Inhibition of HDAC Enzymes Contributes to Differential Expression of Pro-Inflammatory Proteins in the TLR-4 Signaling Cascade


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
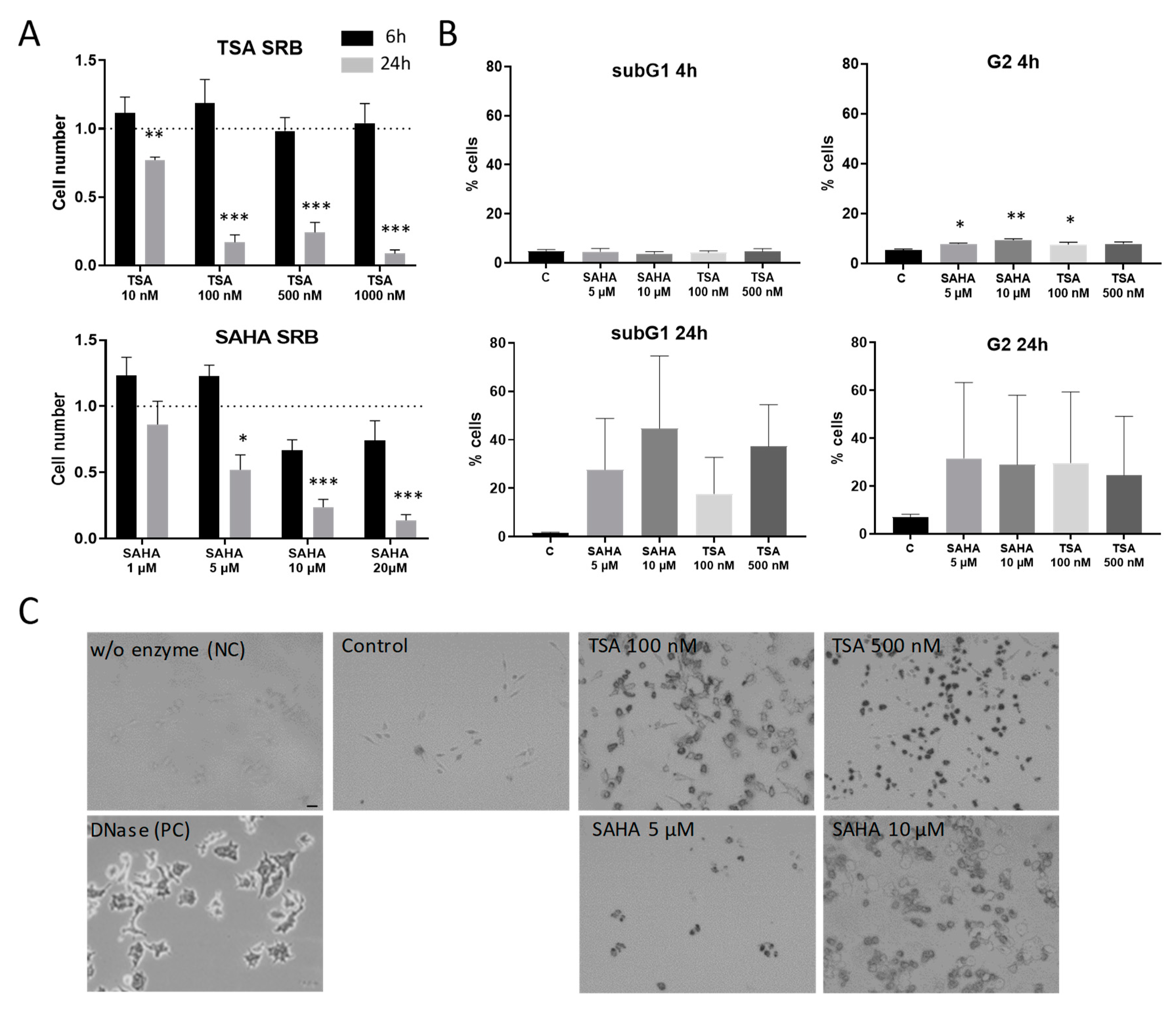
2.1. Effects of HDAC Inhibitors on Cell Proliferation and Apoptosis Induction
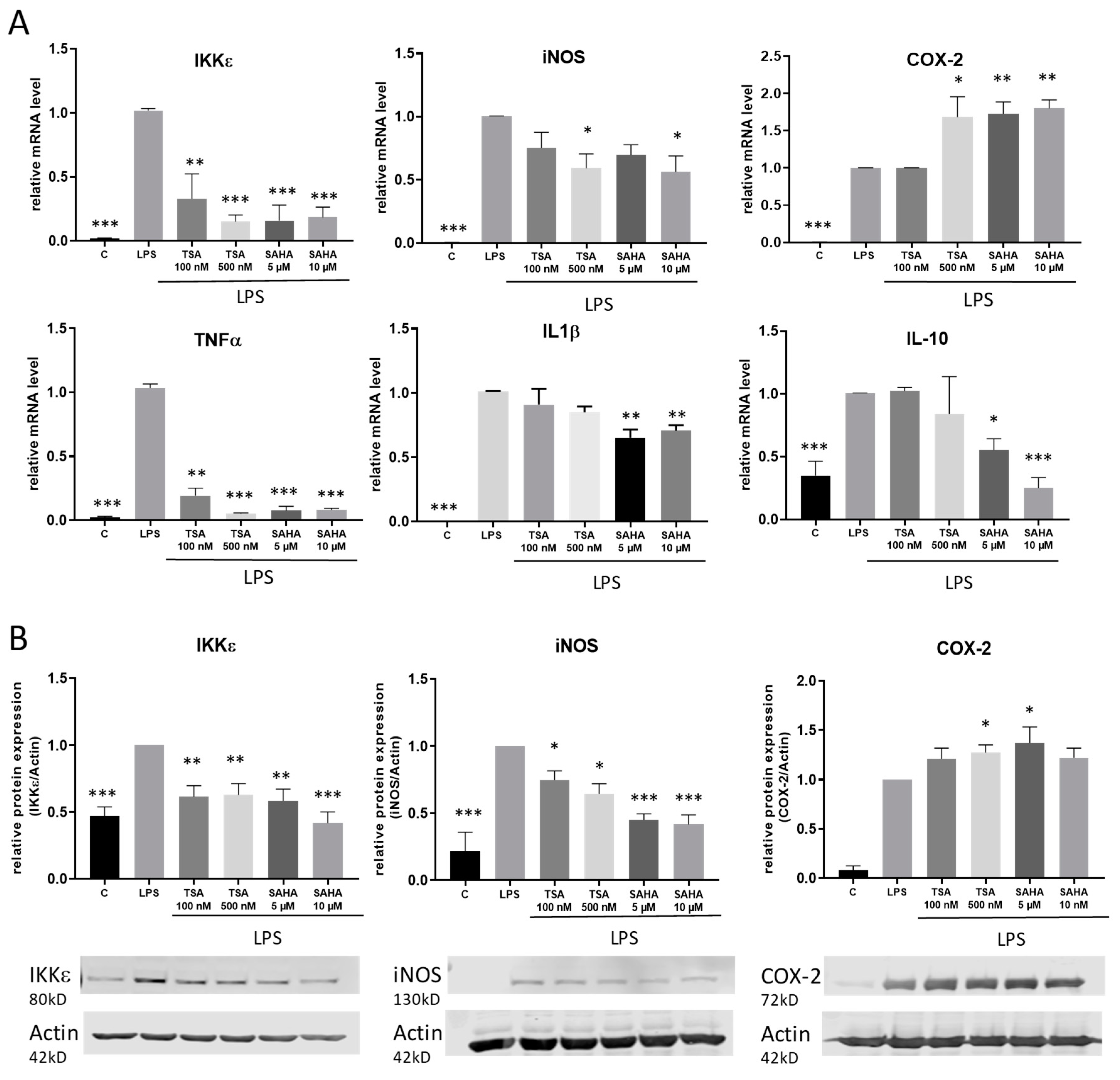
2.2. Impact of TSA and SAHA on the Expression of Pro-Inflammatory Genes
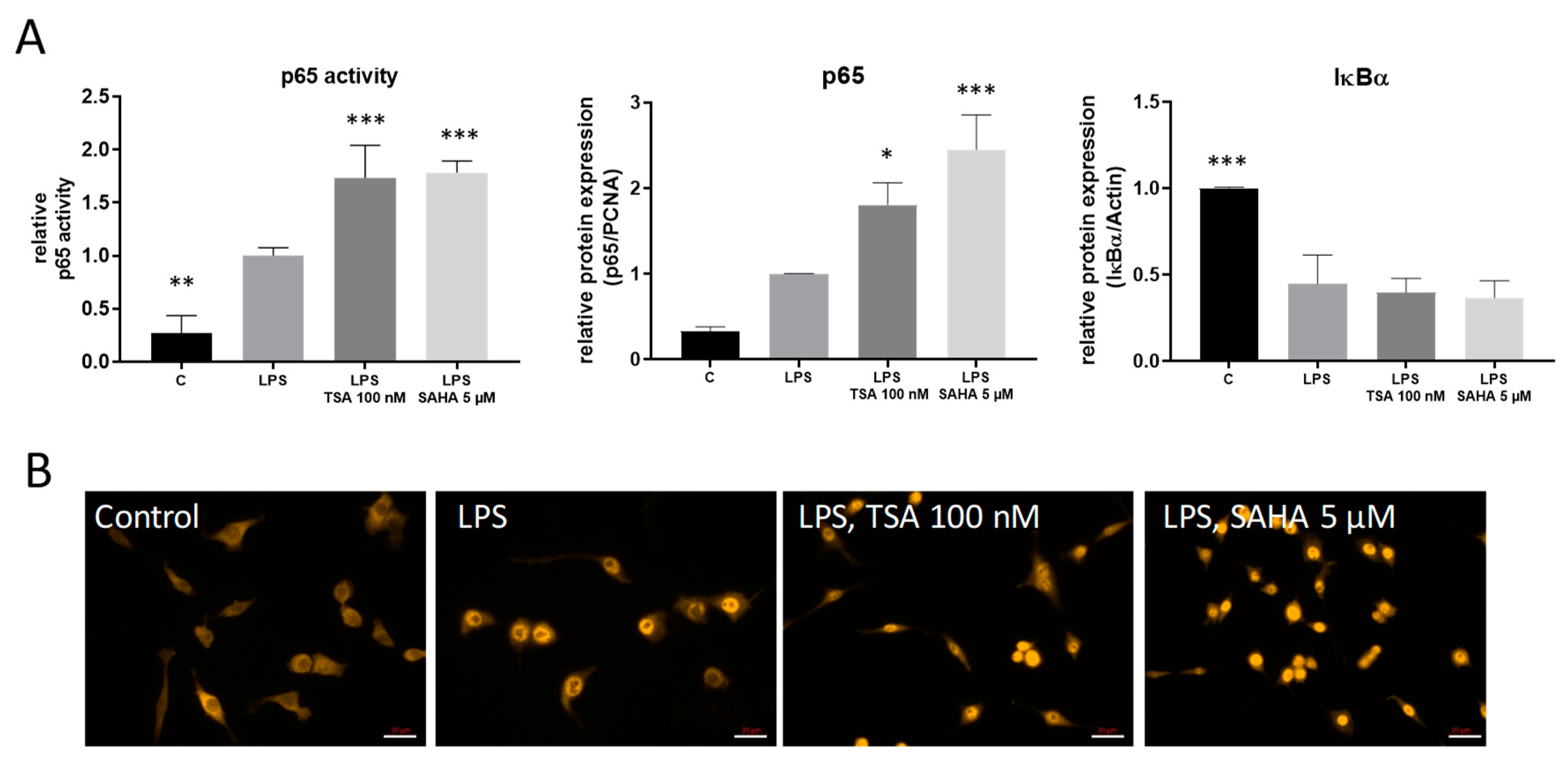
2.3. TSA and SAHA Enhance LPS-Induced NF-κB Activation
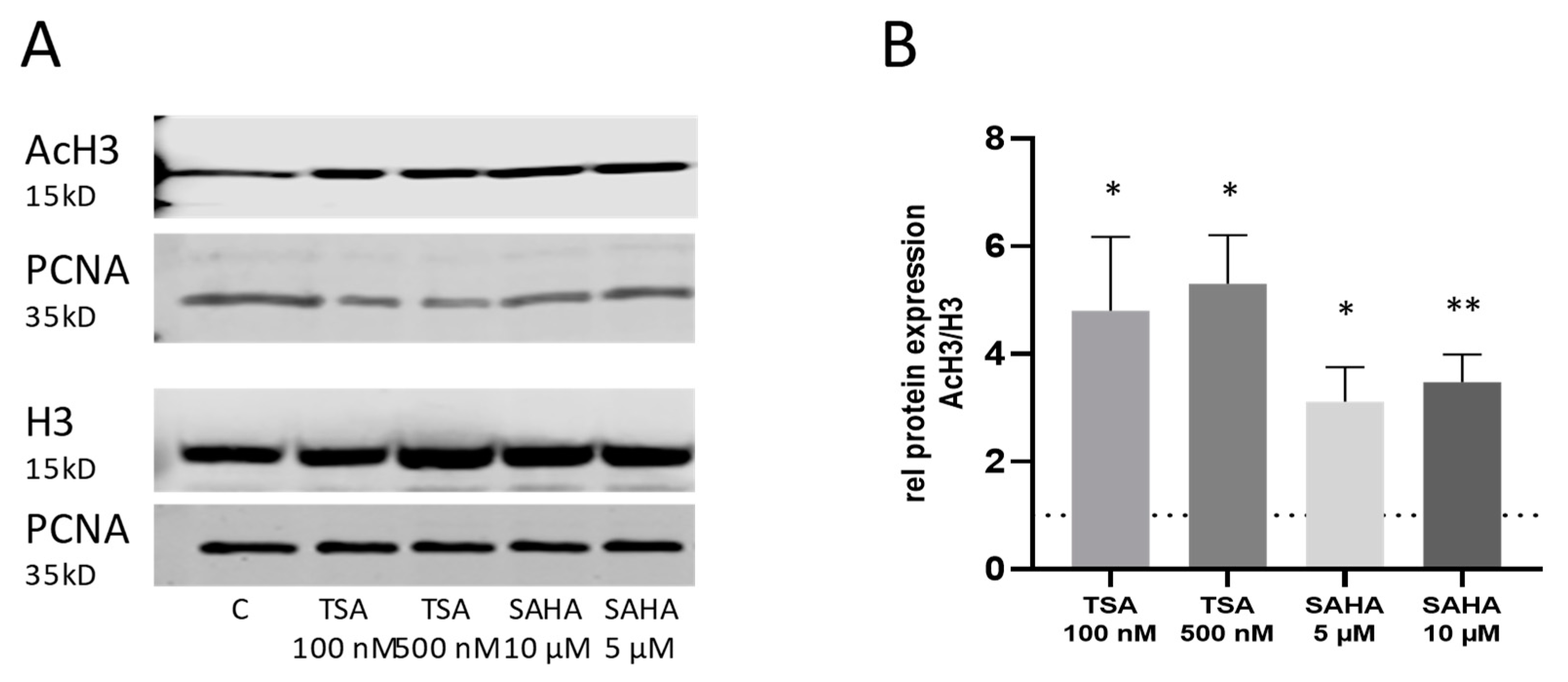
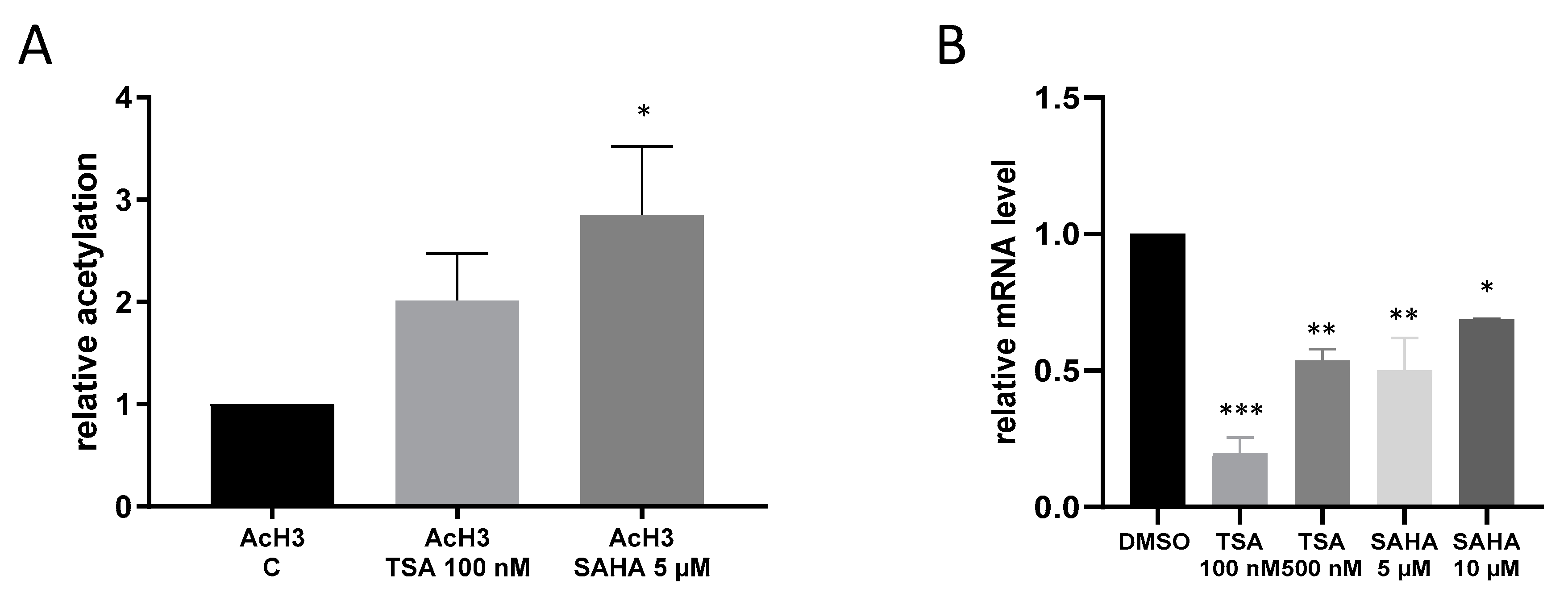
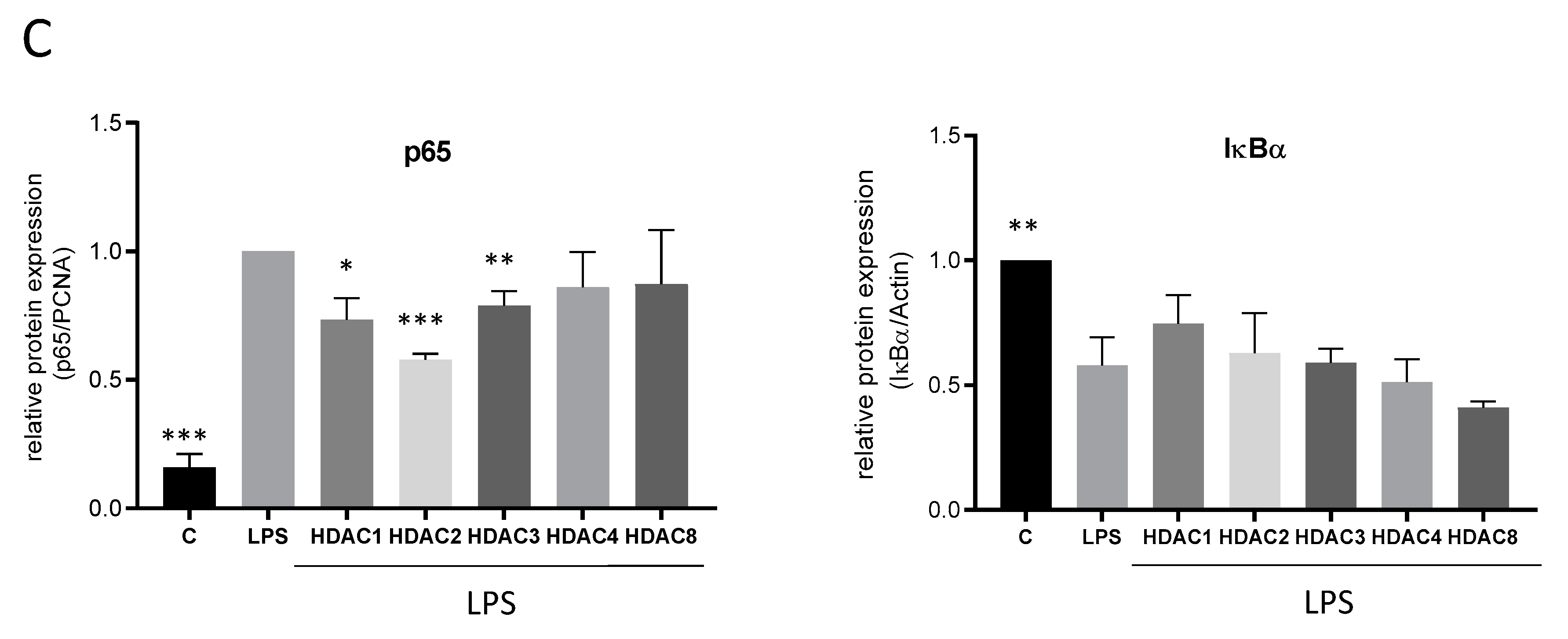
2.4. Effects of TSA and SAHA Are Only Partially Mediated by Histone Modulations
3. Discussion
4. Materials and Methods
4.1. Drugs
4.2. Cells
4.3. siRNA
4.4. Sulforhodamine B Cytotoxicity Assay
4.5. TUNEL Staining
4.6. Polymerase Chain Reactions (PCR)
| COX-2 | FW 5’-AGACACTCAGGTAGACATGATCTACCCT-3′ |
| RV 5′-GGCACCAGACCAAAGACTTCC-3′ | |
| IKKε | FW 5′-GTACAAGGCCCGAAACAAGA-3′ |
| RV 5′-TCCTCCACTGCGAATAGCTT-3′ | |
| IL-1β | FW 5′-CTGGTGTGTGACGTTCCCATTA-3′ |
| RV 5′-CCGACAGCACGAGGCTTT-3′ | |
| IL-10 | FW 5′-GCTCTTACTGACTGGCATGAG-3′ |
| RV 5′-CGCAGCTCTAGGAGCATGTG-3′ | |
| iNOS | FW 5′-ACACAGCGCTACAACATCCT-3′ |
| RV 5′-TGGAGCACAGCCACATTGAT-3′ | |
| TNFα | FW 5′-GCTGAGCTCAAACCCTGGTA-3′ |
| RV 5′-CGGACTCCGCAAAGTCTAAG-3′ | |
| GAPDH | FW 5′-CAATGTGTCCGTCGTGGATCT-3′ |
| RV 5′-GTCCTCAGTGTAGCCCAAGATG-3′ |
4.7. ChIP Assay
- FW
- 5′-GATCGACAAGTGCAATCCCC-3′
- RV
- 5′-AGCCACTCAAATGCACTGTG-3′
4.8. Western Blot Analysis
4.9. p65 TransAM ELISA
4.10. Flow Cytometry
4.11. Data Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AcH3 | Acetyl histone H3 |
| AmD | Actinomycin D |
| BMM | Bone marrow macrophages |
| ChIP | Chromatin immunoprecipitation |
| COX-2 | Cyclooxygenase 2 |
| DMSO | Dimethylsulfoxide |
| ELISA | Enzyme linked immunosorbent assay |
| FACS | Fluorescence activated cell sorting |
| FCS | Fetal calf serum |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| HAT | Histone acetylase |
| HDAC | Histone deacetylase |
| HDACi | Histone deacetylase inhibitor |
| IFN | Interferone |
| IκBa | Inhibitor kappa B protein alpha |
| IKKε | I-κB kinase epsilon |
| IL | Interleukin |
| LPS | lipopolysaccharide |
| M-CSF | murine macrophage colony stimulating factor |
| iNOS | Inducible nitric oxide synthase |
| NFκB | Nuclear factor kappa B |
| PCNA | Proliferating-Cell-Nuclear-Antigen |
| RLP30 | Receptor-like protein 30 |
| qRT-PCR | Quantitative reverse transcription polymerase chain reaction |
| TCA | Trichloroacetic acid |
| TLR | Toll-like receptor |
| TNF | Tumor necrosis factor |
| TSA | Trichostatin A |
| TUNEL | TdT-mediated dUTP nick end labeling |
| SAHA | Suberoylanilide hydroxamic acid |
| SRB | Sulforhodamine B |
References
- Rothbart, S.B.; Strahl, B.D. Interpreting the language of histone and DNA modifications. Biochim. Biophys Acta 2014, 1839, 627–643. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Zentner, G.E.; Henikoff, S. Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol. 2013, 20, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Petty, E.; Pillus, L. Balancing chromatin remodeling and histone modifications in transcription. Trends Genet. 2013, 29, 621–629. [Google Scholar] [CrossRef]
- Schmitz, M.L.; de la Vega, L. New Insights into the Role of Histone Deacetylases as Coactivators of Inflammatory Gene Expression. Antioxid Redox Signal 2015, 23, 85–98. [Google Scholar] [CrossRef]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef]
- Shakespear, M.R.; Halili, M.A.; Irvine, K.M.; Fairlie, D.P.; Sweet, M.J. Histone deacetylases as regulators of inflammation and immunity. Trends Immunol. 2011, 32, 335–343. [Google Scholar] [CrossRef]
- Sweet, M.J.; Shakespear, M.R.; Kamal, N.A.; Fairlie, D.P. HDAC inhibitors: Modulating leukocyte differentiation, survival, proliferation and inflammation. Immunol. Cell Biol. 2012, 90, 14–22. [Google Scholar] [CrossRef]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef]
- Daskalaki, M.G.; Tsatsanis, C.; Kampranis, S.C. Histone methylation and acetylation in macrophages as a mechanism for regulation of inflammatory responses. J. Cell. Physiol. 2018, 233, 6495–6507. [Google Scholar] [CrossRef]
- Schultze, J.L. Chromatin Remodeling in Monocyte and Macrophage Activation. Adv. Protein Chem. Struct. Biol. 2017, 106, 1–15. [Google Scholar] [CrossRef]
- Samanta, S.; Rajasingh, S.; Cao, T.; Dawn, B.; Rajasingh, J. Epigenetic dysfunctional diseases and therapy for infection and inflammation. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Halili, M.A.; Andrews, M.R.; Sweet, M.J.; Fairlie, D.P. Histone deacetylase inhibitors in inflammatory disease. Curr. Top. Med. Chem. 2009, 9, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Adam, E.; Quivy, V.; Bex, F.; Chariot, A.; Collette, Y.; Vanhulle, C.; Schoonbroodt, S.; Goffin, V.; Nguyen, T.L.; Gloire, G.; et al. Potentiation of tumor necrosis factor-induced NF-kappa B activation by deacetylase inhibitors is associated with a delayed cytoplasmic reappearance of I kappa B alpha. Mol. Cell. Biol. 2003, 23, 6200–6209. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Rahmani, M.; Dent, P.; Grant, S. Blockade of histone deacetylase inhibitor-induced RelA/p65 acetylation and NF-kappaB activation potentiates apoptosis in leukemia cells through a process mediated by oxidative damage, XIAP downregulation, and c-Jun N-terminal kinase 1 activation. Mol. Cell. Biol. 2005, 25, 5429–5444. [Google Scholar] [CrossRef]
- Moser, C.V.; Kynast, K.; Baatz, K.; Russe, O.Q.; Ferreiros, N.; Costiuk, H.; Lu, R.; Schmidtko, A.; Tegeder, I.; Geisslinger, G.; et al. The protein kinase IKKepsilon is a potential target for the treatment of inflammatory hyperalgesia. J. Immunol. 2011, 187, 2617–2625. [Google Scholar] [CrossRef] [PubMed]
- Oral, E.A.; Reilly, S.M.; Gomez, A.V.; Meral, R.; Butz, L.; Ajluni, N.; Chenevert, T.L.; Korytnaya, E.; Neidert, A.H.; Hench, R.; et al. Inhibition of IKKvarepsilon and TBK1 Improves Glucose Control in a Subset of Patients with Type 2 Diabetes. Cell Metab. 2017, 26, 157–170.e157. [Google Scholar] [CrossRef]
- Reilly, S.M.; Chiang, S.H.; Decker, S.J.; Chang, L.; Uhm, M.; Larsen, M.J.; Rubin, J.R.; Mowers, J.; White, N.M.; Hochberg, I.; et al. An inhibitor of the protein kinases TBK1 and IKK-varepsilon improves obesity-related metabolic dysfunctions in mice. Nat. Med. 2013, 19, 313–321. [Google Scholar] [CrossRef]
- Aung, H.T.; Schroder, K.; Himes, S.R.; Brion, K.; van Zuylen, W.; Trieu, A.; Suzuki, H.; Hayashizaki, Y.; Hume, D.A.; Sweet, M.J.; et al. LPS regulates proinflammatory gene expression in macrophages by altering histone deacetylase expression. FASEB J. 2006, 20, 1315–1327. [Google Scholar] [CrossRef]
- Halili, M.A.; Andrews, M.R.; Labzin, L.I.; Schroder, K.; Matthias, G.; Cao, C.; Lovelace, E.; Reid, R.C.; Le, G.T.; Hume, D.A.; et al. Differential effects of selective HDAC inhibitors on macrophage inflammatory responses to the Toll-like receptor 4 agonist LPS. J. Leukoc. Biol. 2010, 87, 1103–1114. [Google Scholar] [CrossRef]
- Chong, W.; Li, Y.; Liu, B.; Zhao, T.; Fukudome, E.Y.; Liu, Z.; Smith, W.M.; Velmahos, G.C.; de Moya, M.A.; Alam, H.B. Histone deacetylase inhibitor suberoylanilide hydroxamic acid attenuates Toll-like receptor 4 signaling in lipopolysaccharide-stimulated mouse macrophages. J. Surg. Res. 2012, 178, 851–859. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef] [PubMed]
- Grabiec, A.M.; Krausz, S.; de Jager, W.; Burakowski, T.; Groot, D.; Sanders, M.E.; Prakken, B.J.; Maslinski, W.; Eldering, E.; Tak, P.P.; et al. Histone deacetylase inhibitors suppress inflammatory activation of rheumatoid arthritis patient synovial macrophages and tissue. J. Immunol. 2010, 184, 2718–2728. [Google Scholar] [CrossRef]
- Leoni, F.; Zaliani, A.; Bertolini, G.; Porro, G.; Pagani, P.; Pozzi, P.; Dona, G.; Fossati, G.; Sozzani, S.; Azam, T.; et al. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc. Natl. Acad. Sci. USA 2002, 99, 2995–3000. [Google Scholar] [CrossRef] [PubMed]
- Jiao, F.Z.; Wang, Y.; Zhang, H.Y.; Zhang, W.B.; Wang, L.W.; Gong, Z.J. Histone Deacetylase 2 Inhibitor CAY10683 Alleviates Lipopolysaccharide Induced Neuroinflammation through Attenuating TLR4/NF-kappaB Signaling Pathway. Neurochem. Res. 2018, 43, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Li, A.; Hu, J.; Kang, J. Histone deacetylase 2 is essential for LPS-induced inflammatory responses in macrophages. Immunol. Cell Biol. 2019, 97, 72–84. [Google Scholar] [CrossRef]
- Jeong, Y.; Du, R.; Zhu, X.; Yin, S.; Wang, J.; Cui, H.; Cao, W.; Lowenstein, C.J. Histone deacetylase isoforms regulate innate immune responses by deacetylating mitogen-activated protein kinase phosphatase-1. J. Leukoc. Biol. 2014, 95, 651–659. [Google Scholar] [CrossRef]
- Han, S.B.; Lee, J.K. Anti-inflammatory effect of Trichostatin-A on murine bone marrow-derived macrophages. Arch. Pharmacal Res. 2009, 32, 613–624. [Google Scholar] [CrossRef]
- Niederberger, E.; Moser, C.; Kynast, K.; Geisslinger, G. The non-canonical IkappaB kinases IKKepsilon and TBK1 as potential targets for the development of novel therapeutic drugs. Curr. Mol. Med. 2012, 13, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.G.; Zhu, Y.; Wu, K.K. Role of p300 and PCAF in regulating cyclooxygenase-2 promoter activation by inflammatory mediators. Blood 2004, 103, 2135–2142. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.W.; Wang, S.A.; Hsu, T.Y.; Chen, T.A.; Chang, W.C.; Hung, J.J. Inhibition of LPS-induced C/EBP delta by trichostatin a has a positive effect on LPS-induced cyclooxygenase 2 expression in RAW264.7 cells. J. Cell. Biochem. 2010, 110, 1430–1438. [Google Scholar] [CrossRef] [PubMed]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Ghisletti, S.; Barozzi, I.; Mietton, F.; Polletti, S.; De Santa, F.; Venturini, E.; Gregory, L.; Lonie, L.; Chew, A.; Wei, C.L.; et al. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity 2010, 32, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Karpurapu, M.; Wang, X.; Deng, J.; Park, H.; Xiao, L.; Sadikot, R.T.; Frey, R.S.; Maus, U.A.; Park, G.Y.; Scott, E.W.; et al. Functional PU.1 in macrophages has a pivotal role in NF-kappaB activation and neutrophilic lung inflammation during endotoxemia. Blood 2011, 118, 5255–5266. [Google Scholar] [CrossRef]
- Chen, L.F.; Mu, Y.; Greene, W.C. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO J. 2002, 21, 6539–6548. [Google Scholar] [CrossRef]
- Gupta, S.C.; Sundaram, C.; Reuter, S.; Aggarwal, B.B. Inhibiting NF-kappaB activation by small molecules as a therapeutic strategy. Biochim. Biophys. Acta 2010, 1799, 775–787. [Google Scholar] [CrossRef]
- Bode, K.A.; Schroder, K.; Hume, D.A.; Ravasi, T.; Heeg, K.; Sweet, M.J.; Dalpke, A.H. Histone deacetylase inhibitors decrease Toll-like receptor-mediated activation of proinflammatory gene expression by impairing transcription factor recruitment. Immunology 2007, 122, 596–606. [Google Scholar] [CrossRef]
- Ito, K.; Yamamura, S.; Essilfie-Quaye, S.; Cosio, B.; Ito, M.; Barnes, P.J.; Adcock, I.M. Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression. J. Exp. Med. 2006, 203, 7–13. [Google Scholar] [CrossRef]
- Choo, Q.Y.; Ho, P.C.; Tanaka, Y.; Lin, H.S. Histone deacetylase inhibitors MS-275 and SAHA induced growth arrest and suppressed lipopolysaccharide-stimulated NF-kappaB p65 nuclear accumulation in human rheumatoid arthritis synovial fibroblastic E11 cells. Rheumatology (Oxford) 2010, 49, 1447–1460. [Google Scholar] [CrossRef]
- Usami, M.; Kishimoto, K.; Ohata, A.; Miyoshi, M.; Aoyama, M.; Fueda, Y.; Kotani, J. Butyrate and trichostatin A attenuate nuclear factor kappaB activation and tumor necrosis factor alpha secretion and increase prostaglandin E2 secretion in human peripheral blood mononuclear cells. Nutr. Res. 2008, 28, 321–328. [Google Scholar] [CrossRef]
- Huang, B.; Yang, X.D.; Lamb, A.; Chen, L.F. Posttranslational modifications of NF-kappaB: Another layer of regulation for NF-kappaB signaling pathway. Cell. Signal. 2010, 22, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiss, U.; Möller, M.; Husseini, S.A.; Manderscheid, C.; Häusler, J.; Geisslinger, G.; Niederberger, E. Inhibition of HDAC Enzymes Contributes to Differential Expression of Pro-Inflammatory Proteins in the TLR-4 Signaling Cascade. Int. J. Mol. Sci. 2020, 21, 8943. https://doi.org/10.3390/ijms21238943
Weiss U, Möller M, Husseini SA, Manderscheid C, Häusler J, Geisslinger G, Niederberger E. Inhibition of HDAC Enzymes Contributes to Differential Expression of Pro-Inflammatory Proteins in the TLR-4 Signaling Cascade. International Journal of Molecular Sciences. 2020; 21(23):8943. https://doi.org/10.3390/ijms21238943
Chicago/Turabian StyleWeiss, Ulrike, Moritz Möller, Sayed Adham Husseini, Christine Manderscheid, Julia Häusler, Gerd Geisslinger, and Ellen Niederberger. 2020. "Inhibition of HDAC Enzymes Contributes to Differential Expression of Pro-Inflammatory Proteins in the TLR-4 Signaling Cascade" International Journal of Molecular Sciences 21, no. 23: 8943. https://doi.org/10.3390/ijms21238943
APA StyleWeiss, U., Möller, M., Husseini, S. A., Manderscheid, C., Häusler, J., Geisslinger, G., & Niederberger, E. (2020). Inhibition of HDAC Enzymes Contributes to Differential Expression of Pro-Inflammatory Proteins in the TLR-4 Signaling Cascade. International Journal of Molecular Sciences, 21(23), 8943. https://doi.org/10.3390/ijms21238943