A Long-Read Sequencing Approach for Direct Haplotype Phasing in Clinical Settings

 , , ,
, , ,
Abstract
1. Introduction
2. Results
2.1. Optimization of APOE Locus Amplification
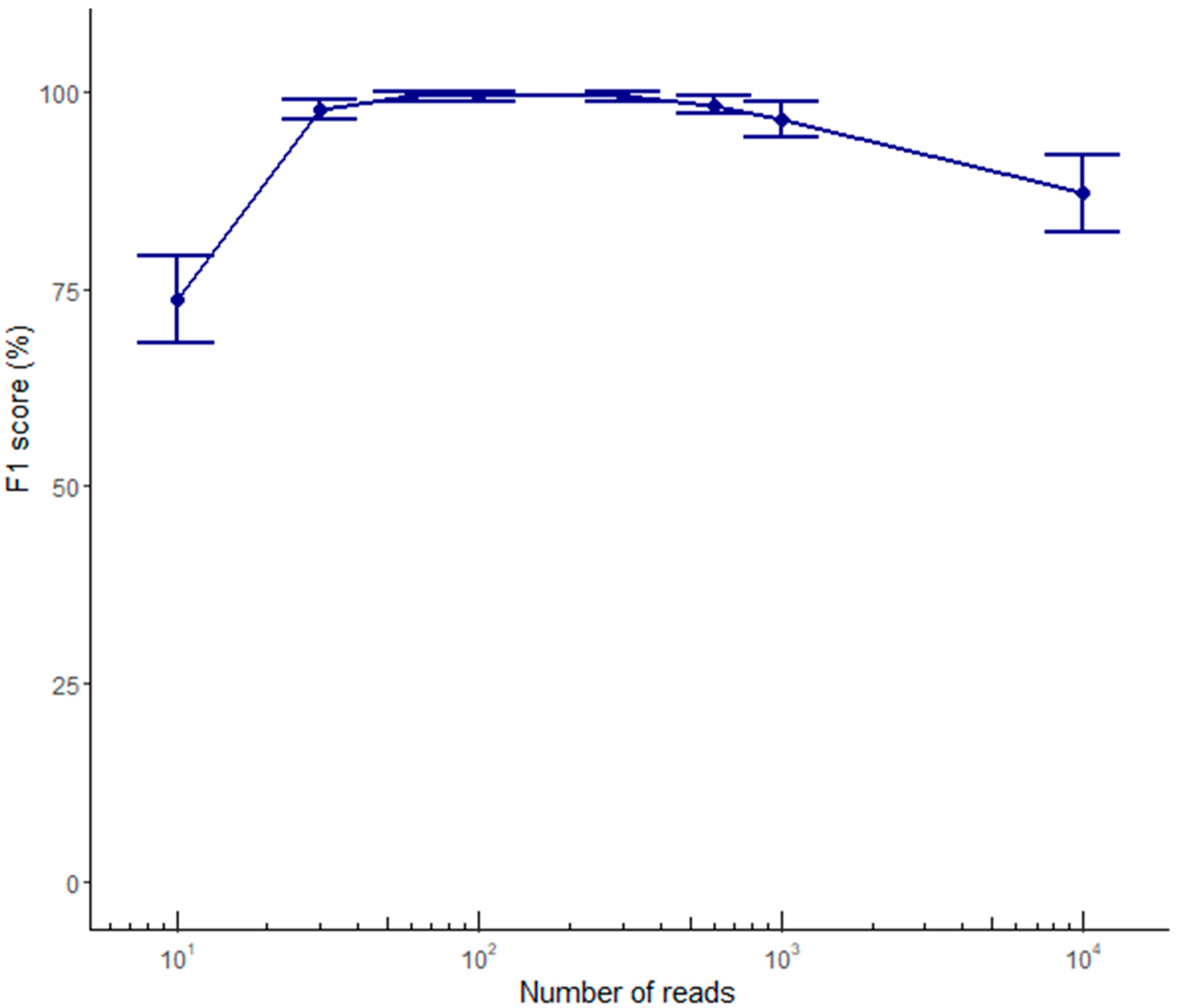
2.2. Variant Calling Performance Using ONT Data
2.3. Optimization of a Haplotype Phasing Pipeline Based on ONT Data
3. Discussion
4. Materials and Methods
4.1. DNA Extraction
4.2. PCR Amplification of The Target Region
4.3. 10× Genomics Library Preparation, Sequencing, and Data Analysis
4.4. ONT Sequencing and Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Scitable by Nature Education. Available online: https://www.nature.com/scitable/definition/haplotype-haplotypes-142/ (accessed on 30 November 2020).
- Allen, M.; Kachadoorian, M.; Quicksall, Z.; Zou, F.; Chai, H.S.; Younkin, C.; E Crook, J.; Pankratz, V.S.; Carrasquillo, M.M.; Krishnan, S.; et al. Association of MAPT haplotypes with Alzheimer’s disease risk and MAPT brain gene expression levels. Alzheimer’s Res. Ther. 2014, 6, 39. [Google Scholar] [CrossRef]
- Williams, M.A.; McKay, G.J.; Carson, R.; Craig, D.; Silvestri, G.; Passmore, P. Age-Related Macular Degeneration-Associated Genes in Alzheimer Disease. Am. J. Geriatr. Psychiatry 2015, 23, 1290–1296. [Google Scholar] [CrossRef]
- Lescai, F.; Chiamenti, A.M.; Codemo, A.; Pirazzini, C.; D’Agostino, G.; Ruaro, C.; Ghidoni, R.; Benussi, L.; Galimberti, D.; Esposito, F.; et al. An APOE Haplotype Associated with Decreased epsilon4 Expression Increases the Risk of Late Onset Alzheimer’s Disease. J. Alzheimer’s Dis. 2011, 24, 235–245. [Google Scholar] [CrossRef]
- Navarro, S.; Medina, P.; Mira, Y.; Estelles, A.; Villa, P.; Ferrando, F.; Vaya, A.; Bertina, R.M.; España, F. Haplotypes of the EPCR gene, prothrombin levels, and the risk of venous thrombosis in carriers of the prothrombin G20210A mutation. Haematol. 2008, 93, 885–891. [Google Scholar] [CrossRef][Green Version]
- Vymetalkova, V.P.; Soucek, P.; Kunická, T.; Jiraskova, K.; Brynychová, V.; Pardini, B.; Novosadova, V.; Polivkova, Z.; Kubáčková, K.; Kozevnikovova, R.; et al. Genotype and Haplotype Analyses of TP53 Gene in Breast Cancer Patients: Association with Risk and Clinical Outcomes. PLOS ONE 2015, 10, e0134463. [Google Scholar] [CrossRef]
- Schächter, F.; Faure-Delanef, L.; Guénot, F.; Rouger, H.; Froguel, P.; Lesueur-Ginot, L.; Cohen, D. Genetic associations with human longevity at the APOE and ACE loci. Nat. Genet. 1994, 6, 29–32. [Google Scholar] [CrossRef]
- Soerensen, M.; Dato, S.; Tan, Q.; Thinggaard, M.; Kleindorp, R.; Beekman, M.; Suchiman, H.E.D.; Jacobsen, R.; McGue, M.; Stevnsner, T.; et al. Evidence from case-control and longitudinal studies supports associations of genetic variation in APOE, CETP, and IL6 with human longevity. AGE 2012, 35, 487–500. [Google Scholar] [CrossRef]
- Ferrari, R.; Wang, Y.; Vandrovcova, J.; Guelfi, S.; Witeolar, A.; Karch, C.M.; Schork, A.J.; Fan, C.C.; Brewer, J.B.; Momeni, P.; et al. Genetic architecture of sporadic frontotemporal dementia and overlap with Alzheimer’s and Parkinson’s diseases. J. Neurol. Neurosurg. Psychiatry 2017, 88, 152–164. [Google Scholar] [CrossRef]
- Coon, K.D.; Myers, A.J.; Craig, D.W.; Webster, J.A.; Pearson, J.V.; Lince, D.H.; Zismann, V.L.; Beach, T.G.; Leung, D.; Bryden, L.; et al. A High-Density Whole-Genome Association Study Reveals That APOE Is the Major Susceptibility Gene for Sporadic Late-Onset Alzheimer’s Disease. J. Clin. Psychiatry 2007, 68, 613–618. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; E Schmechel, D.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; A Pericak-Vance, M. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Babenko, V.N.; Afonnikov, D.A.; Ignatieva, E.V.; Klimov, A.V.; Gusev, F.E.; Rogaev, E.I. Haplotype analysis of APOE intragenic SNPs. BMC Neurosci. 2018, 19, 29–40. [Google Scholar] [CrossRef]
- Xiao, H.; Gao, Y.; Liu, L.; Li, Y. Association between polymorphisms in the promoter region of the apolipoprotein E (APOE) gene and Alzheimer’s disease: A meta-analysis. EXCLI J. 2017, 16, 921–938. [Google Scholar]
- Deelen, J.; Beekman, M.; Uh, H.-W.; Helmer, Q.; Kuningas, M.; Christiansen, L.; Kremer, D.; Van Der Breggen, R.; Suchiman, H.E.D.; Lakenberg, N.; et al. Genome-wide association study identifies a single major locus contributing to survival into old age; the APOE locus revisited. Aging Cell 2011, 10, 686–698. [Google Scholar] [CrossRef]
- Deelen, J.; Beekman, M.; Uh, H.-W.; Broer, L.; Ayers, K.L.; Tan, Q.; Kamatani, Y.; Bennet, A.M.; Tamm, R.; Trompet, S.; et al. Genome-wide association meta-analysis of human longevity identifies a novel locus conferring survival beyond 90 years of age. Hum. Mol. Genet. 2014, 23, 4420–4432. [Google Scholar] [CrossRef]
- Nebel, A.; Kleindorp, R.; Caliebe, A.; Nothnagel, M.; Blanché, H.; Junge, O.; Wittig, M.; Ellinghaus, D.; Flachsbart, F.; Wichmann, H.-E.; et al. A genome-wide association study confirms APOE as the major gene influencing survival in long-lived individuals. Mech. Ageing Dev. 2011, 132, 324–330. [Google Scholar] [CrossRef]
- Lin, R.; Zhang, Y.; Yan, D.; Liao, X.; Gong, G.; Hu, J.; Fu, Y.; Cai, W. Association of common variants in TOMM40/APOE/APOC1 region with human longevity in a Chinese population. J. Hum. Genet. 2016, 61, 323–328. [Google Scholar] [CrossRef]
- Snyder, M.W.; Adey, A.; Kitzman, J.O.; Shendure, J. Haplotype-resolved genome sequencing: Experimental methods and applications. Nat. Rev. Genet. 2015, 16, 344–358. [Google Scholar] [CrossRef]
- Huang, M.; Tu, J.; Lu, Z. Recent Advances in Experimental Whole Genome Haplotyping Methods. Int. J. Mol. Sci. 2017, 18, 1944. [Google Scholar] [CrossRef]
- Porubský, D.; Sanders, A.D.; Van Wietmarschen, N.; Falconer, E.; Hills, M.; Spierings, D.C.; Bevova, M.R.; Guryev, V.; Lansdorp, P.M. Direct chromosome-length haplotyping by single-cell sequencing. Genome Res. 2016, 26, 1565–1574. [Google Scholar] [CrossRef]
- Putnam, N.H.; O’Connell, B.L.; Stites, J.C.; Rice, B.J.; Blanchette, M.; Calef, R.; Troll, C.J.; Fields, A.; Hartley, P.D.; Sugnet, C.W.; et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 2016, 26, 342–350. [Google Scholar] [CrossRef]
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef]
- Zheng, G.X.; Lau, B.T.; Schnall-Levin, M.; Jarosz, M.; Bell, J.M.; Hindson, C.M.; Kyriazopoulou-Panagiotopoulou, S.; Masquelier, D.A.; Merrill, L.; Terry, J.M.; et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat. Biotechnol. 2016, 34, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; A Sasani, T.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Porubsky, D.; Garg, S.; Sanders, A.D.; Korbel, J.O.; Guryev, V.; Lansdorp, P.M.; Marschall, T. Dense and accurate whole-chromosome haplotyping of individual genomes. Nat. Commun. 2017, 8, 1293. [Google Scholar] [CrossRef] [PubMed]
- Rang, F.J.; Kloosterman, W.P.; De Ridder, J. From squiggle to basepair: Computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 2018, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Giordano, F.; Ning, Z. Oxford Nanopore MinION Sequencing and Genome Assembly. Genom. Proteom. Bioinform. 2016, 14, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Laver, T.W.; Caswell, R.C.; Moore, K.A.; Poschmann, J.; Johnson, M.B.; Owens, M.M.; Ellard, S.; Paszkiewicz, K.H.; Weedon, M.N. Pitfalls of haplotype phasing from amplicon-based long-read sequencing. Sci. Rep. 2016, 6, 21746. [Google Scholar] [CrossRef]
- Edge, P.; Bafna, V.; Bansal, V. HapCUT2: Robust and accurate haplotype assembly for diverse sequencing technologies. Genome Res. 2017, 27, 801–812. [Google Scholar] [CrossRef]
- Patterson, M.; Marschall, T.; Pisanti, N.; Van Iersel, L.; Stougie, L.; Klau, G.W.; Hu, Y.-J. WhatsHap: Weighted Haplotype Assembly for Future-Generation Sequencing Reads. J. Comput. Biol. 2015, 22, 498–509. [Google Scholar] [CrossRef]
- Kuleshov, V. Probabilistic single-individual haplotyping. Bioinform. 2014, 30, i379–i385. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinform. 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- 10x Genomics. Discontinuation of Linked-Reads. Available online: https://www.10xgenomics.com/products/linked-reads (accessed on 30 November 2020).
- Gilpatrick, T.; Lee, I.; Graham, J.E.; Raimondeau, E.; Bowen, R.; Heron, A.; Downs, B.; Sukumar, S.; Sedlazeck, F.J.; Timp, W. Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat. Biotechnol. 2020, 38, 433–438. [Google Scholar] [CrossRef]
- Edge, P.; Bansal, V. Longshot enables accurate variant calling in diploid genomes from single-molecule long read sequencing. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Luo, R.; Wong, C.-L.; Wong, Y.-S.; Tang, C.-I.; Liu, C.-M.; Leung, C.-M.; Lam, T.-W. Exploring the limit of using a deep neural network on pileup data for germline variant calling. Nat. Mach. Intell. 2020, 2, 220–227. [Google Scholar] [CrossRef]
- Ammar, R.; Paton, T.A.; Torti, D.; Shlien, A.; Bader, G.D. Long read nanopore sequencing for detection of HLA and CYP2D6 variants and haplotypes. F1000Research 2015, 4, 17. [Google Scholar] [CrossRef]
- Stancu, M.C.; Van Roosmalen, M.J.; Renkens, I.; Nieboer, M.M.; Middelkamp, S.; De Ligt, J.; Pregno, G.; Giachino, D.; Mandrile, G.; Valle-Inclan, J.E.; et al. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Leija-Salazar, M.; Sedlazeck, F.; Toffoli, M.; Mullin, S.; Mokretar, K.; Athanasopoulou, M.; Donald, A.; Sharma, R.; Hughes, D.; Schapira, A.H.V.; et al. Detection of GBA missense mutations and other variants using the Oxford Nanopore MinION. Mol. Genet. Genomic. Med. 2019, 7, 3. [Google Scholar] [CrossRef]
- Maestri, S.; Cosentino, E.; Paterno, M.; Freitag, H.; Garces, J.M.; Marcolungo, L.; Alfano, M.; Njunjić, I.; Schilthuizen, M.; Slik, F.J.; et al. A Rapid and Accurate MinION-Based Workflow for Tracking Species Biodiversity in the Field. Genes 2019, 10, 468. [Google Scholar] [CrossRef]
- Menegon, M.; Cantaloni, C.; Rodriguez-Prieto, A.; Centomo, C.; Abdelfattah, A.; Rossato, M.; Bernardi, M.; Xumerle, L.; Loader, S.; Delledonne, M. On site DNA barcoding by nanopore sequencing. PLoS ONE 2017, 12, e0184741. [Google Scholar] [CrossRef]
- Knot, I.E.; Zouganelis, G.D.; Weedall, G.D.; Wich, S.A.; Rae, R. DNA Barcoding of Nematodes Using the MinION. Front. Ecol. Evol. 2020, 8, 8. [Google Scholar] [CrossRef]
- Karst, S.; Ziels, R.; Kirkegaard, R.; Sørensen, E.; McDonald, D.; Zhu, Q.; Knight, R.; Albertsen, M. Enabling high-accuracy long-read amplicon sequences using unique molecular identifiers with Nanopore or PacBio sequencing. bioRxiv 2020. Available online: https://www.biorxiv.org/content/10.1101/645903v3.full (accessed on 30 November 2020).
- Wick, R.R.; Judd, L.M.; E Holt, K. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 2019, 20, 1–10. [Google Scholar] [CrossRef]
- Vereecke, N.; Bokma, J.; Haesebrouck, F.; Nauwynck, H.; Boyen, F.; Pardon, B.; Theuns, S. High quality genome assemblies of Mycoplasma bovis using a taxon-specific Bonito basecaller for MinION and Flongle long-read nanopore sequencing. BMC Bioinform. 2020, 21, 1–16. [Google Scholar] [CrossRef]
- Tytgat, O.; Gansemans, Y.; Weymaere, J.; Rubben, K.; Deforce, D.; Van Nieuwerburgh, F. Nanopore Sequencing of a Forensic STR Multiplex Reveals Loci Suitable for Single-Contributor STR Profiling. Genes 2020, 11, 381. [Google Scholar] [CrossRef]
- Gabrieli, T.; Sharim, H.; Michaeli, Y.; Ebenstein, Y. Cas9-Assisted Targeting of CHromosome segments (CATCH) for targeted nanopore sequencing and optical genome mapping. BioRxiv 2017. Available online: https://www.biorxiv.org/content/10.1101/110163v3 (accessed on 30 November 2020).
- Madsen, E.B.; Höijer, I.; Kvist, T.; Ameur, A.; Mikkelsen, M.J. Xdrop: Targeted sequencing of long DNA molecules from low input samples using droplet sorting. Hum. Mutat. 2020, 41, 1671–1679. [Google Scholar] [CrossRef]
- Mantere, T.; Kersten, S.; Hoischen, A. Long-Read Sequencing Emerging in Medical Genetics. Front. Genet. 2019, 10, 426. [Google Scholar] [CrossRef]
- Zhao, H.; Sun, Z.; Wang, J.; Huang, H.; Kocher, J.-P.; Wang, L. CrossMap: A versatile tool for coordinate conversion between genome assemblies. Bioinform. 2014, 30, 1006–1007. [Google Scholar] [CrossRef]
- Van Der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinform. 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| dbSNP ID | Pos. | Ref. | Alt. |
|---|---|---|---|
| rs893292251 | chr19:44902764 | C | T |
| rs34215622 | chr19:44903281 | C | CG |
| - | chr19:44903416 | G | A |
| - | chr19:44904398 | C | T |
| rs7259620 | chr19:44904531 | G | A,C |
| rs449647 | chr19:44905307 | A | T |
| rs769446 | chr19:44905371 | T | C |
| rs405509 | chr19:44905579 | T | G |
| - | chr19:44905832 | C | T |
| rs440446 | chr19:44905910 | C | G |
| - | chr19:44906337 | G | A |
| rs769450 | chr19:44907187 | G | A |
| rs429358 | chr19:44908684 | T | C |
| rs7412 | chr19:44908822 | C | T |
| rs747519137 | chr19:44909521 | CT | C |
| - | chr19:44909967 | TG | T |
| rs1065853 | chr19:44909976 | G | C,T |
| rs75627662 | chr19:44910319 | C | T |
| - | chr19:44910393 | A | C |
| - | chr19:44910397 | T | C |
| - | chr19:44910405 | A | C |
| - | chr19:44910410 | A | C |
| rs72654469 | chr19:44910678 | T | C |
| rs72654473 | chr19:44911142 | C | A |
| rs439401 | chr19:44911194 | T | C |
| rs5828224 | chr19:44911609 | AT | A |
| rs445925 | chr19:44912383 | G | A,C |
| rs483082 | chr19:44912921 | G | T |
| rs59325138 | chr19:44913034 | C | T |
| rs584007 | chr19:44913221 | A | G |
| rs438811 | chr19:44913484 | C | T |
| rs390082 | chr19:44913574 | T | A,C,G |
| rs72654445 | chr19:44913943 | G | A |
| - | chr19:44914318 | A | T |
| rs34954997 | chr19:44914381 | C | CTTCG |
| Sample Name | Homozygous Variants | Heterozygous Variants | Heterozygous Phased Variants | |||
|---|---|---|---|---|---|---|
| SNVs | Indels | SNVs | Indels | SNVs | Indels | |
| NA12878 | 0 | 0 | 10 | 1 | 10 | 1 |
| NA12892 | 0 | 0 | 4 | 0 | 4 | 0 |
| V1 | 6 | 3 | 11 | 1 | 11 | 1 |
| V2 | 4 | 0 | 6 | 2 | 6 | 1 |
| V3 | 4 | 1 | 4 | 1 | 4 | 1 |
| Sample Name | Number of Reads | Reads Mean Length | Number of PASS Reads (%) | PASS Reads Mean Length (bp) | Number of PASS Reads Aligned | Number of PASS Reads Covering the Whole Region |
|---|---|---|---|---|---|---|
| NA12878 | 28,251 | 11,871 | 24,335 (86%) | 12,046 | 24,330 (99.99%) | 23,799 (97.82%) |
| NA12892 | 27,410 | 11,710 | 23,541 (86%) | 12,046 | 23,541 (100%) | 23,022 (97.80%) |
| V1 | 39,993 | 11,552 | 33,489 (84%) | 12,042 | 33,486 (99.99%) | 32,941 (98.37%) |
| V2 | 38,501 | 11,690 | 33,373 (87%) | 12,044 | 33,371 (99.99%) | 32,755 (98.15%) |
| V3 | 32,084 | 11,588 | 27,165 (85%) | 12,046 | 27,162 (99.99%) | 26,673 (98.20%) |
| NA12878 | NA12892 | V1 | V2 | V3 | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNVs | Indels | SNVs | Indels | SNVs | Indels | SNVs | Indels | SNVs | Indels | |||||||||||||||||||||
| Number of Reads | TP | FN | FP | TP | FN | FP | TP | FN | FP | TP | FN | FP | TP | FN | FP | TP | FN | FP | TP | FN | FP | TP | FN | FP | TP | FN | FP | TP | FN | FP |
| 10 | 10 | 0 | 9 | 0 | 1 | 1 | 4 | 0 | 3 | 0 | 0 | 0 | 16 | 2 | 0 | 0 | 3 | 3 | 9 | 1 | 10 | 0 | 2 | 2 | 6 | 2 | 3 | 0 | 2 | 3 |
| 30 | 10 | 0 | 0 | 0 | 1 | 1 | 4 | 0 | 0 | 0 | 0 | 3 | 16 | 2 | 0 | 1 | 2 | 2 | 9 | 1 | 0 | 0 | 2 | 4 | 8 | 0 | 0 | 0 | 2 | 2 |
| 60 | 10 | 0 | 0 | 0 | 1 | 2 | 4 | 0 | 0 | 0 | 0 | 3 | 17 | 1 | 0 | 0 | 3 | 2 | 10 | 0 | 0 | 0 | 2 | 2 | 8 | 0 | 0 | 1 | 1 | 2 |
| 100 | 10 | 0 | 0 | 0 | 1 | 2 | 4 | 0 | 0 | 0 | 0 | 2 | 17 | 1 | 0 | 0 | 3 | 2 | 10 | 0 | 0 | 0 | 2 | 2 | 8 | 0 | 0 | 0 | 2 | 2 |
| 300 | 10 | 0 | 0 | 0 | 1 | 1 | 4 | 0 | 0 | 0 | 0 | 0 | 17 | 1 | 0 | 0 | 3 | 2 | 10 | 0 | 0 | 0 | 2 | 0 | 8 | 0 | 0 | 0 | 2 | 0 |
| 600 | 10 | 0 | 0 | 0 | 1 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 17 | 1 | 0 | 0 | 3 | 2 | 9 | 1 | 0 | 0 | 2 | 0 | 8 | 0 | 0 | 0 | 2 | 0 |
| 1000 | 10 | 0 | 0 | 0 | 1 | 0 | 4 | 0 | 0 | 0 | 0 | 0 | 16 | 2 | 0 | 0 | 3 | 1 | 8 | 2 | 0 | 0 | 2 | 0 | 8 | 0 | 0 | 0 | 2 | 1 |
| 10000 | 8 | 2 | 0 | 0 | 1 | 1 | 4 | 0 | 0 | 0 | 0 | 1 | 10 | 8 | 0 | 0 | 3 | 1 | 7 | 3 | 0 | 0 | 2 | 3 | 7 | 1 | 0 | 0 | 2 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maestri, S.; Maturo, M.G.; Cosentino, E.; Marcolungo, L.; Iadarola, B.; Fortunati, E.; Rossato, M.; Delledonne, M. A Long-Read Sequencing Approach for Direct Haplotype Phasing in Clinical Settings. Int. J. Mol. Sci. 2020, 21, 9177. https://doi.org/10.3390/ijms21239177
Maestri S, Maturo MG, Cosentino E, Marcolungo L, Iadarola B, Fortunati E, Rossato M, Delledonne M. A Long-Read Sequencing Approach for Direct Haplotype Phasing in Clinical Settings. International Journal of Molecular Sciences. 2020; 21(23):9177. https://doi.org/10.3390/ijms21239177
Chicago/Turabian StyleMaestri, Simone, Maria Giovanna Maturo, Emanuela Cosentino, Luca Marcolungo, Barbara Iadarola, Elisabetta Fortunati, Marzia Rossato, and Massimo Delledonne. 2020. "A Long-Read Sequencing Approach for Direct Haplotype Phasing in Clinical Settings" International Journal of Molecular Sciences 21, no. 23: 9177. https://doi.org/10.3390/ijms21239177
APA StyleMaestri, S., Maturo, M. G., Cosentino, E., Marcolungo, L., Iadarola, B., Fortunati, E., Rossato, M., & Delledonne, M. (2020). A Long-Read Sequencing Approach for Direct Haplotype Phasing in Clinical Settings. International Journal of Molecular Sciences, 21(23), 9177. https://doi.org/10.3390/ijms21239177