Abstract
Calcium signaling is essential for neuronal function, and its dysregulation has been implicated across neurodegenerative diseases, including Alzheimer’s disease (AD). A close reciprocal relationship exists between calcium signaling and mitochondrial function. Growing evidence in a variety of AD models indicates that calcium dyshomeostasis drastically alters mitochondrial activity which, in turn, drives neurodegeneration. This review discusses the potential pathogenic mechanisms by which calcium impairs mitochondrial function in AD, focusing on the impact of calcium in endoplasmic reticulum (ER)–mitochondrial communication, mitochondrial transport, oxidative stress, and protein homeostasis. This review also summarizes recent data that highlight the need for exploring the mechanisms underlying calcium-mediated mitochondrial dysfunction while suggesting potential targets for modulating mitochondrial calcium levels to treat neurodegenerative diseases such as AD.
1. Introduction
With a rapidly aging population, it is expected that neurodegenerative disorders will become an increasingly pressing health issue, intensifying the urgency for effective treatment. Most neurological disorders are chronic and incurable, and their debilitating effects can continue for decades. There are approximately 50 million people living worldwide with dementia and this number is expected to reach 82 million in 2030 and 152 million by 2050 (WHO.int) [1]. Alzheimer’s disease (AD), the most common neurodegenerative disease [1], is the sixth leading cause of death [2]. In the United States alone, approximately 5 million people currently suffer from the disease, and these numbers are projected to triple by the year 2050 [2]. AD is characterized by gradual cognitive decline and memory loss, synaptic dysfunction, and neuronal death. Despite decades of research, there is no effective therapy for AD and the cause of AD remains unclear, especially due to its complex etiology. The primary histopathological hallmarks of AD are neurofibrillary tangles composed of the microtubule-associated protein tau and extracellular deposition of senile plaques composed of amyloid-beta (Abeta) peptides. Most AD research has focused on investigating the amyloid hypothesis, which proposes that aberrant Abeta production is the core cause of AD, and that all other dysfunctions observed in AD, including hyperphosphorylated tau tangles, are a consequence of Abeta neurotoxicity. Abeta plaques on their own have been shown to interfere with synaptic function and cause extensive neuronal death, especially within the cortical and hippocampal regions of the brain that are responsible for learning and memory [3]. In support of Abeta being the causative agent of AD, genetic mutations in genes encoding presenilin 1 (PSEN1), presenilin 2 (PSEN2), and the amyloid precursor protein (APP), which lead to nearly all early-onset familial AD (FAD) cases, are all involved in Abeta processing. Abeta peptides of varying lengths are generated from APP by the consecutive cleavage by beta-secretase and gamma-secretase. Mutations in the genes encoding PSEN1 and PSEN2, which underlie roughly 70% of all familial AD cases [4], form the catalytic component of the gamma-secretase complex. Increased production of the more toxic, aggregation-prone Abeta42 species relative to Abeta40, which results in the generation of extracellular amyloid fibrils and plaques, is thought to be the primary cause of AD [3]. Therapeutic strategies for treating AD have focused on reducing the burden of Abeta plaques by either preventing Abeta production or by promoting its clearance. However, gamma-secretase inhibitors have been a failure clinically, and Abeta plaque load is not strongly correlated with dementia onset or severity [5], which has prompted reconsideration of the amyloid hypothesis [6,7,8,9]. There is also considerable evidence that Abeta oligomers, as opposed to the more highly aggregated Abeta fibrils, are the primary cytotoxic species and pathological agent of AD. This soluble form has been shown to damage synapses absent Abeta plaque formation [10,11]. Additionally, oligomeric Abeta isolated from AD patients was sufficient to cause synaptic dysfunction and memory loss in mice [12]. It remains unclear whether selectively targeting Abeta oligomers will show more success in clinical trials. Indeed, the use of immunotherapies targeting Abeta, some of which have been shown to bind oligomeric Abeta with high affinity, have no effect on disease progression [8,13]. Other studies have shown that overexpression of Abeta peptides in mice, while simulating the Abeta pathology observed in AD, does not result in similar synaptic loss and memory impairment [14]. Additionally, neurodegeneration induced by presenilin mutations can occur in the absence of Abeta production [15,16]. Mutations in other components of the gamma-secretase complex (APH-1, PEN-2, and nicastrin) have not been implicated in FAD, implying APP and presenilin mutations do not cause FAD merely by altering Abeta production [17]. Furthermore, many other symptoms (e.g., increased inflammation, altered calcium signaling, mitochondrial dysfunction, oxidative damage) appear to arise independently of any Abeta involvement as they often precede Abeta plaque formation, and are also more strongly correlated with cognitive decline [18,19]. Collectively, this suggests that Abeta production is not necessarily the proximate cause of AD and that additional pathological factors must be explored to develop viable therapies.
2. Calcium Dysregulation in AD
The connection between calcium and AD was observed several decades ago [20], but recent data have increased support for this hypothesis, and strongly implicate a role for calcium in AD. The calcium hypothesis of AD states that disruptions to neuronal calcium signaling underlie not only amyloid plaque deposition but a series of molecular changes within the neuron that cause neuronal dysfunction [21]. During neurotransmission, a rise in intracellular calcium following membrane depolarization transmits the signal to synapses. Calcium signaling in neurons is, therefore, crucial for neurotransmission and for maintaining synaptic plasticity and generating long-term potentiation (LTP), which forms the basis of learning and memory through the progressive strengthening of synapses [22,23]. Calcium signaling also regulates neuronal metabolism and energy production which is necessary to sustain synaptic transmission [24]. Unsurprisingly, disruptions to calcium signaling have debilitating consequences on neuronal function. Evidence for calcium dysregulation in AD was initially found over 25 years ago in fibroblast cells isolated from AD patients, which showed enhanced endoplasmic reticulum (ER) calcium uptake and ER calcium release [25,26]. Further studies in AD mouse models have supported the ubiquitous involvement of calcium dysregulation in AD, linking it to memory loss and increased toxicity of Abeta peptides [27,28,29]. Other studies have shown that elevated ER or cytoplasmic calcium increases Abeta production by triggering phosphorylation of APP and tau, indicating intracellular calcium dysregulation exacerbates amyloidosis and tau pathology [30,31]. Emilsson and Jazin also showed that mRNA expression of genes involved in calcium regulation is altered in AD brains [32]. The results from this study further suggest that ER calcium channel activity is elevated in AD [32].
Many familial AD PSEN mutations are also linked to dysregulated calcium signaling [33]. In addition to PSEN1/2′s role as the catalytic subunit of gamma-secretase, PSEN1/2 regulates ER calcium stores, a function that is notably gamma-secretase independent, demonstrating that PSEN1 and PSEN2′s impact on neuronal function extends beyond Abeta generation [34,35]. PSEN1/2 are transmembrane proteins present on most endomembranes but predominate on the ER membrane, where they have been found to physically interact with ER calcium channels, including the two main ER calcium release channels, inositol 1,4,5-trisphosphate receptors (IP3Rs) and ryanodine receptors (RyRs) [28,36,37]. In line with this, mice carrying FAD mutations show altered activity of both IP3Rs and RyRs. Presenilin mutations have been documented to increase either the expression or sensitivity of RyRs and IP3Rs, leading to exaggerated RyR- and IP3R-mediated calcium release in response to various agonists [28,36,38,39,40]. Loss of PSEN1 function has also been shown in Xenopus oocytes to increase the activity of smooth endoplasmic reticulum Ca2+ ATPase (SERCA), which is responsible for pumping calcium into the ER to maintain cytosolic calcium levels [29]. Elevated SERCA activity in turn leads to overloading of ER calcium stores and a compensatory release of ER calcium [29]. PSEN itself may act as a passive ER calcium leak channel, which similarly results in ER calcium overfilling and exaggerated ER calcium release [35]. Notably, this elevated ER calcium release also precedes Abeta pathology and stimulates Abeta formation [29]. This is consistent with a previous study showing that altered calcium signaling in fibroblasts derived from asymptomatic FAD families was a strong predictor of future disease development [41]. Moreover, several other studies demonstrate that FAD mutations in the genes encoding PSEN1 or PSEN2 result in higher basal levels of calcium in cortical and hippocampal neurons due to excessive ER calcium release [29,36,38,39]. Elevated cytosolic calcium, in turn, impedes induction of long-term potentiation (LTP) [42]. There is also evidence that FAD mutations in APP similarly cause increased intracellular calcium concentrations independent of Abeta involvement [43,44]. Abeta itself has also been shown to derive some of its cytotoxic effects from promoting cytosolic calcium influx [45]. Other studies in cultured cortical neurons showed that oligomeric Abeta promoted ER calcium release, which led to cell death [46]. Altogether, the data strongly suggest that exaggerated ER calcium release and disruptions to calcium homeostasis play an important role in AD pathogenesis.
3. Mitochondria and ER Crosstalk in Presenilin Mutants, and in Sporadic AD
Both ER and mitochondria are dynamic organelles that are actively moving within the cell and make transient contacts to facilitate crosstalk between the two organelles [47]. The mitochondria-associated membranes (MAMs) are regions where the ER closely associates with the outer mitochondrial membrane (OMM). These MAMs contain a specialized distribution of phospholipids and proteins to enable ER–mitochondria communication [48]. MAMs regulate a variety of processes including mitochondrial metabolism and energy production, the ER stress response, lipid synthesis, and apoptosis signaling [48]. Calcium homeostasis is also highly dependent on MAM function (Figure 1). IP3R and RyR localization is concentrated at the MAMs to promote the rapid uptake of calcium into the mitochondria [49] (Figure 1). MAMs are also enriched in voltage-dependent anion channels (VDACs), an ion channel on the OMM that regulates the transportation of a variety of ions and metabolites into and out of the mitochondria, and is primarily responsible for calcium uptake into the mitochondria across the OMM [50,51] (Figure 1). There is growing evidence that ER–mitochondrial communication is perturbed in AD. It has been shown in neurons of both sporadic and familial AD patients and an AD mouse model that there are increased ER–mitochondria contact points and expression of MAM-associated proteins, including IP3Rs, RyRs, and VDACs [50]. Abeta exposure to hippocampal neurons was also shown to increase ER–mitochondrial contact and promote transfer of calcium from the ER into the mitochondria [50]. Similarly, cells expressing the ε4 allele of apolipoprotein E (APOE4), which is considered a major risk factor for developing sporadic AD [52], show upregulated MAM activity and ER–mitochondrial communication [53], further suggesting disruption of MAM function is a common characteristic observed in AD. PSEN1/2 localization is also concentrated at the MAMs [54] (Figure 1). Accordingly, PSEN1 and PSEN2 FAD mutations have been shown to alter lipid and phospholipid synthesis, lipid exchange, and calcium transfer between the ER and mitochondria [55,56]. Moreover, presenilin and APP FAD mutations have been shown to increase the number of ER–mitochondria contact sites [57]. In Caenorhabditis elegans, which has a highly conserved presenilin ortholog but does not produce Abeta peptides [58], presenilin loss promotes ER-to-mitochondria calcium uptake, suggesting that presenilin alters ER–mitochondria calcium transfer via an Abeta-independent mechanism [59]. This study further demonstrated that the function of presenilin in mediating ER–mitochondrial calcium signaling is independent of gamma-secretase activity. However, there is also evidence that changes to MAM function or connectivity may alter or increase gamma-secretase activity due to presenilin enrichment at the MAM, and this may in turn promote pathologic Abeta42 generation [60]. Considering MAMs are involved in processes that are frequently disrupted in AD, including calcium homeostasis, it is likely the altered MAM distribution and functions are involved in AD pathogenesis. The increased ER–mitochondrial communication observed across multiple AD models has especially important implications on mitochondrial function. Indubitably, neurons are highly sensitive to mitochondrial defects and alterations in mitochondrial oxidative respiration.
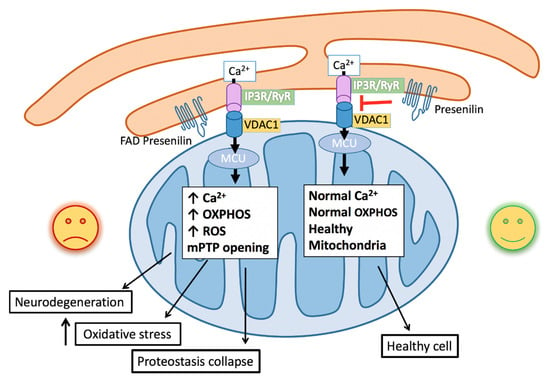
Figure 1.
Schematic of familial Alzheimer’s Disease (FAD) mutations facilitating endoplasmic reticulum (ER)–mitochondrial calcium transfer. Increased calcium release from the IP3 receptor (IP3R) and Ryanodine receptors (RyR) at the ER mitochondrial-associated membranes (MAMs) is taken up through the voltage-dependent ion channel (VDAC) on the mitochondrial outer membrane and the calcium-selective mitochondrial calcium uniporter (MCU) on the mitochondrial inner membrane. Increased mitochondrial calcium stimulates oxidative phosphorylation, leading to increased reactive oxygen species (ROS) generation, which promotes oxidative stress, mitochondrial permeable transition pore (mPTP) opening and apoptosis, protein misfolding and proteostatic collapse, and neurodegeneration.
4. Mitochondrial Calcium and AD
Neurons are particularly reliant on mitochondria for energy generation to sustain synaptic transmission. The brain accounts for only 2% of human body weight, but uses roughly 20% of the body’s oxygen supply [61], The vast majority of neuronal ATP is produced through mitochondrial oxidative respiration [62,63], and neurons use this ATP primarily to generate the ionic gradients necessary for synaptic transmission [64]. Mitochondria are highly mobile organelles, and their subcellular localization impacts their ability to provide ATP to various cellular compartments [65]. In neurons, mitochondria are predominantly localized to the synapses to meet the energy demand at these sites [64]. Preserving mitochondrial quality and the mitochondrial trafficking network is thus especially important for neurons, as both are required to maintain synaptic plasticity and ultimately the learning and memory process [66,67]. Consequently, neurons are highly sensitive to mitochondrial defects and alterations in mitochondrial oxidative respiration [64]. Mitochondrial dysfunction is a common feature across neurodegenerative diseases, including AD [66,67]. In fact, mitochondrial dysfunction is thought to be one of the main drivers of the disease [68,69].
Intracellular calcium greatly impacts mitochondrial function. Indeed, calcium plays a direct role in stimulating enzymes of the tricarboxylic acid (TCA) cycle and electron transport chain leading to increased oxidative phosphorylation [70,71,72,73] (Figure 1). Mitochondria, in turn, regulate cellular calcium signaling by sequestering and buffering cytosolic calcium. As mentioned, the positioning of the ER calcium channels at the MAMs facilitates calcium transfer into the mitochondria. Selective transport of calcium into the matrix across the inner mitochondrial membrane (IMM) is accomplished by the highly calcium selective mitochondrial calcium uniporter (MCU) protein complex [74,75,76] (Figure 1). The MCU complex is composed of four core components: the pore forming MCU protein, an auxiliary subunit EMRE (essential for MCU regulator) and the MICU gatekeepers, MICU1 and MICU2/3 [77,78,79]. The MCU complex regulates calcium uptake into the matrix primarily through the MICU1 and MICU2/3 proteins that sense calcium through their conserved calcium-binding EF hand domains [80]. MICU1 and MICU2 are widely expressed in most mammalian tissues, whereas MICU3 is expressed only in skeletal muscles and the CNS [81]. MICU2 and MICU3 have similar structure and function in regulating MCU complex activity [82,83]. Recently, MICU2 has been shown bind to MICU1 and together these proteins allow for gatekeeper activity. Specifically, elevation in cytosolic calcium promotes calcium binding to the EF hands of the MICU1-MICU2 heterodimer, enabling MICU1 to facilitate MCU activity allowing calcium entry into the mitochondria [84,85]. MICU1 activation is also influenced by the activity and expression of cytosolic calcium binding proteins, which also have EF-hand domains that compete with MICU1 for calcium binding [86]. By promoting calcium uptake when cytosolic levels are high, mitochondria buffer calcium to maintain intracellular calcium homeostasis. This uptake is energetically favorable due to the negative membrane potential generated by transport of H+ across the IMM by the electron transport chain, making mitochondria well suited for this task. The mitochondria at synapses are important not only for ATP delivery but also for tightly regulating calcium concentration at the synapses for effective neurotransmission [87]. Considering the abundant evidence implicating intracellular calcium dysregulation in AD, it is likely that regulation of intracellular calcium through mitochondrial calcium buffering is a factor in this process.
Calcium signaling also plays a direct role in mitochondrial activity [88]. Mitochondrial calcium levels significantly impact mitochondrial activity and cellular ATP supply. Calcium uptake into the mitochondria increases oxidative phosphorylation by stimulating the activity of the F1F0-ATP synthase and enzymes within the TCA cycle, specifically alpha-ketoglutarate dehydrogenase, isocitrate dehydrogenase, and pyruvate dehydrogenase [88]. Mitochondrial activity is thus highly sensitive to calcium levels. Evidence suggests that along with increased ER–mitochondrial communication and elevated cytosolic calcium, mitochondrial calcium is elevated in AD. Due to MICU1 and MICU2/3 calcium-sensing and gating properties, increased cytosolic calcium can promote mitochondrial calcium uptake [74]. Multiple studies have also reported that expression of cytosolic calcium binding proteins calmodulin, calbindin D28K, and parvalbumin is reduced in AD patients and AD models, which would presumably free up calcium to bind MICU1 and MICU2/3 and activate MCU [89,90,91,92]. Therefore, it is unsurprising that mitochondrial calcium is elevated in AD models. Moreover, Abeta exposure has been shown to increase mitochondrial calcium levels in cortical neurons that promotes neurodegeneration, which can be reversed by blocking MCU [50]. There is evidence that the oligomeric form of Abeta produces calcium-permeable pores in the mitochondrial membrane, which promotes calcium uptake, suggesting that Abeta’s toxicity may result in part from its ability to disrupt mitochondrial calcium homeostasis [93,94]. Similarly, an additional study demonstrated that Abeta oligomers increased mitochondrial calcium levels by promoting ER calcium release, resulting in mitochondrial dysfunction [95]. Furthermore, a separate study looking at a mouse model with FAD mutations in PSEN1 and APP showed that these animals had elevated mitochondrial calcium [96]. Mitochondrial calcium levels are also increased in C. elegans presenilin mutants, which can be abrogated by preventing ER calcium release or mitochondrial calcium uptake, suggesting a conserved role for presenilin in maintaining mitochondrial calcium homeostasis by regulating ER calcium release at the MAMs [59]. Importantly, reduction of mitochondrial calcium levels in the C. elegans presenilin mutants restores neuronal function.
What are the consequences of elevated mitochondrial calcium? Although mitochondrial calcium homeostasis can be restored through calcium efflux pathways, which are regulated primarily through the sodium–calcium exchanger NCLX [97], excessive calcium uptake or impairments to calcium efflux can overwhelm mitochondrial calcium capacity. Mitochondrial calcium overload, which when combined with other stressors such as oxidative damage, results in the formation and opening of the mitochondrial permeability transition pore (mPTP) [98] (Figure 1). Although the molecular components of the mPTP are still under debate, the activity of the mPTP is known to span both the OMM and IMM, and its opening induces calcium efflux from the matrix. However, prolonged opening of the mPTP leaves the mitochondria open to the osmotic influx of cytosolic solutes and water, which causes the matrix to swell and rupture. Cytochrome c is also released from the mitochondria from prolonged mPTP opening, leading to the initiation of apoptosis. This process has been observed in several AD mouse models. In the aforementioned study showing elevated mitochondrial calcium in AD mice, high mitochondrial calcium correlated with the induction of apoptosis, whereas neurons containing mitochondria with normal calcium concentrations did not undergo apoptosis [96]. Another study showed that Abeta42 exposure could cause neuronal apoptosis by promoting mitochondrial calcium overload and stimulating the opening of the mPTP [99]. Excessive mitochondrial uptake in such contexts would present a particular problem for aged mitochondria, which are particularly vulnerable to high calcium levels, as they have lower calcium buffering capacities and are more susceptible to calcium overload [100].
5. Calcium-Induced Changes to Mitochondrial Activity Promote ROS Production
Elevated mitochondrial calcium has also been shown to disrupt neuronal function through the elevated production of reactive oxygen species (ROS) [101]. Mitochondrial oxidative respiration is a major source of ROS, as ROS are produced as a byproduct of the reduction of oxygen [102]. The energy used to pump H+ across the mitochondria’s inner membrane space is generated by a series of electron transfer reactions along the electron transport chain where each electron transport chain complex has increasingly greater reduction potential, the final electron acceptor being oxygen. Although electrons transferred sequentially along the electron transport chain will react at the end with oxygen to produce water, electrons may also be passed to oxygen prematurely, resulting in partial reduction of oxygen to superoxide anion (O2•−). This superoxide radical is a major source of oxidants and free radicals in the cell, as it reacts to produce hydrogen peroxide, which again reacts to produce the highly toxic hydroxyl radical (·OH). Although ROS at low levels provide important cellular functions by acting as signaling molecules, excessive ROS are toxic to the cell. As a site of superoxide generation, mitochondria are exposed to an especially high level of ROS, which can damage mitochondrial DNA and proteins over time, leading to severe mitochondrial damage and inefficiency. Dysfunctional mitochondria, in turn, generate greater levels of ROS; in this way, oxidative stress and mitochondrial dysfunction are closely linked. It has long been theorized that the aging process is caused in part by the progressive accumulation of ROS-induced cellular damage, both as a result of increased ROS generated by an inefficient electron transport chain or failure of antioxidant systems [103]. If the collective damage to proteins, DNA, and organelles induced by ROS is too great, the cell will undergo apoptosis.
Like mitochondrial dysfunction, oxidative stress plays a significant role in the pathogenesis of AD [104,105]. In vivo and in vitro studies show a direct relationship between oxidative stress and AD [105,106,107]. Furthermore, elevated ROS is highly correlated with the early stages of AD [108] and precedes Abeta plaque formation [109]. Oxidative stress can also promote tau hyperphosphorylation and fibril formation [110]. In a variety of AD animal models, high ROS levels have been shown to cause extensive neuronal death and cognitive decline [111]. Neurons are especially vulnerable to oxidative damage induced by mitochondrial respiration due to their especially high energy and oxygen demand, which in turn produces greater relative levels of ROS [98]. Their high lipid content also makes them vulnerable to lipid peroxidation by ·OH [98]. It has also been proposed that the regions of the brain that first undergo neurodegeneration in AD occur due to their increased susceptibility to oxidative damage, which can result from greater energy demands or higher basal levels of ROS required for signaling [112]. Elevated mitochondrial calcium levels have been associated with increased oxidative stress in AD models. Impairment to NCLX function that prevents mitochondrial calcium efflux in a mouse AD model resulted in excessive mitochondrial calcium and oxidative stress, which in turn led to amyloid and tau pathology and ultimately neuronal death [113]. Rescue of NCLX function was able to prevent cognitive defects in these animals. Buildup of mitochondrial ROS as a result of mitochondrial calcium overload also triggered apoptosis in a separate AD mouse model [99]. Mitochondrial calcium influx has also been shown to induce mitochondrial damage by stimulating oxidative phosphorylation, increasing the amount of ROS generated as a byproduct (Figure 1). Abeta oligomers also damage neurons by elevating lipid peroxidation and oxidative stress, further supporting the idea that oxidative damage underlies AD pathogenesis [114,115,116]. High mitochondrial calcium, fragmentation of mitochondria, oxidative stress and neuronal death have also been observed in APP/PS1 transgenic mice and in the brains of AD patients prior to Abeta formation [33,99,117]. Fibroblasts isolated from FAD patients showed both elevated oxidative respiration and ROS, which could be blocked either by preventing mitochondrial calcium uptake or by reducing respiration [94]. Similarly, in astrocytes differentiated from iPSCs derived from FAD patients bearing PSEN1 mutations, the mutant astrocytes showed higher oxygen consumption that resulted in greater ROS levels [118]. Notably, this alteration in metabolism was gamma-secretase independent. In C. elegans, neuronal and behavior defects resulting from presenilin dysfunction was dependent on elevated mitochondrial calcium-induced oxidative respiration and concomitant ROS generation [119]. Indeed, by limiting ER calcium release or mitochondrial calcium uptake in the C. elegans presenilin mutants reduced mitochondrial generated ROS and neurodegeneration. Furthermore, the neuronal defects observed in the C. elegans presenilin mutants were suppressed by treating these animals with the mitochondrial directed antioxidant, MitoTEMPO [94]. These studies indicate that aberrant mitochondrial activity induced by altered calcium signaling may be a mechanism by which FAD mutations generate ROS and lead to neuronal dysfunction. Therefore, this initial mitochondrial hyperactivity caused by aberrant mitochondrial calcium influx may in turn accelerate mitochondrial impairment, resulting in reduced ATP generation insufficient for neuronal synaptic transmission. Taken together, these data indicate that altered mitochondrial calcium homeostasis in FAD models leads to mitochondrial dysfunction and ROS production that promotes neurodegeneration.
6. Mitochondrial Function in Protein Homeostasis
The preservation of a highly functional proteome is vital for the aging nervous system. Indeed, protein misfolding and aggregation is a common manifestation observed in neurodegenerative diseases [120,121]. Therefore, the integrity of the protein homeostasis (proteostasis) network is critical for cell function and survival and is particularly important for non-dividing cells such as the neurons of the central nervous system. Insults that affect proteostasis include oxidative stress [122]. Accordingly, perturbations in mitochondrial function resulting in elevated production of ROS will greatly impact the maintenance of the proteome. Proteins that are irreversibly oxidized need to be removed by the proteostasis network; however, as organismal aging occurs, the efficiency of the proteostasis network diminishes, leading to protein misfolding and aggregation. In C. elegans, a clear link between altered calcium uptake into the mitochondria and the impact this has on proteostasis has been demonstrated. Mutations in the gene encoding presenilin in C. elegans result in elevated ER to mitochondrial calcium transfer. The elevation in mitochondrial calcium results in mitochondrial hyperactivity, namely increased oxidative phosphorylation and ROS production that promotes neuronal dysfunction [119]. The elevated oxidative stress caused by increased mitochondrial calcium leads to proteostasis defects in the presenilin mutants that can be rescued by inhibiting mitochondrial calcium uptake or by treating the animals with antioxidants [123] (Figure 1). The two main pathways involved in proteome maintenance are the ubiquitin-proteasome system and the autophagy-lysosome system. While the ubiquitin-proteasome activity appeared normal in the C. elegans presenilin mutants, there was a clear reduction in the formation of autophagosomes suggesting a defect in the autophagy-lysosome system [123]. Findings from FAD mouse and cell culture models also demonstrate that calcium dysregulation is linked to defects in autophagy [124]. Interestingly, the nutrient and energy sensing mechanistic target of rapamycin complex 1 (mTORC1) signaling pathway is a central inhibitor of autophagy and has been reported to be upregulated in AD as well as other neurodegenerative disorders [125,126,127,128,129]. However, the mechanism underlying the upregulation of mTORC1 signaling in these disorders is not clear. Nevertheless, these studies highlight the importance of mitochondrial calcium homeostasis and the impact it can have on proteostasis.
7. Mitochondrial Calcium-Dependent Subcellular Localization and Transport
As mentioned, trafficking and localization of mitochondria is essential for neuronal function. Mitochondria are extremely dynamic organelles undergoing constant fission and fusion events and they migrate along microtubule tracks to ensure their localization throughout the neuron. Principally, the fission and fusion events are regulated by three large GTPases, namely mitofusins (Mfn1/2), optic atrophy1 (OPA1) and dynamin related protein 1 (Drp1) and the trafficking along microtubules is mediated by the kinesin and dynein motors through the action of the adaptor protein Milton/Trak and the atypical Rho GTPase Miro [130,131,132]. These pathways are interconnected and are required to balance mitochondrial shape, morphology and function. Given the critical role of mitochondria in neurons, impaired mitochondrial dynamics is increasingly implicated in neurodegenerative diseases [133,134,135]. Additionally, mitochondrial fission and fusion dynamics have been associated in neurotransmitter release and terminal axon branching by regulating cytosolic calcium levels [136]. Trafficking of mitochondria is regulated in part by calcium and is mediated via Miro 1 and Miro 2 [137]. Miro proteins are transmembrane proteins that are localized to the OMM and contain two-conserved EF-hand calcium binding domains flanked by two Rho-like GTPase domains [133,138,139]. Miro interacts with Milton/Trak to link mitochondria to kinesin and dynein to allow for trafficking along the microtubule cytoskeleton [140,141]. When cytosolic calcium levels increase, calcium binds to the Miro EF-hands creating a conformational change that disengages Miro and the mitochondria from the microtubule network [132,142]. Consequently, an increase in calcium can pause the trafficking of mitochondria along the microtubules. It has also been demonstrated that Miro can influence mitochondrial trafficking by directly mediating mitochondrial calcium uptake via the MCU complex. In this study, the authors found that increased mitochondrial matrix calcium influx inversely correlated with mitochondrial trafficking speed [143]. Thus, cytosolic as well as mitochondrial calcium levels, can influence the localization and transport of mitochondria. In addition to mediating mitochondrial trafficking and mitochondrial calcium uptake, Miro proteins have also been implicated in several other mitochondrial functions, including IMM and OMM organization, mitochondrial fission, ER–mitochondrial contacts, and mitophagy. In both Drosophila and mammalian cells, Miro was shown to promote ER–mitochondrial interactions [144,145]. Moreover, in Drosophila, Miro was shown to facilitate ER to mitochondria calcium signaling [144,146]. In C. elegans and mammalian cells, Miro was shown to mediate mitochondrial fission upon an increase in cytosolic calcium that is independent of the conical Drp1 fission machinery [147,148]. Notably in mammalian cells, this fission event facilitates the turnover of mitochondria through the process of mitophagy, which is the selective removal of defective mitochondria by autophagy [147]. Furthermore, another study using human induced pluripotent stem cell-derived neurons from Parkinson’s disease patients found that targeted loss of Miro enables the removal of damaged mitochondria by mitophagy and promotes neuronal fitness [149]. It is unclear whether the trafficking function of Miro is required to mediate these additional activities of Miro (e.g., ER–mitochondria contact, fission or mitophagy). Nonetheless, these data highlight the pleiotropic role Miro and calcium have on mediating mitochondrial localization and function.
8. Conclusions
The influx of calcium into the mitochondria has long been established as a key regulator of many cellular homeostatic processes that range from energy production to cell death and necrosis [150,151]. The recent molecular discovery of the MCU complex components has provided critical insight into the role mitochondrial calcium influx has in energy production under increased work load but also, paradoxically, in promoting disease, such as neurodegeneration [152,153]. For example, studies in mice and C. elegans AD models have shown that there is an increase in mitochondrial calcium levels that can be reduced by blocking the MCU complex [99,119]. Critically, it was demonstrated in the mouse AD model that the high levels of mitochondrial calcium precede neuronal dysfunction and that this dysfunction can be rescued by inhibiting the MCU complex and blocking mitochondrial calcium uptake [99]. Similar observations have been made studying other neurodegenerative diseases [154,155,156]. For instance, recent work in zebrafish and Drosophila models of Parkinson’s disease have discovered elevated mitochondrial calcium levels that when reduced, could alleviate neurodegeneration [146,157]. These data highlight the significance mitochondrial calcium homeostasis has in neurodegeneration and point to the importance of understanding the mechanisms mediating mitochondrial calcium influx and activity. Indeed several recent review articles have suggested that targeting the MCU complex could be a potential therapeutic target for treating neurodegenerative diseases [158,159,160].
Author Contributions
Conceptualization, K.C.R., Z.A. and K.R.N.; writing—original draft preparation K.C.R. and Z.A.; writing—review and editing, K.C.R., Z.A. and K.R.N. All authors have read and agreed to the published version of the manuscript.
Funding
This work is funded by NIH grants AG064175 and GM088213.
Conflicts of Interest
The authors declare no conflict of interest.
References
- WHO. 10 Facts on Dementia; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Hebert, L.E.; Weuve, J.; Scherr, P.A.; Evans, D.A. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 2013, 80, 1778–1783. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.-H.; Zheng, H.; Zeng, L.-H.; Zhang, Y. The genes associated with early-onset Alzheimer’s disease. Oncotarget 2018, 9, 15132. [Google Scholar] [CrossRef] [PubMed]
- Price, J.L.; Morris, J.C. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1999, 45, 358–368. [Google Scholar] [CrossRef]
- Golde, T.E.; Schneider, L.S.; Koo, E.H. Anti-Aβ therapeutics in Alzheimer’s disease: The need for a paradigm shift. Neuron 2011, 69, 203–213. [Google Scholar] [CrossRef]
- Holmes, C.; Boche, D.; Wilkinson, D.; Yadegarfar, G. Long-term effects of Aβ42 immunisation in Alzheimer’s disease: Follow-up of a randomised, placebo-controlled phase I trial. Lancet 2008, 372, 216–223. [Google Scholar] [CrossRef]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef]
- Chételat, G. Aβ-independent processes—Rethinking preclinical AD. Nat. Rev. Neurol. 2013, 9, 123–124. [Google Scholar] [CrossRef]
- Sengupta, U.; Nilson, A.N.; Kayed, R. The role of amyloid-β oligomers in toxicity, propagation, and immunotherapy. EBioMedicine 2016, 6, 42–49. [Google Scholar] [CrossRef]
- Mucke, L.; Masliah, E.; Yu, G.-Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, H.T.; Gracia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemer, C.A.; et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Panza, F.; Lozupone, M.; Dibello, V.; Greco, A.; Daniele, A.; Serpia, D.; Logroscino, G.; Imbimbo, B.P. Are antibodies directed against amyloid-β (Aβ) oligomers the last call for the Aβ hypothesis of Alzheimer’s disease? Future Med. 2019, 11, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Chakrabarty, P.; Hanna, A.; March, A.; Dickson, D.W.; Borchelt, D.R.; Golde, T.; Janus, C. Normal cognition in transgenic BRI2-Aβ mice. Mol. Neurodegener. 2013, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.E.; Bertram, L. Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell 2005, 120, 545–555. [Google Scholar] [CrossRef]
- Saura, C.A.; Choi, S.-Y.; Beglupoulos, V.; Malkani, S.; Zhang, D.; Shankaranarayana Rao, B.S.; Chatarji, S.; Kelleher, R.J., 3rd; Kendel, E.R.; Duff, K.; et al. Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron 2004, 42, 23–36. [Google Scholar] [CrossRef]
- Wu, L.; Rosa-Neto, P.; Hsiung, G.R.; Sadovnik, D.A.; Masellis, M.; Black, S.E.; Jia, J.; Gauthier, S. Early-onset familial Alzheimer’s disease (EOFAD). Can. J. Neurol. Sci. 2012, 39, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Bekris, L.M.; Yu, C.E.; Bird, T.D.; Tsuang, D.W. Genetics of Alzheimer disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef]
- Morris, G.P.; Clark, I.A.; Vissel, B. Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2, 135. [Google Scholar] [CrossRef]
- Khachaturian, Z.S. Introduction and overview. Ann. N. Y. Acad. Sci. 1989, 568, 1–4. [Google Scholar] [CrossRef]
- Alzheimer’s Association Calcium Hypothesis Workgroup; Khachaturian, Z.S. Calcium hypothesis of Alzheimer’s disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimer’s Dement. 2017, 13, 178–182.e17. [Google Scholar]
- Morris, R.G. Long-term potentiation and memory. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2003, 358, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, E.M.; Vivar, C.; Camandola, S. Physiology and pathology of calcium signaling in the brain. Front. Pharmacol. 2012, 3, 61. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Cali, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell. Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.; Gibson, G.; Blass, J. Altered calcium uptake in cultured skin fibroblasts from patients with Alzheimer’s disease. N. Engl. J. Med. 1985, 312, 1063. [Google Scholar] [PubMed]
- Ito, E.; Oka, K.; Etcheberrigaray, R.; Nelson, T.J.; McPhie, D.L.; Tofel-Grehl, B.; Gibson, G.E.; Alkon, D.L. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc. Batl. Acad. Sci. USA 1994, 91, 534–538. [Google Scholar] [CrossRef]
- LaFerla, F.M. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef]
- Smith, I.F.; Hitt, B.; Green, K.N.; Oddo, S.; Laferla, F.M. Enhanced caffeine-induced Ca2+ release in the 3xTg-AD mouse model of Alzheimer’s disease. J. Neurochem. 2005, 94, 1711–1718. [Google Scholar] [CrossRef]
- Green, K.; Demuro, A.; Akbari, Y.; Hitt, B.D.; Smith, I.F.; Parker, I.; LaFerla, F.M. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J. Cell Biol. 2008, 181, 1107–1116. [Google Scholar] [CrossRef]
- Querfurth, H.W.; Selkoe, D.J. Calcium ionophore increases amyloid. beta. Peptide production by cultured cells. Biochemistry 1994, 33, 4550–4561. [Google Scholar] [CrossRef]
- Pierrot, N.; Ferrao Santos, S.; Feyt, C.H.; Morel, M.; Brion, J.-P.; Octave, J.-N. Calcium-mediated transient phosphorylation of tau and amyloid precursor protein followed by intraneuronal amyloid-β accumulation. J. Biol. Chem. 2006, 281, 39907–39914. [Google Scholar] [CrossRef]
- Emilsson, L.; Saetre, P.; Jazin, E. Alzheimer’s disease: mRNA expression profiles of multiple patients show alterations of genes involved with calcium signaling. Neurobiol. Dis. 2006, 21, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Supnet, C. Bezprozvanny I the dysregulation of intracellular calcium in Alzheimer disease. Cell Calcium. 2010, 47, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Greig, N.H.; Yu, Q.-S.; Mattson, M.P. Presenilin-1 mutation impairs cholinergic modulation of synaptic plasticity and suppresses NMDA currents in hippocampus slices. Neurobiol. Aging 2009, 30, 1061–1068. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tu, H.; Nelson, O.; Bezprozvanny, A.; Wang, Z.; Lee, S.-F.; Hao, Y.-H.; Serneels, L.; De Strooper, B.; Yu, G.; Bezprozvanny, I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 2006, 126, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.-H.; Mei, L.; Mak, D.O.; Hayashi, I.; Iwatsubo, T.; Kang, D.E.; Foskett, J.K. Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer’s disease–linked presenilin mutants in human cells and mouse neurons. Sci. Signal. 2010, 3, ra22. [Google Scholar] [CrossRef]
- Cheung, K.-H.; Shineman, D.; Muller, M.; Cardenas, C.; Mei, L.; Yang, J.; Tomita, T.; Iwatsubo, T.; Lee, V.M.; Foskett, J.K. Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron 2008, 58, 871–883. [Google Scholar] [CrossRef]
- Chan, S.L.; Mayne, M.; Holden, C.P.; Geiger, J.D.; Mattson, M.P. Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J. Biol. Chem. 2000, 275, 18195–18200. [Google Scholar] [CrossRef]
- Lee, S.Y.; Hwang, D.Y.; Kim, Y.K.; Lee, J.W.; Shin, I.C.; Oh, K.W.; Lee, M.K.; Lim, J.S.; Yoon, D.Y.; Hwang, S.J.; et al. PS2 mutation increases neuronal cell vulnerability to neurotoxicants through activation of caspase-3 by enhancing of ryanodine receptor-mediated calcium release. FASEB J. 2006, 20, 151–153. [Google Scholar]
- Stutzmann, G.E.; Caccamo, A.; Laferla, F.M.; Parker, I. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer’s-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J. Neurosci. 2004, 24, 508–513. [Google Scholar] [CrossRef]
- Etcheberrigaray, R.; Hirashima, N.; Nee, L.; Prince, J.; Govoni, S.; Racchi, M.; Tanzi, R.E.; Alkon, D.L. Calcium responses in fibroblasts from asymptomatic members of Alzheimer’s disease families. Neurobiol. Dis. 1998, 5, 37–45. [Google Scholar] [CrossRef]
- Briggs, C.A.; Chakroborty, S.; Stutzmann, G.E. Emerging pathways driving early synaptic pathology in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2017, 483, 988–997. [Google Scholar] [CrossRef] [PubMed]
- Rojas, G.; Cardenas, A.M.; Fernandez-Olivares, P.; Shimahara, T.; Segura-Aguilar, J.; Caviedes, R.; Caviedes, P. Effect of the knockdown of amyloid precursor protein on intracellular calcium increases in a neuronal cell line derived from the cerebral cortex of a trisomy 16 mouse. Exp. Neurol. 2008, 209, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Leissring, M.A.; Murphy, M.P.; Mead, T.R.; Akbari, Y.; Sugarman, M.C.; Jannatipour, P.; Anliker, B.; Muller, S.; Saftig, P.; De Strooper, B.; et al. A physiologic signaling role for the γ-secretase-derived intracellular fragment of APP. Proc. Natl. Acad. Sci. USA 2002, 99, 4697–4702. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, J.; Bali, J.; Rajendran, L.; Nitsch, R.M.; Tackenberg, C. Calcium flux-independent NMDA receptor activity is required for A β oligomer-induced synaptic loss. Cell Death Dis. 2015, 6, e1791. [Google Scholar] [CrossRef] [PubMed]
- Resende, R.; Ferreiro, E.; Pereira, C.; de Oliveira, C.R. Neurotoxic effect of oligomeric and fibrillar species of amyloid-beta peptide 1-42: Involvement of endoplasmic reticulum calcium release in oligomer-induced cell death. Neuroscience 2008, 155, 725–737. [Google Scholar] [CrossRef]
- Lackner, L.L.; Voeltz, G.K. The mechanisms and functions of interorganelle interactions. Mol. Biol. Cell 2017, 28, 703–704. [Google Scholar] [CrossRef]
- Pinton, P. Mitochondria-Associated Membranes (MAMs) and Pathologies. Cell Death Dis. 2018, 9, 413. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Hedskog, L.; Pinho, C.M.; Filadi, R.; Ronnback, A.; Hertwig, L.; Wiehager, B.; Larssen, P.; Gellhaar, S.; Sandebring, A.; Westerlund, M.; et al. Modulation of the endoplasmic reticulum–mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. USA 2013, 110, 7916–7921. [Google Scholar] [CrossRef]
- Fujimoto, M.; Hayashi, T. New insights into the role of mitochondria-associated endoplasmic reticulum membrane. Int. Rev. Cell Mol. Biol. 2011, 73–117. [Google Scholar]
- Roses, M.; Allen, D. Apolipoprotein E alleles as risk factors in Alzheimer’s disease. Annu. Rev. Med. 1996, 47, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Tambini, M.D.; Pera, M.; Kanter, E.; Yang, H.; Guardia-Laguarta, C.; Holtzman, D.; Sulzer, D.; Area-Gomez, E.; Schon, E.A. ApoE4 upregulates the activity of mitochondria-associated ER membranes. EMBO Rep. 2016, 17, 27–36. [Google Scholar] [CrossRef]
- Area-Gomez, E.; de Groof, A.J.; Boldogh, I.; Bird, T.D.; Gibson, G.E.; Koehler, C.M.; Yu, W.H.; Duff, K.E.; Yaffe, M.P.; Pon, L.A.; et al. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 2009, 175, 1810–1816. [Google Scholar] [CrossRef] [PubMed]
- Area-Gomez, E.; Del Camen Lara Castillo, M.; Tambini, M.D.; Guardia-Laguarta, C.; De Groof, A.J.; Madra, M.; Ikenouchi, J.; Umeda, M.; Bird, T.D.; Sturley, S.; et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012, 31, 4106–4123. [Google Scholar] [CrossRef] [PubMed]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)–mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, D.; Suski, J.M.; Oules, B.; Debayle, D.; Gay, A.S.; Lacas-Gervais, S.; Bussiere, R.; Bauer, C.; Pinton, P.; Paterlini-Brechot, P.; et al. Localization and processing of the amyloid-β protein precursor in mitochondria-associated membranes. J. Alzheimer’s Dis. 2017, 55, 1549–1570. [Google Scholar] [CrossRef] [PubMed]
- Daigle, I.; Li, C. Apl-1, a Caenorhabditis elegans gene encoding a protein related to the human beta-amyloid protein precursor. Proc. Natl. Acad. Sci. USA 1993, 90, 12045–12049. [Google Scholar] [CrossRef]
- Sarasija, S.; Norman, K.R. A γ-secretase independent role for presenilin in calcium homeostasis impacts mitochondrial function and morphology in Caenorhabditis elegans. Genetics 2015, 201, 1453–1466. [Google Scholar] [CrossRef]
- Schreiner, B.; Hedskog, L.; Weihager, H.; Ankarcrona, M. Amyloid-β peptides are generated in mitochondria-associated endoplasmic reticulum membranes. J. Alzheimer’s Dis. 2015, 43, 369–374. [Google Scholar] [CrossRef]
- Mink, J.W.; Blumenschine, R.J.; Adams, D.B. Ratio of central nervous system to body metabolism in vertebrates: Its constancy and functional basis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1981, 241, R203–R212. [Google Scholar] [CrossRef]
- Ames, A., III. CNS energy metabolism as related to function. Brain Res. Rev. 2000, 34, 42–68. [Google Scholar] [CrossRef]
- Sokoloff, L. The metabolism of the central nervous system in vivo. In Handbook of Physiology, Section I; Neurophysiology: Washington, DC, USA, 1960; Volume 3, pp. 1843–1864. [Google Scholar]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic energy use and supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, T.L. Mitochondrial trafficking in neurons. Cold Spring Harb. Perspect. Biol. 2013, 5, a011304. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Todorova, V.; Blokland, A. Mitochondria and synaptic plasticity in the mature and aging nervous system. Curr. Neuropharmacol. 2017, 15, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Cenini, G.; Voos, W. Mitochondria as potential targets in Alzheimer disease therapy: An update. Front. Pharmacol. 2019, 10, 902. [Google Scholar] [CrossRef]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 2–10. [Google Scholar] [CrossRef]
- Balaban, R.S. The role of Ca2+ signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim. Biophys. Acta (BBA) Bioenerg. 2009, 1787, 1334–1341. [Google Scholar] [CrossRef]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta (BBA) Bioenerg. 2009, 1787, 1309–1316. [Google Scholar] [CrossRef]
- Carafoli, E. The fateful encounter of mitochondria with calcium: How did it happen? Biochim. Biophys. Acta (BBA) Bioenerg. 2010, 1797, 595–606. [Google Scholar] [CrossRef]
- Ivannikov, M.V.; Macleod, G.T. Mitochondrial free Ca2+ levels and their effects on energy metabolism in Drosophila motor nerve terminals. Biophys. J. 2013, 104, 2353–2361. [Google Scholar] [CrossRef] [PubMed]
- Mishra, J.; Jhun, B.S.; Hurst, S.; O-Uchi, J.; Csordas, G.; Sheu, S.-S. The Mitochondrial Ca 2+ uniporter: Structure, function, and pharmacology. In Pharmacology of Mitochondria; Springer: Berlin/Heidelberg, Germany, 2017; pp. 129–156. [Google Scholar]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Trimme, C.A.; Sancak, Y.; Bao, X.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Kamer, K.J.; Mootha, V.K. The molecular era of the mitochondrial calcium uniporter. Nat. Rev. Mol. Cell Biol. 2015, 16, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Pallafacchina, G.; Zanin, S.; Rizzuto, R. Recent advances in the molecular mechanism of mitochondrial calcium uptake. F1000Research 2018, 7. [Google Scholar] [CrossRef]
- Patron, M.; Checchetto, V.; Raffaello, A.; Teardo, E.; Vecellio Reane, D.; Mantoan, M.; Granatiero, V.; Szabo, I.; De Stefani, D.; Rizzuto, R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 2014, 53, 726–737. [Google Scholar] [CrossRef]
- Perocchi, F.; Gohil, V.M.; Girgis, H.S.; Bao, X.R.; McCombs, J.E.; Palmer, A.E.; Mootha, V.K. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 2010, 467, 291–296. [Google Scholar] [CrossRef]
- Plovanich, M.; Bogorad, R.L.; Sancak, Y.; Kamer, K.J.; Strittmatter, L.; Li, A.A.; Girgis, H.S.; Kuchimanchi, S.; De Groot, J.; Speciner, L.; et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS ONE 2013, 8, e55785. [Google Scholar] [CrossRef]
- Xing, Y.; Wang, M.; Wang, J.; Nie, Z.; Wu, G.; Yang, X.; Shen, Y. Dimerization of MICU proteins controls Ca2+ influx through the mitochondrial Ca2+ uniporter. Cell Rep. 2019, 26, 1203–1212.e4. [Google Scholar] [CrossRef]
- Patron, M.; Granatiero, V.; Espino, J.; Rizzuto, R.; De Stefani, D. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 2019, 26, 179–195. [Google Scholar] [CrossRef]
- Wang, C.; Jacewicz, A.; Delgado, B.D.; Baradaran, R.; Long, S.B. Structures reveal gatekeeping of the mitochondrial Ca2+ uniporter by MICU1-MICU2. eLife 2020, 9, e59991. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Zhang, J.; Tsai, C.-W.; Orlando, B.J.; Rodriguez, M.; Xu, Y.; Liao, M.; Tsao, M.-F.; Feng, L. Structure and mechanism of the mitochondrial Ca2+ uniporter holocomplex. Nature 2020, 582, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Schwaller, B. Cytosolic Ca2+ buffers. Cold Spring Harb. Perspect. Biol. 2010, 2, a004051. [Google Scholar] [CrossRef] [PubMed]
- Pivovarova, N.B.; Andrews, S.B. Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 2010, 277, 3622–3636. [Google Scholar] [CrossRef]
- Jouaville, L.; Pinton, P.; Bastianutto, C.; Rutter, G.A.; Rizzuto, R. Regulation of mitochondrial ATP synthesis by calcium: Evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. USA 1999, 96, 13807–13812. [Google Scholar] [CrossRef]
- Riascos, D.; de Leon, D.; Baker-Neigh, A.; Nicholas, A.; Yukhananov, R.; Bu, J.; Wu, C.-K.; Geula, C. Age-related loss of calcium buffering and selective neuronal vulnerability in Alzheimer’s disease. Acta Neuropathol. 2011, 122, 565. [Google Scholar] [CrossRef]
- McLachlan, D.R.; Wong, L.; Bergeron, C.; Baimbridge, K.G. Calmodulin and calbindin D28K in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1987, 1, 171–179. [Google Scholar] [CrossRef]
- Ahmadian, S.S.; Rezvanian, A.; Peterson, M.; Weintraub, S.; Bigio, E.H.; Mesulam, M.M.; Geula, C. Loss of calbindin-D28K is associated with the full range of tangle pathology within basal forebrain cholinergic neurons in Alzheimer’s disease. Neurobiol. Aging 2015, 36, 3163–3170. [Google Scholar] [CrossRef]
- Ali, F.; Baringer, S.L.; Neal, A.; Cho, E.Y.; Kwan, A.C. Parvalbumin-Positive Neuron Loss and Amyloid-β Deposits in the Frontal Cortex of Alzheimer’s Disease-Related Mice. J. Alzheimer’s Dis. 2019, 1–17. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Hartley, D.; Petre, B.M.; Walz, T.; Lansbuty, P.T., Jr. Neurodegenerative disease: Amyloid pores from pathogenic mutations. Nature 2002, 418, 291. [Google Scholar] [CrossRef]
- Shirwany, N.A.; Payette, D.; Xie, J.; Guo, Q. The amyloid beta ion channel hypothesis of Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 2007, 3, 597. [Google Scholar] [PubMed]
- Ferreira, I.L.; Ferreiro, E.; Schmidt, J.; Cardoso, J.M.; Pereira, C.M.; Carvalho, A.L.; Oliveira, C.R.; Rego, A.C. Aβ and NMDAR activation cause mitochondrial dysfunction involving ER calcium release. Neurobiol. Aging 2015, 36, 680–692. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Blasco, S.; Valero, R.A.; Rodriguez-Crespo, I.; Villalobos, C.; Nunez, L. Mitochondrial Ca2+ overload underlies Aβ oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS ONE 2008, 3, e2718. [Google Scholar] [CrossRef] [PubMed]
- Boyman, L.; Williams, G.S.; Khananshvili, D.; Sekler, I.; Lederer, W.J. NCLX: The mitochondrial sodium calcium exchanger. J. Mol. Cell. Cardiol. 2013, 59, 205–213. [Google Scholar] [CrossRef]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The role of oxidative stress in neurodegenerative diseases. Exp. Neurobiol. 2015, 24, 325. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Hou, S.H.; Synder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; Fan, Z.; Muzikansky, A.; Gracia-Alloza, M.; Serrano-Pozo, A.; et al. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Panel, M.; Ghaleh, B.; Morin, D. Mitochondria and aging: A role for the mitochondrial transition pore? Aging Cell 2018, 17, e12793. [Google Scholar] [CrossRef]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.-S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Balaban, R.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- Birnbaum, J.H.; Wanner, D.; Geitl, A.F.; Saake, A.; Kundig, T.; Hock, C.; Nitsch, R.M.; Tackenberg, C. Oxidative stress and altered mitochondrial protein expression in the absence of amyloid-β and tau pathology in iPSC-derived neurons from sporadic Alzheimer’s disease patients. Stem Cell Res. 2018, 27, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Pohanka, M. Alzheimer s disease and oxidative stress: A review. Curr. Med. Chem. 2014, 21, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; Allard, J.; Bixier, R.; Usoh, C.; Li, L.; May, J.M.; McDonald, M.P. Antioxidants and cognitive training interact to affect oxidative stress and memory in APP/PSEN1 mice. Nutr. Neurosci. 2009, 12, 203–218. [Google Scholar] [CrossRef] [PubMed]
- Tönnies, E.; Trushina, E. Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [PubMed]
- de Leeuw, F.A.; Peeters, C.F.W.; Kester, M.I.; Harms, A.C.; Struys, E.A.; Hankemeier, T.; van Vlijmen, H.; van der Lee, S.; van Dujin, C.M.; Scheltens, P.; et al. Blood-based metabolic signatures in Alzheimer’s disease. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2017, 8, 196–207. [Google Scholar] [CrossRef]
- Su, B.; Wang, X.; Nunomura, A.; Moreira, P.I.; Lee, H.G.; Perry, G.; Smith, M.A.; Zhu, X. Oxidative stress signaling in Alzheimer’s disease. Curr. Alzheimer Res. 2008, 5, 525–532. [Google Scholar] [CrossRef]
- Liu, Q.; Smith, M.A.; Avila, J.; DeBernardis, J.; Kansal, M.; Takeda, A.; Zhu, X.; Nunomura, A.; Honda, K.; Moreira, P.; et al. Alzheimer-specific epitopes of tau represent lipid peroxidation-induced conformations. Free Radic. Biol. Med. 2005, 38, 746–754. [Google Scholar] [CrossRef]
- LaFerla, F.M.; Green, K.N. Animal models of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006320. [Google Scholar] [CrossRef]
- Wang, X.; Michaelis, E. Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2010, 2, 1–13. [Google Scholar] [CrossRef]
- Jadiya, P.; Kolmetzky, D.W.; Tomar, D.; Di Meco, A.; Lombardi, A.A.; Lambert, J.P.; Luongo, T.S.; Ludtmann, M.H.; Pratico, D.; Elord, J.W. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 2019, 10, 3885. [Google Scholar] [CrossRef]
- Tamagno, E.; Bardini, P.; Guglielmotto, M.; Danni, O.; Tabaton, M. The various aggregation states of β-amyloid 1–42 mediate different effects on oxidative stress, neurodegeneration, and BACE-1 expression. Free Radic. Biol. Med. 2006, 41, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Castegna, A.; Lauderback, C.M.; Drake, J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiol. Aging 2002, 23, 655–664. [Google Scholar] [CrossRef]
- Alberdi, E.; Sanchez-Gomez, M.V.; Cavaliere, F.; Perez-Samartin, A.; Zugaza, J.L.; Trullas, R.; Domercq, M.; Matute, C. Amyloid β oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium 2010, 47, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Rodriguez, M.; Bacskai, B.J. High mitochondrial calcium levels precede neuronal death in vivo in Alzheimer’s disease. Cell Stress 2020, 4, 187. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, M.; Peterson, A.J.; Naumenko, N.; Puttonen, K.; Lehtonen, S.; Gubert Olive, M.; Shakrizyanova, A.; Leskela, S.; Sarajarvi, T.; Viitanen, M.; et al. PSEN1 mutant iPSC-derived model reveals severe astrocyte pathology in Alzheimer’s disease. Stem Cell Rep. 2017, 9, 1885–1897. [Google Scholar] [CrossRef]
- Sarasija, S.; Norman, K.R. Role of presenilin in mitochondrial oxidative stress and neurodegeneration in Caenorhabditis elegans. Antioxidants 2018, 7, 111. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef]
- Di Domenico, F.; Head, E.; Butterfield, D.A.; Perluigi, M. Oxidative stress and proteostasis network: Culprit and casualty of Alzheimer’s-like neurodegeneration. Adv. Geriatr. 2014, 2014, 527518. [Google Scholar] [CrossRef]
- Ashkavand, Z.; Sarasija, S.; Ryan, K.C.; Laboy, J.T.; Norman, K.R. Corrupted ER-mitochondrial calcium homeostasis promotes the collapse of proteostasis. Aging Cell 2020, 19, e13065. [Google Scholar] [CrossRef]
- Fedeli, C.; Filadi, R.; Rossi, A.; Mammucari, C.; Pizzo, P. PSEN2 (presenilin 2) mutants linked to familial Alzheimer disease impair autophagy by altering Ca2+ homeostasis. Autophagy 2019, 15, 2044–2062. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, K.; Miller, M.; Chludzinski, A.; Herrup, K.; Zagorski, M.; Lamb, B.T. The PI3K-Akt-mTOR pathway regulates Aβ oligomer induced neuronal cell cycle events. Mol. Neurodegener. 2009, 4, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; Maldonado, M.A.; Majumder, S.; Medina, D.X.; Holbein, W.; Magri, A.; Oddo, S. Naturally secreted amyloid-β increases mammalian target of rapamycin (mTOR) activity via a PRAS40-mediated mechanism. J. Biol. Chem. 2011, 286, 8924–8932. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Guan, K.-L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Sabatini, D.M. Author Correction: mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 246. [Google Scholar] [CrossRef]
- Tramutola, A.; Lanzillota, C.; Perluigi, M.; Butterfield, D.A. Oxidative stress, protein modification and Alzheimer disease. Brain Res. Bull. 2017, 133, 88–96. [Google Scholar] [CrossRef]
- Hirokawa, N.; Niwa, S.; Tanaka, Y. Mwolecular motors in neurons: Transport mechanisms and roles in brain function, development, and disease. Neuron 2010, 68, 610–638. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 2006, 22, 79–99. [Google Scholar] [CrossRef]
- MacAskill, A.F.; Rinholm, J.E.; Twelevetrees, A.E.; Arancibia-Carcamo, I.L.; Muir, J.; Fransson, A.; Aspenstrom, P.; Atwell, D.; Kittler, J.T. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 2009, 61, 541–555. [Google Scholar] [CrossRef]
- Kerr, J.S.; Adriaanese, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s disease: Cellular and molecular mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Schon, E.A.; Przedborski, S. Mitochondria: The next (neurode)generation. Neuron 2011, 70, 1033–1053. [Google Scholar] [CrossRef] [PubMed]
- Lewis, T.L.; Kwon, S.-K.; Lee, A.; Shaw, R.; Polleux, F. MFF-dependent mitochondrial fission regulates presynaptic release and axon branching by limiting axonal mitochondria size. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Saotome, M.; Safiliulina, D.; Szabadkai, G.; Das, S.; Fransson, A.; Aspenstrom, P.; Rizzuto, R.; Hajnoczky, G. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc. Natl. Acad. Sci. USA 2008, 105, 20728–20733. [Google Scholar] [CrossRef]
- Fransson, Å.; Ruusala, A.; Aspenström, P. The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem. Biophys. Res. Commun. 2006, 344, 500–510. [Google Scholar] [CrossRef]
- Fransson, Å.; Ruusala, A.; Aspenström, P. Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis. J. Biol. Chem. 2003, 278, 6495–6502. [Google Scholar] [CrossRef]
- Brickley, K.; Stephenson, F.A. Trafficking kinesin protein (TRAK)-mediated transport of mitochondria in axons of hippocampal neurons. J. Biol. Chem. 2011, 286, 18079–18092. [Google Scholar] [CrossRef]
- Glater, E.E.; Megeath, L.J.; Stowers, R.S.; Schawaz, T.L. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J. Cell Biol. 2006, 173, 545–557. [Google Scholar] [CrossRef]
- Wang, X.; Schwarz, T.L. The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell 2009, 136, 163–174. [Google Scholar] [CrossRef]
- Chang, K.T.; RNiescier, F.; Min, K.-T. Mitochondrial matrix Ca2+ as an intrinsic signal regulating mitochondrial motility in axons. Proc. Natl. Acad. Sci. USA 2011, 108, 15456–15461. [Google Scholar] [CrossRef]
- Lee, S.; Lee, K.-S.; Huh, S.; Liu, S.; Lee, D.-Y.; Hong, S.H.; Yu, K.; Lu, B. Polo kinase phosphorylates miro to control ER-mitochondria contact sites and mitochondrial Ca2+ homeostasis in neural stem cell development. Dev. Cell 2016, 37, 174–189. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Lopez-Domenech, G.; Halff, E.F.; Covill-Cooke, C.; Ivankovic, D.; Melandri, D.; Arancibia- Carcamo, I.L.; Burden, J.J.; Lowe, A.R.; Kittler, J.T. Miro clusters regulate ER-mitochondria contact sites and link cristae organization to the mitochondrial transport machinery. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-S.; Huh, S.; Lee, S.; Wu, Z.; Kim, A.-K.; Kang, H.-Y.; Lu, B. Altered ER–mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc. Natl. Acad. Sci. USA 2018, 115, E8844–E8853. [Google Scholar] [CrossRef] [PubMed]
- Nemani, N.; Carvalho, E.; Tomar, D.; Dong, Z.; Ketschek, A.; Breves, S.L.; Jana, F.; Worth, A.M.; Heffler, J.; Palaniappan, P.; et al. MIRO-1 determines mitochondrial shape transition upon GPCR activation and Ca2+ stress. Cell Rep. 2018, 23, 1005–1019. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Zhou, H.; Yu, X.; Xu, J.; Zhou, J.; Meng, X.; Zhao, J.; Zhou, Y.; Chisholm, A.D.; XU, S. Wounding triggers MIRO-1 dependent mitochondrial fragmentation that accelerates epidermal wound closure through oxidative signaling. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Hsieh, C.-H.; Shaltouki, A.; Gonzalez, A.E.; Battencourt da Cruz, A.; Burbulla, L.F.; St Lawrence, E.; Schule, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional impairment in miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson’s disease. Cell Stem Cell 2016, 19, 709–724. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Pinton, P. Publisher Correction: The machineries, regulation and cellular functions of mitochondrial calcium. Nature reviews. Mol. Cell Biol. 2018, 19, 746. [Google Scholar] [CrossRef]
- Granatiero, V.; de Stefani, D.; Rizzuto, R. Mitochondrial calcium handling in physiology and disease. In Mitochondrial Dynamics in Cardiovascular Medicine; Springer: Berlin/Heidelberg, Germany, 2017; pp. 25–47. [Google Scholar]
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1464–1472. [Google Scholar] [CrossRef]
- Liao, Y.; Dong, Y.; Cheng, J. The function of the mitochondrial calcium uniporter in neurodegenerative disorders. Int. J. Mol. Sci. 2017, 18, 248. [Google Scholar] [CrossRef]
- Ham, S.J.; Lee, D.; Yoo, H.; Jun, K.; Shin, H.; Chung, J. Decision between mitophagy and apoptosis by Parkin via VDAC1 ubiquitination. Proc. Natl. Acad. Sci. USA 2020, 117, 4281–4291. [Google Scholar] [CrossRef]
- Ludtmann, M.H.R.; Kostik, M.; Horne, A.; Gandhi, S.; Sekler, I.; Abramov, A.Y. LRRK2 deficiency induced mitochondrial Ca 2+ efflux inhibition can be rescued by Na+/Ca2+/Li+ exchanger upregulation. Cell Death Dis. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Callio, J.; Otero, P.A.; Sekler, I.; Wills, Z.P.; Chu, C.T. Mitochondrial calcium dysregulation contributes to dendrite degeneration mediated by PD/LBD-associated LRRK2 mutants. J. Neurosci. 2017, 37, 11151–11165. [Google Scholar] [CrossRef] [PubMed]
- Soman, S.K.; Bazala, M.; Keatinge, M.; Bandmann, O.; Kuznicki, J. Restriction of mitochondrial calcium overload by mcu inactivation renders a neuroprotective effect in zebrafish models of Parkinson’s disease. Biol. Open 2019, 8, bio044347. [Google Scholar] [CrossRef] [PubMed]
- Woods, J.J.; Wilson, J.J. Inhibitors of the mitochondrial calcium uniporter for the treatment of disease. Curr. Opin. Chem. Biol. 2020, 55, 9–18. [Google Scholar] [CrossRef]
- Dey, K.; Bazala, M.A.; Kuznicki, J. Targeting mitochondrial calcium pathways as a potential treatment against Parkinson’s disease. Cell Calcium 2020, 16, 102216. [Google Scholar] [CrossRef]
- Venugopal, A.; Iyer, M.; Balasubramanian, V.; Vellingiri, B. Mitochondrial calcium uniporter as a potential therapeutic strategy for Alzheimer’s disease. Acta Neuropsychiatr. 2020, 32, 65–71. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).