Danger-Sensing/Patten Recognition Receptors and Neuroinflammation in Alzheimer’s Disease



Abstract
1. Alzheimer’s Disease (AD)
2. Glia Roles in AD
3. Danger-Sensing/Pattern Recognition Receptors (PRRs)
3.1. Scavenger Receptors (SRs)
3.1.1. Triggering Receptor Expressed on Myeloid Cells 2 (TREM2) and 1 (TREM1)
3.1.2. Receptor for Advanced Glycation Endproducts (RAGE)
3.1.3. CD36 Superfamily SRs
3.2. Toll-Like Receptors (TLRs)
3.3. NOD (Nucleotide-Binding Oligomerization Domain) Receptors and (NOD)-Like Leucine-Rich Repeat Receptors (NLRs) Inflammasomes
3.3.1. NLRP1 Inflammasome
3.3.2. NLRP2 Inflammasome
3.3.3. NLRP3 Inflammasome
3.3.4. AIM2-Like Receptors
3.3.5. NLRC4 Inflammasome
3.3.6. IPAF Inflammasome
3.3.7. NOD Inflammasomes
3.3.8. NLRP10/PYNOD Inflammasome
3.4. C-Type Lectin (Dectin-1, CLEC-2) Receptors (CLRs)
3.5. RIG-I (Retinoid Acid-Inducible Gene-I)-Like Receptors
3.6. Calcium-Sensing Receptor (CaSR)
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Aβs | Amyloid-β peptides |
| Ab(RAGE) | anti-RAGE antibody |
| AD | Alzheimer’s disease |
| AGEs | advanced glycation endproducts |
| AIM2 | absent in melanoma 2 protein |
| APOE | Apolipoprotein E |
| APP | amyloid precursor protein |
| ASC | apoptosis-associated speck-like adaptor protein containing a CARD |
| Aβ-os | Aβ oligomers |
| BBB | blood-brain barrier |
| BECs/CECs | brain/cerebral endothelial cells |
| C1q | complement component 1q |
| CAA | cerebral amyloid angiopathy |
| cAMP | 3′,5′-cyclic adenosine monophosphate |
| CARD | caspase activation and recruitment domain |
| CaSR | calcium-sensing receptor |
| CCL | C-C Motif Chemokine Ligand |
| CLEC-2 | C-type lectin-like immune receptor 2 |
| CLRs | C-type lectin receptors |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| DAMPs | damage-associated molecular patterns |
| DAP12/TYROBP | DNAX activating protein |
| Dectin-1 | Dendritic cell (DC)-associated C-type lectin-1 cluster |
| DS | Down’s syndrome |
| ERK1/2 | extracellular signal-regulated kinase 1/2 |
| GdCl3 | Gadolinium trichloride |
| GFAP | glial fibrillar acidic protein |
| GMF | glia maturation factor |
| GPCRs | G-protein-coupled receptors |
| GSK-3β | glycogen synthase kinase-3β |
| HDL | high-density lipoprotein |
| HMGB1 | high-mobility group box B1 |
| hNSCs | human neural stem cells |
| ICAM-1 | Intercellular Adhesion Molecule-1 |
| IPAF | ICE protease-activating factor |
| ITAM | immune-receptor tyrosine-based activation motif |
| JEV | Japanese encephalitis virus |
| JNK | c-JUN terminal kinase |
| LPS | lipopolysaccharide |
| MAPK | mitogen activated protein kinase |
| MCI | mild cognitive impairment |
| MCP | Monocyte Chemotactic Protein |
| MDP | muramyl dipeptide |
| mTOR | mammalian target of rapamycin |
| NACHT | NAIP plus CIITA (MHC class II transcription activator) plus HET-E (incompatibility locus protein from Podospora anserina) and TP1 (telomerase-associated protein) |
| NAHAs | nontumorigenic adult human astrocytes |
| NAIP | neuronal apoptosis inhibitor protein |
| NAM | negative allosteric modulator |
| NF-κB | nuclear factor-κB |
| NFT | neurofibrillary tangle |
| NLR | NOD-like receptor |
| NLRP | Nucleotide-binding domain, leucine-rich repeat and PYRIN domain containing protein |
| NO | nitric oxide |
| NOD | nucleotide-binding oligomerization domain |
| NOS-2 | nitric oxide synthase-2 |
| oxLDLs | oxidized low-density lipoproteins |
| p-Tau-es | hyperphosphorylated Tau proteins |
| p-Tau-os | p-Tau oligomers |
| PAMPs | pathogen-associated molecular patterns |
| PAMs | positive allosteric modulators |
| Panx-1 | Pannexin 1 protein |
| PI3K | Phosphoinositide 3-kinase |
| PICs | proinflammatory cytokines |
| PKR | double-stranded RNA-activated protein kinase |
| PPRs | pattern recognition receptors |
| PYHIN | N-terminal pyrin domain (PYD) plus hemopoietic inducible nuclear (HIN) proteins |
| PYRIN/DAPIN | Domain in Apoptosis and Interferon response |
| RAGE | receptor for AGEs |
| RANTES | Regulated upon Activation, normal T cell Expressed and presumably Secreted |
| RCT | reverse cholesterol transport |
| RIG | retinoic acid-inducible gene |
| ROS | reactive oxygen species |
| SRs | scavenger receptors |
| sTREM2 | soluble TREM2 |
| Syk | spleen tyrosine kinase |
| TAM | Tyro3, Axl, and Mer receptor |
| TLRs | Toll-like receptors |
| Toll-like | similar to Drosophila Toll gene |
| TREM | triggering receptor expressed on myeloid cells |
| VEGF-A | vascular endothelial growth factor-A |
References
- Prince, M.J.; Wimo, A.; Guerchet, M.M.; Ali, G.C.; Wu, Y.-T.; Prina, M. World Alzheimer Report 2015. The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and Trends. Available online: https://www.alzint.org (accessed on 21 September 2015).
- Knopman, D.S.; Haeberlein, S.B.; Carrillo, M.C.; Hendrix, J.A.; Kerchner, G.; Margolin, R.; Maruff, P.; Miller, D.S.; Tong, G.; Tome, M.B.; et al. The National Institute on Aging and the Alzheimer’s Association Research framework for Alzheimer’s disease: Perspectives from the research roundtable. Alzheimers Dement. 2018, 14, 563–575. [Google Scholar] [CrossRef]
- Labzin, L.I.; Heneka, M.T.; Latz, E. Innate immunity and neurodegeneration. Annu. Rev. Med. 2018, 69, 437–449. [Google Scholar] [CrossRef]
- Becher, B.; Spath, S.; Goverman, J. Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 2017, 17, 49–59. [Google Scholar] [CrossRef]
- Gouras, G.K.; Olsson, T.T.; Hansson, O. β-Amyloid pep–tides and amyloid plaques in Alzheimer’s disease. Neurotherapeutics 2015, 12, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Della Bianca, V.; Dusi, S.; Bianchini, E.; Dal Pra, I.; Rossi, F. Beta-amyloid activates the O−2 forming NADPH oxidase in microglia, monocytes, and neutrophils: A possible inflammatory mechanism of neuronal damage in Alzheimer’s disease. J. Biol Chem. 1999, 274, 15493–15499. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Boyd-Kimball, D. Oxidative stress, amyloid-β peptide, and altered key molecular pathways in the pathogenesis and progression of Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1345–1367. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, A.; Dal Pra, I.; Menapace, L.; Pacchiana, R.; Whitfield, J.F.; Armato, U. Soluble amyloid β-peptide and myelin basic protein strongly stimulate, alone and in synergism with joint proinflammatory cytokines, the expression of functional nitric oxide synthase-2 in normal adult human astrocytes. Int. J. Mol. Med. 2005, 16, 801–807. [Google Scholar] [CrossRef]
- Dal Prà, I.; Armato, U.; Chioffi, F.; Pacchiana, R.; Whitfield, J.F.; Chakravarthy, B.; Gui, L.; Chiarini, A. The Aβ peptides-activated calcium-sensing receptor stimulates the production and secretion of vascular endothelial growth factor-A by normoxic adult human cortical astrocytes. Neuromol. Med. 2014, 16, 645–657. [Google Scholar] [CrossRef]
- Dal Prà, I.; Chiarini, A.; Pacchiana, R.; Gardenal, E.; Chakravarthy, B.; Whitfield, J.F.; Armato, U. Calcium-Sensing Receptors of Human Astrocyte-Neuron Teams: Amyloid-β-Driven Mediators and Therapeutic Targets of Alzheimer’s Disease. Curr. Neuropharmacol. 2014, 12, 353–364. [Google Scholar] [CrossRef]
- Chiarini, A.; Armato, U.; Liu, D.; Dal Prà, I. Calcium-sensing receptors of human neural cells play crucial roles in Alzheimer’s disease. Front. Physiol. 2016, 7, 134. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, A.; Armato, U.; Hu, P.; Dal Prà, I. CaSR Antagonist (Calcilytic) NPS 2143 Hinders the Release of Neuroinflammatory IL-6, Soluble ICAM-1, RANTES, and MCP-2 from Aβ-Exposed Human Cortical Astrocytes. Cells 2020, 9, 1386. [Google Scholar] [CrossRef] [PubMed]
- Dal Prà, I.; Armato, U.; Chiarini, A. Specific interactions of calcium-sensing receptors (CaSRs) with soluble amyloid-β peptides—A study using cultured normofunctioning adult human astrocytes. In Proceedings of the 2nd International Symposium on the Calcium-sensing Receptor, San Diego, CA, USA, 3–4 March 2015; pp. 90–91. [Google Scholar]
- Dal Prà, I.; Chiarini, A.; Gui, L.; Chakravarthy, B.; Pacchiana, R.; Gardenal, E.; Armato, U. Do astrocytes collaborate with neurons in spreading the “infectious” Aβ and Tau drivers of Alzheimer’s disease? Neuroscientist 2015, 21, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. The preclinical phase of the pathological process underlying sporadic Alzheimer’s disease. Brain 2015, 138, 2814–2833. [Google Scholar] [CrossRef]
- Calhoun, A.; King, C.; Khoury, R.; Grossberg, G.T. An evaluation of memantine ER + donepezil for the treatment of Alzheimer’s disease. Expert Opin. Pharmacother. 2018, 19, 1711–1717. [Google Scholar] [CrossRef]
- Davies, P.; Maloney, A.J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 2, 1403. [Google Scholar] [CrossRef]
- Selkoe, D.J. The molecular pathology of Alzheimer’s disease. Neuron 1991, 6, 487–498. [Google Scholar] [CrossRef]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Mattson, M.P.; Cheng, B.; Davis, D.; Bryant, K.; Lieberburg, I.; Rydel, R.E. beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. 1992, 12, 376–389. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Rogers, J. Anti-inflammatory agents as a therapeutic approach to Alzheimer’s disease. Neurology 1992, 42, 447–449. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I.; Pettingell, W.H.; Multhaup, G.; d Paradis, M.; Vonsattel, J.P.; Gusella, J.F.; Beyreuther, K.; Masters, C.L.; Tanzi, R.E. Rapid induction of Alzheimer A beta amyloid formation by zinc. Science 1994, 265, 1464–1467. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Ho-Pao, C.L.; Kanazirska, M.; Quinn, S.; Rogers, K.; Seidman, C.E.; Seidman, J.G.; Brown, E.M.; Vassilev, P.M. Amyloid-beta proteins activate Ca(2+)-permeable channels through calcium-sensing receptors. J. Neurosci. Res. 1997, 47, 547–554. [Google Scholar] [CrossRef]
- Kalaria, R.N. The role of cerebral ischemia in Alzheimer’s disease. Neurobiol. Aging 2000, 21, 321–330. [Google Scholar] [CrossRef]
- De la Torre, J.C. Alzheimer disease as a vascular disorder: Nosological evidence. Stroke 2002, 33, 1152–1162. [Google Scholar] [CrossRef]
- Iadecola, C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 2004, 5, 347–360. [Google Scholar] [CrossRef]
- Swerdlow, R.H.; Khan, S.M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 2004, 63, 8–20. [Google Scholar] [CrossRef]
- Deane, R.; Wu, Z.; Sagare, A.; Davis, J.; Du Yan, S.; Hamm, K.; Xu, F.; Parisi, M.; LaRue, B.; Hu, H.W.; et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 2004, 43, 333–344. [Google Scholar] [CrossRef]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef]
- Armato, U.; Chiarini, A.; Chakravarthy, B.; Chioffi, F.; Pacchiana, R.; Colarusso, E.; Whitfield, J.F.; Dal Prà, I. Calcium-sensing receptor antagonist (calcilytic) NPS 2143 specifically blocks the increased secretion of endogenous Aβ42 prompted by exogenous fibrillary or soluble Aβ25-35 in human cortical astrocytes and neurons—Therapeutic relevance to Alzheimer’s disease. Biochim. Biophys. Acta 2013, 1832, 1634–1652. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Petersen, R.C. Cellular senescence in brain aging and neurodegenerative diseases: Evidence and perspectives. J. Clin. Investig. 2018, 128, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Maccioni, R.B.; Navarrete, L.P.; González, A.; González-Canacer, A.; Guzmán-Martínez, L.; Cortés, N. Inflammation: A major target for compounds to control Alzheimer’s Disease. J. Alzheimers Dis. 2020, 76, 1199–1213. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.M.; Raiford, K.; Edland, S.; Fillenbaum, G.; Morris, J.C.; Clark, C.M.; Kukull, W.; Heyman, A. The consortium to establish a registry for Alzheimer’s disease (CERAD). Part VI. Family history assessment: A multicenter study of first-degree relatives of Alzheimer’s disease probands and nondemented spouse controls. Neurology 1994, 44, 1253–1259. [Google Scholar] [CrossRef]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Razay, G.; Vreugdenhil, A.; Wilcock, G. The Metabolic Syndrome and Alzheimer Disease. Arch. Neurol. 2007, 64, 93–96. [Google Scholar] [CrossRef]
- Dias, H.K.; Brown, C.L.; Polidori, M.C.; Lip, G.Y.; Griffiths, H.R. LDL-lipids from patients with hypercholesterolaemia and Alzheimer’s disease are inflammatory to microvascular endothelial cells: Mitigation by statin intervention. Clin. Sci. 2015, 129, 1195–1206. [Google Scholar] [CrossRef]
- Whitmer, R.A.; Gunderson, E.P.; Barrett-Connor, E.; Quesenberry, C.P., Jr.; Yaffe, K. Obesity in middle age and future risk of dementia: A 27 year longitudinal population based study. BMJ 2005, 330, 1360. [Google Scholar] [CrossRef]
- Zhuo, J.M.; Wang, H.; Praticò, D. Is hyperhomocysteinemia an Alzheimer’s disease (AD) risk factor, an AD marker, or neither? Trends Pharmacol. Sci. 2011, 32, 562–571. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef]
- Launer, L.J.; Ross, G.W.; Petrovitch, H.; Masaki, K.; Foley, D.; White, L.R.; Havlik, R.J. Midlife blood pressure and dementia: The Honolulu-Asia aging study. Neurobiol. Aging 2000, 21, 49–55. [Google Scholar] [CrossRef]
- Osorio, R.S.; Pirraglia, E.; Agüera-Ortiz, L.F.; During, E.H.; Sacks, H.; Ayappa, I.; Walsleben, J.; Mooney, A.; Hussain, A.; Glodzik, L.; et al. Greater risk of Alzheimer’s disease in older adults with insomnia. J. Am. Geriatr. Soc. 2011, 59, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Olsen, I.; Singhrao, S.K. Can oral infection be a risk factor for Alzheimer’s disease? J. Oral Mcrobiol. 2015, 7, 29143. [Google Scholar] [CrossRef] [PubMed]
- Vogt, N.M.; Kerby, R.L.; Dill-McFarland, K.A.; Harding, S.J.; Merluzzi, A.P.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Zetterberg, H.; Blennow, K.; et al. Gut microbiome alterations in Alzheimer’s disease. Sci. Rep. 2017, 7, 13537. [Google Scholar] [CrossRef] [PubMed]
- Canet, G.; Dias, C.; Gabelle, A.; Simonin, Y.; Gosselet, F.; Marchi, N.; Makinson, A.; Tuaillon, E.; Van de Perre, P.; Givalois, L.; et al. HIV Neuroinfection and Alzheimer’s Disease: Similarities and Potential Links? Front. Cell. Neurosci. 2018, 12, 307. [Google Scholar] [CrossRef]
- Lövheim, H.; Gilthorpe, J.; Adolfsson, R.; Nilsson, L.G.; Elgh, F. Reactivated herpes simplex infection increases the risk of Alzheimer’s disease. Alzheimers Demen. 2015, 11, 593–599. [Google Scholar] [CrossRef]
- Saez-Atienzar, S.; Masliah, E. Cellular senescence and Alzheimer disease: The egg and the chicken scenario. Nat. Rev. Neurosci. 2020, 21, 433–444. [Google Scholar] [CrossRef]
- Han, X.; Zhang, T.; Liu, H.; Mi, Y.; Gou, X. Astrocyte senescence and Alzheimer’s Disease: A review. Front. Aging Neurosci. 2020, 12, 148. [Google Scholar] [CrossRef]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and Future Treatments in Alzheimer Disease: An Update. J. Cent. Nerv. Syst. Dis. 2020, 12, 1179573520907397. [Google Scholar] [CrossRef]
- Selkoe, D.J. Biochemistry and molecular biology of amyloid β-protein and the mechanism of Alzheimer’s disease. Handb. Clin. Neurol. 2008, 89, 245–260. [Google Scholar] [CrossRef]
- Yin, J.; Valin, K.L.; Dixon, M.L.; Leavenworth, J.W. The role of microglia and macrophages in CNS homeostasis, autoimmunity, and cancer. J. Immunol. Res. 2017, 2017, 5150678. [Google Scholar] [CrossRef] [PubMed]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef] [PubMed]
- Hemonnot, A.L.; Hua, J.; Ulmann, L.; Hirbec, H. Microglia in Alzheimer Disease: Well-known targets and new opportunities. Front. Aging Neurosci. 2019, 11, 233. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Lin, C.; Yan, F.; Yu, G.Q.; Rohde, M.; McConlogue, L.; Masliah, E.; Mucke, L. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat. Med. 2001, 7, 612–618. [Google Scholar] [CrossRef]
- Mandrekar-Colucci, S.; Landreth, G.E. Microglia and inflammation in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2010, 9, 156–167. [Google Scholar] [CrossRef]
- Araque, A.; Navarrete, M. Glial cells in neuronal network function. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 2375–2381. [Google Scholar] [CrossRef]
- Medeiros, R.; LaFerla, F.M. Astrocytes: Conductors of the Alzheimer disease neuroinflammatory symphony. Exp. Neurol. 2013, 239, 133–138. [Google Scholar] [CrossRef]
- Rodríguez-Arellano, J.J.; Parpura, V.; Zorec, R.; Verkhratsky, A. Astrocytes in physiological aging and Alzheimer’s disease. Neuroscience 2016, 323, 170–182. [Google Scholar] [CrossRef]
- Kato, S.; Gondo, T.; Hoshii, Y.; Takahashi, M.; Yamada, M.; Ishihara, T. Confocal observation of senile plaques in Alzheimer’s disease: Senile plaque morphology and relationship between senile plaques and astrocytes. Pathol. Int. 1998, 48, 332–340. [Google Scholar] [CrossRef]
- Von Bernhardi, R.; Ramirez, G. Microglia-astrocyte interaction in Alzheimer’s disease: Friends or foes for the nervous system? Biol. Res. 2001, 34, 123–128. [Google Scholar] [CrossRef]
- Nicoll, J.A.; Weller, R.O. A new role for astrocytes: β-amyloid homeostasis and degradation. Trends Mol. Med. 2003, 9, 281–282. [Google Scholar] [CrossRef]
- Liu, C.C.; Hu, J.; Zhao, N.; Wang, J.; Wang, N.; Cirrito, J.R.; Kanekiyo, T.; Holtzman, D.M.; Bu, G. Astrocytic LRP1 mediates brain Aβ clearance and impacts amyloid deposition. J. Neurosci. 2017, 37, 4023–4031. [Google Scholar] [CrossRef] [PubMed]
- Maragakis, N.J.; Rothstein, J.D. Mechanisms of Disease: Astrocytes in neurodegenerative disease. Nat. Clin. Pract. Neurol. 2006, 2, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, A.; Armato, U.; Liu, D.; Dal Prà, I. Calcium-Sensing Receptor Antagonist NPS 2143 Restores Amyloid Precursor Protein Physiological Non-Amyloidogenic Processing in Aβ-Exposed Adult Human Astrocytes. Sci. Rep. 2017, 7, 1277. [Google Scholar] [CrossRef]
- Bushong, E.A.; Martone, M.E.; Jones, Y.Z.; Ellisman, M.H. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J. Neurosci. 2002, 22, 183–192. [Google Scholar] [CrossRef]
- McIver, S.R.; Faideau, M.; Haydon, P.G. Astrocyte-neuron communications. In Neural-Immune Interactions in Brain Function and Alcohol-Related Disorders; Cui, C., Grandison, L., Noronha, A., Eds.; Springer: Boston, MA, USA, 2013; pp. 31–64. [Google Scholar] [CrossRef]
- Antanitus, D.S. A theory of cortical neuron-astrocyte interaction. Neuroscientist 1998, 4, 154–159. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef]
- Kettenmann, H.; Ransom, B.R. (Eds.) Neuroglia, 3rd ed.; Oxford University Press: New York, NY, USA, 2013. [Google Scholar]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of astrocytes in Alzheimer’s Disease from a neuroinflammatory and oxidative stress perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef]
- Kigerl, K.A.; Lai, W.; Rivest, S.; Hart, R.P.; Satoskar, A.R.; Popovich, P.G. Toll-like receptor (TLR)-2 and TLR-4 regulate inflammation, gliosis, and myelin sparing after spinal cord injury. J. Neurochem. 2007, 102, 37–50. [Google Scholar] [CrossRef]
- Lee, G.S.; Subramanian, N.; Kim, A.I.; Aksentijevich, I.; Goldbach-Mansky, R.; Sacks, D.B.; Germain, R.N.; Kastner, D.L.; Chae, J.J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 2012, 492, 123–127. [Google Scholar] [CrossRef]
- Labbé, K.; Saleh, M. Pyroptosis: A Caspase-1-dependent programmed cell death and a barrier to infection. In The Inflammasomes. Progress in Inflammation Research; Couillin, I., Pétrilli, V., Martinon, F., Eds.; Springer: Basel, Switzerland, 2011; pp. 17–36. [Google Scholar] [CrossRef]
- Lee, M.S.; Kim, Y.J. Pattern-recognition receptor signaling initiated from extracellular, membrane, and cytoplasmic space. Mol. Cells. 2007, 23, 1–10. [Google Scholar] [PubMed]
- Thundyil, J.; Lim, K.L. DAMPs and neurodegeneration. Ageing Res Rev. 2015, 24, 17–28. [Google Scholar] [CrossRef]
- Schaefer, L. Complexity of danger: The diverse nature of damage-associated molecular patterns. J. Biol. Chem. 2014, 289, 35237–35245. [Google Scholar] [CrossRef] [PubMed]
- Santoni, G.; Cardinali, C.; Morelli, M.B.; Santoni, M.; Nabissi, M.; Amantini, C. Danger- and pathogen-associated molecular patterns recognition by pattern-recognition receptors and ion channels of the transient receptor potential family triggers the inflammasome activation in immune cells and sensory neurons. J. Neuroinflammation 2015, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.S.; Suh, H.W.; Kim, S.J.; Jo, E.K. Mitochondrial control of innate immunity and inflammation. Immune Netw. 2017, 17, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Frevert, C.W.; Felgenhauer, J.; Wygrecka, M.; Nastase, M.V.; Schaefer, L. Danger-associated molecular patterns derived from the extracellular matrix provide temporal control of innate immunity. J. Histochem. Cytochem. 2018, 66, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Verdier, Y.; Penke, B. Binding sites of amyloid β-peptide in cell plasma membrane and implications for Alzheimer’s disease. Curr. Protein Pept. Sci. 2004, 5, 19–31. [Google Scholar] [CrossRef]
- Prabhudas, M.; Bowdish, D.; Drickamer, K.; Febbraio, M.; Herz, J.; Kobzik, L.; Krieger, M.; Loike, J.; Means, T.K.; Moestrup, S.K.; et al. Standardizing scavenger receptor nomenclature. J. Immunol. 2014, 192, 1997–2006. [Google Scholar] [CrossRef]
- Colonna, M. TREMs in the immune system and beyond. Nat. Rev. Immunol. 2003, 3, 445–453. [Google Scholar] [CrossRef]
- Hickman, S.E.; El Khoury, J. TREM2 and the neuroimmunology of Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Klesney-Tait, J.; Turnbull, I.R.; Colonna, M. The TREM receptor family and signal integration. Nat. Immunol. 2006, 7, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.P.; O’Driscoll, M.; Litman, G.W. Specific lipid recognition is a general feature of CD300 and TREM molecules. Immunogenetics 2012, 64, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Daws, M.R.; Sullam, P.M.; Niemi, E.C.; Chen, T.T.; Tchao, N.K.; Seaman, W.E. Pattern recognition by TREM-2: Binding of anionic ligands. J. Immunol. 2003, 171, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Rochford, C.D.; Neumann, H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 2005, 201, 647–657. [Google Scholar] [CrossRef]
- Jiang, T.; Tan, L.; Zhu, X.C.; Zhang, Q.Q.; Cao, L.; Tan, M.S.; Gu, L.Z.; Wang, H.F.; Ding, Z.Z.; Zhang, Y.D.; et al. Upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology 2014, 39, 2949–2962. [Google Scholar] [CrossRef]
- Kawabori, M.; Kacimi, R.; Kauppinen, T.; Calosing, C.; Kim, J.Y.; Hsieh, C.L.; Nakamura, M.C.; Yenari, M.A. Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J. Neurosci. 2015, 35, 3384–3396. [Google Scholar] [CrossRef]
- Bisht, K.; Sharma, K.P.; Lecours, C.; Sánchez, M.G.; El Hajj, H.; Milior, G.; Olmos-Alonso, A.; Gómez-Nicola, D.; Luheshi, G.; Vallières, L.; et al. Dark microglia: A new phenotype predominantly associated with pathological states. Glia 2016, 64, 826–839. [Google Scholar] [CrossRef]
- Mazaheri, F.; Snaidero, N.; Kleinberger, G.; Madore, C.; Daria, A.; Werner, G.; Krasemann, S.; Capell, A.; Trümbach, D.; Wurst, W.; et al. TREM2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep. 2017, 18, 1186–1198. [Google Scholar] [CrossRef]
- Ulland, T.K.; Song, W.M.; Huang, S.C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell 2017, 170, 649–663.e13. [Google Scholar] [CrossRef] [PubMed]
- Poliani, P.L.; Wang, Y.; Fontana, E.; Robinette, M.L.; Yamanishi, Y.; Gilfillan, S.; Colonna, M. TREM2 sustains microglial expansion during aging and response to demyelination. J. Clin. Investig. 2015, 125, 2161–2170. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Condello, C.; Keene, C.D.; Wang, Y.; Bird, T.D.; Paul, S.M.; Luo, W.; Colonna, M.; Baddeley, D.; Grutzendler, J. TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 2016, 92, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef]
- Jay, T.R.; Hirsch, A.M.; Broihier, M.L.; Miller, C.M.; Neilson, L.E.; Ransohoff, R.M.; Lamb, B.T.; Landreth, G.E. Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J. Neurosci. 2017, 37, 637–647. [Google Scholar] [CrossRef]
- Jiang, T.; Zhang, Y.D.; Chen, Q.; Gao, Q.; Zhu, X.C.; Zhou, J.S.; Shi, J.Q.; Lu, H.; Tan, L.; Yu, J.T. TREM2 modifies microglial phenotype and provides neuroprotection in P301S tau transgenic mice. Neuropharmacology 2016, 105, 196–206. [Google Scholar] [CrossRef]
- Jiang, T.; Wan, Y.; Zhang, Y.D.; Zhou, J.S.; Gao, Q.; Zhu, X.C.; Shi, J.Q.; Lu, H.; Tan, L.; Yu, J.T. TREM2 Overexpression has no improvement on neuropathology and cognitive impairment in aging APPswe/PS1dE9 mice. Mol. Neurobiol. 2017, 54, 855–865. [Google Scholar] [CrossRef]
- Yeh, F.L.; Wang, Y.; Tom, I.; Gonzalez, L.C.; Sheng, M. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron 2016, 91, 328–340. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A unique microglia type associated with restricting development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Wunderlich, P.; Glebov, K.; Kemmerling, N.; Tien, N.T.; Neumann, H.; Walter, J. Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and γ-secretase-dependent intramembranous cleavage. J. Biol. Chem. 2013, 288, 33027–33036. [Google Scholar] [CrossRef] [PubMed]
- Schmid, C.D.; Sautkulis, L.N.; Danielson, P.E.; Cooper, J.; Hasel, K.W.; Hilbush, B.S.; Sutcliffe, J.G.; Carson, M.J. Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J. Neurochem. 2002, 83, 1309–1320. [Google Scholar] [CrossRef] [PubMed]
- Piccio, L.; Deming, Y.; Del-Águila, J.L.; Ghezzi, L.; Holtzman, D.M.; Fagan, A.M.; Fenoglio, C.; Galimberti, D.; Borroni, B.; Cruchaga, C. Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. 2016, 131, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Heslegrave, A.; Heywood, W.; Paterson, R.; Magdalinou, N.; Svensson, J.; Johansson, P.; Öhrfelt, A.; Blennow, K.; Hardy, J.; Schott, J.; et al. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Chen, X.F.; Wang, T.; Wang, Z.; Liao, C.; Wang, Z.; Huang, R.; Wang, D.; Li, X.; Wu, L.; et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J. Exp. Med. 2017, 214, 597–607. [Google Scholar] [CrossRef]
- Bemiller, S.M.; McCray, T.J.; Allan, K.; Formica, S.V.; Xu, G.; Wilson, G.; Kokiko-Cochran, O.N.; Crish, S.D.; Lasagna-Reeves, C.A.; Ransohoff, R.M.; et al. TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol. Neurodegener. 2017, 12, 74. [Google Scholar] [CrossRef]
- Lill, C.M.; Rengmark, A.; Pihlstrom, L.; Fogh, I.; Shatunov, A.; Sleiman, P.M.; Wang, L.S.; Liu, T.; Lassen, C.F.; Meissner, E.; et al. The role of TREM2 R47H as a risk factor for Alzheimer’s disease, frontotemporal lobar degeneration, amyotrophic lateral sclerosis, and Parkinson’s disease. Alzheimers Dement. 2015, 11, 1407–1416. [Google Scholar] [CrossRef]
- Ayer, A.H.; Wojta, K.; Ramos, E.M.; Dokuru, D.; Chen, J.A.; Karydas, A.M.; Papatriantafyllou, J.D.; Agiomyrgiannakis, D.; Kamtsadeli, V.; Tsinia, N.; et al. Frequency of the TREM2 R47H variant in various neurodegenerative disorders. Alzheimer Dis. Assoc. Disord. 2019, 33, 327–330. [Google Scholar] [CrossRef]
- Ghezzi, L.; Carandini, T.; Arighi, A.; Fenoglio, C.; Arcaro, M.; De Riz, M.; Pietroboni, A.M.; Fumagalli, G.G.; Basilico, P.; Calvi, A.; et al. Evidence of CNS β-amyloid deposition in Nasu-Hakola disease due to the TREM2 Q33X mutation. Neurology 2017, 89, 2503–2505. [Google Scholar] [CrossRef]
- Borroni, B.; Ferrari, F.; Galimberti, D.; Nacmias, B.; Barone, C.; Bagnoli, S.; Fenoglio, C.; Piaceri, I.; Archetti, S.; Bonvicini, C.; et al. Heterozygous TREM2 mutations in frontotemporal dementia. Neurobiol. Aging 2014, 35, e937-10. [Google Scholar] [CrossRef]
- Rayaprolu, S.; Mullen, B.; Baker, M.; Lynch, T.; Finger, E.; Seeley, W.W.; Hatanpaa, K.J.; Lomen-Hoerth, C.; Kertesz, A.; Bigio, E.H.; et al. TREM2 in neurodegeneration: Evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol. Neurodegener. 2013, 8, 19. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Zhang, Y.D.; Gao, Q.; Zhou, J.S.; Zhu, X.C.; Lu, H.; Shi, J.Q.; Tan, L.; Chen, Q.; Yu, J.T. TREM1 facilitates microglial phagocytosis of amyloid beta. Acta Neuropathol. 2016, 132, 667–683. [Google Scholar] [CrossRef] [PubMed]
- Maillard-Lefebvre, H.; Boulanger, E.; Daroux, M.; Gaxatte, C.; Hudson, B.I.; Lambert, M. Soluble receptor for advanced glycation end products: A new biomarker in diagnosis and prognosis of chronic inflammatory diseases. Rheumatology 2009, 48, 1190–1196. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Kuja-Panula, J.; Sorci, G.; Agneletti, A.L.; Donato, R.; Rauvala, H. Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through receptor for advanced glycation end products (RAGE) activation. J. Biol. Chem. 2000, 275, 40096–40105. [Google Scholar] [CrossRef] [PubMed]
- Chuah, Y.K.; Basir, R.; Talib, H.; Tie, T.H.; Nordin, N. Receptor for advanced glycation end products and its involvement in inflammatory diseases. Int. J Inflam. 2013, 2013, 403460. [Google Scholar] [CrossRef]
- MacLean, M.; Derk, J.; Ruiz, H.H.; Juranek, J.K.; Ramasamy, R.; Schmidt, A.M. The Receptor for Advanced Glycation End Products (RAGE) and DIAPH1: Implications for vascular and neuroinflammatory dysfunction in disorders of the central nervous system. Neurochem. Int. 2019, 126, 154–164. [Google Scholar] [CrossRef]
- Ibrahim, Z.A.; Armour, C.L.; Phipps, S.; Sukkar, M.B. RAGE and TLRs: Relatives, friends or neighbours? Mol. Immunol. 2013, 56, 739–744. [Google Scholar] [CrossRef]
- Shekhtman, A.; Ramasamy, R.; Schmidt, A.M. Glycation & the RAGE axis: Targeting signal transduction through DIAPH1. Expert Rev. Proteom. 2017, 14, 147–156. [Google Scholar] [CrossRef]
- Chen, G.Y.; Nuñez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef]
- Vénéreau, E.; Ceriotti, C.; Bianchi, M.E. DAMPs from cell death to new life. Front. Immunol. 2015, 6, 422. [Google Scholar] [CrossRef]
- Land, W.G. The role of Damage-Associated Molecular Patterns in human diseases: Part I—Promoting inflammation and immunity. Sultan Qaboos Univ. Med. J. 2015, 15, e9–e21. [Google Scholar] [PubMed]
- Festoff, B.W.; Sajja, R.K.; van Dreden, P.; Cucullo, L. HMGB1 and thrombin mediate the blood-brain barrier dysfunction acting as biomarkers of neuroinflammation and progression to neurodegeneration in Alzheimer’s disease. J. Neuroinflammation 2016, 13, 194. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Méndez, J.D.; Méndez-Valenzuela, V.; Aguilar-Hernández, M.M. Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell Signal. 2013, 25, 2185–2197. [Google Scholar] [CrossRef] [PubMed]
- Gasparotto, J.; Girardi, C.S.; Somensi, N.; Ribeiro, C.T.; Moreira, J.; Michels, M.; Sonai, B.; Rocha, M.; Steckert, A.V.; Barichello, T.; et al. Receptor for advanced glycation end products mediates sepsis-triggered amyloid-β accumulation, Tau phosphorylation, and cognitive impairment. J. Biol. Chem. 2018, 293, 226–244. [Google Scholar] [CrossRef] [PubMed]
- Fournet, M.; Bonté, F.; Desmoulière, A. Glycation damage: A possible hub for major pathophysiological disorders and aging. Aging Dis. 2018, 9, 880–900. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.M.; Nicol, C.J.; Cheng, Y.C.; Lin, K.H.; Chen, Y.L.; Pei, D.; Lin, C.H.; Shih, Y.N.; Yen, C.H.; Chen, S.J.; et al. Metformin activation of AMPK suppresses AGE-induced inflammatory response in hNSCs. Exp. Cell Res. 2017, 352, 75–83. [Google Scholar] [CrossRef]
- Sawikr, Y.; Yarla, N.S.; Peluso, I.; Kamal, M.A.; Aliev, G.; Bishayee, A. Neuroinflammation in Alzheimer’s Disease: The preventive and therapeutic potential of polyphenolic nutraceuticals. Adv. Protein Chem. Struct. Biol. 2017, 108, 33–57. [Google Scholar] [CrossRef]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The biology of the receptor for advanced glycation end products and its ligands. Biochim. Biophys. Acta 2000, 1498, 99–111. [Google Scholar] [CrossRef]
- Barnes, P.J.; Karin, M. Nuclear factor-kappaB: A pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 1997, 336, 1066–1071. [Google Scholar] [CrossRef]
- Fann, D.Y.; Lim, Y.A.; Cheng, Y.L.; Lok, K.Z.; Chunduri, P.; Baik, S.H.; Drummond, G.R.; Dheen, S.T.; Sobey, C.G.; Jo, D.G.; et al. Evidence that NF-κB and MAPK signaling promotes NLRP inflammasome activation in neurons following ischemic stroke. Mol. Neurobiol. 2018, 55, 1082–1096. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Brownlee, M. Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (RAGE) and RAGE ligands. Diabetes 2010, 59, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Reyes, R.E.; Rubiano, M.G. Astrocyte’s RAGE: More than just a question of mood. Cent. Nerv. Syst. Agents Med. Chem. 2018, 18, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Ponath, G.; Schettler, C.; Kaestner, F.; Voigt, B.; Wentker, D.; Arolt, V.; Rothermundt, M. Autocrine S100B effects on astrocytes are mediated via RAGE. J. Neuroimmunol. 2007, 184, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, D.D.; Liang, Y.Y.; Wang, D.S.; Cai, N.S. Activation of astrocytes by advanced glycation end products: Cytokines induction and nitric oxide release. Acta Pharmacol. Sin. 2002, 23, 974–980. [Google Scholar]
- Sparvero, L.J.; Asafu-Adjei, D.; Kang, R.; Tang, D.; Amin, N.; Im, J.; Rutledge, R.; Lin, B.; Amoscato, A.A.; Zeh, H.J.; et al. RAGE (Receptor for Advanced Glycation Endproducts), RAGE ligands, and their role in cancer and inflammation. J. Transl. Med. 2009, 7, 17. [Google Scholar] [CrossRef]
- Villarreal, A.; Seoane, R.; González Torres, A.; Rosciszewski, G.; Angelo, M.F.; Rossi, A.; Barker, P.A.; Ramos, A.J. S100B protein activates a RAGE-dependent autocrine loop in astrocytes: Implications for its role in the propagation of reactive gliosis. J. Neurochem. 2014, 131, 190–205. [Google Scholar] [CrossRef]
- Kaur, D.; Sharma, V.; Deshmukh, R. Activation of microglia and astrocytes: A roadway to neuroinflammation and Alzheimer’s disease. Inflammopharmacology 2019, 27, 663–677. [Google Scholar] [CrossRef]
- Nonaka, S.; Nakanishi, H. Microglial clearance of focal apoptotic synapses. Neurosci. Lett. 2019, 707, 134317. [Google Scholar] [CrossRef]
- Sasaki, N.; Toki, S.; Chowei, H.; Saito, T.; Nakano, N.; Hayashi, Y.; Takeuchi, M.; Makita, Z. Immunohistochemical distribution of the receptor for advanced glycation end products in neurons and astrocytes in Alzheimer’s disease. Brain Res. 2001, 888, 256–262. [Google Scholar] [CrossRef]
- Criscuolo, C.; Fontebasso, V.; Middei, S.; Stazi, M.; Ammassari-Teule, M.; Yan, S.S.; Origlia, N. Entorhinal Cortex dysfunction can be rescued by inhibition of microglial RAGE in an Alzheimer’s disease mouse model. Sci. Rep. 2017, 7, 42370. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K. AGE-RAGE stress: A changing landscape in pathology and treatment of Alzheimer’s disease. Mol. Cell. Biochem. 2019, 459, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.R.; Cho, W.H.; Kim, J.; Lee, H.J.; Chung, C.; Jeon, W.K.; Han, J.S. Increased expression of the receptor for advanced glycation end products in neurons and astrocytes in a triple transgenic mouse model of Alzheimer’s disease. Exp. Mol. Med. 2014, 46, e75. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Yu, Q.; Arancio, O.; Chen, D.; Gore, S.S.; Yan, S.S.; Yan, S.F. RAGE mediates Aβ accumulation in a mouse model of Alzheimer’s disease via modulation of β- and γ-secretase activity. Hum. Mol. Genet. 2018, 27, 1002–1014. [Google Scholar] [CrossRef] [PubMed]
- Batkulwar, K.; Godbole, R.; Banarjee, R.; Kassaar, O.; Williams, R.J.; Kulkarni, M.J. Advanced Glycation End products modulate amyloidogenic APP processing and Tau phosphorylation: A mechanistic link between glycation and the development of Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 988–1000. [Google Scholar] [CrossRef]
- Du, H.; Li, P.; Wang, J.; Qing, X.; Li, W. The interaction of amyloid β and the receptor for advanced glycation endproducts induces matrix metalloproteinase-2 expression in brain endothelial cells. Cell Mol. Neurobiol. 2012, 32, 141–147. [Google Scholar] [CrossRef]
- Yan, S.D.; Bierhaus, A.; Nawroth, P.P.; Stern, D.M. RAGE and Alzheimer’s disease: A progression factor for amyloid-β-induced cellular perturbation? J. Alzheimers Dis. 2009, 16, 833–843. [Google Scholar] [CrossRef]
- Askarova, S.; Yang, X.; Sheng, W.; Sun, G.Y.; Lee, J.C. Role of Aβ-receptor for advanced glycation endproducts interaction in oxidative stress and cytosolic phospholipase A₂ activation in astrocytes and cerebral endothelial cells. Neuroscience 2011, 199, 375–385. [Google Scholar] [CrossRef]
- Emi, M.; Asaoka, H.; Matsumoto, A.; Itakura, H.; Kurihara, Y.; Wada, Y.; Kanamori, H.; Yazaki, Y.; Takahashi, E.; Lepert, M. Structure, organization, and chromosomal mapping of the human macrophage scavenger receptor gene. J. Biol. Chem. 1993, 268, 2120–2125. [Google Scholar]
- Kelley, J.L.; Ozment, T.R.; Li, C.; Schweitzer, J.B.; Williams, D.L. Scavenger receptor-A (CD204): A two-edged sword in health and disease. Crit. Rev. Immunol. 2014, 34, 241–261. [Google Scholar] [CrossRef]
- Gough, P.J.; Greaves, D.R.; Gordon, S. A naturally occurring isoform of the human macrophage scavenger receptor (SR-A) gene generated by alternative splicing blocks modified LDL uptake. J. Lipid Res. 1998, 39, 531–543. [Google Scholar]
- Yu, B.; Cheng, C.; Wu, Y.; Guo, L.; Kong, D.; Zhang, Z.; Wang, Y.; Zheng, E.; Liu, Y.; He, Y. Interactions of ferritin with scavenger receptor class A members. J. Biol. Chem. 2020, 295, 15727–15741. [Google Scholar] [CrossRef]
- El Khoury, J.B.; Moore, K.J.; Means, T.K.; Leung, J.; Terada, K.; Toft, M.; Freeman, M.W.; Luster, A.D. CD36 mediates the innate host response to beta-amyloid. J. Exp. Med. 2003, 197, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Su, Y.J.; Zhou, W.W.; Wang, S.W.; Xu, P.X.; Yu, X.L.; Liu, R.T. Activated scavenger receptor A promotes glial internalization of aβ. PLoS ONE 2014, 9, e94197. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef] [PubMed]
- Christie, R.H.; Freeman, M.; Hyman, B.T. Expression of the macrophage scavenger receptor, a multifunctional lipoprotein receptor, in microglia associated with senile plaques in Alzheimer’s disease. Am. J Pathol. 1996, 148, 399–403. [Google Scholar]
- Yu, Y.; Ye, R.D. Microglial Aβ receptors in Alzheimer’s disease. Cell. Mol. Neurobiol. 2015, 35, 71–83. [Google Scholar] [CrossRef]
- Plüddemann, A.; Neyen, C.; Gordon, S. Macrophage scavenger receptors and host-derived ligands. Methods 2007, 43, 207–217. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Varin, A.; Chen, Y.; Liu, B.; Tryggvason, K.; Gordon, S. SR-A/MARCO-mediated ligand delivery enhances intracellular TLR and NLR function, but ligand scavenging from cell surface limits TLR4 response to pathogens. Blood 2011, 117, 1319–1328. [Google Scholar] [CrossRef]
- Novakowski, K.E.; Huynh, A.; Han, S.; Dorrington, M.G.; Yin, C.; Tu, Z.; Pelka, P.; Whyte, P.; Guarné, A.; Sakamoto, K.; et al. A naturally occurring transcript variant of MARCO reveals the SRCR domain is critical for function. Immunol. Cell Biol. 2016, 94, 646–655. [Google Scholar] [CrossRef]
- Xu, J.; Flaczyk, A.; Neal, L.M.; Fa, Z.; Eastman, A.J.; Malachowski, A.N.; Cheng, D.; Moore, B.B.; Curtis, J.L.; Osterholzer, J.J.; et al. Scavenger Receptor MARCO Orchestrates Early Defenses and Contributes to Fungal Containment during Cryptococcal Infection. J. Immunol. 2017, 198, 3548–3557. [Google Scholar] [CrossRef] [PubMed]
- Pittman, R.C.; Knecht, T.P.; Rosenbaum, M.S.; Taylor, C.A., Jr. A non endocytotic mechanism for the selective uptake of high density lipoprotein-associated cholesterol esters. J. Biol. Chem. 1987, 262, 2443–2450. [Google Scholar] [PubMed]
- Shen, W.J.; Asthana, S.; Kraemer, F.B.; Azhar, S. Scavenger receptor B type 1: Expression, molecular regulation, and cholesterol transport function. J. Lipid Res. 2018, 59, 1114–1131. [Google Scholar] [CrossRef]
- Shen, W.J.; Azhar, S.; Kraemer, F.B. SR-B1: A unique multifunctional receptor for cholesterol influx and efflux. Annu. Rev. Physiol. 2018, 80, 95–116. [Google Scholar] [CrossRef]
- Reina, S.A.; Llabre, M.M.; Allison, M.A.; Wilkins, J.T.; Mendez, A.J.; Arnan, M.K.; Schneiderman, N.; Sacco, R.L.; Carnethon, M.; Delaney, J.A.C. HDL cholesterol and stroke risk: The multi-ethnic study of atherosclerosis. Atherosclerosis 2015, 243, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Linton, M.F.; Tao, H.; Linton, E.F.; Yancey, P.G. SR-BI: A Multifunctional Receptor in Cholesterol Homeostasis and Atherosclerosis. Trends Endocrinol. Metab. 2017, 28, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Ohgami, N.; Miyazaki, A.; Sakai, M.; Kuniyasu, A.; Nakayama, H.; Horiuchi, S. Advanced glycation end products (AGE) inhibit scavenger receptor class B type I-mediated reverse cholesterol transport: A new crossroad of AGE to cholesterol metabolism. J. Atheroscler. Thromb. 2003, 10, 1–6. [Google Scholar] [CrossRef]
- Lescoat, A.; Ballerie, A.; Lelong, M.; Augagneur, Y.; Morzadec, C.; Jouneau, S.; Jégo, P.; Fardel, O.; Vernhet, L.; Lecureur, V. Crystalline silica impairs efferocytosis abilities of human and mouse macrophages: Implication for silica-associated systemic sclerosis. Front. Immunol. 2020, 11, 219. [Google Scholar] [CrossRef]
- Sekhar, V.; Pollicino, T.; Diaz, G.; Engle, R.E.; Alayli, F.; Melis, M.; Kabat, J.; Tice, A.; Pomerenke, A.; Altan-Bonnet, N.; et al. Infection with hepatitis C virus depends on TACSTD2, a regulator of claudin-1 and occludin highly downregulated in hepatocellular carcinoma. PLoS Pathog. 2018, 14, e1006916. [Google Scholar] [CrossRef]
- Iram, T.; Trudler, D.; Kain, D.; Kanner, S.; Galron, R.; Vassar, R.; Barzilai, A.; Blinder, P.; Fishelson, Z.; Frenkel, D. Astrocytes from old Alzheimer’s disease mice are impaired in Aβ uptake and in neuroprotection. Neurobiol. Dis. 2016, 96, 84–94. [Google Scholar] [CrossRef]
- Thanopoulou, K.; Fragkouli, A.; Stylianopoulou, F.; Georgopoulos, S. Scavenger receptor class B type I (SR-BI) regulates perivascular macrophages and modifies amyloid pathology in an Alzheimer mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 20816–20821. [Google Scholar] [CrossRef] [PubMed]
- Febbraio, M.; Hajjar, D.P.; Silverstein, R.L. CD36: A class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation and lipid metabolism. J. Clin. Investig. 2001, 108, 785–791. [Google Scholar] [CrossRef]
- Leitinger, N. Oxidized phospholipids as modulators of inflammation in atherosclerosis. Curr. Opin. Lipidol. 2003, 14, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Coraci, I.S.; Husemann, J.; Berman, J.W.; Hulette, C.; Dufour, J.H.; Campanella, G.K.; Luster, A.D.; Silverstein, S.C.; El-Khoury, J.B. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer’s disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am. J. Pathol. 2002, 160, 101–112. [Google Scholar] [CrossRef]
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T.; et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013, 14, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Bamberger, M.E.; Harris, M.E.; McDonald, D.R.; Husemann, J.; Landreth, G.E. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J. Neurosci. 2003, 23, 2665–2674. [Google Scholar] [CrossRef] [PubMed]
- Husemann, J.; Silverstein, S.C. Expression of scavenger receptor class B, type I, by astrocytes and vascular smooth muscle cells in normal adult mouse and human brain and in Alzheimer’s disease brain. Am. J. Pathol. 2001, 158, 825–832. [Google Scholar] [CrossRef]
- Husemann, J.; Loike, J.D.; Anankov, R.; Febbraio, M.; Silverstein, S.C. Scavenger receptors in neurobiology and neuropathology: Their role on microglia and other cells of the nervous system. Glia 2002, 40, 195–205. [Google Scholar] [CrossRef]
- Alarcón, R.; Fuenzalida, C.; Santibáñez, M.; von Bernhardi, R. Expression of scavenger receptors in glial cells. Comparing the adhesion of astrocytes and microglia from neonatal rats to surface-bound β-amyloid. J. Biol. Chem. 2005, 280, 30406–30415. [Google Scholar] [CrossRef]
- Mulder, S.D.; Veerhuis, R.; Blankenstein, M.A.; Nielsen, H.M. The effect of amyloid associated proteins on the expression of genes involved in amyloid-β clearance by adult human astrocytes. Exp. Neurol. 2012, 233, 373–379. [Google Scholar] [CrossRef]
- Thal, D.R. The role of astrocytes in amyloid β-protein toxicity and clearance. Exp. Neurol. 2012, 236, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Eugenín, J.; Vecchiola, A.; Murgas, P.; Arroyo, P.; Cornejo, F.; von Bernhardi, R. Expression pattern of scavenger receptors and amyloid-β phagocytosis of astrocytes and microglia in culture are modified by acidosis: Implications for Alzheimer’s Disease. J. Alzheimers Dis. 2016, 53, 857–873. [Google Scholar] [CrossRef] [PubMed]
- Barton, G.M.; Medzhitov, R. Toll-like receptor signaling pathways. Science 2003, 300, 1524–1525. [Google Scholar] [CrossRef] [PubMed]
- Crack, P.J.; Bray, P.J. Toll-like receptors in the brain and their potential roles in neuropathology. Immunol. Cell Biol. 2007, 85, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, P.A.; Sriskandan, S. Mammalian Toll-like receptors: To immunity and beyond. Clin. Exp. Immunol. 2005, 140, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Jana, M.; Palencia, C.A.; Pahan, K. Fibrillar amyloid-beta peptides activate microglia via TLR2: Implications for Alzheimer’s disease. J. Immunol. 2008, 181, 7254–7262. [Google Scholar] [CrossRef] [PubMed]
- Letiembre, M.; Liu, Y.; Walter, S.; Hao, W.; Pfander, T.; Wrede, A.; Schulz-Schaeffer, W.; Fassbender, K. Screening of innate immune receptors in neurodegenerative diseases: A similar pattern. Neurobiol. Aging 2009, 30, 759–768. [Google Scholar] [CrossRef]
- Liu, Y.; Walter, S.; Stagi, M.; Cherny, D.; Letiembre, M.; Schulz-Schaeffer, W.; Heine, H.; Penke, B.; Neumann, H.; Fassbender, K. LPS receptor (CD14): A receptor for phagocytosis of Alzheimer’s amyloid peptide. Brain 2005, 128, 1778–1789. [Google Scholar] [CrossRef]
- Walter, S.; Letiembre, M.; Liu, Y.; Heine, H.; Penke, B.; Hao, W.; Bode, B.; Manietta, N.; Walter, J.; Schulz-Schuffer, W.; et al. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell. Physiol. Biochem. 2007, 20, 947–956. [Google Scholar] [CrossRef]
- Liu, S.; Liu, Y.; Hao, W.; Wolf, L.; Kiliaan, A.J.; Penke, B.; Rübe, C.E.; Walter, J.; Heneka, M.T.; Hartmann, T.; et al. TLR2 is a primary receptor for Alzheimer’s amyloid β peptide to trigger neuroinflammatory activation. J. Immunol. 2012, 188, 1098–1107. [Google Scholar] [CrossRef]
- Balducci, C.; Frasca, A.; Zotti, M.; La Vitola, P.; Mhillaj, E.; Grigoli, E.; Iacobellis, M.; Grandi, F.; Messa, M.; Colombo, L.; et al. Toll-like receptor 4-dependent glial cell activation mediates the impairment in memory establishment induced by β-amyloid oligomers in an acute mouse model of Alzheimer’s disease. Brain Behav. Immun. 2017, 60, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Tahara, K.; Kim, H.D.; Jin, J.J.; Maxwell, J.A.; Li, L.; Fukuchi, K. Role of toll-like receptor signalling in Abeta uptake and clearance. Brain 2006, 129, 3006–3019. [Google Scholar] [CrossRef]
- Tauber, S.C.; Ebert, S.; Weishaupt, J.H.; Reich, A.; Nau, R.; Gerber, J. Stimulation of Toll-like receptor 9 by chronic intraventricular unmethylated cytosine-guanine DNA infusion causes neuroinflammation and impaired spatial memory. J. Neuropathol. Exp. Neurol. 2009, 68, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Kufer, T.A.; Sansonetti, P.J. Sensing of bacteria: NOD a lonely job. Curr. Opin. Microbiol. 2007, 10, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Ting, J.P. NLR, the nucleotide-binding domain leucine-rich repeat containing gene family. Curr. Opin. Immunol. 2008, 20, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Boivin, A.; Pineau, I.; Barrette, B.; Filali, M.; Vallières, N.; Rivest, S.; Lacroix, S. Toll-like receptor signaling is critical for Wallerian degeneration and functional recovery after peripheral nerve injury. J. Neurosci. 2007, 27, 12565–12576. [Google Scholar] [CrossRef] [PubMed]
- Kigerl, K.A.; de Rivero Vaccari, J.P.; Dietrich, W.D.; Popovich, P.G.; Keane, R.W. Pattern recognition receptors and central nervous system repair. Exp. Neurol. 2014, 258, 5–16. [Google Scholar] [CrossRef] [PubMed]
- De Rivero Vaccari, J.P.; Minkiewicz, J.; Wang, X.; de Rivero Vaccari, J.C.; German, R.; Marcillo, A.E.; Dietrich, W.D.; Keane, R.W. Astrogliosis involves activation of retinoic acid-inducible gene-like signaling in the innate immune response after spinal cord injury. Glia 2012, 60, 414–421. [Google Scholar] [CrossRef]
- Kufer, T.A.; Sansonetti, P.J. NLR functions beyond pathogen recognition. Nat. Immunol. 2011, 12, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.G.; Eibl, C.; Fuchs, J.E. Intrinsic flexibility of NLRP pyrin domains is a key factor in their conformational dynamics, fold stability, and dimerization. Protein Sci. 2015, 24, 174–181. [Google Scholar] [CrossRef]
- Kufer, T.A.; Kremmer, E.; Banks, D.J.; Philpott, D.J. Role for Erbin in Bacterial Activation of Nod2. Infect. Immun. 2006, 74, 3115–3124. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Gross, O.; Thomas, C.J.; Guarda, G.; Tschopp, J. The inflammasome: An integrated view. Immunol. Rev. 2011, 243, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.K.; Wen, H.; Ting, J.P. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef] [PubMed]
- Minkiewicz, J.; de Rivero Vaccari, J.P.; Keane, R.W. Human astrocytes express a novel NLRP2 inflammasome. Glia 2013, 61, 1113–1121. [Google Scholar] [CrossRef]
- Vladimer, G.I.; Weng, D.; Paquette, S.W.; Vanaja, S.K.; Rathinam, V.A.; Aune, M.H.; Conlon, J.E.; Burbage, J.J.; Proulx, M.K.; Liu, Q.; et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity 2012, 37, 96–107. [Google Scholar] [CrossRef]
- Walsh, J.G.; Muruve, D.A.; Power, C. Inflammasomes in the CNS. Nat. Rev. Neurosci. 2014, 15, 84–97. [Google Scholar] [CrossRef]
- Vande Walle, L.; Lamkanfi, M. Pyroptosis. Curr. Biol. 2016, 26, R568–R572. [Google Scholar] [CrossRef]
- McKenzie, B.A.; Mamik, M.K.; Saito, L.B.; Boghozian, R.; Monaco, M.C.; Major, E.O.; Lu, J.Q.; Branton, W.G.; Power, C. Caspase-1 inhibition prevents glial inflammasome activation and pyroptosis in models of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, E6065–E6074. [Google Scholar] [CrossRef]
- Kaushal, V.; Dye, R.; Pakavathkumar, P.; Foveau, B.; Flores, J.; Hyman, B.; Ghetti, B.; Koller, B.H.; LeBlanc, A.C. Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ. 2015, 22, 1676–1686. [Google Scholar] [CrossRef]
- Freeman, L.; Guo, H.; David, C.N.; Brickey, W.J.; Jha, S.; Ting, J.P. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J. Exp. Med. 2017, 214, 1351–1370. [Google Scholar] [CrossRef] [PubMed]
- Yap, J.K.Y.; Pickard, B.S.; Chan, E.W.L.; Gan, S.Y. The role of neuronal NLRP1 inflammasome in Alzheimer’s disease: Bringing neurons into the neuroinflammation game. Mol. Neurobiol. 2019, 56, 7741–7753. [Google Scholar] [CrossRef] [PubMed]
- Nyúl-Tóth, Á.; Kozma, M.; Nagyőszi, P.; Nagy, K.; Fazakas, C.; Haskó, J.; Molnár, K.; Farkas, A.E.; Végh, A.G.; Váró, G.; et al. Expression of pattern recognition receptors and activation of the non-canonical inflammasome pathway in brain pericytes. Brain Behav. Immun. 2017, 64, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Nagyőszi, P.; Nyúl-Tóth, Á.; Fazakas, C.; Wilhelm, I.; Kozma, M.; Molnár, J.; Haskó, J.; Krizbai, I.A. Regulation of NOD-like receptors and inflammasome activation in cerebral endothelial cells. J. Neurochem. 2015, 135, 551–564. [Google Scholar] [CrossRef]
- Abulafia, D.P.; de Rivero Vaccari, J.P.; Lozano, J.D.; Lotocki, G.; Keane, R.W.; Dietrich, W.D. Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. J. Cereb. Blood Flow Metab. 2009, 29, 534–544. [Google Scholar] [CrossRef]
- De Rivero Vaccari, J.P.; Lotocki, G.; Marcillo, A.E.; Dietrich, W.D.; Keane, R.W. A molecular platform in neurons regulates inflammation after spinal cord injury. J. Neurosci. 2008, 28, 3404–3414. [Google Scholar] [CrossRef]
- Lechan, R.M.; Toni, R.; Clark, B.D.; Cannon, J.G.; Shaw, A.R.; Dinarello, C.A.; Reichlin, S. Immunoreactive interleukin-1 beta localization in the rat forebrain. Brain Res. 1990, 514, 135–140. [Google Scholar] [CrossRef]
- Heneka, M.T. Inflammasome activation and innate immunity in Alzheimer’s disease. Brain Pathol. 2017, 27, 220–222. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Finger, J.N.; Lich, J.D.; Dare, L.C.; Cook, M.N.; Brown, K.K.; Duraiswami, C.; Bertin, J.; Gough, P.J. Autolytic proteolysis within the function to find domain (FIIND) is required for NLRP1 inflammasome activity. J. Biol. Chem. 2012, 287, 25030–25037. [Google Scholar] [CrossRef]
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Nuñez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar] [CrossRef] [PubMed]
- White, C.S.; Lawrence, C.B.; Brough, D.; Rivers-Auty, J. Inflammasomes as therapeutic targets for Alzheimer’s disease. Brain Pathol. 2017, 27, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Fann, D.Y.; Lee, S.Y.; Manzanero, S.; Tang, S.C.; Gelderblom, M.; Chunduri, P.; Bernreuther, C.; Glatzel, M.; Cheng, Y.L.; Thundyil, J.; et al. Intravenous immunoglobulin suppresses NLRP1 and NLRP3 inflammasome-mediated neuronal death in ischemic stroke. Cell Death Dis. 2013, 4, e790. [Google Scholar] [CrossRef]
- Tan, M.S.; Tan, L.; Jiang, T.; Zhu, X.C.; Wang, H.F.; Jia, C.D.; Yu, J.T. Amyloid-β induces NLRP1-dependent neuronal pyroptosis in models of Alzheimer’s disease. Cell Death Dis. 2014, 5, e1382. [Google Scholar] [CrossRef] [PubMed]
- Saadi, M.; Karkhah, A.; Pourabdolhossein, F.; Ataie, A.; Monif, M.; Nouri, H.R. Involvement of NLRC4 inflammasome through caspase-1 and IL-1β augments neuroinflammation and contributes to memory impairment in an experimental model of Alzheimer’s like disease. Brain Res. Bull. 2020, 154, 81–90. [Google Scholar] [CrossRef]
- Healy, L.M.; Yaqubi, M.; Ludwin, S.; Antel, J.P. Species differences in immune-mediated CNS tissue injury and repair: A (neuro)inflammatory topic. Glia 2020, 68, 811–829. [Google Scholar] [CrossRef]
- Kinoshita, T.; Wang, Y.; Hasegawa, M.; Imamura, R.; Suda, T. PYPAF3, a PYRIN-containing APAF-1-like protein, is a feedback regulator of caspase-1-dependent interleukin-1beta secretion. J. Biol. Chem. 2005, 280, 21720–21725. [Google Scholar] [CrossRef]
- Vizlin-Hodzic, D.; Zhai, Q.; Illes, S.; Södersten, K.; Truvé, K.; Parris, T.Z.; Sobhan, P.K.; Salmela, S.; Kosalai, S.T.; Kanduri, C.; et al. Early onset of inflammation during ontogeny of bipolar disorder: The NLRP2 inflammasome gene distinctly differentiates between patients and healthy controls in the transition between iPS cell and neural stem cell stages. Transl. Psychiatry 2017, 7, e1010. [Google Scholar] [CrossRef]
- Peng, H.; Chang, B.; Lu, C.; Su, J.; Wu, Y.; Lv, P.; Wang, Y.; Liu, J.; Zhang, B.; Quan, F.; et al. Nlrp2, a maternal effect gene required for early embryonic development in the mouse. PLoS ONE 2012, 7, e30344. [Google Scholar] [CrossRef]
- Sun, X.; Song, X.; Zhang, L.; Sun, J.; Wei, X.; Meng, L.; An, J. NLRP2 is highly expressed in a mouse model of ischemic stroke. Biochem. Biophys. Res. Commun. 2016, 479, 656–662. [Google Scholar] [CrossRef]
- Zhang, Q.; Sun, Y.; He, Z.; Xu, Y.; Li, X.; Ding, J.; Lu, M.; Hu, G. Kynurenine regulates NLRP2 inflammasome in astrocytes and its implications in depression. Brain Behav. Immun. 2020, 88, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Bruey, J.M.; Bruey-Sedano, N.; Newman, R.; Chandler, S.; Stehlik, C.; Reed, J.C. PAN1/NALP2/PYPAF2, an inducible inflammatory mediator that regulates NF-kappaB and caspase-1 activation in macrophages. J. Biol. Chem. 2004, 279, 51897–51907. [Google Scholar] [CrossRef] [PubMed]
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606. [Google Scholar] [CrossRef] [PubMed]
- Leemans, J.C.; Cassel, S.L.; Sutterwala, F.S. Sensing damage by the NLRP3 inflammasome. Immunol. Rev. 2011, 243, 152–162. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef]
- Katsnelson, M.A.; Rucker, L.G.; Russo, H.M.; Dubyak, G.R. K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J. Immunol. 2015, 194, 3937–3952. [Google Scholar] [CrossRef]
- Gong, T.; Jiang, W.; Zhou, R. Control of inflammasome activation by phosphorylation. Trends Biochem. Sci. 2018, 43, 685–699. [Google Scholar] [CrossRef]
- Choi, A.J.; Ryter, S.W. Inflammasomes: Molecular regulation and implications for metabolic and cognitive diseases. Mol. Cells 2014, 37, 441–448. [Google Scholar] [CrossRef]
- Elliott, E.I.; Sutterwala, F.S. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev. 2015, 265, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef] [PubMed]
- Rossol, M.; Pierer, M.; Raulien, N.; Quandt, D.; Meusch, U.; Rothe, K.; Schubert, K.; Schöneberg, T.; Schaefer, M.; Krügel, U.; et al. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat. Commun. 2012, 3, 1329. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, Z.J. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 2018, 564, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Sokolowska, M.; Chen, L.Y.; Liu, Y.; Martinez-Anton, A.; Qi, H.Y.; Logun, C.; Alsaaty, S.; Park, Y.H.; Kastner, D.L.; Chae, J.J.; et al. Prostaglandin E2 Inhibits NLRP3 Inflammasome Activation through EP4 Receptor and Intracellular Cyclic AMP in Human Macrophages. J. Immunol. 2015, 194, 5472–5487. [Google Scholar] [CrossRef]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Thangavel, R.; Stolmeier, D.; Yang, X.; Anantharam, P.; Zaheer, A. Expression of glia maturation factor in neuropathological lesions of Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2012, 38, 572–581. [Google Scholar] [CrossRef]
- Thangavel, R.; Bhagavan, S.M.; Ramaswamy, S.B.; Surpur, S.; Govindarajan, R.; Kempuraj, D.; Zaheer, S.; Raikwar, S.; Ahmed, M.E.; Selvakumar, G.P.; et al. Co-Expression of Glia Maturation factor and Apolipoprotein E4 in Alzheimer’s Disease Brain. J. Alzheimers Dis. 2018, 61, 553–560. [Google Scholar] [CrossRef]
- Ahmed, M.E.; Iyer, S.; Thangavel, R.; Kempuraj, D.; Selvakumar, G.P.; Raikwar, S.P.; Zaheer, S.; Zaheer, A. Co-Localization of Glia Maturation Factor with NLRP3 Inflammasome and Autophagosome Markers in Human Alzheimer’s Disease Brain. J. Alzheimers Dis. 2017, 60, 1143–1160. [Google Scholar] [CrossRef]
- Ramaswamy, S.B.; Bhagavan, S.M.; Kaur, H.; Giler, G.E.; Kempuraj, D.; Thangavel, R.; Ahmed, M.E.; Selvakumar, G.P.; Raikwar, S.P.; Zaheer, S.; et al. Glia Maturation Factor in the Pathogenesis of Alzheimer’s disease. Open Access J. Neurol. Neurosurg. 2019, 12, 79–82. [Google Scholar] [PubMed]
- Couturier, J.; Stancu, I.C.; Schakman, O.; Pierrot, N.; Huaux, F.; Kienlen-Campard, P.; Dewachter, I.; Octave, J.N. Activation of phagocytic activity in astrocytes by reduced expression of the inflammasome component ASC and its implication in a mouse model of Alzheimer disease. J. Neuroinflammation 2016, 13, 20. [Google Scholar] [CrossRef] [PubMed]
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Wang, J.X.; Du, Y.H.; Liu, Y.; Zhang, W.; Chen, J.F.; Liu, Y.J.; Zheng, M.; Wang, K.J.; He, G.Q. Dihydromyricetin inhibits microglial activation and neuroinflammation by suppressing NLRP3 inflammasome activation in APP/PS1 transgenic mice. CNS Neurosci. Ther. 2018, 24, 1207–1218. [Google Scholar] [CrossRef]
- Dempsey, C.; Rubio Araiz, A.; Bryson, K.J.; Finucane, O.; Larkin, C.; Mills, E.L.; Robertson, A.; Cooper, M.A.; O’Neill, L.; Lynch, M.A. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain Behav. Immun. 2017, 61, 306–316. [Google Scholar] [CrossRef]
- Ebrahimi, T.; Rust, M.; Kaiser, S.N.; Slowik, A.; Beyer, C.; Koczulla, A.R.; Schulz, J.B.; Habib, P.; Bach, J.P. α1-antitrypsin mitigates NLRP3-inflammasome activation in amyloid β1-42-stimulated murine astrocytes. J. Neuroinflammation 2018, 15, 282. [Google Scholar] [CrossRef]
- Stancu, I.C.; Cremers, N.; Vanrusselt, H.; Couturier, J.; Vanoosthuyse, A.; Kessels, S.; Lodder, C.; Brône, B.; Huaux, F.; Octave, J.N.; et al. Aggregated Tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 2019, 137, 599–617. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Murphy, N.; Grehan, B.; Lynch, M.A. Glial uptake of amyloid beta induces NLRP3 inflammasome formation via cathepsin-dependent degradation of NLRP10. Neuromolecular Med. 2014, 16, 205–215. [Google Scholar] [CrossRef]
- Lu, B.; Nakamura, T.; Inouye, K.; Li, J.; Tang, Y.; Lundbäck, P.; Valdes-Ferrer, S.I.; Olofsson, P.S.; Kalb, T.; Roth, J.; et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 2012, 488, 670–674. [Google Scholar] [CrossRef]
- He, Y.; Franchi, L.; Núñez, G. The protein kinase PKR is critical for LPS-induced iNOS production but dispensable for inflammasome activation in macrophages. Eur. J. Immunol. 2013, 43, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Matyszewski, M.; Morrone, S.R.; Sohn, J. Digital signaling network drives the assembly of the AIM2-ASC inflammasome. Proc. Natl. Acad. Sci. USA 2018, 115, E1963–E1972. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.J.; Hung, Y.F.; Liu, H.Y.; Hsueh, Y.P. Deletion of the inflammasome sensor Aim2 mitigates Aβ deposition and microglial activation but increases inflammatory cytokine expression in an Alzheimer Disease mouse model. Neuroimmunomodulation 2017, 24, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, J.; Shi, J.; Gong, Y.N.; Lu, Q.; Xu, H.; Liu, L.; Shao, F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 2011, 477, 596–600. [Google Scholar] [CrossRef]
- Bauer, R.; Rauch, I. The NAIP/NLRC4 inflammasome in infection and pathology. Mol. Aspects Med. 2020, 100863. [Google Scholar] [CrossRef]
- Mariathasan, S.; Newton, K.; Monack, D.M.; Vucic, D.; French, D.M.; Lee, W.; Roose-Girma, M.; Erickson, S.; Dixit, V.M. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004, 430, 213–218. [Google Scholar] [CrossRef]
- Qu, Y.; Misaghi, S.; Izrael-Tomasevic, A.; Newton, K.; Gilmour, L.L.; Lamkanfi, M.; Louie, S.; Kayagaki, N.; Liu, J.; Kömüves, L.; et al. Phosphorylation of NLRC4 is critical for inflammasome activation. Nature 2012, 490, 539–542. [Google Scholar] [CrossRef]
- Christie, L.A.; Su, J.H.; Tu, C.H.; Dick, M.C.; Zhou, J.; Cotman, C.W. Differential regulation of inhibitors of apoptosis proteins in Alzheimer’s disease brains. Neurobiol. Dis. 2007, 26, 165–173. [Google Scholar] [CrossRef]
- Lesné, S.; Gabriel, C.; Nelson, D.A.; White, E.; Mackenzie, E.T.; Vivien, D.; Buisson, A. Akt-dependent expression of NAIP-1 protects neurons against amyloid-{beta} toxicity. J. Biol. Chem. 2005, 280, 24941–24947. [Google Scholar] [CrossRef]
- Seidl, R.; Bajo, M.; Böhm, K.; LaCasse, E.C.; MacKenzie, A.E.; Cairns, N.; Lubec, G. Neuronal apoptosis inhibitory protein (NAIP)-like immunoreactivity in brains of adult patients with Down syndrome. J. Neural Transm. Suppl. 1999, 57, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Chan, C. IPAF inflammasome is involved in interleukin-1β production from astrocytes, induced by palmitate; implications for Alzheimer’s Disease. Neurobiol. Aging. 2014, 35, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Marwarha, G.; Schommer, J.; Lund, J.; Schommer, T.; Ghribi, O. Palmitate-induced C/EBP homologous protein activation leads to NF-κB-mediated increase in BACE1 activity and amyloid beta genesis. J. Neurochem. 2018, 144, 761–779. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zhang, L.; Huang, J.; Gong, W.; Dunlop, N.M.; Wang, J.M. Cooperation between NOD2 and Toll-like receptor 2 ligands in the up-regulation of mouse mFPR2, a G-protein-coupled Abeta42 peptide receptor, in microglial cells. J. Leukoc. Biol. 2008, 83, 1467–1475. [Google Scholar] [CrossRef]
- Fani Maleki, A.; Cisbani, G.; Plante, M.M.; Préfontaine, P.; Laflamme, N.; Gosselin, J.; Rivest, S. Muramyl dipeptide-mediated immunomodulation on monocyte subsets exerts therapeutic effects in a mouse model of Alzheimer’s disease. J. Neuroinflammation 2020, 17, 218. [Google Scholar] [CrossRef]
- Li, Z.G.; Shui, S.F.; Han, X.W.; Yan, L. NLRP10 ablation protects against ischemia/reperfusion-associated brain injury by suppression of neuroinflammation. Exp. Cell Res. 2020, 389, 111912. [Google Scholar] [CrossRef]
- Zeng, Q.; Hu, C.; Qi, R.; Lu, D. PYNOD reduces microglial inflammation and consequent neurotoxicity upon lipopolysaccharides stimulation. Exp. Ther. Med. 2018, 15, 5337–5343. [Google Scholar] [CrossRef]
- Shichita, T.; Hasegawa, E.; Kimura, A.; Morita, R.; Sakaguchi, R.; Takada, I.; Sekiya, T.; Ooboshi, H.; Kitazono, T.; Yanagawa, T.; et al. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat. Med. 2012, 18, 911–917. [Google Scholar] [CrossRef]
- Gringhuis, S.I.; Kaptein, T.M.; Wevers, B.A.; Theelen, B.; van der Vlist, M.; Boekhout, T.; Geijtenbeek, T.B. Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1β via a noncanonical caspase-8 inflammasome. Nat. Immunol. 2012, 13, 246–254. [Google Scholar] [CrossRef]
- Gensel, J.C.; Wang, Y.; Guan, Z.; Beckwith, K.A.; Braun, K.J.; Wei, P.; McTigue, D.M.; Popovich, P.G. Toll-Like Receptors and Dectin-1, a C-Type Lectin Receptor, Trigger Divergent Functions in CNS Macrophages. J. Neurosci. 2015, 35, 9966–9976. [Google Scholar] [CrossRef]
- Ye, X.C.; Hao, Q.; Ma, W.J.; Zhao, Q.C.; Wang, W.W.; Yin, H.H.; Zhang, T.; Wang, M.; Zan, K.; Yang, X.X.; et al. Dectin-1/Syk signaling triggers neuroinflammation after ischemic stroke in mice. J. Neuroinflammation 2020, 17, 17. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, K.T.; Carbajal, K.S.; Segal, B.M.; Giger, R.J. Neuroinflammation triggered by β-glucan/dectin-1 signaling enables CNS axon regeneration. Proc. Natl. Acad. Sci. USA 2015, 112, 2581–2586. [Google Scholar] [CrossRef] [PubMed]
- Webster, J.A.; Gibbs, J.R.; Clarke, J.; Ray, M.; Zhang, W.; Holmans, P.; Rohrer, K.; Zhao, A.; Marlowe, L.; Kaleem, M.; et al. Genetic control of human brain transcript expression in Alzheimer disease. Am. J. Hum. Genet. 2009, 84, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.H.; Haddick, P.; Ngu, H.; et al. Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s Disease not evident in mouse models. Cell Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef]
- Wes, P.D.; Easton, A.; Corradi, J.; Barten, D.M.; Devidze, N.; DeCarr, L.B.; Truong, A.; He, A.; Barrezueta, N.X.; Polson, C.; et al. Tau overexpression impacts a neuroinflammation gene expression network perturbed in Alzheimer’s disease. PLoS ONE 2014, 9, e106050. [Google Scholar] [CrossRef]
- Holtman, I.R.; Raj, D.D.; Miller, J.A.; Schaafsma, W.; Yin, Z.; Brouwer, N.; Wes, P.D.; Möller, T.; Orre, M.; Kamphuis, W.; et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: A co-expression meta-analysis. Acta Neuropathol. Commun. 2015, 3, 31. [Google Scholar] [CrossRef]
- Wang, X.; Liu, G.J.; Gao, Q.; Li, N.; Wang, R.T. C-type lectin-like receptor 2 and zonulin are associated with mild cognitive impairment and Alzheimer’s disease. Acta Neurol. Scand. 2020, 141, 250–255. [Google Scholar] [CrossRef]
- Patel, J.R.; García-Sastre, A. Activation and regulation of pathogen sensor RIG-I. Cytokine Growth Factor Rev. 2014, 25, 513–523. [Google Scholar] [CrossRef]
- De Rivero Vaccari, J.P.; Brand, F.J., 3rd; Sedaghat, C.; Mash, D.C.; Dietrich, W.D.; Keane, R.W. RIG-1 receptor expression in the pathology of Alzheimer’s disease. J. Neuroinflammation 2014, 11, 67. [Google Scholar] [CrossRef]
- Chazal, M.; Beauclair, G.; Gracias, S.; Najburg, V.; Simon-Lorière, E.; Tangy, F.; Komarova, A.V.; Jouvenet, N. RIG-I recognizes the 5’ Region of Dengue and Zika virus genomes. Cell Rep. 2018, 24, 320–328. [Google Scholar] [CrossRef]
- Johnson, M.B.; Halman, J.R.; Burmeister, A.R.; Currin, S.; Khisamutdinov, E.F.; Afonin, K.A.; Marriott, I. Retinoic acid inducible gene-I mediated detection of bacterial nucleic acids in human microglial cells. J. Neuroinflammation 2020, 17, 139. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Wang, X.; Liu, Y.; Li, Y.; Long, S.; Gu, C.; Ye, J.; Xie, S.; Cao, S. Japanese Encephalitis Virus infection induces inflammation of swine testis through RIG-I-NF-ĸB signaling pathway. Vet. Microbiol. 2019, 238, 108430. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yang, R.; Feng, M.; Guo, Y.; Wang, Y.; Guo, J.; Lu, X. Rig-I is involved in inflammation through the IPS-1/TRAF6 pathway in astrocytes under chemical hypoxia. Neurosci. Lett. 2018, 672, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Chiarini, A.; Armato, U.; Dal Prà, I. Family C G-Protein-Coupled Receptors in Alzheimer’s Disease and Therapeutic Implications. Front. Pharmacol. 2019, 10, 1282. [Google Scholar] [CrossRef]
- Brown, E.M. The pathophysiology of primary hyperparathyroidism. J. Bone Miner. Res. 2002, 17 (Suppl. 2), N24–N29. [Google Scholar]
- Breitwieser, G.E. The calcium sensing receptor life cycle: Trafficking, cell surface expression, and degradation. Best Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 303–313. [Google Scholar] [CrossRef]
- Chiarini, A.; Armato, U.; Gardenal, E.; Gui, L.; Dal Prà, I. Amyloid β-Exposed Human Astrocytes Overproduce Phospho-Tau and Overrelease It within Exosomes, Effects Suppressed by Calcilytic NPS 2143-Further Implications for Alzheimer’s Therapy. Front. Neurosci. 2017, 11, 217. [Google Scholar] [CrossRef]
- Leach, K.; Gregory, K.J.; Kufareva, I.; Khajehali, E.; Cook, A.E.; Abagyan, R.; Conigrave, A.D.; Sexton, P.M.; Christopoulos, A. Towards a structural understanding of allosteric drugs at the human calcium-sensing receptor. Cell Res. 2016, 26, 574–592. [Google Scholar] [CrossRef]
- Nemeth, E.F.; Goodman, W.G. Calcimimetic and calcilytic drugs: Feats, flops, and futures. Calcif. Tissue Int. 2016, 98, 341–358. [Google Scholar] [CrossRef]
- Chakravarty, B.; Chattopadhyay, N.; Brown, E.M. Signaling through the extracellular calcium-sensing receptor (CaSR). Adv. Exp. Med. Biol. 2012, 740, 103–142. [Google Scholar] [CrossRef]
- Zhang, C.; Miller, C.L.; Brown, E.M.; Yang, J.J. The calcium sensing receptor: From calcium sensing to signaling. Sci. China Life Sci. 2015, 58, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Hofer, A.M.; Brown, E.M. Extracellular calcium sensing and signalling. Nat. Rev. Mol. Cell Biol. 2003, 4, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Klein, G.L.; Castro, S.M.; Garofalo, R.P. The calcium-sensing receptor as a mediator of inflammation. Semin. Cell Dev. Biol. 2016, 49, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Yarova, P.L.; Stewart, A.L.; Sathish, V.; Britt, R.D., Jr.; Thompson, M.A.P.; Lowe, A.P.; Freeman, M.; Aravamudan, B.; Kita, H.; Brennan, S.C.; et al. Calcium-sensing receptor antagonists abrogate airway hyperresponsiveness and inflammation in allergic asthma. Sci. Transl. Med. 2015, 7, 284ra60. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hong, S.; Qi, S.; Liu, W.; Zhang, X.; Shi, Z.; Chen, W.; Zhao, M.; Yin, X. NLRP3 Inflammasome is involved in Calcium-Sensing Receptor-induced aortic remodeling in SHRs. Mediat. Inflamm. 2019, 2019, 6847087. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Park, H.A.; Kwon, O.K.; Park, J.W.; Lee, G.; Lee, H.J.; Lee, S.J.; Oh, S.R.; Ahn, K.S. NPS 2143, a selective calcium-sensing receptor antagonist inhibits lipopolysaccharide-induced pulmonary inflammation. Mol. Immunol. 2017, 90, 150–157. [Google Scholar] [CrossRef]
- Jäger, E.; Murthy, S.; Schmidt, C.; Hahn, M.; Strobel, S.; Peters, A.; Stäubert, C.; Sungur, P.; Venus, T.; Geisler, M.; et al. Calcium-sensing receptor-mediated NLRP3 inflammasome response to calciprotein particles drives inflammation in rheumatoid arthritis. Nat. Commun. 2020, 11, 4243. [Google Scholar] [CrossRef] [PubMed]
- Mattar, P.; Bravo-Sagua, R.; Tobar, N.; Fuentes, C.; Troncoso, R.; Breitwieser, G.; Lavandero, S.; Cifuentes, M. Autophagy mediates calcium-sensing receptor-induced TNFα production in human preadipocytes. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3585–3594. [Google Scholar] [CrossRef]
- Hu, B.; Tong, F.; Xu, L.; Shen, Z.; Yan, L.; Xu, G.; Shen, R. Role of Calcium Sensing Receptor in streptozotocin-induced diabetic rats exposed to renal ischemia reperfusion injury. Kidney Blood Press. Res. 2018, 43, 276–286. [Google Scholar] [CrossRef]
- Iamartino, L.; Elajnaf, T.; Kallay, E.; Schepelmann, M. Calcium-sensing receptor in colorectal inflammation and cancer: Current insights and future perspectives. World J. Gastroenterol. 2018, 24, 4119–4131. [Google Scholar] [CrossRef]
- Bernichtein, S.; Pigat, N.; Barry Delongchamps, N.; Boutillon, F.; Verkarre, V.; Camparo, P.; Reyes-Gomez, E.; Méjean, A.; Oudard, S.M.; Lepicard, E.M.; et al. Vitamin D3 prevents calcium-induced progression of early-stage prostate tumors by counteracting TRPC6 and Calcium Sensing Receptor upregulation. Cancer Res. 2017, 77, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Olszak, I.T.; Poznansky, M.C.; Evans, R.H.; Olson, D.; Kos, C.; Pollak, M.R.; Brown, E.M.; Scadden, D.T. Extracellular calcium elicits a chemokinetic response from monocytes in vitro and in vivo. J. Clin. Investig. 2000, 105, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Hendy, G.N.; Canaff, L. Calcium-Sensing Receptor Gene: Regulation of Expression. Front. Physiol. 2016, 7, 394. [Google Scholar] [CrossRef] [PubMed]
- Bagur, R.; Hajnoczky, G. Intracellular Ca(2+) sensing: Its role in calcium homeostasis and signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Hendy, G.N.; Canaff, L. Calcium-sensing receptor, proinflammatory cytokines and calcium homeostasis. Semin. Cell. Dev. Biol. 2016, 49, 37–43. [Google Scholar] [CrossRef]
- Canton, J.; Schlam, D.; Breuer, C.; Gütschow, M.; Glogauer, M.; Grinstein, S. Calcium-sensing receptors signal constitutive macropinocytosis and facilitate the uptake of NOD2 ligands in macrophages. Nat. Commun. 2016, 7, 11284. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Tfelt-Hansen, J.; Chattopadhyay, N. Diverse roles of extracellular calcium-sensing receptor in the central nervous system. J. Neurosci. Res. 2010, 88, 2073–2082. [Google Scholar] [CrossRef]
- Yano, S.; Brown, E.M.; Chattopadhyay, N. Calcium-sensing receptor in the brain. Cell Calcium 2004, 35, 257–264. [Google Scholar] [CrossRef]
- Chattopadhyay, N.; Ye, C.; Yamaguchi, T.; Nakai, M.; Kifor, O.; Vassilev, P.M.; Nishimura, R.N.; Brown, E.M. The extracellular calcium-sensing receptor is expressed in rat microglia and modulates an outward K+ channel. J. Neurochem. 1999, 72, 1915–1922. [Google Scholar] [CrossRef]
- Riccardi, D.; Kemp, P.J. The calcium-sensing receptor beyond extracellular calcium homeostasis: Conception, development, adult physiology, and disease. Annu. Rev. Physyiol. 2012, 74, 271–297. [Google Scholar] [CrossRef]
- Ruat, M.; Traiffort, E. Roles of the calcium sensing receptor in the central nervous system. Best Pract. Res. Clin. Endocrinol. Metab. 2013, 27, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Noh, J.S.; Pak, H.J.; Shin, Y.J.; Riew, T.R.; Park, J.H.; Moon, Y.W.; Lee, M.Y. Differential expression of the calcium-sensing receptor in the ischemic and border zones after transient focal cerebral ischemia in rats. J. Chem. Neuroanat. 2015, 66–67, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Yang, X.; He, J.; Liu, J.; Yang, S.; Dong, H. Important roles of the Ca2+-sensing receptor in vascular health and disease. Life Sci. 2018, 209, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Conigrave, A.D.; Hampson, D.R. Broad-spectrum L-amino acid sensing by class 3 G-protein-coupled receptors. Trends Endocrinol. Metab. 2006, 17, 398–407. [Google Scholar] [CrossRef]
- Zhen, Y.; Ding, C.; Sun, J.; Wang, Y.; Li, S.; Dong, L. Activation of the calcium-sensing receptor promotes apoptosis by modulating the JNK/p38 MAPK pathway in focal cerebral ischemia-reperfusion in mice. Am. J. Transl. Res. 2016, 8, 911–921. [Google Scholar]
- Wang, C.; Jia, Q.; Sun, C.; Jing, C. Calcium sensing receptor contribute to early brain injury through the CaMKII/NLRP3 pathway after subarachnoid hemorrhage in mice. Biochem. Biophys. Res. Commun. 2020, 530, 651–657. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, N.; Yenari, M.A.; Chang, W. Hypothermia and pharmacological regimens that prevent overexpression and overactivity of the extracellular calcium-sensing receptor protect neurons against traumatic brain injury. J. Neurotrauma 2013, 30, 1170–1176. [Google Scholar] [CrossRef]
- Bai, S.; Mao, M.; Tian, L.; Yu, Y.; Zeng, J.; Ouyang, K.; Yu, L.; Li, L.; Wang, D.; Deng, X.; et al. Calcium sensing receptor mediated the excessive generation of β-amyloid peptide induced by hypoxia in vivo and in vitro. Biochem. Biophys. Res. Commun. 2015, 459, 568–573. [Google Scholar] [CrossRef]
- Gardenal, E.; Chiarini, A.; Armato, U.; Dal Prà, I.; Verkhratsky, A.; Rodríguez, J.J. Increased Calcium-Sensing Receptor Immunoreactivity in the Hippocampus of a Triple Transgenic Mouse Model of Alzheimer’s Disease. Front. Neurosci. 2017, 11, 81. [Google Scholar] [CrossRef]
- Feng, C.; Bao, X.; Shan, L.; Ling, Y.; Ding, Y.; Wang, J.; Cao, Y.; Wang, Q.; Cui, W.; Xu, S. Calcium-Sensing Receptor Mediates β-amyloid-induced synaptic formation impairment and cognitive deficits via regulation of cytosolic Phospholipase A2/Prostaglandin E2 metabolic pathway. Front. Aging Neurosci. 2020, 12, 144. [Google Scholar] [CrossRef]
- Dal Prà, I.; Chiarini, A.; Nemeth, E.F.; Armato, U.; Whitfield, J.F. Roles of Ca2+ and the Ca2+-sensing receptor (CaSR) in the expression of inducible NOS (nitric oxide synthase)-2 and its BH4 (tetrahydrobiopterin)-dependent activation in cytokine-stimulated adult human astrocytes. J. Cell. Biochem. 2005, 96, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.N.; Pan, R.; Qin, X.J.; Yang, W.L.; Qi, Z.; Liu, W.; Liu, K.J. Ischemic neurons activate astrocytes to disrupt endothelial barrier via increasing VEGF expression. J. Neurochem. 2014, 129, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, M.N.; Agro, A.; Bell, J.; Aisen, P.S.; Schweizer, E.; Galasko, D. PF-04494700, an oral inhibitor of receptor for advanced glycation end products (RAGE), in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2011, 25, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mustafa, M.; Yuede, C.M.; Salazar, S.V.; Kong, P.; Long, H.; Ward, M.; Siddiqui, O.; Paul, R.; Gilfillan, S.; et al. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J. Exp. Med. 2020, 217, e20200785. [Google Scholar] [CrossRef]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef]
- Yamanaka, M.; Ishikawa, T.; Griep, A.; Axt, D.; Kummer, M.P.; Heneka, M.T. PPARγ/RXRα-induced and CD36-mediated microglial amyloid-β phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J. Neurosci. 2012, 32, 17321–17331. [Google Scholar] [CrossRef]
- Mattson, M.P. ER calcium and Alzheimer’s disease: In a state of flux. Sci. Signal. 2010, 3, pe10. [Google Scholar] [CrossRef]



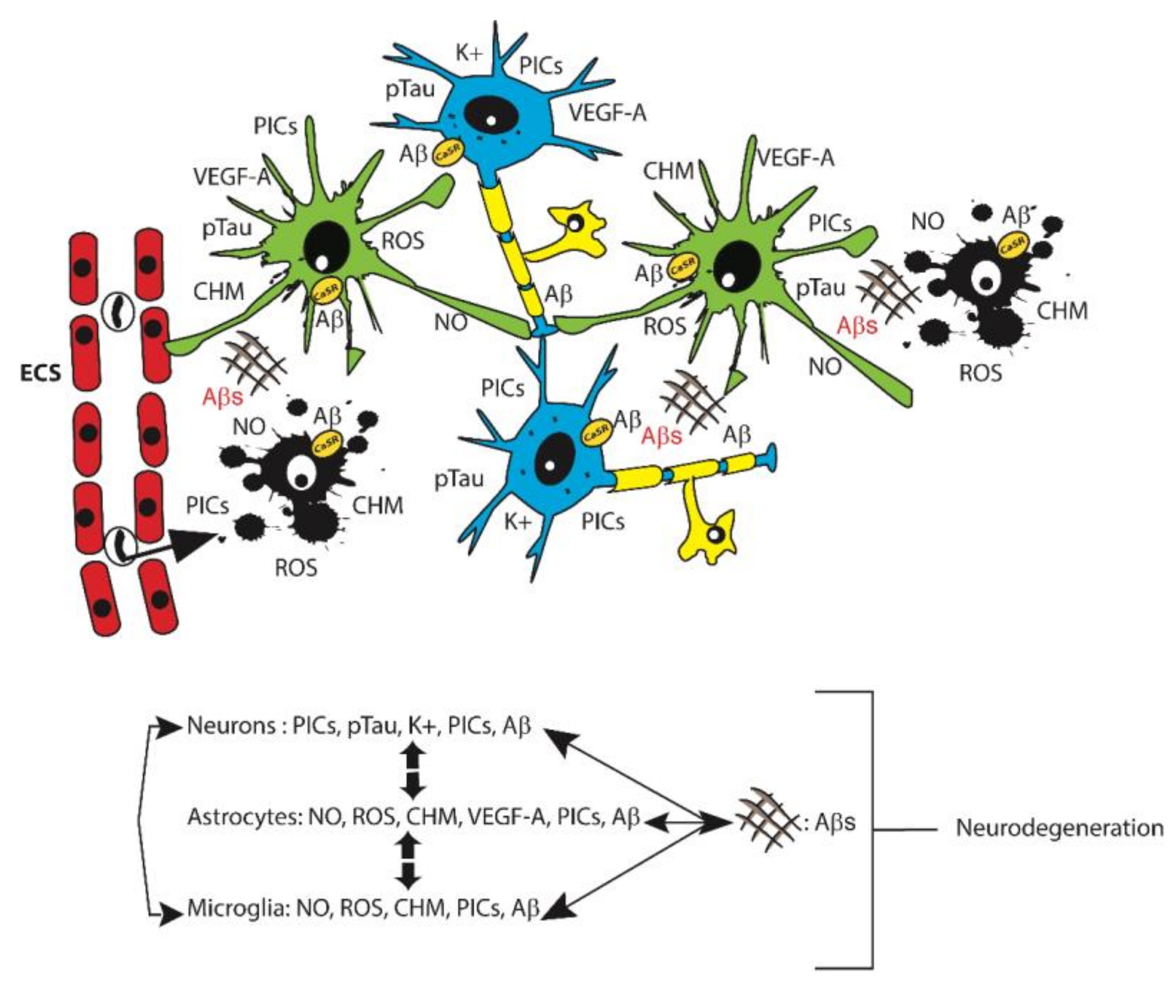{kind=link}
{kind=link}
| Year * | Refs. | |
|---|---|---|
| 1976 | Cholinergic hypothesis | [20] |
| 1991 | Amyloid-β hypothesis | [21,22] |
| 1992 | Calcium dyshomeostasis hypothesis | [23] |
| 1992 | Inflammation hypothesis | [24] |
| 1994 | Metal ions hypothesis | [25] |
| 1997 | Aβ•CaSR activating Ca2+ channels hypothesis | [26] |
| 2000–2004 | Neurovascular hypothesis | [27,28,29] |
| 2004 | Mitochondrial hypothesis | [30] |
| 2004 | Glymphatic system hypothesis | [31] |
| 2009 | Tau propagation hypothesis | [32] |
| 2013–2020 | Aβ•CaSR driving AD progression hypothesis | [33] |
| 2018 | Cellular senescence | [34] |
| 2020 | Neuroimmunomodulation hypothesis | [35] |
| Family history | [36] |
| Apolipoprotein-ε4 genotype | [37] |
| Metabolic syndrome Midlife obesity Hypercholesterolemia Hyperhomocysteinemia Type 2 Diabetes | [38,39,40,41,42] |
| Oxidative stress | [42] |
| Midlife hypertension | [43] |
| Sleep disorders | [44] |
| Oral infections | [45] |
| Gut microbiome dysbiosis | [46] |
| Human immunodeficiency virus (HIV) | [47] |
| Herpes simplex virus type 1 (HSV-1) | [48] |
| NLRP1 | NLRP2 | NLRP3 | NAIP/ NLRC4 | AIM2 | IPAF | NOD1 | NOD2 | NLRP10/ PYNOD | Refs. # | |
|---|---|---|---|---|---|---|---|---|---|---|
| Neurons | + ‡ | + | + | [212,215,217] | ||||||
| Astrocytes | + | + | + | + | [210,212,216] | |||||
| Microglia | + | + | [212] | |||||||
| Oligo-dendrocytes | + | + | [214] | |||||||
| Pericytes | + | + | + | + | + | + | [218] | |||
| Endothelial cells | + | + | + | + | + | [219] | ||||
| Postmortem AD human brain | + | + | + | + | [215,226,256,272,274,275] | |||||
| AD animal models | + | + | + | + | + | + | [227,228,229, 249,250,255,257,258,259,260,261,262,267,273,278] |
| Target | Treatment | U.S. FDA Status: | Study Status | Refs. |
|---|---|---|---|---|
| RAGE | Azeliragon/PF-04494700 (oral RAGE inhibitor) | AD (Phase 2/3) | Active | [338] |
| TREM2 | AL002 (anti-human TREM2) | AD (Phase 1/2) | Active | [339] |
| NLRP3 | Inzomelid (oral, brain-penetrant inhibitor of inflammasomes containing NLRP3) | AD (Phase 1) | Completed in March 2020 | [340] |
| CD36 | Pioglitazone (thiazolidinedione peroxisome-proliferator activated receptor γ [PPARγ] agonist) | Mild Cognitive Impairment (Phase 3) | Terminated (Lack of efficacy of the drug) | [341] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiarini, A.; Armato, U.; Hu, P.; Dal Prà, I. Danger-Sensing/Patten Recognition Receptors and Neuroinflammation in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 9036. https://doi.org/10.3390/ijms21239036
Chiarini A, Armato U, Hu P, Dal Prà I. Danger-Sensing/Patten Recognition Receptors and Neuroinflammation in Alzheimer’s Disease. International Journal of Molecular Sciences. 2020; 21(23):9036. https://doi.org/10.3390/ijms21239036
Chicago/Turabian StyleChiarini, Anna, Ubaldo Armato, Peng Hu, and Ilaria Dal Prà. 2020. "Danger-Sensing/Patten Recognition Receptors and Neuroinflammation in Alzheimer’s Disease" International Journal of Molecular Sciences 21, no. 23: 9036. https://doi.org/10.3390/ijms21239036
APA StyleChiarini, A., Armato, U., Hu, P., & Dal Prà, I. (2020). Danger-Sensing/Patten Recognition Receptors and Neuroinflammation in Alzheimer’s Disease. International Journal of Molecular Sciences, 21(23), 9036. https://doi.org/10.3390/ijms21239036