Liver Growth Factor “LGF” as a Therapeutic Agent for Alzheimer’s Disease

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
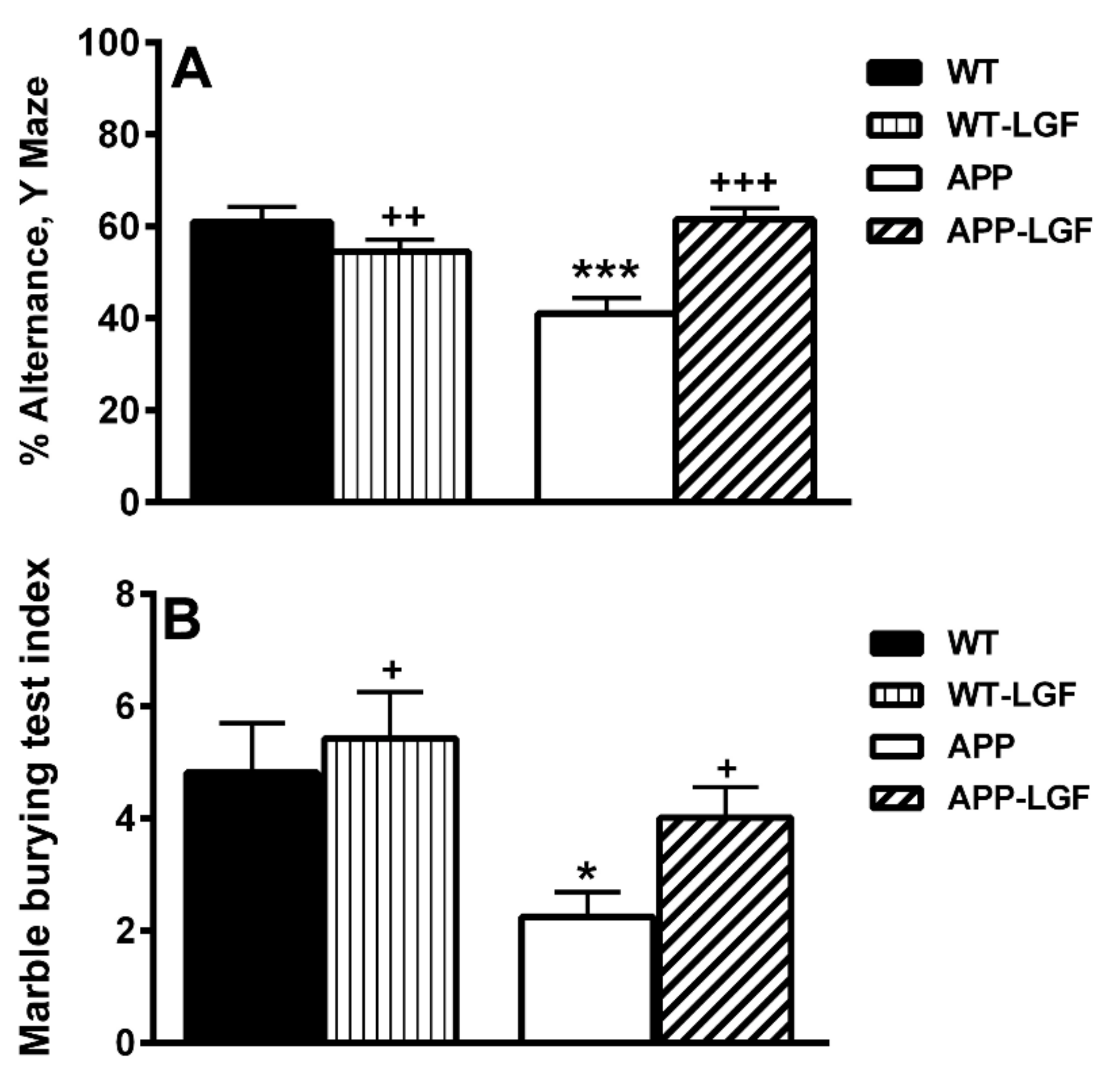
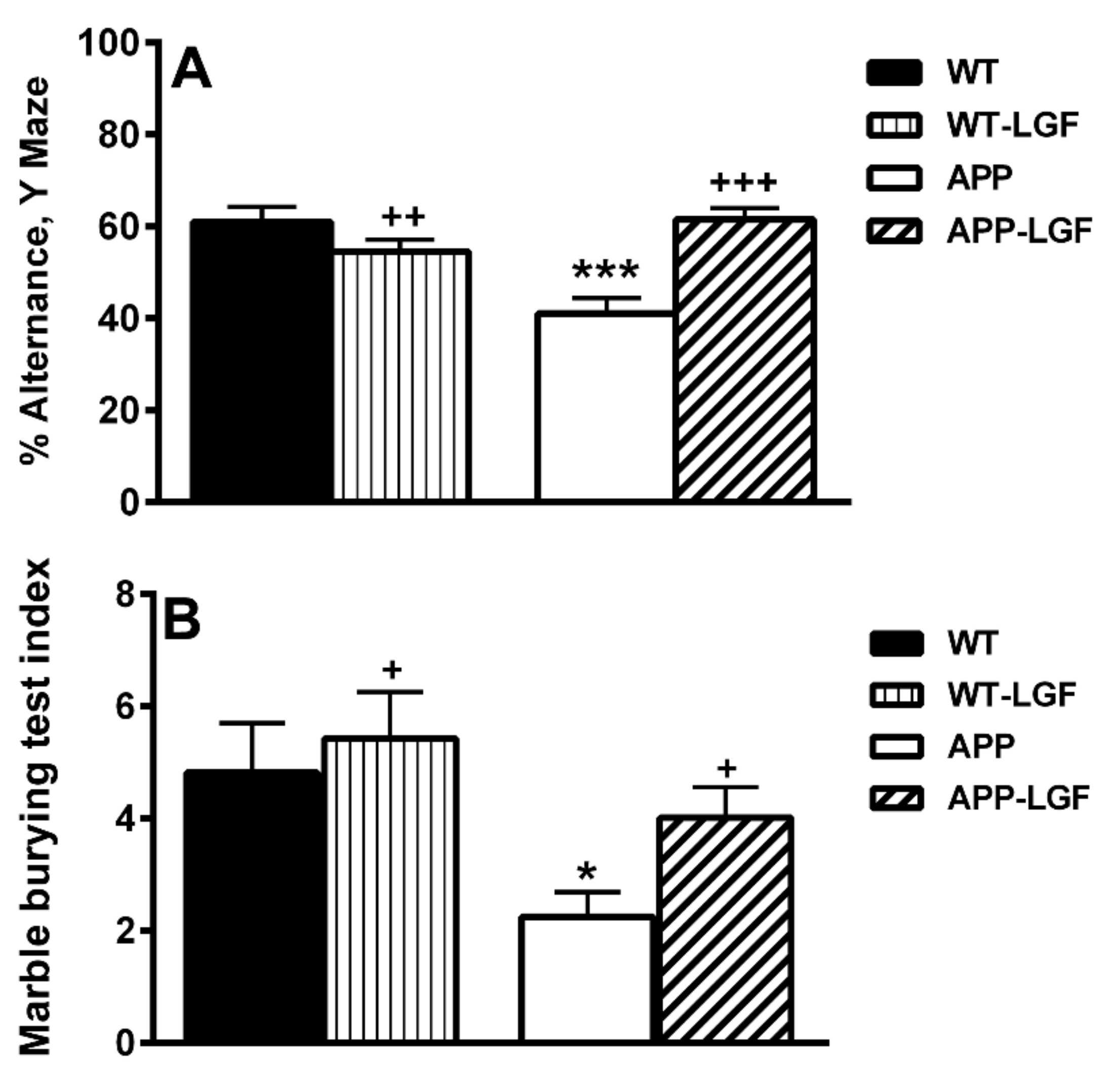
2.1. LGF Improves Behavioral Working Memory in APPswe Mice
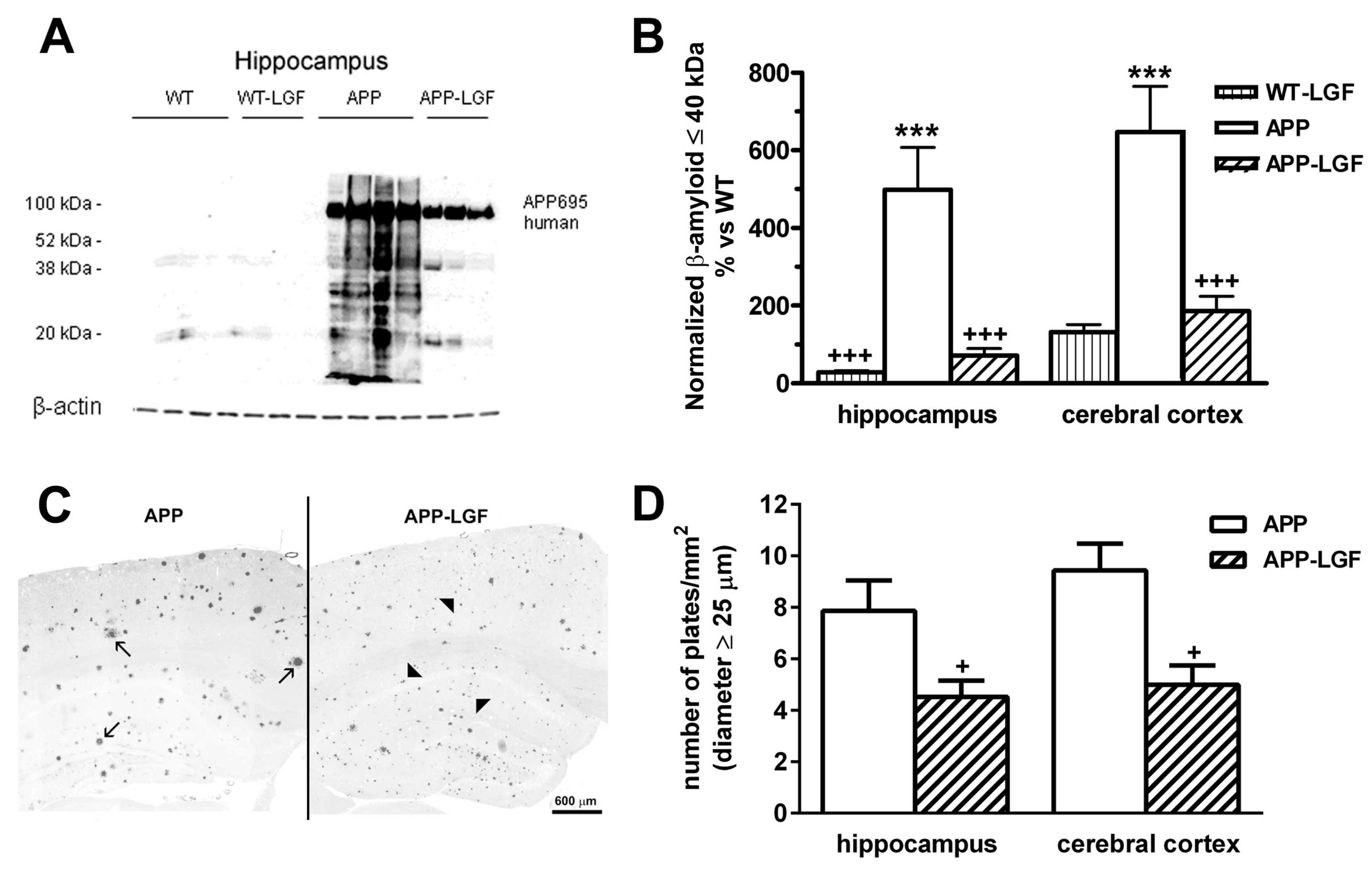
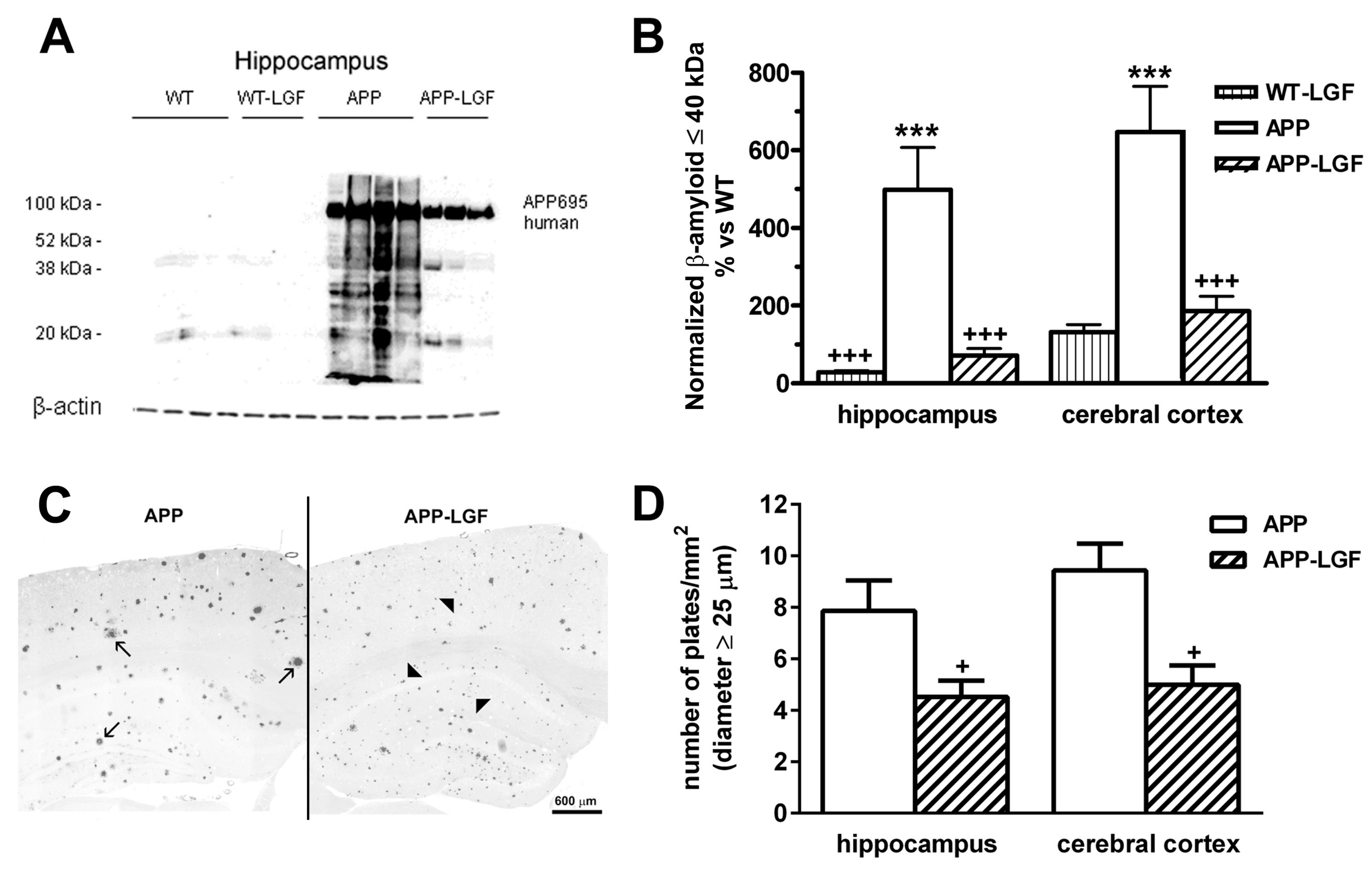
2.2. LGF Modulatesβ-Amyloid Protein Expression in Hippocampus and Cerebral Cortex of APPswe Mice
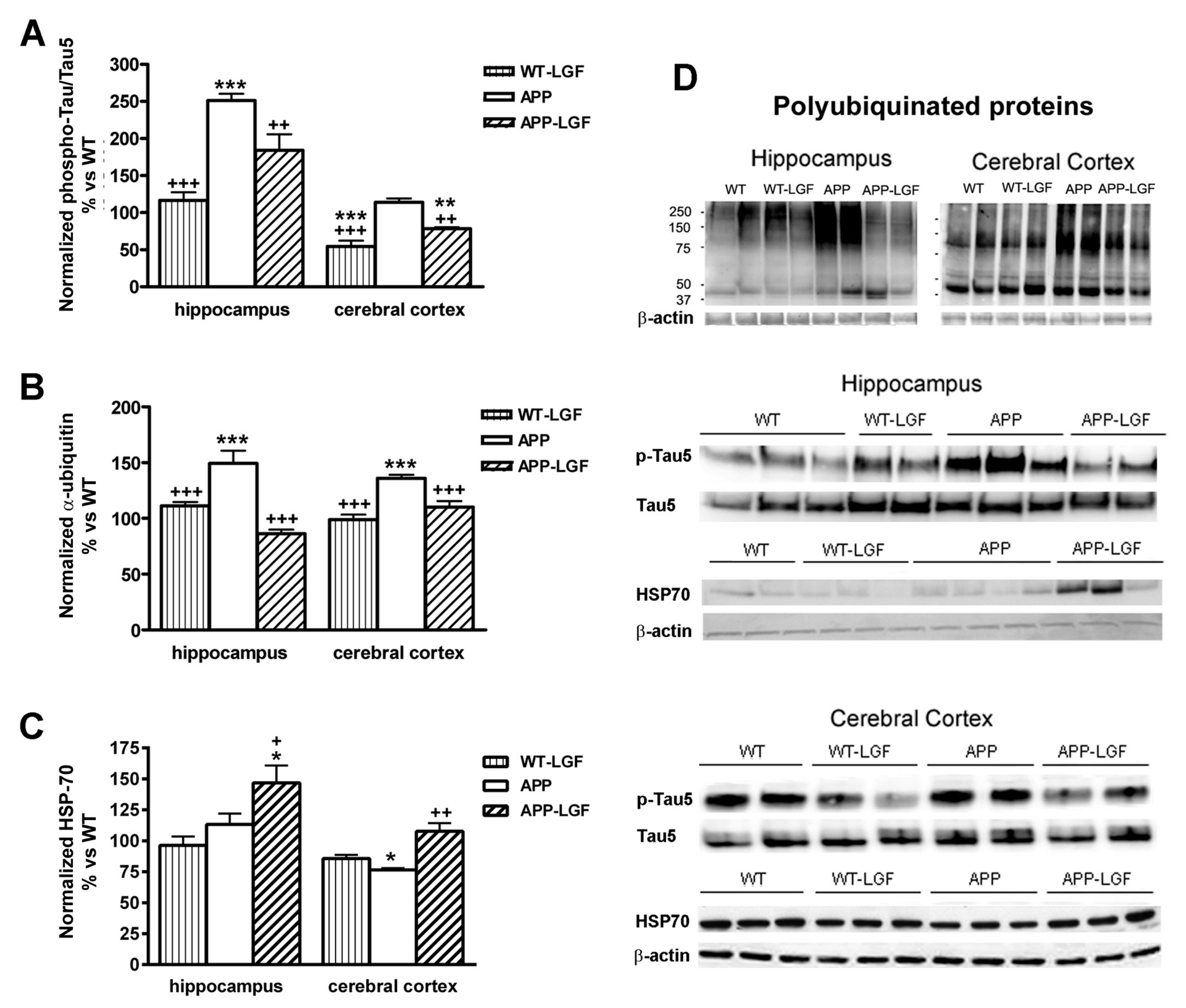
2.3. Effects of LGF in Tau Phosphorylation and Protein Ubiquitination
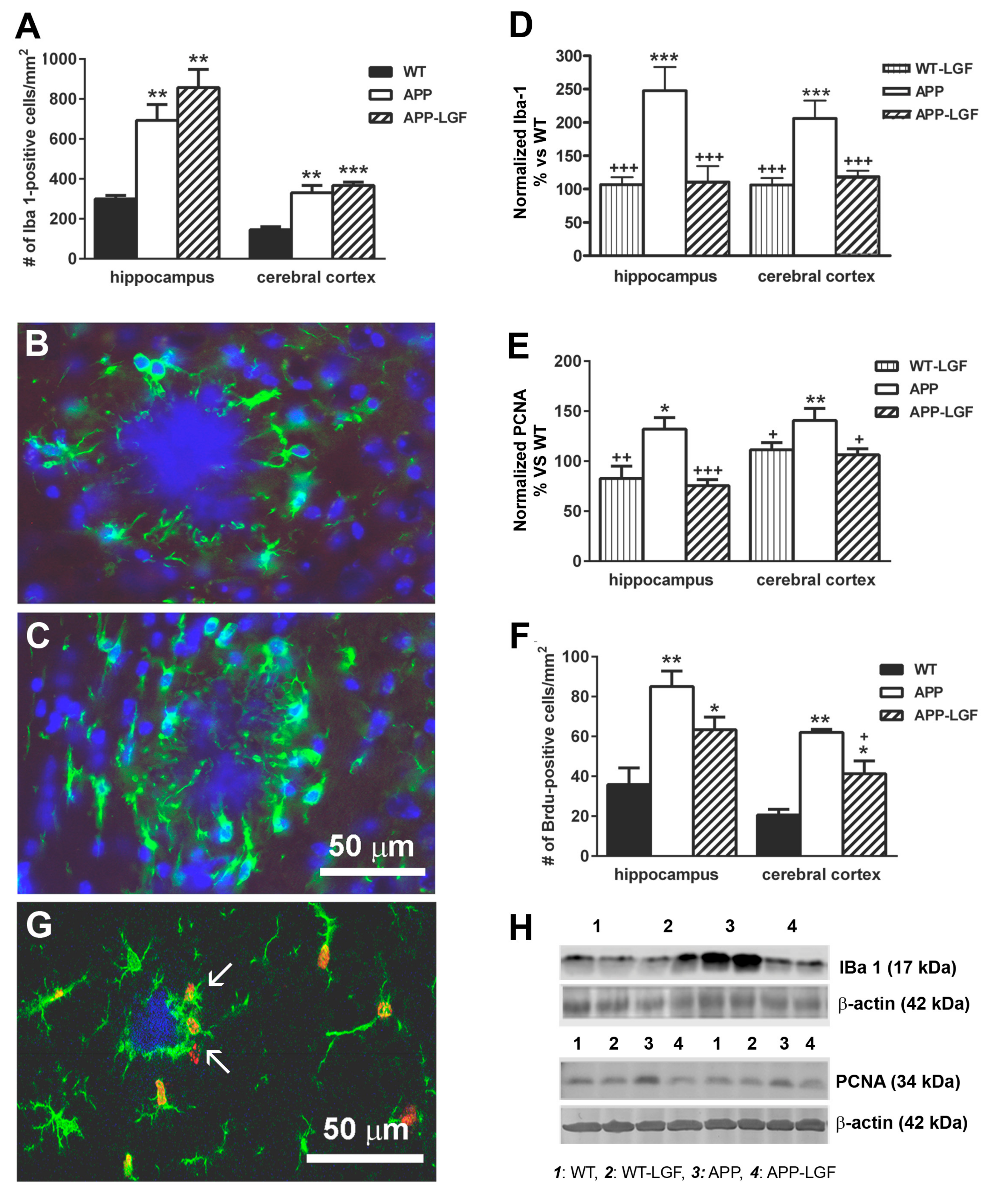
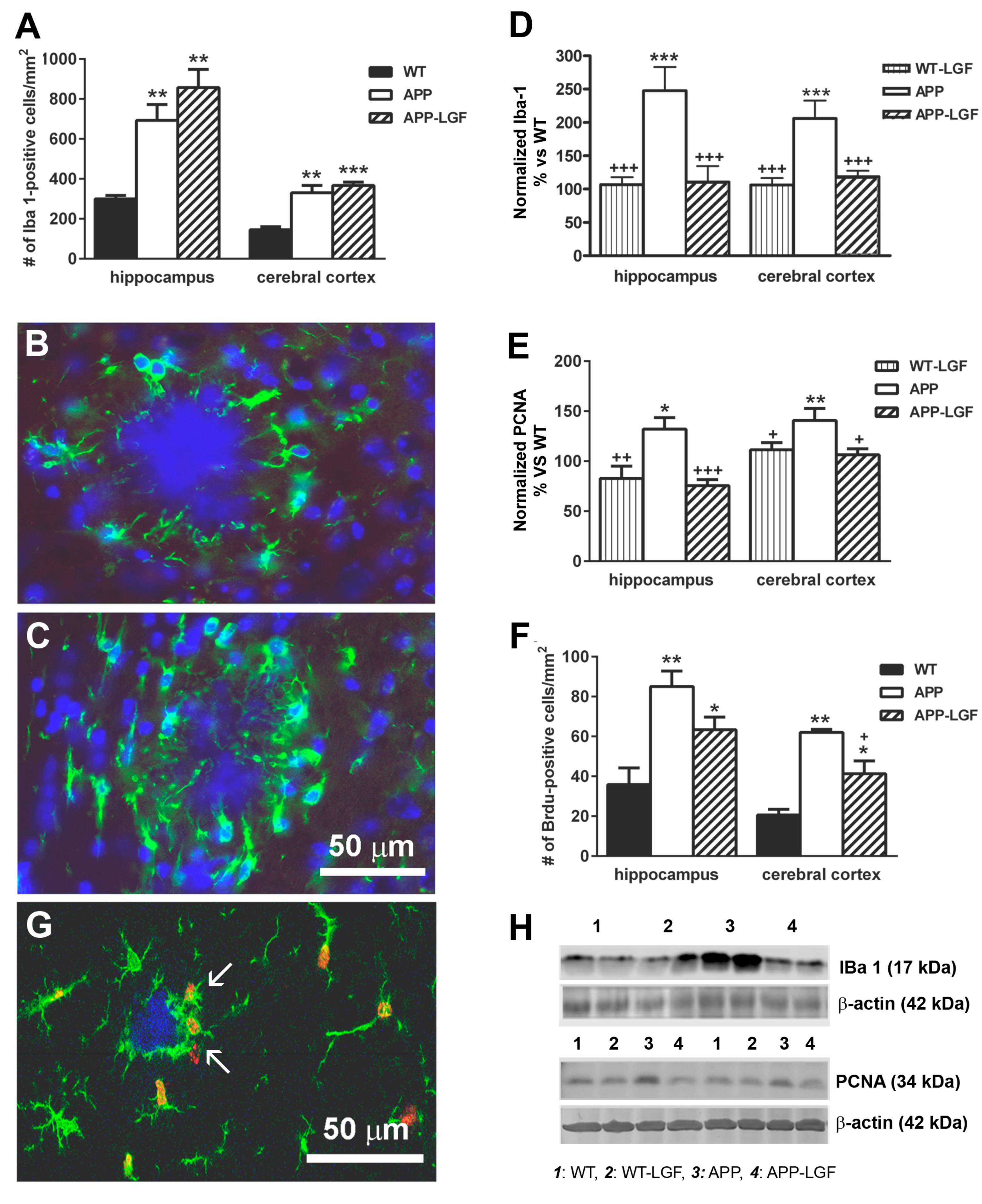
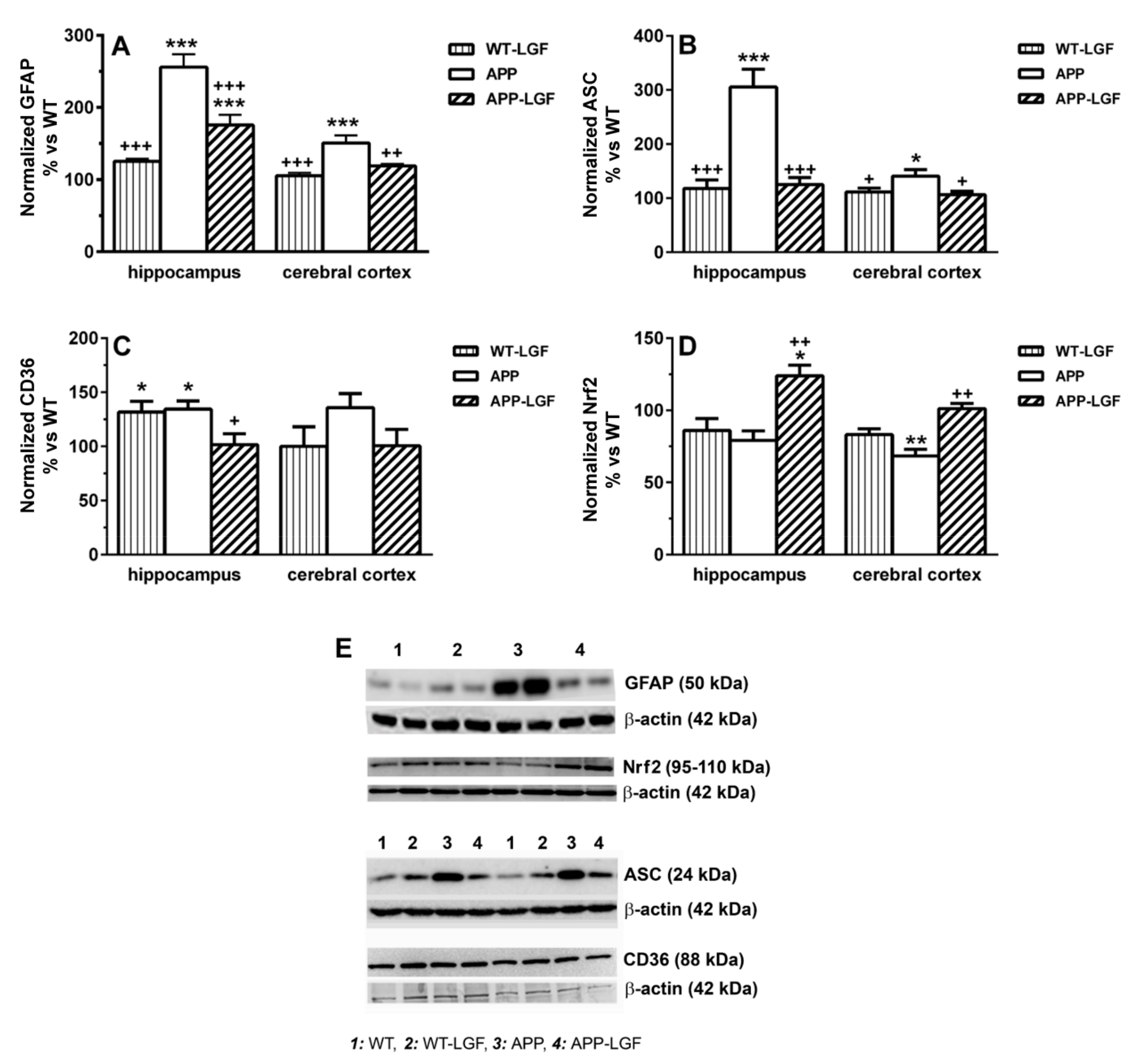
2.4. LGF Modulates Microglia Activation and Reduces Inflammation in APPswe Mice
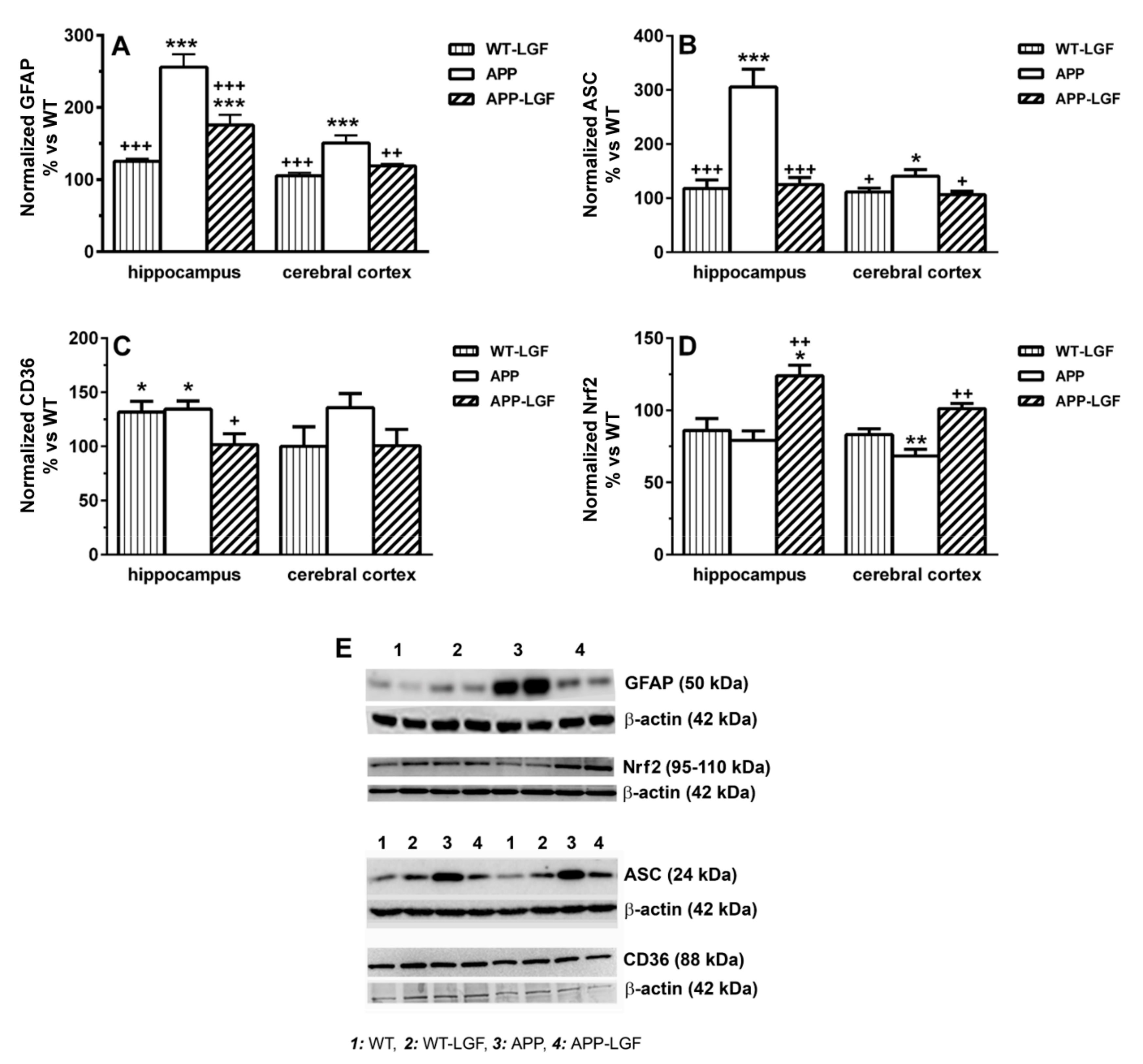
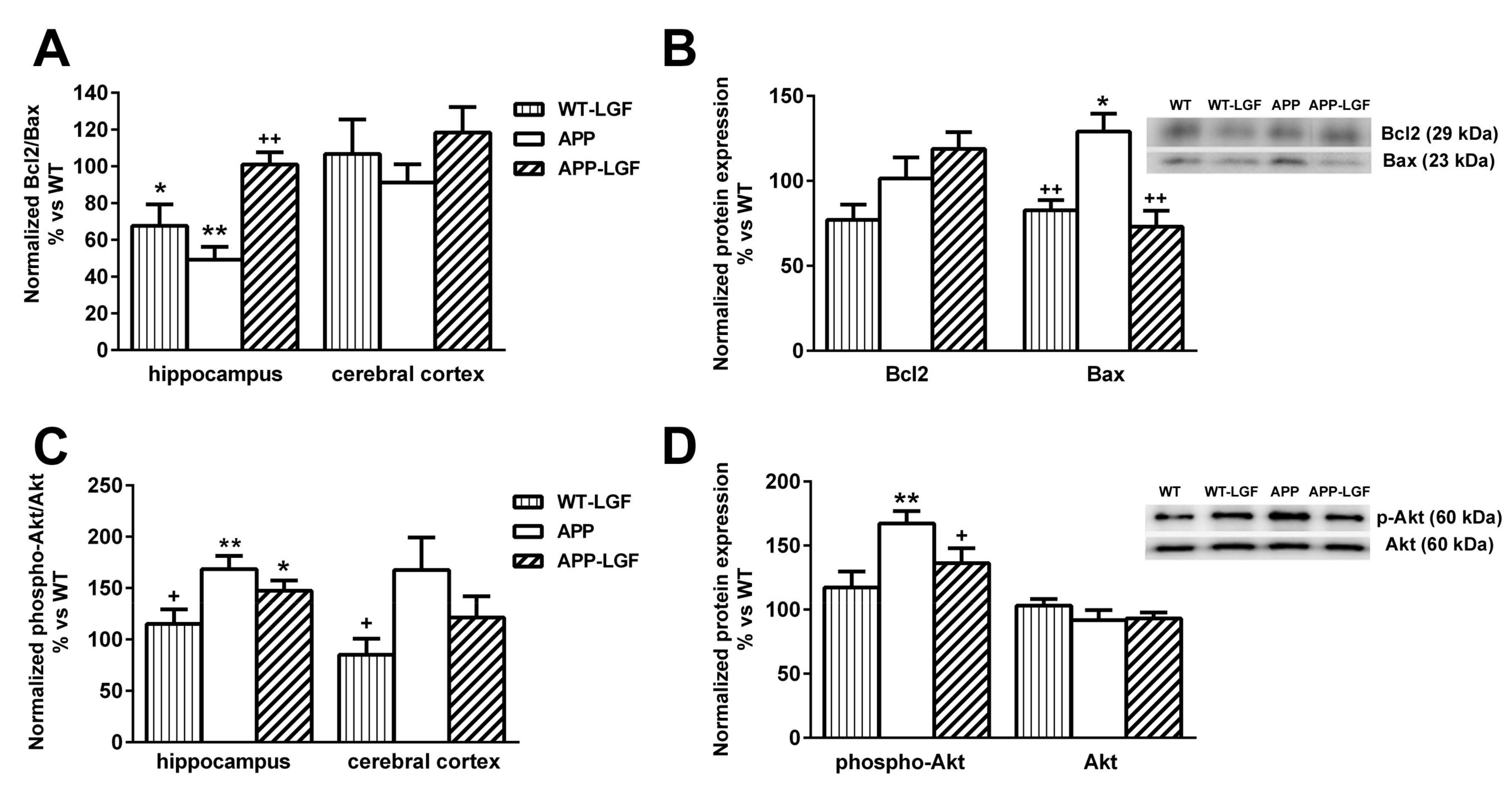
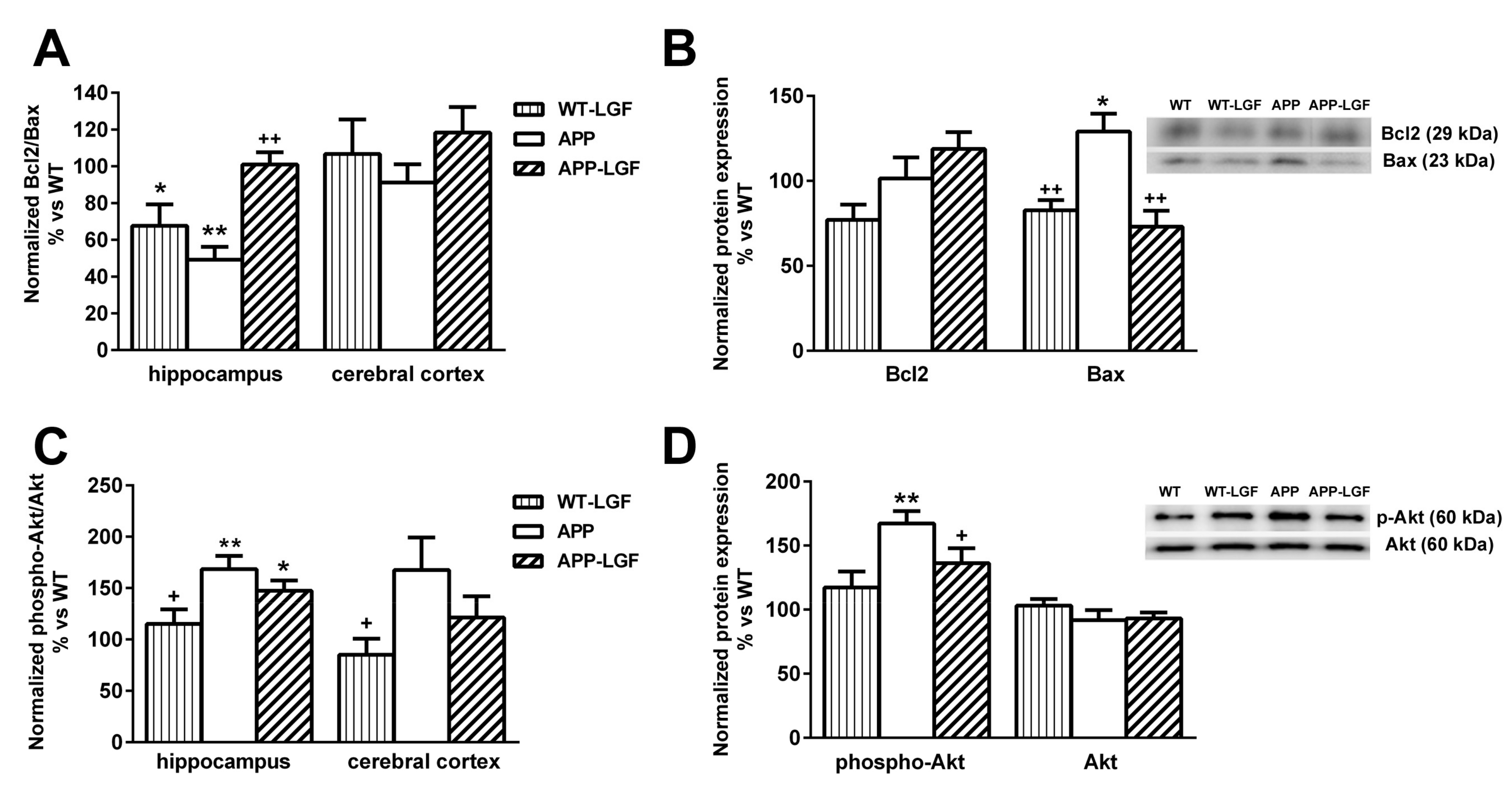
2.5. LGF Modulates the Expression of Proteins Involved in Cell Survival
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. LGF Purification
4.3. Transgenic Animals
4.4. Genotype by PCR
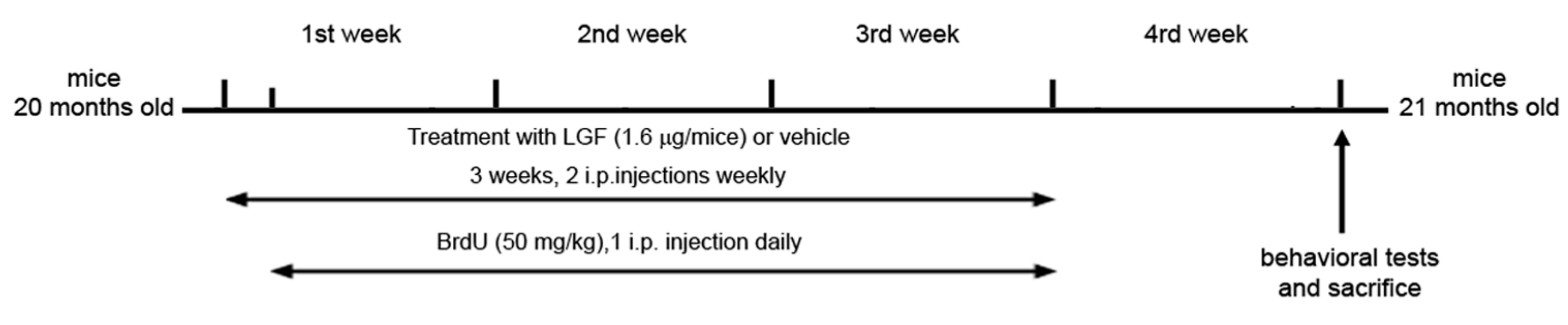
4.5. LGF and BrdU Administration
4.6. Behavioral Studies
4.6.1. Y-Maze
4.6.2. Marble Burying Test
4.7. Antibodies and Immunochemicals for Immunohistochemistry
4.8. Tissue Processing, Immunohistochemistry and Morphometric Analyses
4.9. Brain Regions and Tissue Preparation for Biochemical Analysis
4.10. Protein Analysis
4.10.1. Primary Antibodies
4.10.2. Secondary Antibodies
4.11. Statistical Analysis
5. Conclusions
6. Patents
- US 8,642,551 B2, 4 February 2014
- CE, num. 09732019.6-1456, Ref. EP-883, April 2015
- US 14/140.014, 26 May 2015
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bird, T.D. Alzheimer Disease Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA; pp. 1993–2020.
- De-Paula, V.J.; Radanovic, M.; Diniz, B.S.; Forlenza, O.V. Alzheimer’s disease. Subcell. Biochem. 2012, 65, 329–352. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Gandy, S.; DeKosky, S.T. Toward the treatment and prevention of Alzheimer’s disease: Rational strategies and recent progress. Annu. Rev. Med. 2013, 64, 367–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díaz-Gil, J.J.; Escartin, P.; Garcia-Canero, R.; Trilla, C.; Veloso, J.J.; Sanchez, G.; Moreno-Caparros, A.; Enrique de Salamanca, C.; Lozano, R.; Gavilanes, J.G.; et al. Purification of a liver DNA-synthesis promoter from plasma of partially hepatectomized rats. Biochem. J. 1986, 235, 49–55. [Google Scholar] [CrossRef]
- Gonzalo-Gobernado, R.; Calatrava-Ferreras, L.; Perucho, J.; Reimers, D.; Casarejos, M.J.; Herranz, A.S.; Jimenez-Escrig, A.; Diaz-Gil, J.J.; Bazan, E. Liver growth factor as a tissue regenerating factor in neurodegenerative diseases. Recent Pat. CNS Drug Discov. 2014, 9, 173–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalo-Gobernado, R.; Reimers, D.; Herranz, A.S.; Diaz-Gil, J.J.; Osuna, C.; Asensio, M.J.; Baena, S.; Rodriguez-Serrano, M.; Bazan, E. Mobilization of neural stem cells and generation of new neurons in 6-OHDA-lesioned rats by intracerebroventricular infusion of liver growth factor. J. Histochem. Cytochem. 2009, 57, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Reimers, D.; Herranz, A.S.; Diaz-Gil, J.J.; Lobo, M.V.; Paino, C.L.; Alonso, R.; Asensio, M.J.; Gonzalo-Gobernado, R.; Bazan, E. Intrastriatal infusion of liver growth factor stimulates dopamine terminal sprouting and partially restores motor function in 6-hydroxydopamine-lesioned rats. J. Histochem. Cytochem. 2006, 54, 457–465. [Google Scholar] [CrossRef]
- Gonzalo-Gobernado, R.; Calatrava-Ferreras, L.; Reimers, D.; Herranz, A.S.; Rodriguez-Serrano, M.; Miranda, C.; Jimenez-Escrig, A.; Diaz-Gil, J.J.; Bazan, E. Neuroprotective activity of peripherally administered liver growth factor in a rat model of Parkinson’s disease. PLoS ONE 2013, 8, e67771. [Google Scholar] [CrossRef] [Green Version]
- Reimers, D.; Osuna, C.; Gonzalo-Gobernado, R.; Herranz, A.S.; Diaz-Gil, J.J.; Jimenez-Escrig, A.; Asensio, M.J.; Miranda, C.; Rodriguez-Serrano, M.; Bazan, E. Liver growth factor promotes the survival of grafted neural stem cells in a rat model of Parkinson’s disease. Curr. Stem. Cell Res. Ther. 2012, 7, 15–25. [Google Scholar] [CrossRef]
- Díaz-Gil, J.J.; Garcia-Monzon, C.; Rua, C.; Martin-Sanz, P.; Cereceda, R.M.; Miquilena-Colina, M.E.; Machin, C.; Fernandez-Martinez, A.; Garcia-Canero, R. The anti-fibrotic effect of liver growth factor is associated with decreased intrahepatic levels of matrix metalloproteinases 2 and 9 and transforming growth factor beta 1 in bile duct-ligated rats. Histol. Histopathol. 2008, 23, 583–591. [Google Scholar] [CrossRef]
- Díaz-Gil, J.J.; Munoz, J.; Albillos, A.; Rua, C.; Machin, C.; Garcia-Canero, R.; Cereceda, R.M.; Guijarro, M.C.; Trilla, C.; Escartin, P. Improvement in liver fibrosis, functionality and hemodynamics in CCI4-cirrhotic rats after injection of the Liver Growth Factor. J. Hepatol. 1999, 30, 1065–1072. [Google Scholar] [CrossRef]
- Martinez-Galan, L.; del Puerto-Nevado, L.; Perez-Rial, S.; Diaz-Gil, J.J.; Gonzalez-Mangado, N.; Peces-Barba, G. Liver growth factor improves pulmonary fibrosis secondary to cadmium administration in mice. Arch. Bronconeumol. 2010, 46, 20–26. [Google Scholar] [CrossRef]
- Calatrava-Ferreras, L.; Gonzalo-Gobernado, R.; Reimers, D.; Herranz, A.S.; Casarejos, M.J.; Jimenez-Escrig, A.; Regadera, J.; Velasco-Martin, J.; Vallejo-Munoz, M.; Diaz-Gil, J.J.; et al. Liver Growth Factor (LGF) Upregulates Frataxin Protein Expression and Reduces Oxidative Stress in Friedreich’s Ataxia Transgenic Mice. Int. J. Mol. Sci. 2016, 17, 2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calatrava-Ferreras, L.; Gonzalo-Gobernado, R.; Reimers, D.; Herranz, A.S.; Jimenez-Escrig, A.; Diaz-Gil, J.J.; Casarejos, M.J.; Montero-Vega, M.T.; Bazan, E. Neuroprotective Role of Liver Growth Factor “LGF” in an Experimental Model of Cerebellar Ataxia. Int. J. Mol. Sci. 2014, 15, 19056–19073. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; O’Banion, M.K.; Terwel, D.; Kummer, M.P. Neuroinflammatory processes in Alzheimer’s disease. J. Neural. Transm. (Vienna) 2010, 117, 919–947. [Google Scholar] [CrossRef]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef]
- Lee, Y.W.; Kim, D.H.; Jeon, S.J.; Park, S.J.; Kim, J.M.; Jung, J.M.; Lee, H.E.; Bae, S.G.; Oh, H.K.; Son, K.H.; et al. Neuroprotective effects of salvianolic acid B on an Abeta25-35 peptide-induced mouse model of Alzheimer’s disease. Eur. J. Pharmacol. 2013, 704, 70–77. [Google Scholar] [CrossRef]
- Gonzalo-Gobernado, R.; Reimers, D.; Casarejos, M.J.; Calatrava Ferreras, L.; Vallejo-Munoz, M.; Jimenez-Escrig, A.; Diaz-Gil, J.J.; Ulzurrun de Asanza, G.M.; Bazan, E. Liver Growth Factor Induces Glia-Associated Neuroprotection in an in Vitro Model of Parkinson’s Disease. Brain Sci. 2020, 10, 315. [Google Scholar] [CrossRef]
- Arbor, S.C.; LaFontaine, M.; Cumbay, M. Amyloid-beta Alzheimer targets—Protein processing, lipid rafts, and amyloid-beta pores. Yale J. Biol. Med. 2016, 89, 5–21. [Google Scholar] [PubMed]
- Butterfield, D.A.; Swomley, A.M.; Sultana, R. Amyloid beta-peptide (1-42)-induced oxidative stress in Alzheimer disease: Importance in disease pathogenesis and progression. Antioxid. Redox Signal. 2013, 19, 823–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, R.C.; Tan, M.S.; Wang, H.; Xie, A.M.; Yu, J.T.; Tan, L. Heat shock protein 70 in Alzheimer’s disease. Biomed. Res. Int. 2014, 2014, 435203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, T.; Murao, N.; Namba, T.; Takehara, M.; Adachi, H.; Katsuno, M.; Sobue, G.; Matsushima, T.; Suzuki, T.; Mizushima, T. Suppression of Alzheimer’s disease-related phenotypes by expression of heat shock protein 70 in mice. J. Neurosci. 2011, 31, 5225–5234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surra, J.C.; Guillen, N.; Barranquero, C.; Arbones-Mainar, J.M.; Navarro, M.A.; Gascon, S.; Arnal, C.; Godino, J.; Guzman, M.A.; Diaz-Gil, J.J.; et al. Sex-dependent effect of liver growth factor on atherosclerotic lesions and fatty liver disease in apolipoprotein E knockout mice. Histol. Histopathol. 2010, 25, 609–618. [Google Scholar] [PubMed]
- Avila, J. Alzheimer disease: Caspases first. Nat. Rev. Neurol. 2010, 6, 587–588. [Google Scholar] [CrossRef]
- Hernandez, F.; Avila, J. Tauopathies. Cell Mol. Life Sci. 2007, 64, 2219–2233. [Google Scholar] [CrossRef] [PubMed]
- Dou, F.; Netzer, W.J.; Tanemura, K.; Li, F.; Hartl, F.U.; Takashima, A.; Gouras, G.K.; Greengard, P.; Xu, H. Chaperones increase association of tau protein with microtubules. Proc. Natl. Acad. Sci. USA 2003, 100, 721–726. [Google Scholar] [CrossRef] [Green Version]
- Miyata, Y.; Koren, J.; Kiray, J.; Dickey, C.A.; Gestwicki, J.E. Molecular chaperones and regulation of tau quality control: Strategies for drug discovery in tauopathies. Future Med. Chem. 2011, 3, 1523–1537. [Google Scholar] [CrossRef] [Green Version]
- Dossi, E.; Vasile, F.; Rouach, N. Human astrocytes in the diseased brain. Brain Res. Bull. 2018, 136, 139–156. [Google Scholar] [CrossRef]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef]
- Taipa, R.; Ferreira, V.; Brochado, P.; Robinson, A.; Reis, I.; Marques, F.; Mann, D.M.; Melo-Pires, M.; Sousa, N. Inflammatory pathology markers (activated microglia and reactive astrocytes) in early and late onset Alzheimer disease: A post mortem study. Neuropathol. Appl. Neurobiol. 2018, 44, 298–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Bernhardi, R.; Eugenin-von Bernhardi, L.; Eugenin, J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015, 7, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakimura, J.; Kitamura, Y.; Takata, K.; Umeki, M.; Suzuki, S.; Shibagaki, K.; Taniguchi, T.; Nomura, Y.; Gebicke-Haerter, P.J.; Smith, M.A.; et al. Microglial activation and amyloid-beta clearance induced by exogenous heat-shock proteins. FASEB J. 2002, 16, 601–603. [Google Scholar] [CrossRef] [PubMed]
- Von Bernhardi, R.; Ramirez, G. Microglia-astrocyte interaction in Alzheimer’s disease: Friends or foes for the nervous system? Biol. Res. 2001, 34, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Franklin, B.S.; Bossaller, L.; de Nardo, D.; Ratter, J.M.; Stutz, A.; Engels, G.; Brenker, C.; Nordhoff, M.; Mirandola, S.R.; Al-Amoudi, A.; et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat. Immunol. 2014, 15, 727–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friker, L.L.; Scheiblich, H.; Hochheiser, I.V.; Brinkschulte, R.; Riedel, D.; Latz, E.; Geyer, M.; Heneka, M.T. beta-Amyloid Clustering around ASC Fibrils Boosts Its Toxicity in Microglia. Cell Rep. 2020, 30, 3743–3754. [Google Scholar] [CrossRef]
- Scott, X.O.; Stephens, M.E.; Desir, M.C.; Dietrich, W.D.; Keane, R.W.; de Rivero Vaccari, J.P. The Inflammasome Adaptor Protein ASC in Mild Cognitive Impairment and Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 4674. [Google Scholar] [CrossRef]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef]
- Couturier, J.; Stancu, I.C.; Schakman, O.; Pierrot, N.; Huaux, F.; Kienlen-Campard, P.; Dewachter, I.; Octave, J.N. Activation of phagocytic activity in astrocytes by reduced expression of the inflammasome component ASC and its implication in a mouse model of Alzheimer disease. J. Neuroinflamm. 2016, 13, 20. [Google Scholar] [CrossRef] [Green Version]
- Doens, D.; Fernandez, P.L. Microglia receptors and their implications in the response to amyloid beta for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2014, 11, 48. [Google Scholar] [CrossRef] [Green Version]
- Martin, E.; Boucher, C.; Fontaine, B.; Delarasse, C. Distinct inflammatory phenotypes of microglia and monocyte-derived macrophages in Alzheimer’s disease models: Effects of aging and amyloid pathology. Aging Cell 2017, 16, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Coraci, I.S.; Husemann, J.; Berman, J.W.; Hulette, C.; Dufour, J.H.; Campanella, G.K.; Luster, A.D.; Silverstein, S.C.; El-Khoury, J.B. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer’s disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am. J. Pathol. 2002, 160, 101–112. [Google Scholar] [CrossRef]
- Ricciarelli, R.; D’Abramo, C.; Zingg, J.M.; Giliberto, L.; Markesbery, W.; Azzi, A.; Marinari, U.M.; Pronzato, M.A.; Tabaton, M. CD36 overexpression in human brain correlates with beta-amyloid deposition but not with Alzheimer’s disease. Free Radic. Biol. Med. 2004, 36, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, K.; Boyd, J.D.; Glicksman, M.; Moore, K.J.; El Khoury, J. A high content drug screen identifies ursolic acid as an inhibitor of amyloid beta protein interactions with its receptor CD36. J. Biol. Chem. 2011, 286, 34914–34922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conde, J.R.; Streit, W.J. Microglia in the aging brain. J. Neuropathol. Exp. Neurol. 2006, 65, 199–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosher, K.I.; Wyss-Coray, T. Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Suzuki, T.; Muramatsu, A.; Saito, R.; Iso, T.; Shibata, T.; Kuwata, K.; Kawaguchi, S.I.; Iwawaki, T.; Adachi, S.; Suda, H.; et al. Molecular Mechanism of Cellular Oxidative Stress Sensing by Keap1. Cell Rep. 2019, 28, 746–758. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Mochizuki, M.; Ishii, Y.; Ishii, T.; Shibata, T.; Kawamoto, Y.; Kelly, V.; Sekizawa, K.; Uchida, K.; Yamamoto, M. Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-deoxy-Delta(12,14)-prostaglandin j(2). Mol. Cell Biol. 2004, 24, 36–45. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef]
- Kerr, F.; Sofola-Adesakin, O.; Ivanov, D.K.; Gatliff, J.; Gomez Perez-Nievas, B.; Bertrand, H.C.; Martinez, P.; Callard, R.; Snoeren, I.; Cocheme, H.M.; et al. Direct Keap1-Nrf2 disruption as a potential therapeutic target for Alzheimer’s disease. PLoS Genet 2017, 13, e1006593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youssef, P.; Chami, B.; Lim, J.; Middleton, T.; Sutherland, G.T.; Witting, P.K. Evidence supporting oxidative stress in a moderately affected area of the brain in Alzheimer’s disease. Sci. Rep. 2018, 8, 11553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, G.; Gan, K.A.; Johnson, D.A.; Johnson, J.A. Increased Alzheimer’s disease-like pathology in the APP/PS1DeltaE9 mouse model lacking Nrf2 through modulation of autophagy. Neurobiol. Aging. 2015, 36, 664–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojo, A.I.; Pajares, M.; Garcia-Yague, A.J.; Buendia, I.; van Leuven, F.; Yamamoto, M.; Lopez, M.G.; Cuadrado, A. Deficiency in the transcription factor NRF2 worsens inflammatory parameters in a mouse model with combined tauopathy and amyloidopathy. Redox. Biol. 2018, 18, 173–180. [Google Scholar] [CrossRef]
- Uruno, A.; Matsumaru, D.; Ryoke, R.; Saito, R.; Kadoguchi, S.; Saigusa, D.; Saito, T.; Saido, T.C.; Kawashima, R.; Yamamoto, M. Nrf2 Suppresses Oxidative Stress and Inflammation in App Knock-In Alzheimer’s Disease Model Mice. Mol. Cell Biol. 2020, 40. [Google Scholar] [CrossRef]
- Wang, C.Y.; Wang, Z.Y.; Xie, J.W.; Cai, J.H.; Wang, T.; Xu, Y.; Wang, X.; An, L. CD36 upregulation mediated by intranasal LV-NRF2 treatment mitigates hypoxia-induced progression of Alzheimer’s-like pathogenesis. Antioxid. Redox Signal. 2014, 21, 2208–2230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giron-Martinez, A.; Perez-Rial, S.; Terron-Exposito, R.; Diaz-Gil, J.J.; Gonzalez-Mangado, N.; Peces-Barba, G. Proliferative Activity of Liver Growth Factor is Associated with an Improvement of Cigarette Smoke-Induced Emphysema in Mice. PLoS ONE 2014, 9, e112995. [Google Scholar] [CrossRef] [Green Version]
- Frebel, K.; Wiese, S. Signalling molecules essential for neuronal survival and differentiation. Biochem. Soc. Trans. 2006, 34, 1287–1290. [Google Scholar] [CrossRef] [Green Version]
- Eckert, A.; Marques, C.A.; Keil, U.; Schussel, K.; Muller, W.E. Increased apoptotic cell death in sporadic and genetic Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2003, 1010, 604–609. [Google Scholar] [CrossRef]
- Feng, Z.; Qin, C.; Chang, Y.; Zhang, J.T. Early melatonin supplementation alleviates oxidative stress in a transgenic mouse model of Alzheimer’s disease. Free Radic. Biol. Med. 2006, 40, 101–109. [Google Scholar] [CrossRef]
- Yan, Y.; Gong, K.; Ma, T.; Zhang, L.; Zhao, N.; Zhang, X.; Tang, P.; Gong, Y. Protective effect of edaravone against Alzheimer’s disease-relevant insults in neuroblastoma N2a cells. Neurosci. Lett. 2012, 531, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Obulesu, M.; Lakshmi, M.J. Apoptosis in Alzheimer’s disease: An understanding of the physiology, pathology and therapeutic avenues. Neurochem. Res. 2014, 39, 2301–2312. [Google Scholar] [CrossRef] [PubMed]
- King, D.L.; Arendash, G.W. Behavioral characterization of the Tg2576 transgenic model of Alzheimer’s disease through 19 months. Physiol. Behav. 2002, 75, 627–642. [Google Scholar] [CrossRef]
- Takeuchi, A.; Irizarry, M.C.; Duff, K.; Saido, T.C.; Hsiao Ashe, K.; Hasegawa, M.; Mann, D.M.; Hyman, B.T.; Iwatsubo, T. Age-related amyloid beta deposition in transgenic mice overexpressing both Alzheimer mutant presenilin 1 and amyloid beta precursor protein Swedish mutant is not associated with global neuronal loss. Am. J. Pathol. 2000, 157, 331–339. [Google Scholar] [CrossRef]
- Noshita, N.; Lewen, A.; Sugawara, T.; Chan, P.H. Evidence of phosphorylation of Akt and neuronal survival after transient focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 2001, 21, 1442–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Sapolsky, R.M.; Steinberg, G.K. Phosphoinositide-3-kinase/akt survival signal pathways are implicated in neuronal survival after stroke. Mol. Neurobiol. 2006, 34, 249–270. [Google Scholar] [CrossRef]
- Matsuda, S.; Nakagawa, Y.; Tsuji, A.; Kitagishi, Y.; Nakanishi, A.; Murai, T. Implications of PI3K/AKT/PTEN Signaling on Superoxide Dismutases Expression and in the Pathogenesis of Alzheimer’s Disease. Diseases 2018, 6, 28. [Google Scholar] [CrossRef] [Green Version]
- Chong, Z.Z.; Li, F.; Maiese, K. Activating Akt and the brain’s resources to drive cellular survival and prevent inflammatory injury. Histol. Histopathol. 2005, 20, 299–315. [Google Scholar] [CrossRef]
- Gardai, S.J.; Hildeman, D.A.; Frankel, S.K.; Whitlock, B.B.; Frasch, S.C.; Borregaard, N.; Marrack, P.; Bratton, D.L.; Henson, P.M. Phosphorylation of Bax Ser184 by Akt regulates its activity and apoptosis in neutrophils. J. Biol. Chem. 2004, 279, 21085–21095. [Google Scholar] [CrossRef] [Green Version]
- Sagare, A.P.; Winkler, E.A.; Bell, R.D.; Deane, R.; Zlokovic, B.V. From the liver to the blood-brain barrier: An interconnected system regulating brain amyloid-beta levels. J. Neurosci. Res. 2011, 89, 967–968. [Google Scholar] [CrossRef]
- Thornalley, P.J. Cell activation by glycated proteins. AGE receptors, receptor recognition factors and functional classification of AGEs. Cell Mol. Biol. (Noisy-Le-Grand) 1998, 44, 1013–1023. [Google Scholar] [PubMed]
- Park, I.H.; Yeon, S.I.; Youn, J.H.; Choi, J.E.; Sasaki, N.; Choi, I.H.; Shin, J.S. Expression of a novel secreted splice variant of the receptor for advanced glycation end products (RAGE) in human brain astrocytes and peripheral blood mononuclear cells. Mol. Immunol. 2004, 40, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Ameen-Ali, K.E.; Wharton, S.B.; Simpson, J.E.; Heath, P.R.; Sharp, P.; Berwick, J. Review: Neuropathology and behavioural features of transgenic murine models of Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2017, 43, 553–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deacon, R.M. Digging and marble burying in mice: Simple methods for in vivo identification of biological impacts. Nat. Protoc. 2006, 1, 122–124. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.K.; Han, H.E.; Kim, H.; Lee, J.E.; Choi, D.; Park, W.J.; Han, P.L. Expression of the plant viral protease NIa in the brain of a mouse model of Alzheimer’s disease mitigates Abeta pathology and improves cognitive function. Exp. Mol. Med. 2012, 44, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Gil, J.J.; Rua, C.; Machin, C.; Cereceda, R.M.; Garcia-Canero, R.; de Foronda, M.; Perez de Diego, J.; Trilla, C.; Escartin, P. Hepatic growth induced by injection of the liver growth factor into normal rats. Growth Regul. 1994, 4, 113–122. [Google Scholar] [PubMed]
- Singh, J.; Bowers, L.D. Quantitative fractionation of serum bilirubin species by reversed-phase high-performance liquid chromatography. J. Chromatogr. 1986, 380, 321–330. [Google Scholar] [CrossRef]
- Díaz-Gil, J.J.; Sanchez, G.; Trilla, C.; Escartin, P. Identification of biliprotein as a liver growth factor. Hepatology 1988, 8, 484–486. [Google Scholar] [CrossRef]
- Carro, E.; Trejo, J.L.; Gomez-Isla, T.; LeRoith, D.; Torres-Aleman, I. Serum insulin-like growth factor I regulates brain amyloid-beta levels. Nat. Med. 2002, 8, 1390–1397. [Google Scholar] [CrossRef]
- Perucho, J.; Casarejos, M.J.; Gomez, A.; Solano, R.M.; de Yebenes, J.G.; Mena, M.A. Trehalose protects from aggravation of amyloid pathology induced by isoflurane anesthesia in APP(swe) mutant mice. Curr. Alzheimer Res. 2012, 9, 334–343. [Google Scholar] [CrossRef]
- Perucho, J.; Casarejos, M.J.; Rubio, I.; Rodriguez-Navarro, J.A.; Gomez, A.; Ampuero, I.; Rodal, I.; Solano, R.M.; Carro, E.; Garcia de Yebenes, J.; et al. The effects of parkin suppression on the behaviour, amyloid processing, and cell survival in APP mutant transgenic mice. Exp. Neurol. 2010, 221, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Casarejos, M.J.; Perucho, J.; Gomez, A.; Munoz, M.P.; Fernandez-Estevez, M.; Sagredo, O.; Fernandez Ruiz, J.; Guzman, M.; de Yebenes, J.G.; Mena, M.A. Natural cannabinoids improve dopamine neurotransmission and tau and amyloid pathology in a mouse model of tauopathy. J. Alzheimers Dis. 2013, 35, 525–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin, K.; Paxinos, G. The Mouse Brain in Stereotaxic Coordinates, 3rd ed.; Academic Press: San Diego, CA, USA, 2007. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalo-Gobernado, R.; Perucho, J.; Vallejo-Muñoz, M.; Casarejos, M.J.; Reimers, D.; Jiménez-Escrig, A.; Gómez, A.; Ulzurrun de Asanza, G.M.; Bazán, E. Liver Growth Factor “LGF” as a Therapeutic Agent for Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 9201. https://doi.org/10.3390/ijms21239201
Gonzalo-Gobernado R, Perucho J, Vallejo-Muñoz M, Casarejos MJ, Reimers D, Jiménez-Escrig A, Gómez A, Ulzurrun de Asanza GM, Bazán E. Liver Growth Factor “LGF” as a Therapeutic Agent for Alzheimer’s Disease. International Journal of Molecular Sciences. 2020; 21(23):9201. https://doi.org/10.3390/ijms21239201
Chicago/Turabian StyleGonzalo-Gobernado, Rafael, Juan Perucho, Manuela Vallejo-Muñoz, Maria José Casarejos, Diana Reimers, Adriano Jiménez-Escrig, Ana Gómez, Gonzalo M. Ulzurrun de Asanza, and Eulalia Bazán. 2020. "Liver Growth Factor “LGF” as a Therapeutic Agent for Alzheimer’s Disease" International Journal of Molecular Sciences 21, no. 23: 9201. https://doi.org/10.3390/ijms21239201