Impact of AHR Ligand TCDD on Human Embryonic Stem Cells and Early Differentiation

 , , , ,
, , , ,
Abstract
1. Introduction
2. Results
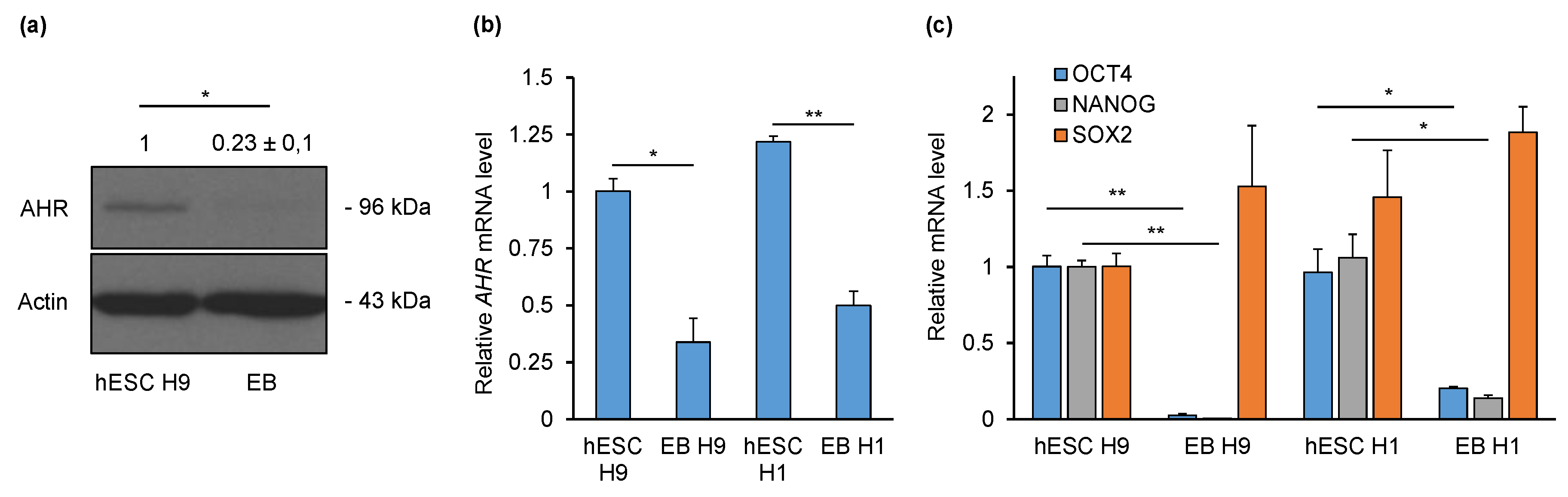
2.1. AHR Is Downregulated during Non-Directed Differentiation into Embryoid Bodies (EBs)
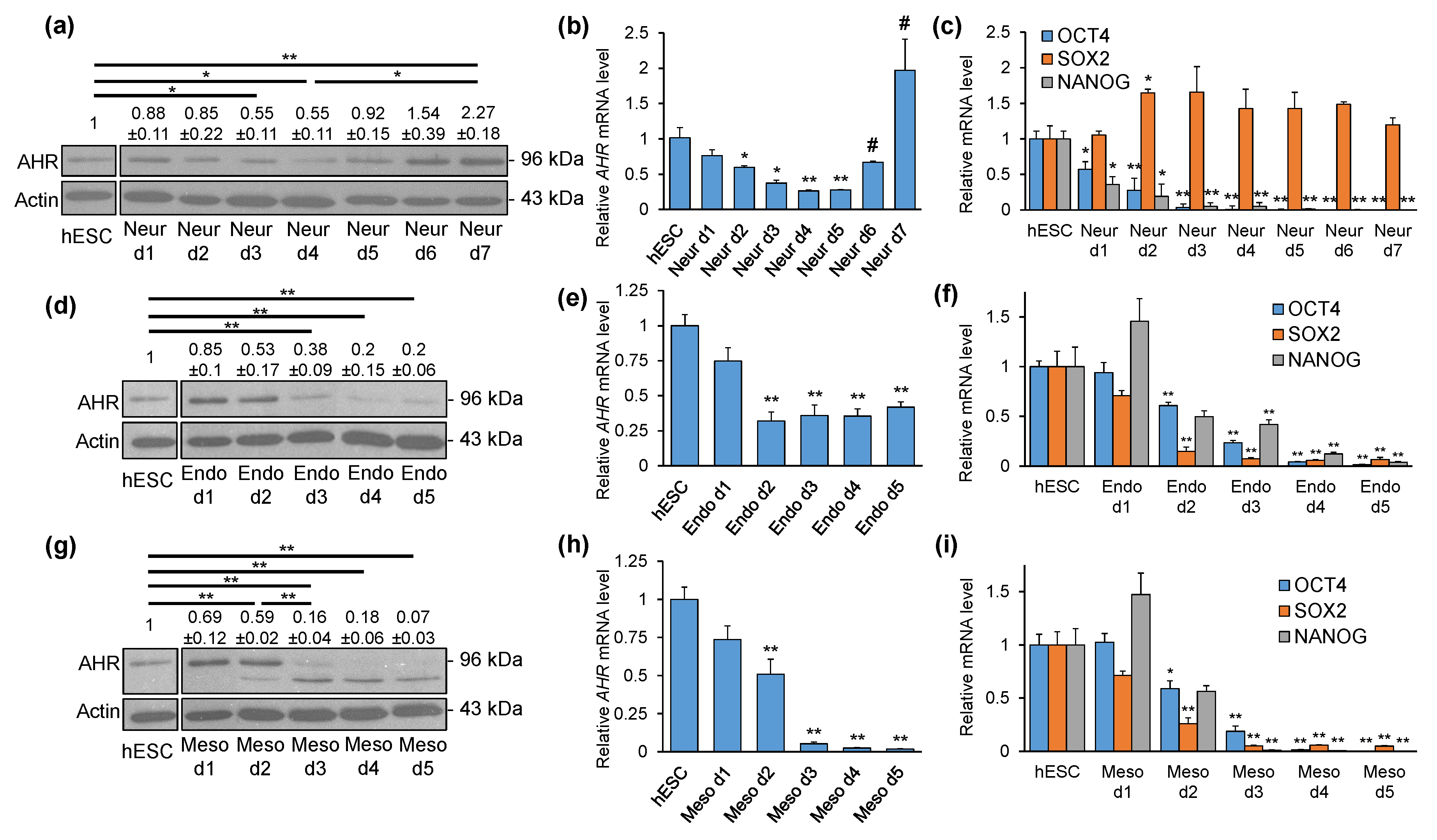
2.2. AHR Expression Shows Distinct Patterns during Directed Differentiation into Three Lineages
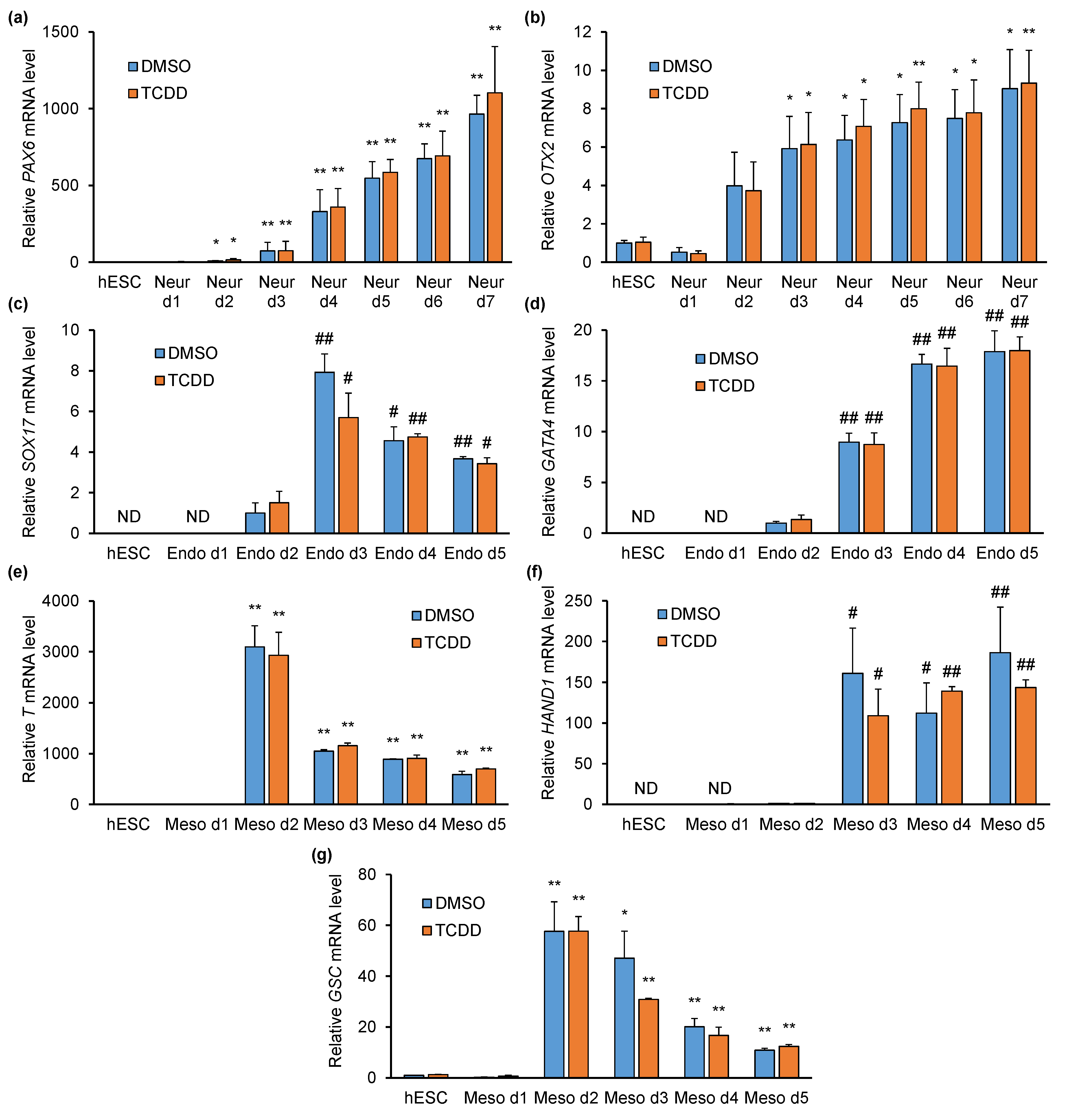
2.3. TCDD Does Not Affect Pluripotency- or Differentiation-Specific Marker Gene Expression
2.4. Impact of TCDD on hESCs and Early Differentiation
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Embryoid Body Formation
4.3. Directed Differentiation of hESCs
4.4. RNA Isolation and Measurement of mRNA Levels
4.5. Western Blotting
4.6. Flow Cytometry
4.7. RNA-seq
4.8. ChIP-seq
4.9. ATAC-seq
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AHR | Aryl hydrocarbon receptor |
| AIP | Aryl hydrocarbon receptor interacting protein |
| ARNT | Aryl hydrocarbon receptor nuclear translocator |
| bHLH/PAS | Basic helix-loop-helix/PER-ARNT-SIM |
| DMSO | Dimethyl sulfoxide |
| EB | Embryoid body |
| EXOC2 | Exocyst complex component 2 |
| hESC | Human embryonic stem cell |
| HSP90 | Heat shock protein 90 |
| LRAT | Lecithin retinol acyltransferase |
| MAP2 | Microtubule-associated protein 2 |
| RORA | Retinoic acid receptor-related orphan receptor alpha |
| TCDD | 2,3,7,8-tetrachlorodibenzo-p-dioxin |
| TH | Tyrosine hydroxylase |
| TIPARP | TCDD-inducible poly(ADP-ribose) polymerase |
References
- Heid, S.E.; Pollenz, R.S.; Swanson, H.I. Role of heat shock protein 90 dissociation in mediating agonist-induced activation of the aryl hydrocarbon receptor. Mol. Pharmacol. 2000, 57, 82–92. [Google Scholar] [PubMed]
- Lees, M.J.; Whitelaw, M.L. Multiple Roles of Ligand in Transforming the Dioxin Receptor to an Active Basic Helix-Loop-Helix/PAS Transcription Factor Complex with the Nuclear Protein Arnt. Mol. Cell. Biol. 1999, 19, 5811–5822. [Google Scholar] [CrossRef] [PubMed]
- Poland, A.; Glover, E.; Kende, A.S. Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. Evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. J. Biol. Chem. 1976, 251, 4936–4946. [Google Scholar] [PubMed]
- Lu, P.; Yan, J.; Liu, K.; Garbacz, W.G.; Wang, P.; Xu, M.; Ma, X.; Xie, W. Activation of aryl hydrocarbon receptor dissociates fatty liver from insulin resistance by inducing fibroblast growth factor 21. Hepatology 2015, 61, 1908–1919. [Google Scholar] [CrossRef] [PubMed]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef]
- Barnett, K.R.; Tomic, D.; Gupta, R.K.; Babus, J.K.; Roby, K.F.; Terranova, P.F.; Flaws, J.A. The aryl hydrocarbon receptor is required for normal gonadotropin responsiveness in the mouse ovary. Toxicol. Appl. Pharmacol. 2007, 223, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Boitano, A.E.; Wang, J.; Romeo, R.; Bouchez, L.C.; Parker, A.E.; Sutton, S.E.; Walker, J.R.; Flaveny, C.A.; Perdew, G.H.; Denison, M.S.; et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science 2010, 329, 1345–1348. [Google Scholar] [CrossRef]
- Hope, K.J.; Cellot, S.; Ting, S.B.; MacRae, T.; Mayotte, N.; Iscove, N.N.; Sauvageau, G. An RNAi Screen Identifies Msi2 and Prox1 as Having Opposite Roles in the Regulation of Hematopoietic Stem Cell Activity. Cell Stem Cell 2010, 7, 101–113. [Google Scholar] [CrossRef]
- Rentas, S.; Holzapfel, N.T.; Belew, M.S.; Pratt, G.A.; Voisin, V.; Wilhelm, B.T.; Bader, G.D.; Yeo, G.W.; Hope, K.J. Musashi-2 attenuates AHR signalling to expand human haematopoietic stem cells. Nature 2016, 532, 508–511. [Google Scholar] [CrossRef]
- Neri, T.; Merico, V.; Fiordaliso, F.; Salio, M.; Rebuzzini, P.; Sacchi, L.; Bellazzi, R.; Redi, C.A.; Zuccotti, M.; Garagna, S. The differentiation of cardiomyocytes from mouse embryonic stem cells is altered by dioxin. Toxicol. Lett. 2011, 202, 226–236. [Google Scholar] [CrossRef]
- Wang, Q.; Hisaka, K.; Carreira, V.; Ko, C.I.; Fan, Y.; Zhang, X.; Biesiada, J.; Medvedovic, M.; Puga, A. Ah receptor activation by dioxin disrupts activin, BMP, and WNT signals during the early differentiation of mouse embryonic stem cells and inhibits cardiomyocyte functions. Toxicol. Sci. 2016, 149, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Wang, L.; Wang, J.; Bennett, B.D.; Li, J.L.; Zhao, B.; Hu, G. Dioxin and AHR impairs mesoderm gene expression and cardiac differentiation in human embryonic stem cells. Sci. Total Environ. 2019, 651, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Latchney, S.E.; Lioy, D.T.; Henry, E.C.; Gasiewicz, T.A.; Strathmann, F.G.; Mayer-Pröschel, M.; Opanashuk, L.A. Neural precursor cell proliferation is disrupted through activation of the aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Stem Cells Dev. 2011, 20, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Kubi, J.A.; Chen, A.C.H.; Fong, S.W.; Lai, K.P.; Wong, C.K.C.; Yeung, W.S.B.; Lee, K.F.; Lee, Y.L. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on the differentiation of embryonic stem cells towards pancreatic lineage and pancreatic beta cell function. Environ. Int. 2019, 130, 104885. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Wiley, L.M. Evidence That Murine Preimplantation Embryos Express Aryl Hydrocarbon Receptor. Toxicol. Appl. Pharmacol. 1995, 134, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Ohsako, S.; Baba, T.; Miyamoto, K.; Tohyama, C. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on preimplantation mouse embryos. Toxicology 2002, 174, 119–129. [Google Scholar] [CrossRef]
- Ko, C.I.; Wang, Q.; Fan, Y.; Xia, Y.; Puga, A. Pluripotency factors and Polycomb Group proteins repress aryl hydrocarbon receptor expression in murine embryonic stem cells. Stem Cell Res. 2014, 12, 296–308. [Google Scholar] [CrossRef]
- Ko, C.-I.; Fan, Y.; de Gannes, M.; Wang, Q.; Xia, Y.; Puga, A. Repression of the Aryl Hydrocarbon Receptor Is Required to Maintain Mitotic Progression and Prevent Loss of Pluripotency of Embryonic Stem Cells. Stem Cells 2016, 34, 2825–2839. [Google Scholar] [CrossRef]
- Yamamoto, T.; Hatabayashi, K.; Arita, M.; Yajima, N.; Takenaka, C.; Suzuki, T.; Takahashi, M.; Oshima, Y.; Hara, K.; Kagawa, K.; et al. Kynurenine signaling through the aryl hydrocarbon receptor maintains the undifferentiated state of human embryonic stem cells. Sci. Signal. 2019, 12, eaaw3306. [Google Scholar] [CrossRef]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef]
- Fong, H.; Hohenstein, K.A.; Donovan, P.J. Regulation of Self-Renewal and Pluripotency by Sox2 in Human Embryonic Stem Cells. Stem Cells 2008, 26, 1931–1938. [Google Scholar] [CrossRef] [PubMed]
- Diacou, R.; Zhao, Y.; Zheng, D.; Cvekl, A.; Liu, W. Six3 and Six6 Are Jointly Required for the Maintenance of Multipotent Retinal Progenitors through Both Positive and Negative Regulation. Cell Rep. 2018, 25, 2510–2523. [Google Scholar] [CrossRef] [PubMed]
- Sears, A.E.; Palczewski, K. Lecithin:Retinol Acyltransferase: A Key Enzyme Involved in the Retinoid (visual) Cycle. Biochemistry 2016, 55, 3082–3091. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.N.; Kang, H.S.; Jetten, A.M. Retinoic Acid-Related Orphan Receptors (RORs): Regulatory Functions in Immunity, Development, Circadian Rhythm, and Metabolism. Nucl. Recept. Res. 2015. [Google Scholar] [CrossRef]
- Mullen, R.D.; Colvin, S.C.; Hunter, C.S.; Savage, J.J.; Walvoord, E.C.; Bhangoo, A.P.S.; Ten, S.; Weigel, J.; Pfäffle, R.W.; Rhodes, S.J. Roles of the LHX3 and LHX4 LIM-homeodomain factors in pituitary development. Mol. Cell. Endocrinol. 2007, 265, 190–195. [Google Scholar] [CrossRef]
- Bai, Q.; Assou, S.; Haouzi, D.; Ramirez, J.-M.; Monzo, C.; Becker, F.; Gerbal-Chaloin, S.; Hamamah, S.; De Vos, J. Dissecting the First Transcriptional Divergence During Human Embryonic Development. Stem Cell Rev. Rep. 2012, 8, 150–162. [Google Scholar] [CrossRef]
- Sarma, S.; Nagano, R.; Ohsako, S. Tyroxine Hydroxylase-Positive Neuronal Cell Population is Increased by Temporal Dioxin Exposure at Early Stage of Differentiation from Human Embryonic Stem Cells. Int. J. Mol. Sci. 2019, 20, 2687. [Google Scholar] [CrossRef]
- Lipchina, I.; Studer, L.; Betel, D. The expanding role of miR-302-367 in pluripotency and reprogramming. Cell Cycle 2012, 11, 1517–1523. [Google Scholar] [CrossRef]
- Balzano, F.; Cruciani, S.; Basoli, V.; Santaniello, S.; Facchin, F.; Ventura, C.; Maioli, M. MiR200 and MiR302: Two big families influencing stem cell behavior. Molecules 2018, 23, 282. [Google Scholar] [CrossRef]
- Mimura, J.; Yamashita, K.; Nakamura, K. Loss of teratogenic response to 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin (TCDD) in mice lacking the Ah (dioxin) receptor. Genes Cells 1997, 2, 645–654. [Google Scholar] [CrossRef]
- Thomae, T.L.; Stevens, E.A.; Bradfield, C.A. Transforming Growth Factor-β3 Restores Fusion in Palatal Shelves Exposed to 2,3,7,8-Tetrachlorodibenzo-p-dioxin. J. Biol. Chem. 2005, 280, 12742–12746. [Google Scholar] [CrossRef] [PubMed]
- Revich, B.; Aksel, E.; Ushakova, T.; Ivanova, I.; Zhuchenko, N.; Klyuev, N.; Brodsky, B.; Sotskov, Y. Dioxin exposure and public health in Chapaevsk, Russia. Chemosphere 2001, 3, 951–966. [Google Scholar] [CrossRef]
- Carreira, V.S.; Fan, Y.; Kurita, H.; Wang, Q.; Ko, C.-I.; Naticchioni, M.; Jiang, M.; Koch, S.; Zhang, X.; Biesiada, J.; et al. Disruption of Ah Receptor Signaling during Mouse Development Leads to Abnormal Cardiac Structure and Function in the Adult. PLoS ONE 2015, 10, e0142440. [Google Scholar] [CrossRef] [PubMed]
- Ngwa, E.N.; Kengne, A.-P.; Tiedeu-Atogho, B.; Mofo-Mato, E.-P.; Sobngwi, E. Persistent organic pollutants as risk factors for type 2 diabetes. Diabetol. Metab. Syndr. 2015, 7, 41. [Google Scholar] [CrossRef]
- Lee, D.H.; Porta, M.; Jacobs, D.R.; Vandenberg, L.N. Chlorinated persistent organic pollutants, obesity, and type 2 diabetes. Endocr. Rev. 2014, 35, 557–601. [Google Scholar] [CrossRef]
- Thiel, R.; Koch, E.; Ulbrich, B.; Chahoud, I. Peri- and postnatal exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin: Effects on physiological development, reflexes, locomotor activity and learning behaviour in Wistar rats. Arch. Toxicol. 1994, 69, 79. [Google Scholar] [CrossRef]
- Markowski, V.P.; Cox, C.; Preston, R.; Weiss, B. Impaired cued delayed alternation behavior in adult rat offspring following exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin on gestation day 15. Neurotoxicol. Teratol. 2002, 24, 209–218. [Google Scholar] [CrossRef]
- Boersma, E.R.; Lanting, C.I. Environmental exposure to polychlorinated biphenyls (PCBs) and dioxins: Consequences for longterm neurological and cognitive development of the child lactation. Adv. Exp. Med. Biol. 2000, 478, 271–287. [Google Scholar] [CrossRef]
- Cheng, J.; Li, W.; Kang, B.; Zhou, Y.; Song, J.; Dan, S.; Yang, Y.; Zhang, X.; Li, J.; Yin, S.; et al. Tryptophan derivatives regulate the transcription of Oct4 in stem-like cancer cells. Nat. Commun. 2015, 6, 7209. [Google Scholar] [CrossRef]
- Stanford, E.A.; Wang, Z.; Novikov, O.; Mulas, F.; Landesman-Bollag, E.; Monti, S.; Smith, B.W.; Seldin, D.C.; Murphy, G.J.; Sherr, D.H. The role of the aryl hydrocarbon receptor in the development of cells with the molecular and functional characteristics of cancer stem-like cells. BMC Biol. 2016, 14, 20. [Google Scholar] [CrossRef]
- Ozturk, F.; Li, Y.; Zhu, X.; Guda, C.; Nawshad, A. Systematic analysis of palatal transcriptome to identify cleft palate genes within TGFβ3-knockout mice alleles: RNA-Seq analysis of TGFβ3 Mice. BMC Genom. 2013, 14, 113. [Google Scholar] [CrossRef] [PubMed]
- Rochette, C.; Jullien, N.; Saveanu, A.; Caldagues, E.; Bergada, I.; Braslavsky, D.; Pfeifer, M.; Reynaud, R.; Herman, J.P.; Barlier, A.; et al. Identifying the deleterious effect of rare LHX4 allelic variants, a challenging issue. PLoS ONE 2015, 10, e0126648. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, W.; Tohyama, C. Mechanisms of Developmental Toxicity of Dioxins and Related Compounds. Int. J. Mol. Sci. 2019, 20, 617. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Valen, E.; Sandelin, A.; Matthews, J. Dioxin increases the interaction between aryl hydrocarbon receptor and estrogen receptor alpha at human promoters. Toxicol. Sci. 2009, 111, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Lo, R.; Celius, T.; Forgacs, A.L.; Dere, E.; MacPherson, L.; Harper, P.; Zacharewski, T.; Matthews, J. Identification of aryl hydrocarbon receptor binding targets in mouse hepatic tissue treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Appl. Pharmacol. 2011, 257, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Lo, R.; Matthews, J. High-Resolution Genome-wide Mapping of AHR and ARNT Binding Sites by ChIP-Seq. Toxicol. Sci. 2012, 130, 349–361. [Google Scholar] [CrossRef]
- Teino, I.; Kuuse, S.; Ingerpuu, S.; Maimets, T.; Tiido, T. The Aryl Hydrocarbon Receptor Regulates Mouse Fshr Promoter Activity Through an E-Box Binding Site1. Biol. Reprod. 2012. [Google Scholar] [CrossRef]
- Huang, G.; Elferink, C.J. A Novel Nonconsensus Xenobiotic Response Element Capable of Mediating Aryl Hydrocarbon Receptor-Dependent Gene Expression. Mol. Pharmacol. 2012, 81, 338–347. [Google Scholar] [CrossRef]
- DuSell, C.D.; Nelson, E.R.; Wittmann, B.M.; Fretz, J.A.; Kazmin, D.; Thomas, R.S.; Pike, J.W.; McDonnell, D.P. Regulation of aryl hydrocarbon receptor function by selective estrogen receptor modulators. Mol. Endocrinol. 2010, 24, 33–46. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, Y.; Puga, A. Dioxin exposure disrupts the differentiation of mouse embryonic stem cells into cardiomyocytes. Toxicol. Sci. 2010, 115, 225–237. [Google Scholar] [CrossRef]
- Henry, E.C.; Welle, S.L.; Gasiewicz, T.A. TCDD and a putative endogenous AhR Ligand, ITE, elicit the same immediate changes in gene expression in mouse lung fibroblasts. Toxicol. Sci. 2009, 114, 90–100. [Google Scholar] [CrossRef] [PubMed]
- McMillan, B.J.; McMillan, S.N.; Glover, E.; Bradfield, C.A. 2,3,7,8-Tetrachlorodibenzo-p-dioxin induces premature activation of the KLF2 regulon during thymocyte development. J. Biol. Chem. 2007, 282, 12590–12597. [Google Scholar] [CrossRef] [PubMed]
- Portal-Nuñez, S.; Shankavaram, U.T.; Rao, M.; Datrice, N.; Atay, S.; Aparicio, M.; Camphausen, K.A.; Fernández-Salguero, P.M.; Chang, H.; Lin, P.; et al. Aryl hydrocarbon receptor-induced adrenomedullin mediates cigarette smoke carcinogenicity in humans and mice. Cancer Res. 2012, 72, 5790–5800. [Google Scholar] [CrossRef] [PubMed]
- Boverhof, D.R.; Burgoon, L.D.; Tashiro, C.; Sharratt, B.; Chittim, B.; Harkema, J.R.; Mendrick, D.L.; Zacharewski, T.R. Comparative toxicogenomic analysis of the hepatotoxic effects of TCDD in Sprague Dawley rats and C57BL/6 mice. Toxicol. Sci. 2006, 94, 398–416. [Google Scholar] [CrossRef]
- Nilsson, C.B.; Hoegberg, P.; Trossvik, C.; Azaïs-Bræsco, V.; Blaner, W.S.; Fex, G.; Harrison, E.H.; Nau, H.; Schmidt, C.K.; van Bennekum, A.M.; et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin Increases Serum and Kidney Retinoic Acid Levels and Kidney Retinol Esterification in the Rat. Toxicol. Appl. Pharmacol. 2000, 169, 121–131. [Google Scholar] [CrossRef]
- Hamilton, B.A.; Frankel, W.N.; Kerrebrock, A.W.; Hawkins, T.L.; FitzHugh, W.; Kusumi, K.; Russell, L.B.; Mueller, K.L.; van Berkel, V.; Birren, B.W.; et al. Disruption of the nuclear hormone receptor RORalpha in staggerer mice. Nature 1996, 379, 736–739. [Google Scholar] [CrossRef]
- Bergen, N.J.V.; Ahmed, S.M.; Collins, F.; Cowley, M.; Vetro, A.; Dale, R.C.; Hock, D.H.; De Caestecker, C.; Minal, M.; Massey, S.; et al. Mutations in the exocyst component EXOC2 cause severe defects in human brain development. J. Exp. Med. 2020. [Google Scholar] [CrossRef]
- Giovannoni, F.; Bosch, I.; Polonio, C.M.; Torti, M.F.; Wheeler, M.A.; Li, Z.; Romorini, L.; Rodriguez Varela, M.S.; Rothhammer, V.; Barroso, A.; et al. AHR is a Zika virus host factor and a candidate target for antiviral therapy. Nat. Neurosci. 2020, 23, 939–951. [Google Scholar] [CrossRef]
- Thompson, D.A.; Li, Y.; McHenry, C.L.; Carlson, T.J.; Ding, X.; Sieving, P.A.; Apfelstedt-Sylla, E.; Gal, A. Mutations in the gene encoding lecithin retinol acyltransferase are associated with early-onset severe retinal dystrophy. Nat. Genet. 2001, 28, 123–124. [Google Scholar] [CrossRef]
- Chevallier, A.; Mialot, A.; Petit, J.M.; Fernandez-Salguero, P.; Barouki, R.; Coumoul, X.; Beraneck, M. Oculomotor Deficits in Aryl Hydrocarbon Receptor Null Mouse. PLoS ONE 2013, 8, e53520. [Google Scholar] [CrossRef]
- Oliver, G.; Mailhos, A.; Wehr, R.; Copeland, N.G.; Jenkins, N.A.; Gruss, P. Six3, a murine homologue of the sine oculis gene, demarcates the most anterior border of the developing neural plate and is expressed during eye development. Development 1995, 121, 4045–4055. [Google Scholar] [PubMed]
- Jean, D.; Bernier, G.; Gruss, P. Six6 (Optx2) is a novel murine Six3-related homeobox gene that demarcates the presumptive pituitary/hypothalamic axis and the ventral optic stalk. Mech. Dev. 1999, 84, 31–40. [Google Scholar] [CrossRef]
- Toy, J.; Sundin, O.H. Expression of the Optx2 homeobox gene during mouse development. Mech. Dev. 1999, 83, 183–186. [Google Scholar] [CrossRef]
- Teijeiro, V.; Yang, D.; Majumdar, S.; González, F.; Rickert, R.W.; Xu, C.; Koche, R.; Verma, N.; Lai, E.C.; Huangfu, D. DICER1 Is Essential for Self-Renewal of Human Embryonic Stem Cells. Stem Cell Rep. 2018, 11, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Houbaviy, H.B.; Murray, M.F.; Sharp, P.A. Embryonic stem cell-specific microRNAs. Dev. Cell 2003, 5, 351–358. [Google Scholar] [CrossRef]
- Suh, M.-R.; Lee, Y.; Kim, J.Y.; Kim, S.-K.; Moon, S.-H.; Lee, J.Y.; Cha, K.-Y.; Chung, H.M.; Yoon, H.S.; Moon, S.Y.; et al. Human embryonic stem cells express a unique set of microRNAs. Dev. Biol. 2004, 270, 488–498. [Google Scholar] [CrossRef]
- Hu, W.; Zhao, J.; Pei, G. Activation of aryl hydrocarbon receptor (AhR) by tranilast, an anti-allergy drug, promotes miR-302 expression and cell reprogramming. J. Biol. Chem. 2013, 288, 22972–22984. [Google Scholar] [CrossRef]
- Alarcón, C.R.; Goodarzi, H.; Lee, H.; Liu, X.; Tavazoie, S.; Tavazoie, S.F. HNRNPA2B1 Is a Mediator of m6A-Dependent Nuclear RNA Processing Events. Cell 2015, 162, 1299–1308. [Google Scholar] [CrossRef]
- Villarroya-Beltri, C.; Gutiérrez-Vázquez, C.; Sánchez-Cabo, F.; Pérez-Hernández, D.; Vázquez, J.; Martin-Cofreces, N.; Martinez-Herrera, D.J.; Pascual-Montano, A.; Mittelbrunn, M.; Sánchez-Madrid, F. Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat. Commun. 2013, 4, 2980. [Google Scholar] [CrossRef]
- Grudzien-Nogalska, E.; Jiao, X.; Song, M.G.; Hart, R.P.; Kiledjian, M. Nudt3 is an mRNA decapping enzyme that modulates cell migration. RNA 2016, 22, 773–781. [Google Scholar] [CrossRef]
- Ngo, T.D.; Partin, A.C.; Nam, Y. RNA Specificity and Autoregulation of DDX17, a Modulator of MicroRNA Biogenesis. Cell Rep. 2019, 29, 4024–4035. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-T.; Chiou, S.-S.; Chai, C.-Y.; Hsi, E.; Yokoyama, K.K.; Wang, S.-N.; Huang, S.-K.; Hsu, S.-H. Intestine-Specific Homeobox Gene ISX Integrates IL6 Signaling, Tryptophan Catabolism, and Immune Suppression. Cancer Res. 2017, 77, 4065–4077. [Google Scholar] [CrossRef] [PubMed]
- Matvere, A.; Teino, I.; Varik, I.; Kuuse, S.; Tiido, T.; Kristjuhan, A.; Maimets, T. Fsh/lh-dependent upregulation of ahr in murine granulosa cells is controlled by pka signaling and involves epigenetic regulation. Int. J. Mol. Sci. 2019, 20, 3068. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Teino, I.; Matvere, A.; Kuuse, S.; Ingerpuu, S.; Maimets, T.; Kristjuhan, A.; Tiido, T. Transcriptional repression of the Ahr gene by LHCGR signaling in preovulatory granulosa cells is controlled by chromatin accessibility. Mol. Cell. Endocrinol. 2014, 382, 292–301. [Google Scholar] [CrossRef]
- Ervin, E.H.; Pook, M.; Teino, I.; Kasuk, V.; Trei, A.; Pooga, M.; Maimets, T. Targeted gene silencing in human embryonic stem cells using cell-penetrating peptide PepFect 14 06 Biological Sciences 0604 Genetics. Stem Cell Res. Ther. 2019, 10, 43. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Dahl, J.A.; Collas, P. A quick and quantitative chromatin immunoprecipitation assay for small cell samples. Front. Biosci. 2007, 12, 4925–4931. [Google Scholar] [CrossRef][Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; Kaul, R.; et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, a.D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nussbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based Analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- McLean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Ortiz, J.A.; Taing, L.; Meyer, C.A.; Lee, B.; Zhang, Y.; Shin, H.; Wong, S.S.; Ma, J.; Lei, Y.; et al. Cistrome: An integrative platform for transcriptional regulation studies. Genome Biol. 2011, 12, R83. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21–29. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RNA-seq | ChIP-seq | ||||
|---|---|---|---|---|---|
| Gene | Fold Change | p-Value | AHR Binding | Distance Relative to TSS | |
| DMSO | TCDD | ||||
| AHRR | 2.26 | 3.50 × 10−3 | + | +69,749 | |
| + | +95,863 | ||||
| APOL3 | 3.6 | 2.40 × 10−2 | + | +15,455 | |
| CASQ1 | 1.94 | 3.50 × 10−2 | + | −18,394 | |
| CCKBR | 1.64 | 1.00 × 10−2 | + | +44,133 | |
| CTIF | 1.41 | 2.60 × 10e−2 | + | −539 | |
| CUZD1 | 2.54 | 1.70 × 10e−6 | + | + | −25,162 |
| CYP1A1 | 10 | 8.40 × 10−11 | + | −702 | |
| CYP1B1 | 2.73 | 2.00 × 10−3 | + | −779 | |
| EXOC2 | 1.38 | 1.10 × 10−3 | + | + | +9040 |
| GALNT5 | 4.04 | 2.10 × 10−3 | + | −11,039 | |
| LHX1 | 2.1 | 4.00 × 10−2 | + | + | +129,491 |
| LHX4 | 3.64 | 3.20 × 10−5 | + | −168 | |
| + | +912 | ||||
| LINC00886 | 4.21 | 4.5 × 10−6 | + | + | 0 |
| LRAT | 8.57 | 7.70 × 10−7 | + | −2640 | |
| + | + | −1296 | |||
| + | −212 | ||||
| LRP4 | 1.3 | 2.80 × 10−2 | + | +14,565 | |
| LRRTM3 | 2.58 | 3.00 × 10−2 | + | −32,899 | |
| MYADML2 | 5.4 | 3.70 × 10−6 | + | +5254 | |
| PCDH8 | 3.02 | 5.50× 10−3 | + | −3158 | |
| PDP1 | 1.66 | 1.90 × 10−3 | + | −70,169 | |
| + | −448 | ||||
| PKNOX2 | 1.52 | 3.30 × 10−2 | + | +98,739 | |
| PXDNL | 4.5 | 1.4 × 10−4 | + | +191,524 | |
| RORA | 1.9 | 2.48 × 10−5 | + | + | −177,729 |
| SIX3 | 2.46 | 6.10 × 10−3 | + | −11,295 | |
| SIX6 | 9.43 | 1.50 × 10−10 | + | +4316 | |
| SLC16A12 | 2.41 | 6.00 × 10−3 | + | +1255 | |
| SLC27A2 | 1.6 | 2.10 × 10−2 | + | +47 | |
| TIPARP | 4.82 | 1.34 × 10−34 | + | + | +142,217 |
| TESC | 1.66 | 3.60 × 10−3 | + | +46,627 | |
| RNA-seq | ChIP-seq | ||||
|---|---|---|---|---|---|
| Gene | Fold Change | p-Value | AHR Binding (hESC) | Distance Relative to TSS | |
| DMSO | TCDD | ||||
| CDCA7L | 1.39 | 2.9 × 10−2 | + | + | −120,620 |
| CUZD1 | −10.16 | 4.2 × 10−2 | + | + | −25,162 |
| MYADML2 | −28.61 | 7.4 × 10−4 | + | +5254 | |
| S1PR1 | 2.55 | 3.0 × 10−2 | + | +73,148 | |
| RNA-seq | ChIP-seq | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Gene | Fold Change | p-Value | AHR Binding | Distance Relative to TSS | |||||
| hESC | Endo | Neur | |||||||
| DMSO | TCDD | DMSO | TCDD | DMSO | TCDD | ||||
| ADM | −2.59 | 4.9 × 10−2 | + | +46,975 | |||||
| AK4 | 1.75 | 2.9 × 10−2 | + | +131,446 | |||||
| AOAH | −3.9 | 3.2 × 10−2 | + | +65,017 | |||||
| CBX4 | −4.6 | 2.8 × 10−5 | + | −110,655 | |||||
| CCNG1 | 1.32 | 3.6 × 10−2 | + | +387 | |||||
| CRABP1 | 3.1 | 3.8 × 10−2 | + | +3175 | |||||
| CYP1A1 | −9.07 | 4.1 × 10−2 | + | + | −702 | ||||
| EPC2 | −1.43 | 1.3 × 10−2 | + | + | + | + | + | +450 | |
| FGFR3 | 1.71 | 1.8 × 10−2 | + | −15,013 | |||||
| GADD45G | −4.29 | 2.0 × 10−3 | + | +72,997 | |||||
| GSE1 | −1.70 | 6.9 × 10−3 | + | + | −29,402 | ||||
| HSPB8 | −5.34 | 1.5 × 10−3 | +124,278 | ||||||
| LRAT | 3.79 | 1.6 × 10−3 | + | −2640 | |||||
| + | + | + | −1296 | ||||||
| + | −212 | ||||||||
| MAP3K8 | 1.51 | 2.2 × 10−2 | + | + | +2436 | ||||
| SLC7A5 | −1.91 | 4.4 × 10−2 | + | −1151 | |||||
| SLC25A25 | −1.46 | 3.2 × 10−2 | + | + | + | −896 | |||
| RNA-seq | ChIP-seq | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Gene | Fold Change | p-Value | AHR Binding | Distance Relative to TSS | |||||
| hESC | Endo | Neur | |||||||
| DMSO | TCDD | DMSO | TCDD | DMSO | TCDD | ||||
| AHRR | 3.95 | 2.2 × 10−6 | + | + | +69,749 | ||||
| + | + | +95,863 | |||||||
| C3orf80 | −9.2 | 4.7 × 10−3 | + | −186,661 | |||||
| CCDC60 | 1.84 | 2.7 × 10−2 | + | −31,792 | |||||
| CCKBR | 2.45 | 2.3 × 10−6 | + | + | +44,133 | ||||
| CRYBB1 | −2.84 | 4.8 × 10−2 | + | + | + | −30,267 | |||
| CYP1A1 | 34.78 | 1.4 × 10−30 | + | + | −702 | ||||
| CYP1B1 | 7.6 | 1.2 × 10−9 | + | −20,263 | |||||
| + | + | −779 | |||||||
| CYP27A1 | 1.65 | 2.2 × 10−2 | + | +24,031 | |||||
| GRB7 | 2.84 | 2.0 × 10−5 | + | +62,663 | |||||
| HS3ST5 | 3.07 | 2.6 × 10−6 | + | + | −20,093 | ||||
| IKZF3 | 7.62 | 1.0 × 10−3 | + | +62,753 | |||||
| LINC00886 | 2.02 | 7.5 × 10−3 | + | + | + | + | + | 0 | |
| LRAT | 3.85 | 1.3 × 10−3 | + | −2640 | |||||
| + | + | + | −1296 | ||||||
| + | −212 | ||||||||
| PDP1 | 1.49 | 1.5 × 10−2 | + | + | −70,169 | ||||
| + | −448 | ||||||||
| TPRA1 | 1.44 | 2.4 × 10−3 | + | + | + | + | + | −376 | |
| RUNX1T1 | 3.38 | 1.3 × 10−2 | + | + | + | −8068 | |||
| TIPARP | 2.27 | 6.96 × 10−9 | + | + | + | + | + | +142,217 | |
| TSC22D1 | 1.34 | 4.0 × 10−2 | + | +179,610 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teino, I.; Matvere, A.; Pook, M.; Varik, I.; Pajusaar, L.; Uudeküll, K.; Vaher, H.; Trei, A.; Kristjuhan, A.; Org, T.; et al. Impact of AHR Ligand TCDD on Human Embryonic Stem Cells and Early Differentiation. Int. J. Mol. Sci. 2020, 21, 9052. https://doi.org/10.3390/ijms21239052
Teino I, Matvere A, Pook M, Varik I, Pajusaar L, Uudeküll K, Vaher H, Trei A, Kristjuhan A, Org T, et al. Impact of AHR Ligand TCDD on Human Embryonic Stem Cells and Early Differentiation. International Journal of Molecular Sciences. 2020; 21(23):9052. https://doi.org/10.3390/ijms21239052
Chicago/Turabian StyleTeino, Indrek, Antti Matvere, Martin Pook, Inge Varik, Laura Pajusaar, Keyt Uudeküll, Helen Vaher, Annika Trei, Arnold Kristjuhan, Tõnis Org, and et al. 2020. "Impact of AHR Ligand TCDD on Human Embryonic Stem Cells and Early Differentiation" International Journal of Molecular Sciences 21, no. 23: 9052. https://doi.org/10.3390/ijms21239052
APA StyleTeino, I., Matvere, A., Pook, M., Varik, I., Pajusaar, L., Uudeküll, K., Vaher, H., Trei, A., Kristjuhan, A., Org, T., & Maimets, T. (2020). Impact of AHR Ligand TCDD on Human Embryonic Stem Cells and Early Differentiation. International Journal of Molecular Sciences, 21(23), 9052. https://doi.org/10.3390/ijms21239052