HSP70-Homolog DnaK of Pseudomonas aeruginosa Increases the Production of IL-27 through Expression of EBI3 via TLR4-Dependent NF-κB and TLR4-Independent Akt Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
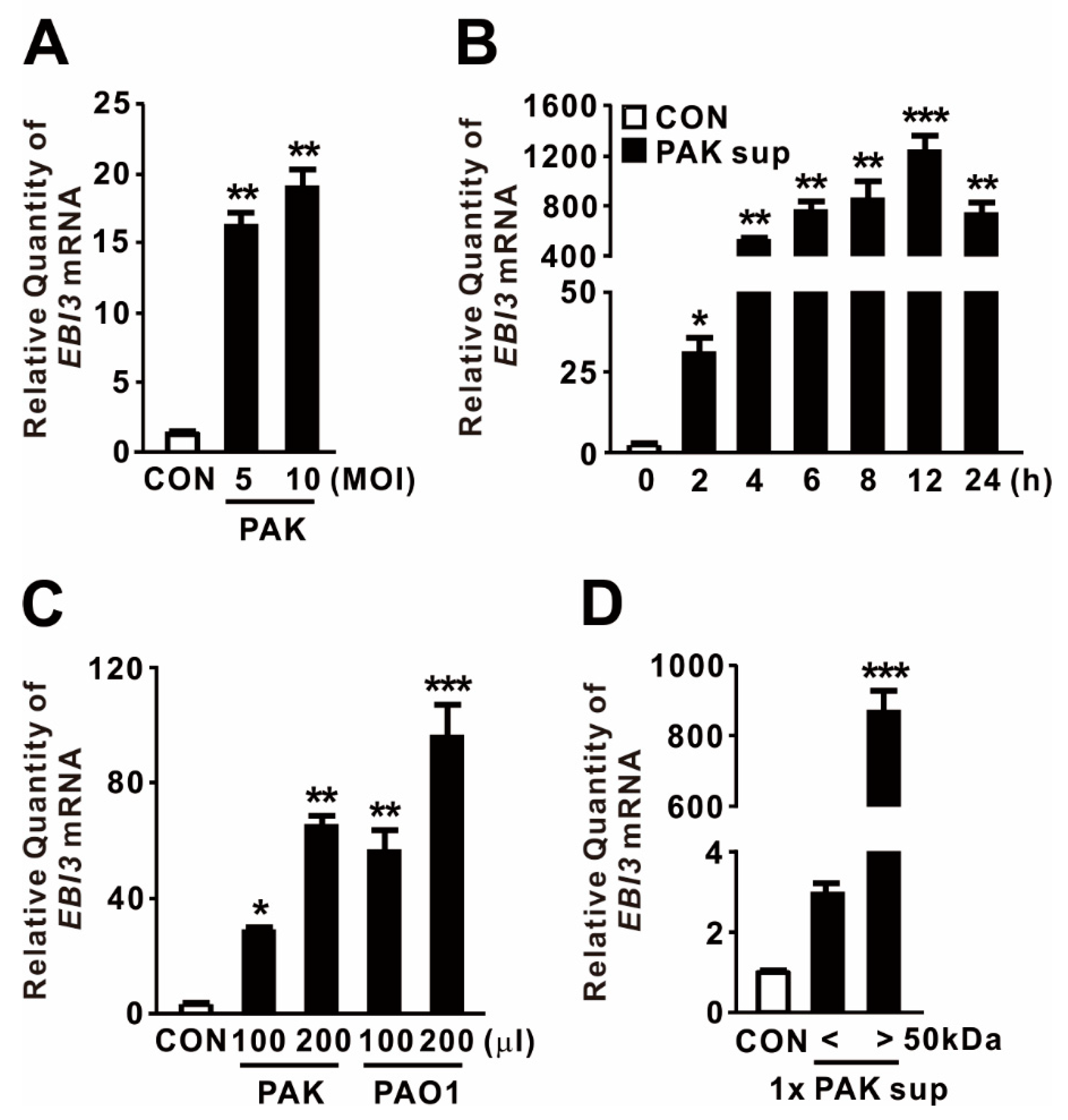
2.1. P. aeruginosa-Mediated Induction of EBI3 Expression
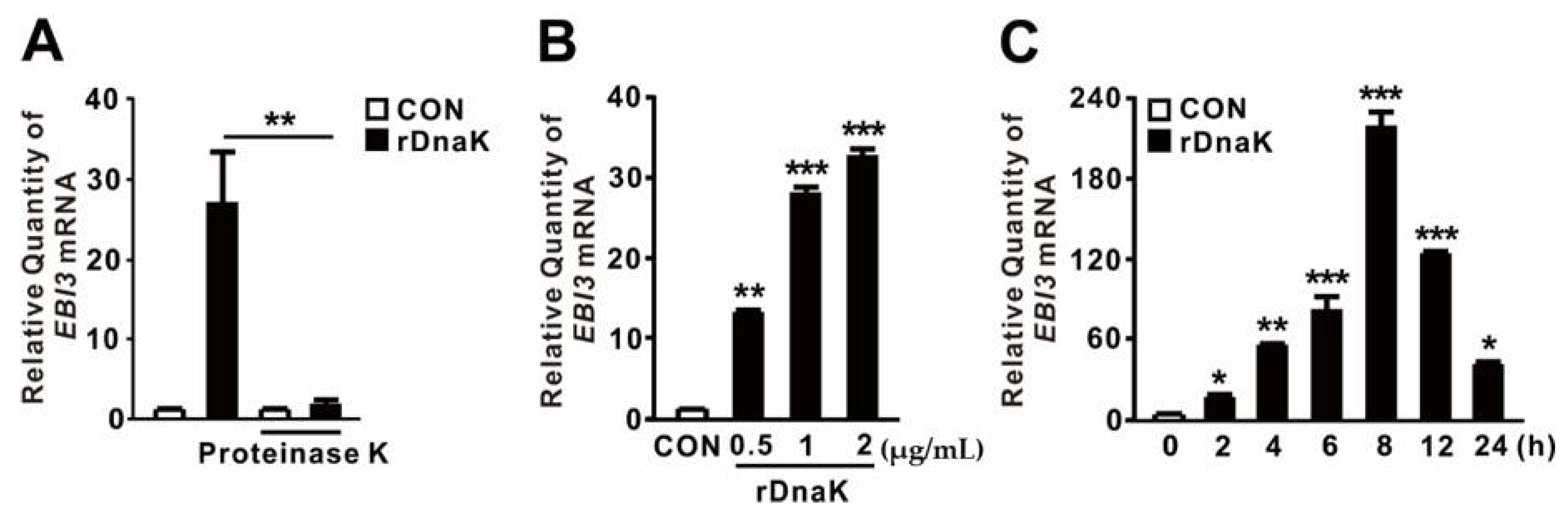
2.2. P. aeruginosa DnaK Induces Expression of EBI3
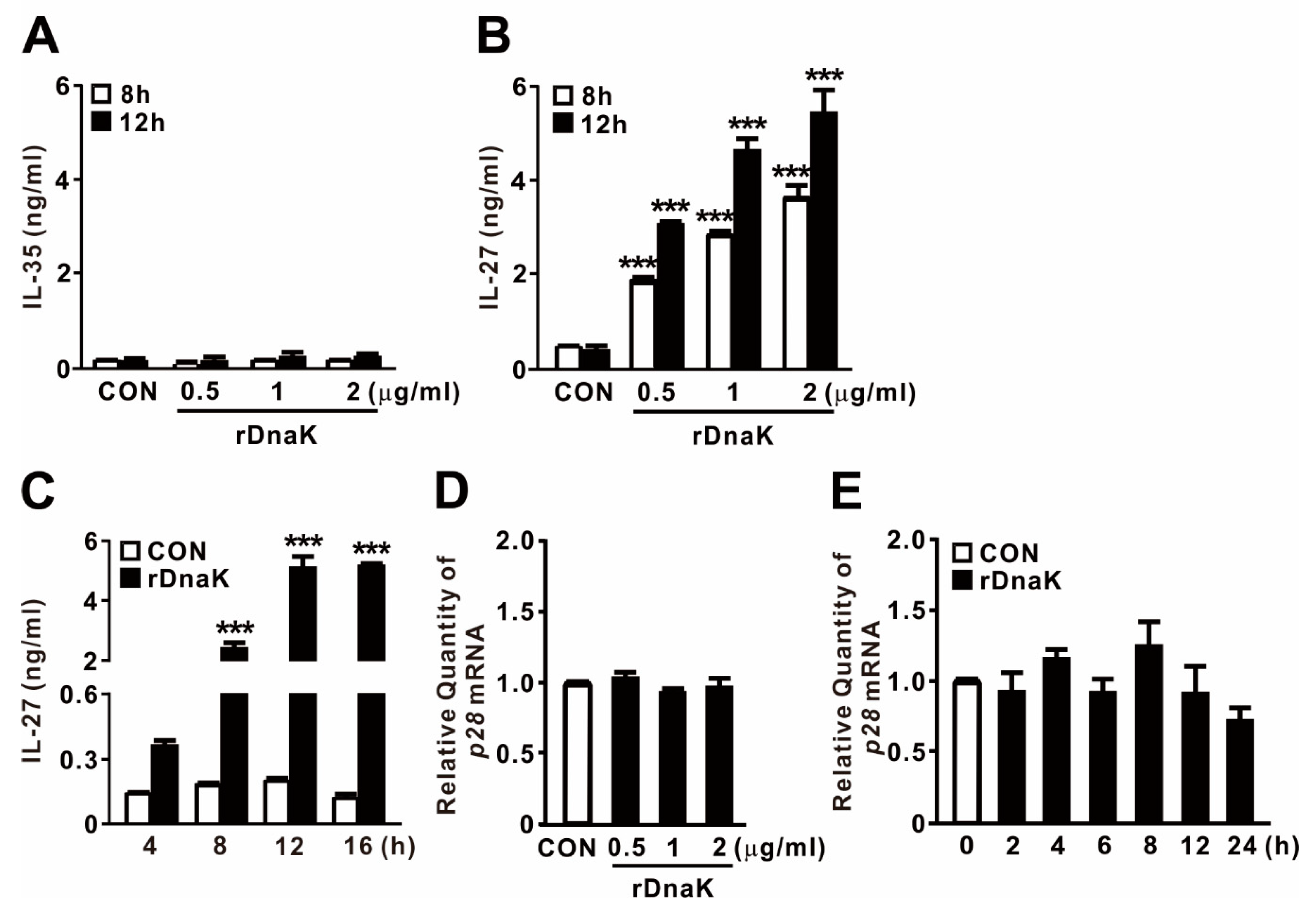
2.3. DnaK-Induced EBI3 Is Involved in the Formation of IL-27
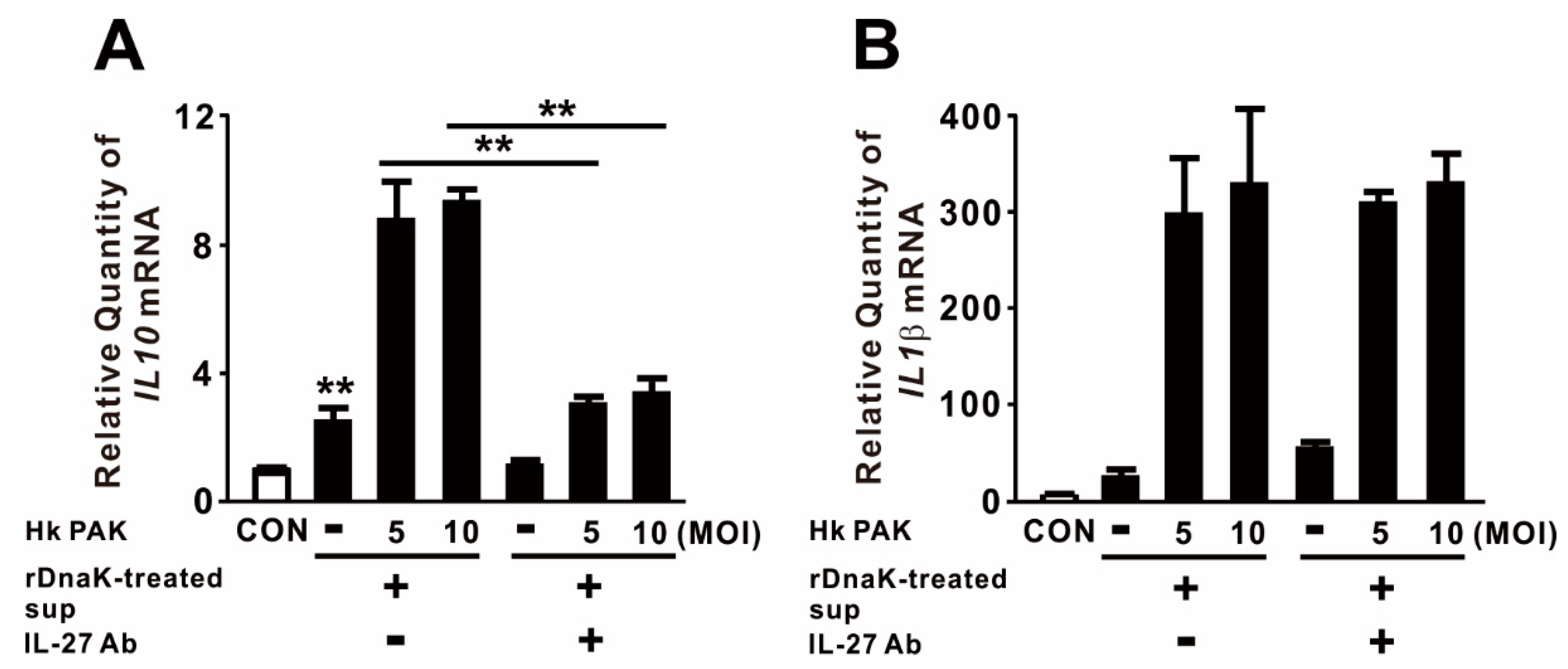
2.4. IL-27 Induced by DnaK Promotes Expression of IL10
2.5. DnaK-Induced Expression of EBI3 Is under the Control of the NF-κB and Akt Signaling Pathways
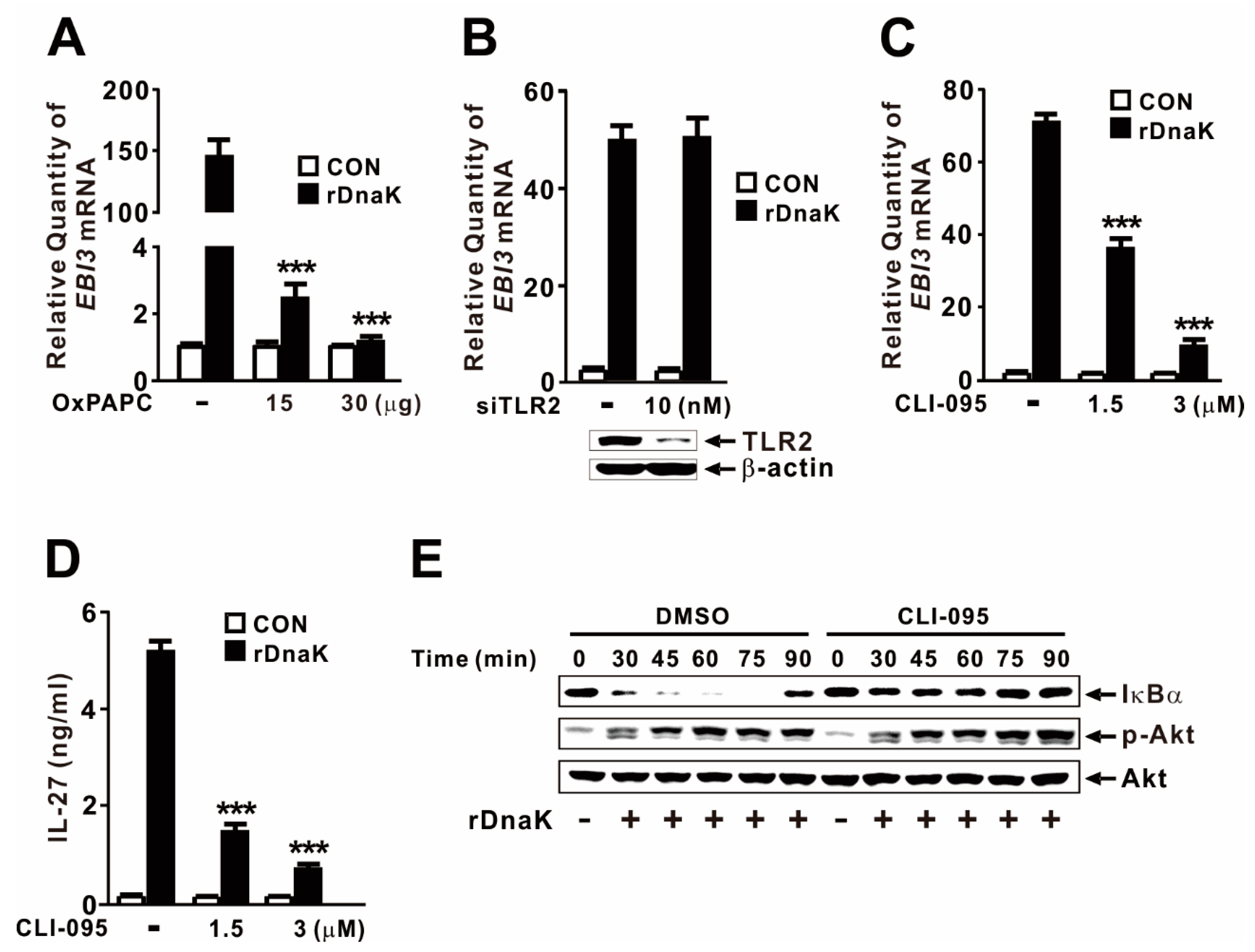
2.6. DnaK-Mediated Induction of EBI3 Expression Is Partly under the Control of TLR4
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Culture Conditions
4.2. Cell Culture
4.3. Construction and Purification of rDnaK
4.4. Quantitative Real-Time PCR (qRT-PCR)
4.5. Immunoblotting Analysis
4.6. Enzyme-Linked Immunosorbent Assay (ELISA)
4.7. Transfection of siRNA
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lyczak, J.B.; Cannon, C.L.; Pier, G.B. Establishment of Pseudomonas aeruginosa infection: Lessons from a versatile opportunist. Microbes Infect. 2000, 2, 1051–1060. [Google Scholar] [CrossRef]
- Bassetti, M.; Vena, A.; Croxatto, A.; Righi, E.; Guery, B. How to manage Pseudomonas aeruginosa infections. Drugs Context 2018, 7, 212527. [Google Scholar] [CrossRef]
- Gellatly, S.L.; Hancock, R.E. Pseudomonas aeruginosa: New insights into pathogenesis and host defenses. Pathog. Dis. 2013, 67, 159–173. [Google Scholar] [CrossRef]
- Lau, G.W.; Hassett, D.J.; Britigan, B.E. Modulation of lung epithelial functions by Pseudomonas aeruginosa. Trends Microbiol. 2005, 13, 389–397. [Google Scholar] [CrossRef]
- Mehrad, B.; Standiford, T.J. Role of cytokines in pulmonary antimicrobial host defense. Immunol. Res. 1999, 20, 15–27. [Google Scholar] [CrossRef]
- Baggiolini, M. Chemokines and leukocyte traffic. Nature 1998, 392, 565–568. [Google Scholar] [CrossRef]
- Flannagan, R.S.; Cosio, G.; Grinstein, S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat. Rev. Microbiol. 2009, 7, 355–366. [Google Scholar] [CrossRef]
- Diacovich, L.; Gorvel, J.P. Bacterial manipulation of innate immunity to promote infection. Nat. Rev. Microbiol. 2010, 8, 117–128. [Google Scholar] [CrossRef]
- Freitas do Rosario, A.P.; Lamb, T.; Spence, P.; Stephens, R.; Lang, A.; Roers, A.; Muller, W.; O’Garra, A.; Langhorne, J. IL-27 promotes IL-10 production by effector Th1 CD4+ T cells: A critical mechanism for protection from severe immunopathology during malaria infection. J. Immunol. 2012, 188, 1178–1190. [Google Scholar] [CrossRef]
- Bosmann, M.; Ward, P.A. Modulation of inflammation by interleukin-27. J. Leukoc. Biol. 2013, 94, 1159–1165. [Google Scholar] [CrossRef]
- Yoshida, H.; Hunter, C.A. The immunobiology of interleukin-27. Annu. Rev. Immunol. 2015, 33, 417–443. [Google Scholar] [CrossRef]
- Iyer, S.S.; Ghaffari, A.A.; Cheng, G. Lipopolysaccharide-mediated IL-10 transcriptional regulation requires sequential induction of type I IFNs and IL-27 in macrophages. J. Immunol. 2010, 185, 6599–6607. [Google Scholar] [CrossRef] [PubMed]
- Steinhauser, M.L.; Hogaboam, C.M.; Kunkel, S.L.; Lukacs, N.W.; Strieter, R.M.; Standiford, T.J. IL-10 is a major mediator of sepsis-induced impairment in lung antibacterial host defense. J. Immunol. 1999, 162, 392–399. [Google Scholar]
- McGuirk, P.; Mills, K.H. Direct anti-inflammatory effect of a bacterial virulence factor: IL-10-dependent suppression of IL-12 production by filamentous hemagglutinin from Bordetella pertussis. Eur. J. Immunol. 2000, 30, 415–422. [Google Scholar] [CrossRef]
- Seman, B.G.; Vance, J.K.; Rawson, T.W.; Witt, M.R.; Huckaby, A.B.; Povroznik, J.M.; Bradford, S.D.; Barbier, M.; Robinson, C.M. Elevated Levels of Interleukin-27 in Early Life Compromise Protective Immunity in a Mouse Model of Gram-Negative Neonatal Sepsis. Infect. Immun. 2020, 88, e00828-19. [Google Scholar] [CrossRef]
- Cao, J.; Xu, F.; Lin, S.; Song, Z.; Zhang, L.; Luo, P.; Xu, H.; Li, D.; Zheng, K.; Ren, G.; et al. IL-27 controls sepsis-induced impairment of lung antibacterial host defence. Thorax 2014, 69, 926–937. [Google Scholar] [CrossRef]
- He, Y.; Du, W.X.; Jiang, H.Y.; Ai, Q.; Feng, J.; Liu, Z.; Yu, J.L. Multiplex Cytokine Profiling Identifies Interleukin-27 as a Novel Biomarker For Neonatal Early Onset Sepsis. Shock 2017, 47, 140–147. [Google Scholar] [CrossRef]
- The INIS Study. International Neonatal Immunotherapy Study: Non-specific intravenous immunoglobulin therapy for suspected or proven neonatal sepsis: An international, placebo controlled, multicentre randomised trial. BMC Pregnancy Childbirth 2008, 8, 52. [Google Scholar] [CrossRef]
- Zugel, U.; Kaufmann, S.H. Role of heat shock proteins in protection from and pathogenesis of infectious diseases. Clin. Microbiol. Rev. 1999, 12, 19–39. [Google Scholar] [CrossRef]
- Srivastava, P. Roles of heat-shock proteins in innate and adaptive immunity. Nat. Rev. Immunol. 2002, 2, 185–194. [Google Scholar] [CrossRef]
- Ferrero, R.L.; Thiberge, J.M.; Kansau, I.; Wuscher, N.; Huerre, M.; Labigne, A. The GroES homolog of Helicobacter pylori confers protective immunity against mucosal infection in mice. Proc. Natl. Acad. Sci. USA 1995, 92, 6499–6503. [Google Scholar] [CrossRef] [PubMed]
- Osterloh, A.; Breloer, M. Heat shock proteins: Linking danger and pathogen recognition. Med. Microbiol. Immunol. 2008, 197, 1–8. [Google Scholar] [CrossRef]
- Silva, C.L.; Silva, M.F.; Pietro, R.C.; Lowrie, D.B. Protection against tuberculosis by passive transfer with T-cell clones recognizing mycobacterial heat-shock protein 65. Immunology 1994, 83, 341–346. [Google Scholar]
- van Eden, W.; van der Zee, R.; Prakken, B. Heat-shock proteins induce T-cell regulation of chronic inflammation. Nat. Rev. Immunol. 2005, 5, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Craig, E.A.; Gambill, B.D.; Nelson, R.J. Heat shock proteins: Molecular chaperones of protein biogenesis. Microbiol. Rev. 1993, 57, 402–414. [Google Scholar] [CrossRef]
- Houry, W.A.; Frishman, D.; Eckerskorn, C.; Lottspeich, F.; Hartl, F.U. Identification of in vivo substrates of the chaperonin GroEL. Nature 1999, 402, 147–154. [Google Scholar] [CrossRef]
- Borrero-de Acuna, J.M.; Molinari, G.; Rohde, M.; Dammeyer, T.; Wissing, J.; Jansch, L.; Arias, S.; Jahn, M.; Schobert, M.; Timmis, K.N.; et al. A Periplasmic Complex of the Nitrite Reductase NirS, the Chaperone DnaK, and the Flagellum Protein FliC Is Essential for Flagellum Assembly and Motility in Pseudomonas aeruginosa. J. Bacteriol. 2015, 197, 3066–3075. [Google Scholar] [CrossRef]
- Nouwens, A.S.; Willcox, M.D.; Walsh, B.J.; Cordwell, S.J. Proteomic comparison of membrane and extracellular proteins from invasive (PAO1) and cytotoxic (6206) strains of Pseudomonas aeruginosa. Proteomics 2002, 2, 1325–1346. [Google Scholar] [CrossRef]
- Martine, P.; Chevriaux, A.; Derangere, V.; Apetoh, L.; Garrido, C.; Ghiringhelli, F.; Rebe, C. HSP70 is a negative regulator of NLRP3 inflammasome activation. Cell Death Dis. 2019, 10, 256. [Google Scholar] [CrossRef]
- Asea, A.; Kraeft, S.K.; Kurt-Jones, E.A.; Stevenson, M.A.; Chen, L.B.; Finberg, R.W.; Koo, G.C.; Calderwood, S.K. HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat. Med. 2000, 6, 435–442. [Google Scholar] [CrossRef]
- Shevtsov, M.; Multhoff, G. Heat Shock Protein-Peptide and HSP-Based Immunotherapies for the Treatment of Cancer. Front. Immunol. 2016, 7, 171. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Jeon, J.; Bai, F.; Jin, S.; Wu, W.; Ha, U.H. The Pseudomonas aeruginosa HSP70-like protein DnaK induces IL-1beta expression via TLR4-dependent activation of the NF-kappaB and JNK signaling pathways. Comp. Immunol. Microbiol. Infect. Dis. 2019, 67, 101373. [Google Scholar] [CrossRef]
- Lee, J.H.; Jeon, J.; Bai, F.; Wu, W.; Ha, U.H. Negative regulation of interleukin 1beta expression in response to DnaK from Pseudomonas aeruginosa via the PI3K/PDK1/FoxO1 pathways. Comp. Immunol. Microbiol. Infect. Dis. 2020, 73, 101543. [Google Scholar] [CrossRef]
- Johnson, J.D.; Fleshner, M. Releasing signals, secretory pathways, and immune function of endogenous extracellular heat shock protein 72. J. Leukoc. Biol. 2006, 79, 425–434. [Google Scholar] [CrossRef]
- Rozhkova, E.; Yurinskaya, M.; Zatsepina, O.; Garbuz, D.; Karpov, V.; Surkov, S.; Murashev, A.; Ostrov, V.; Margulis, B.; Evgen’ev, M.; et al. Exogenous mammalian extracellular HSP70 reduces endotoxin manifestations at the cellular and organism levels. Ann. N. Y. Acad. Sci. 2010, 1197, 94–107. [Google Scholar] [CrossRef]
- Borges, T.J.; Wieten, L.; van Herwijnen, M.J.; Broere, F.; van der Zee, R.; Bonorino, C.; van Eden, W. The anti-inflammatory mechanisms of Hsp70. Front. Immunol. 2012, 3, 95. [Google Scholar] [CrossRef]
- Lee, C.T.; Repasky, E.A. Opposing roles for heat and heat shock proteins in macrophage functions during inflammation: A function of cell activation state? Front. Immunol. 2012, 3, 140. [Google Scholar] [CrossRef]
- Devergne, O.; Hummel, M.; Koeppen, H.; Le Beau, M.M.; Nathanson, E.C.; Kieff, E.; Birkenbach, M. A novel interleukin-12 p40-related protein induced by latent Epstein-Barr virus infection in B lymphocytes. J. Virol. 1996, 70, 1143–1153. [Google Scholar] [CrossRef]
- Ellis, R.J. The molecular chaperone concept. Semin. Cell Biol. 1990, 1, 1–9. [Google Scholar]
- Young, D.B. Chaperonins and the immune response. Semin. Cell Biol. 1990, 1, 27–35. [Google Scholar]
- Chang, W.C.; Wu, S.L.; Huang, W.C.; Hsu, J.Y.; Chan, S.H.; Wang, J.M.; Tsai, J.P.; Chen, B.K. PTX3 gene activation in EGF-induced head and neck cancer cell metastasis. Oncotarget 2015, 6, 7741–7757. [Google Scholar] [CrossRef]
- Asea, A.; Rehli, M.; Kabingu, E.; Boch, J.A.; Bare, O.; Auron, P.E.; Stevenson, M.A.; Calderwood, S.K. Novel signal transduction pathway utilized by extracellular HSP70: Role of toll-like receptor (TLR) 2 and TLR4. J. Biol. Chem. 2002, 277, 15028–15034. [Google Scholar] [CrossRef]
- Zhe, Y.; Li, Y.; Liu, D.; Su, D.M.; Liu, J.G.; Li, H.Y. Extracellular HSP70-peptide complexes promote the proliferation of hepatocellular carcinoma cells via TLR2/4/JNK1/2MAPK pathway. Tumour Biol. 2016, 37, 13951–13959. [Google Scholar] [CrossRef]
- Shin, H.; Jeon, J.; Lee, J.H.; Jin, S.; Ha, U.H. Pseudomonas aeruginosa GroEL Stimulates Production of PTX3 by Activating the NF-kappaB Pathway and Simultaneously Downregulating MicroRNA-9. Infect. Immun. 2017, 85, e00935-16. [Google Scholar] [CrossRef]
- Lee, M.K.; Lee, Y.; Huh, J.W.; Chen, H.; Wu, W.; Ha, U.H. The Pseudomonas aeruginosa HSP90-like protein HtpG regulates IL-8 expression through NF-kappaB/p38 MAPK and CYLD signaling triggered by TLR4 and CD91. Microbes Infect. 2020. [Google Scholar] [CrossRef]
- Awasthi, A.; Carrier, Y.; Peron, J.P.; Bettelli, E.; Kamanaka, M.; Flavell, R.A.; Kuchroo, V.K.; Oukka, M.; Weiner, H.L. A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat. Immunol. 2007, 8, 1380–1389. [Google Scholar] [CrossRef]
- Stumhofer, J.S.; Silver, J.S.; Laurence, A.; Porrett, P.M.; Harris, T.H.; Turka, L.A.; Ernst, M.; Saris, C.J.; O’Shea, J.J.; Hunter, C.A. Interleukins 27 and 6 induce STAT3-mediated T cell production of interleukin 10. Nat. Immunol. 2007, 8, 1363–1371. [Google Scholar] [CrossRef]
- Li, K.; Xu, C.; Jin, Y.; Sun, Z.; Liu, C.; Shi, J.; Chen, G.; Chen, R.; Jin, S.; Wu, W. SuhB is a regulator of multiple virulence genes and essential for pathogenesis of Pseudomonas aeruginosa. mBio 2013, 4, e00419-13. [Google Scholar] [CrossRef]
- Okuda, J.; Yamane, S.; Nagata, S.; Kunikata, C.; Suezawa, C.; Yasuda, M. The Pseudomonas aeruginosa dnaK gene is involved in bacterial translocation across the intestinal epithelial cell barrier. Microbiology 2017, 163, 1208–1216. [Google Scholar] [CrossRef]
- Bulut, Y.; Michelsen, K.S.; Hayrapetian, L.; Naiki, Y.; Spallek, R.; Singh, M.; Arditi, M. Mycobacterium tuberculosis heat shock proteins use diverse Toll-like receptor pathways to activate pro-inflammatory signals. J. Biol. Chem. 2005, 280, 20961–20967. [Google Scholar] [CrossRef]
- Wirtz, S.; Becker, C.; Fantini, M.C.; Nieuwenhuis, E.E.; Tubbe, I.; Galle, P.R.; Schild, H.J.; Birkenbach, M.; Blumberg, R.S.; Neurath, M.F. EBV-induced gene 3 transcription is induced by TLR signaling in primary dendritic cells via NF-kappa B activation. J. Immunol. 2005, 174, 2814–2824. [Google Scholar] [CrossRef]
- Aosai, F.; Rodriguez Pena, M.S.; Mun, H.S.; Fang, H.; Mitsunaga, T.; Norose, K.; Kang, H.K.; Bae, Y.S.; Yano, A. Toxoplasma gondii-derived heat shock protein 70 stimulates maturation of murine bone marrow-derived dendritic cells via Toll-like receptor 4. Cell Stress Chaperones 2006, 11, 13–22. [Google Scholar] [CrossRef]
- Erridge, C. Endogenous ligands of TLR2 and TLR4: Agonists or assistants? J. Leukoc. Biol. 2010, 87, 989–999. [Google Scholar] [CrossRef]
- Fong, J.J.; Sreedhara, K.; Deng, L.; Varki, N.M.; Angata, T.; Liu, Q.; Nizet, V.; Varki, A. Immunomodulatory activity of extracellular Hsp70 mediated via paired receptors Siglec-5 and Siglec-14. EMBO J. 2015, 34, 2775–2788. [Google Scholar] [CrossRef]
- Pawaria, S.; Binder, R.J. CD91-dependent programming of T-helper cell responses following heat shock protein immunization. Nat. Commun. 2011, 2, 521. [Google Scholar] [CrossRef]
- Ha, U.; Jin, S. Growth phase-dependent invasion of Pseudomonas aeruginosa and its survival within HeLa cells. Infect. Immun. 2001, 69, 4398–4406. [Google Scholar] [CrossRef]
- Kaufman, M.R.; Jia, J.; Zeng, L.; Ha, U.; Chow, M.; Jin, S. Pseudomonas aeruginosa mediated apoptosis requires the ADP-ribosylating activity of exoS. Microbiology 2000, 146, 2531–2541. [Google Scholar] [CrossRef]
- Davis, B.D.; Mingioli, E.S. Mutants of Escherichia coli requiring vitamin B12. J. Bacteriol. 1950, 60, 17–28. [Google Scholar] [CrossRef]
- Aida, Y.; Pabst, M.J. Removal of endotoxin from protein solutions by phase separation using Triton X-114. J. Immunol. Methods 1990, 132, 191–195. [Google Scholar] [CrossRef]
- Liu, S.; Tobias, R.; McClure, S.; Styba, G.; Shi, Q.; Jackowski, G. Removal of endotoxin from recombinant protein preparations. Clin. Biochem. 1997, 30, 455–463. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeon, J.; Lee, Y.; Yu, H.; Ha, U.-H. HSP70-Homolog DnaK of Pseudomonas aeruginosa Increases the Production of IL-27 through Expression of EBI3 via TLR4-Dependent NF-κB and TLR4-Independent Akt Signaling. Int. J. Mol. Sci. 2020, 21, 9194. https://doi.org/10.3390/ijms21239194
Jeon J, Lee Y, Yu H, Ha U-H. HSP70-Homolog DnaK of Pseudomonas aeruginosa Increases the Production of IL-27 through Expression of EBI3 via TLR4-Dependent NF-κB and TLR4-Independent Akt Signaling. International Journal of Molecular Sciences. 2020; 21(23):9194. https://doi.org/10.3390/ijms21239194
Chicago/Turabian StyleJeon, Jisu, Yeji Lee, Hyeonseung Yu, and Un-Hwan Ha. 2020. "HSP70-Homolog DnaK of Pseudomonas aeruginosa Increases the Production of IL-27 through Expression of EBI3 via TLR4-Dependent NF-κB and TLR4-Independent Akt Signaling" International Journal of Molecular Sciences 21, no. 23: 9194. https://doi.org/10.3390/ijms21239194
APA StyleJeon, J., Lee, Y., Yu, H., & Ha, U.-H. (2020). HSP70-Homolog DnaK of Pseudomonas aeruginosa Increases the Production of IL-27 through Expression of EBI3 via TLR4-Dependent NF-κB and TLR4-Independent Akt Signaling. International Journal of Molecular Sciences, 21(23), 9194. https://doi.org/10.3390/ijms21239194