar] [CrossRef]
- Cao, X.-X.; Xu, J.-D.; Xu, J.-W.; Liu, X.-L.; Cheng, Y.-Y.; Li, Q.-Q.; Xu, Z.-D.; Liu, X.-P. RACK1 promotes breast carcinoma migration/metastasis via activation of the RhoA/Rho kinase pathway. Breast Cancer Res. Treat. 2011, 126, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, Y.; Qi, H.; Kanazawa, R.; Sugamata, M.; Suzuki, K.; Kobayashi, A.; Shindo, K.; Matsuzawa, A.; Shibata, S.; Endo, S.; et al. RACK1 regulates centriole duplication by controlling localization of BRCA1 to the centrosome in mammary tissue-derived cells. Oncogene 2019, 38, 3077–3092. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Cheng, G.Z.; Gong, J.; Hermanto, U.; Zong, C.S.; Chan, J.; Cheng, J.Q.; Wang, L.-H. RACK1 and CIS Mediate the Degradation of BimEL in Cancer Cells. J. Biol. Chem. 2008, 283, 16416–16426. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Sanchez, R.; Gomez, R.; Salazar, E.P.; Gomez-Ortega, R. Bisphenol A Induces Migration through a GPER-, FAK-, Src-, and ERK2-Dependent Pathway in MDA-MB-231 Breast Cancer Cells. Chem. Res. Toxicol. 2016, 29, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.-L. Integrin signaling through FAK in the regulation of mammary stem cells and breast cancer. IUBMB Life 2010, 62, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Kiely, P.A.; Baillie, G.S.; Barrett, R.; Buckley, D.A.; Adams, D.R.; Houslay, M.D.; Oconnor, R. Phosphorylation of RACK1 on Tyrosine 52 by c-Abl Is Required for Insulin-like Growth Factor I-mediated Regulation of Focal Adhesion Kinase. J. Biol. Chem. 2009, 284, 20263–20274. [Google Scholar] [CrossRef] [PubMed]
- Shen, F.; Yan, C.; Liu, M.; Feng, Y.; Chen, Y. RACK1 promotes prostate cancer cell proliferation, invasion and metastasis. Mol. Med. Rep. 2013, 8, 999–1004. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Xu, Z.; Zou, C.; Wu, D.; Wang, Y.; Yao, X.; Ng, C.F.; Chan, F.L. Ion channel TRPM8 promotes hypoxic growth of prostate cancer cells via an O2-independent and RACK1-mediated mechanism of HIF-1α stabilization. J. Pathol. 2014, 234, 514–525. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buoso, E.; Masi, M.; Racchi, M.; Corsini, E. Endocrine-Disrupting Chemicals’ (EDCs) Effects on Tumour Microenvironment and Cancer Progression: Emerging Contribution of RACK1. Int. J. Mol. Sci. 2020, 21, 9229. https://doi.org/10.3390/ijms21239229
Buoso E, Masi M, Racchi M, Corsini E. Endocrine-Disrupting Chemicals’ (EDCs) Effects on Tumour Microenvironment and Cancer Progression: Emerging Contribution of RACK1. International Journal of Molecular Sciences. 2020; 21(23):9229. https://doi.org/10.3390/ijms21239229
Chicago/Turabian StyleBuoso, Erica, Mirco Masi, Marco Racchi, and Emanuela Corsini. 2020. "Endocrine-Disrupting Chemicals’ (EDCs) Effects on Tumour Microenvironment and Cancer Progression: Emerging Contribution of RACK1" International Journal of Molecular Sciences 21, no. 23: 9229. https://doi.org/10.3390/ijms21239229
APA StyleBuoso, E., Masi, M., Racchi, M., & Corsini, E. (2020). Endocrine-Disrupting Chemicals’ (EDCs) Effects on Tumour Microenvironment and Cancer Progression: Emerging Contribution of RACK1. International Journal of Molecular Sciences, 21(23), 9229. https://doi.org/10.3390/ijms21239229