Abstract
Asthma is a chronic inflammatory airway disease characterized by variable airflow obstruction in response to a wide range of exogenous stimuli. The airway epithelium is the first line of defense and plays an important role in initiating host defense and controlling immune responses. Indeed, increasing evidence indicates a range of abnormalities in various aspects of epithelial barrier function in asthma. A central part of this impairment is a disruption of the airway epithelial layer, allowing inhaled substances to pass more easily into the submucosa where they may interact with immune cells. Furthermore, many of the identified susceptibility genes for asthma are expressed in the airway epithelium. This review focuses on the biology of the airway epithelium in health and its pathobiology in asthma. We will specifically discuss external triggers such as allergens, viruses and alarmins and the effect of type 2 inflammatory responses on airway epithelial function in asthma. We will also discuss epigenetic mechanisms responding to external stimuli on the level of transcriptional and posttranscriptional regulation of gene expression, as well the airway epithelium as a potential treatment target in asthma.
1. Introduction
Affecting more than 300 children and adults worldwide [1], asthma is a chronic inflammatory disease characterized by chest tightness, variable airflow limitation, coughing, wheezing and airway hyperresponsiveness to environmental triggers (i.e., allergens, pollen, animal dander, tobacco smoke and air pollution) [2,3]. Asthma symptoms are a result of an ongoing chronic airway inflammation. Allergic asthma is the most common type, where reversible airway limitation is caused by allergic airway inflammation and allergic sensitization is the major risk factor. Allergen exposure in sensitized individuals typically triggers a type 2 (T2)-biased inflammatory response. In the sensitization phase, inhaled allergens are captured by dendritic cells (DCs) and presented to naive CD4+ T cells in the presence of coactivators, including epithelial-derived cytokines, which promotes activation and polarization of T helper 2 (Th2) cells that produce IL-4, IL-5, and IL-13 [3,4]. These T2 cytokines are also produced by type 2 innate lymphoid cells (ILC2s) and are prominent orchestrators of the allergic inflammatory cascade that occurs in asthma. IL-4 drives isotype switching of B cells and production of IgE, which binds to the high affinity IgE receptor on mast cells. Allergen re-exposure results in allergen-mediated IgE cross-linking, which causes rapid mast cell activation and degranulation. IL-5 promotes airway eosinophilia, IL-4 and IL-13 act directly on the airway epithelium to induce goblet cell metaplasia and mucus hypersecretion, and IL-13 mediates airway hyperresponsiveness by effects on airway smooth muscle cells [4].
While allergic and non-allergic asthma are the most common asthma phenotypes, these can be further divided into a variety of subgroups, including eosinophilic or non-eosinophilic asthma, as well as late-onset asthma [5]. Adding to the complexity, asthma phenotypes are driven by different immunological mechanisms, so-called endotypes. Thus, to date it is clear that asthma is a heterogeneous disease [6,7]. However, current knowledge of the underlying molecular mechanisms in asthma subgroups is limited, and more information is needed in order to improve disease diagnosis and treatment regimes.
The airway epithelium is the first line of defense against pathogenic environmental factors such as allergens, pollution, viruses, fungi, and bacterial infections [8]. Hence, the airway epithelium plays an important role in initiating host defense and controlling immune responses and plays a key role in disease development and progression in asthma [8,9].
2. The Structure and Function of the Airway Epithelium
All surfaces of the mammalian body are covered with epithelial cells, including the skin, the gastrointestinal tract, and the airways from the nose and mouth all the way down to the alveoli. Though the structure and functions of epithelial cells differ depending on their location, they all are tightly interconnected through epithelial junctions. This reveals one key role of the epithelium in serving as a physical barrier against the environment.
The focus of this review is the lower conducting airways, or lower respiratory tract, which includes the trachea and the bronchi that branch out throughout each lung and end as terminal bronchioles just before the alveoli where gas exchange occurs. In the trachea and bronchi, the epithelium is pseudostratified with a clear apical-basolateral orientation and consists of ciliated cells, goblet cells, club cells, and the underlying basal cells (Figure 1). The ciliated cells together with goblet and club cells make up the mucociliary escalator on the apical side in which inhaled particles are trapped in secreted mucus and the beating cilia transport it upwards to the mouth where it is then swallowed or expectorated [10,11]. The basal cells comprise the stem-cell niche of the conducting airways by having the ability to differentiate into the other cell types mentioned above [12,13,14,15].
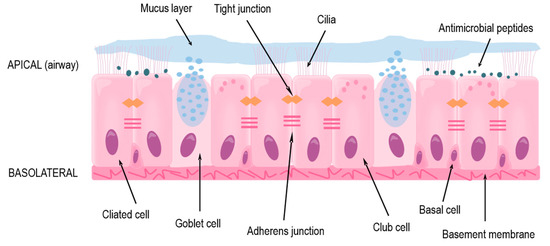
Figure 1.
The structure and protective functions of the human airway epithelium in the lower respiratory tract.
Under normal conditions, the mucociliary escalator ensures homeostasis by preventing any possible irritants or large pathogens from accessing the epithelial cells or underlying systemic circulation [11,16]. Additionally, airway epithelial cells produce various antimicrobial peptides (AMPs) which prevent microbes from colonizing the airways [17,18,19]. In asthma, both of these secretion-based protections are usually impaired, either due to endogenous genetic variations [20] or as an effect of an ongoing inflammation [9,21,22].
If inhaled agents escape the mucus or AMPs, through inherent properties or impairment of these defenses, they reach and can affect or infect the epithelial cells themselves [11,16]. Due to the tight barrier formed by the cells, many pathogens and allergens are prevented from reaching the circulation and accessing other cells and organs. The barrier is maintained by anchoring of the extracellular domains of proteins found near the cellular membrane and is organized into tight and adherens junctions, as well as desmosomes [23]. Tight junctions are located close to the apical side of the cells and are made up of transmembrane proteins occludin and members of the claudin and junctional adhesion molecule (JAM) families, which anchor to cytoplasmic proteins cingulin and members of the zonula occludens (ZO) family [23]. These proteins also interact with polarity proteins, which aid in the correct localization of the tight junctions; the tight junctions are then involved in maintaining the apical-basolateral polarity of the cells [24], which ensures the functionality of the epithelium. Beneath the tight junctions are adherens junctions, where transmembrane-spanning E-cadherin binds to intracellular p120-, β-, and α-catenin [25]. Several junctional proteins have been found to be disorganized or dysregulated in asthma [26,27], leading to an impaired barrier and dysfunction of the epithelium.
On the basolateral side of the airway epithelium is the basement membrane, consisting of extracellular matrix, to which the epithelial cells are strongly anchored through hemidesmosomes [28,29]. Farther below, there are airway smooth muscle cells, fibroblasts, and blood vessels, as well as cartilage rings around the trachea and first-generation bronchi [30]. These cells and structures, together with the epithelium, all contribute to the functionality of the airways and lungs through providing structure, contraction, and nutrients. There is also crosstalk between these underlying cells and the epithelium, mediated mainly through soluble factors [28,30,31].
On each side of the epithelial cells, and reaching between them, are both innate and adaptive immune cells. These cells include ILCs, DCs, mast cells, eosinophils, and T cells. In asthma, both the type and number of these cells are altered. Secreted factors produced by these cells, such as cytokines, proteases, and lipid mediators, affect the airway epithelium which in turn release alarmins and chemokines that affect the immune cells, creating a bridge between innate and adaptive immunity.
3. Genetic Associations with Asthma Linked to the Airway Epithelium
Several genomic screens and genome-wide association (GWA) studies have found genes and genetic loci associated with asthma that are expressed by the epithelium [20,32]. These findings highlight the importance of the airway epithelium in healthy individuals as well as in the pathology of asthma. One group of genes associated with asthma are related to the epithelial barrier function (see summary in ref. [33]); these include PCDH1 (protocadherin-1), which is involved in cell adhesion and epithelial barrier formation [34] and CDHR3 (cadherin-related family member 3), which is also involved in cell adhesion as well as epithelial polarity and is the receptor for rhinovirus (RV) C, where the risk variant could increase susceptibility to infection [35,36,37]. Additionally, ORMDL3 (orosomucoid-like 3) has been linked to asthma in several populations [38,39,40], and its corresponding protein may be involved in cell adhesion and integrity; when increased, it has been shown to promote airway remodeling and hyperresponsiveness [41]. However, deletion of ORMDL3 was also associated with increased airway hyperresponsiveness and remodeling [42], indicating that its mechanistic link to asthma risk is yet unknown. Other asthma-associated genes with possible roles in barrier function are DPP10 (dipeptidyl peptidase 10) [43] and GPRA (G protein–coupled receptor for asthma susceptibility) [44].
Associations have also been found between variants of mucin-encoding genes (MUC5AC and MUC5B) and the risk for asthma [45,46], where the variants are predicted to cause increased mucin production. CLCA1 (calcium-activated chloride channel regulator 1), which is involved in mucus secretion, has also been linked to asthma [47]. Lastly, several studies have linked polymorphisms in epithelial alarmins TSLP and IL33 with the risk for asthma [36,39,46,48,49,50,51]. These findings indicate that genetic defects or variations within the airway epithelium can cause, drive, or worsen asthma, most likely driven through interactions with the environment.
4. Impairment of the Airway Epithelial Barrier in Asthma
Compelling evidence indicates a range of abnormalities in various aspects of epithelial barrier function in asthma. Part of this impairment is a disruption of the airway epithelial layer, which may facilitate submucosal infiltration of inhaled substances and consequently their interaction with immune cells. In situ observations of the airway epithelium have revealed structural changes in asthmatic individuals, including patchy disruption of tight junctions, detachment of ciliated cells, and reduced expression of E-cadherin as well as other cell-cell adhesion molecules [26,27,52,53]. In line with these findings, functional studies of airway epithelial cells cultured at air-liquid interface (ALI) indicate increased permeability and sensitivity to environmental insults in cells from individuals with asthma compared with healthy controls [27,54,55]. Although the mechanisms contributing to loss of airway epithelial barrier function in asthma have not been fully elucidated, a combination of different extrinsic and intrinsic factors are likely to play a role.
Allergens, viral infections, and T2 inflammation are strongly associated with the pathogenesis of allergic asthma and are all considered to have detrimental effects on the barrier integrity of the airway epithelium. A number of in vitro studies have demonstrated the ability of various protease-containing allergens to disrupt the airway epithelial barrier, either directly or indirectly via activation of protease-activated receptor (PAR)-2, a proinflammatory innate immune receptor on epithelial cells [56]. The latter has been shown to cause loss of barrier integrity in house dust mite (HDM)-treated airway epithelial cells through a mechanism of epidermal growth factor receptor (EGFR) transactivation and subsequent E-cadherin destabilization [57]. The major allergen from HDM, Dermatophagoides pteronyssinus antigen P1 (Der p1), can also directly cleave the tight junction proteins occludin and ZO-1 [58,59]. In accordance, HDM extracts and Der p1 cause increased permeability and decreased transepithelial electrical resistance (TEER) in cultured airway epithelial cells [55,59]. Similar effects have been reported for other allergens, including the fungi Alternaria alternata (Alternaria) [53] and various pollen allergens [60,61,62].
In addition to promoting allergic sensitization, increased barrier permeability may also lower the threshold for epithelial damage and activation of a T2 response, which itself may affect barrier function, thus generating a positive feedback loop of increased epithelial permeability. Indeed, the two central T2 cytokines IL-4 and IL-13 have been found to induce barrier disruption by inhibiting the surface expression of ZO-1, occludin, E-cadherin, and β-catenin in bronchial epithelial cells [63,64]. Recently, ILC2s, which constitute an early source of T2 cytokines in asthma, were shown to induce increased epithelial barrier permeability and reduced expression of epithelial tight junction proteins via secretion of IL-13 in an ALI-coculture model of human bronchial epithelial cells (HBECs) and ILC2s [65]. IL-13 has also been found to decrease epithelial expression of claudin-18, the only known lung-specific tight junction protein [66]. Interestingly, lower expression of claudin-18 was identified in epithelial brushings from asthmatic individuals compared to healthy controls and loss of claudin-18 impaired epithelial barrier function both in vitro and in vivo [66].
Furthermore, infections with some respiratory viruses, such as influenza virus, can cause epithelial barrier dysfunction as a result of direct cytopathic effects. RV on the other hand cause little cell death, but have been shown to disrupt epithelial tight junctions by reducing occludin expression in a NADPH-oxidase-dependent manner, leading to increased airway epithelial permeability [67]. A recent study further demonstrated that RV infection caused loss of ZO-1 from tight junctions in ALI-cultured HBECs from asthmatic and healthy children, and that the effect was more pronounced and sustained in cells from children with asthma [68]. Moreover, infection with respiratory syncytial virus (RSV) has been found to cause adverse effects on airway epithelial junctional complexes through sustained activation of protein kinase D [69].
In addition to the impact of different environmental risk factors, the genetic background of the individual may also influence epithelial barrier properties. As described above, several susceptibility genes with potential implications in epithelial barrier function have been identified through GWA studies. Furthermore, epigenetic modifications serve as a secondary level of gene regulation that is likely to effect the translation of disease susceptibility into transformed airway epithelial biology. A more detailed discussion of epigenetic mechanisms in relation to airway epithelial barrier function will be given later in this review.
5. Airway Epithelial Responses to Inhaled Agents
It is now evident that the airway epithelium plays a key role in the initiation and orchestration of the immune response to various environmental factors. Inhaled agents such as aeroallergens, pollutants, and respiratory viruses are sensed by the airway epithelium via a diverse set of pattern recognition receptors (PRRs) like the toll-like receptors (TLRs), retinoic acid-inducible gene (RIG)-I-like receptors (RLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), C-type lectin receptors (CLRs), and PARs. Following activation of these receptors, airway epithelial cells release various inflammatory cytokines, chemokines, endogenous danger signals, and other mediators alarming and activating a variety of immune cells, importantly DCs and ILC2s. In the following sections, we will focus on some aspects of the interactions of airway epithelial cells with common aeroallergens and respiratory viruses and the effects on the airway epithelium in asthma.
5.1. Allergen-Airway Epithelial Interactions
As already highlighted, the airway epithelium does not simply act as a passive barrier hindering allergens from penetrating the mucosal surface, but is highly active in the recognition of allergens and initiation of innate immune responses that are critical for influencing the outcome of allergen inhalation. Allergens commonly involved in asthma development and exacerbation include dust mites, grass and tree pollen, animal dander, and fungi. These aeroallergens are complex mixtures of various constituents, including proteins with different structures and activities, which can interact with epithelial cells through diverse mechanisms. Importantly, repeated or sustained activation of epithelial PRRs, either by allergens themselves or by contaminating microbial pathogen-associated molecular patterns (PAMPs), has been proposed as one of the key steps in the modulation of DC-driven adaptive immune responses and the allergen sensitization process [70].
We have previously discussed the ability of certain protease-containing allergens to disrupt the airway epithelial barrier by acting on tight junctions. In addition, sensing of this protease activity by airway epithelial cells may also induce the release of various inflammatory mediators. For example, Alternaria has been shown to trigger protease-dependent PAR-2-mediated release of IL-6, CXCL8, and GM-CSF from HBECs in vitro [71], and similar effects have been found with cockroach proteases [72,73].
Furthermore, allergens can also activate airway epithelial cells via protease-independent mechanisms. Accordingly, HDM was reported to trigger protease-independent release of the DC-chemoattractant CCL20 via the interaction of HDM-derived β-glucan with the CLR dectin-1 in a bronchial epithelial cell line [74]. Additionally, the non-proteolytic HDM allergen Der p2, has been shown to induce airway epithelial release of CCL20, IL-6, CXCL8, GM-CSF, and MCP-1 via activation of NF-κB and MAPK pathways [75]. Surface expression of the intracellular adhesion molecule (ICAM)-1 was also upregulated on the same cells in response to Der p2, and this was associated with increased adhesion of monocytes to the epithelial cells [75]. Of note, ICAM-1 is also used as the receptor for cellular internalization by the major group RVs [76]. It has been suggested that at least part of the effects of Der p2 on airway epithelial cells could be due to TLR4 activation, since Der p2 shows high sequence homology with myeloid differentiation factor 2 (MD2), a TLR4 co-signaling molecule required for optimal TLR4 activation by LPS, and LPS is a known contaminating factor in extracts from HDM [77].
Thymic stromal lymphopoietin (TSLP), IL-33, and IL-25 are three epithelial-derived cytokines with critical roles in asthma pathogenesis as they are potent activators of DCs and ILC2s, which act upstream in the T2 immune response cascade. Exposure to aeroallergens in vitro has been shown to trigger epithelial release of all three cytokines [78,79,80,81]. Importantly, increased expression of TSLP, IL-33, and IL-25 was recently demonstrated in the airway epithelium of allergen-challenged individuals with mild atopic asthma and correlated with increased airway obstruction [82]. In addition, various allergens have been reported to trigger epithelial release of endogenous danger signals, such as ATP and uric acid [80,83], which may further influence DC and ILC2 behavior as well as amplifying the production of epithelial-derived T2-promoting cytokines [80].
Several in vitro studies have demonstrated synergy between allergens and different inflammatory mediators. For example, a study using HDM allergens showed that HDM acts synergistically with IL-4 and TGF-β, two mediators found to be increased in asthmatic airways [84,85], to trigger airway epithelial release of the Th2 cell chemoattractant CCL17 [86]. A further study found that IL-4 also increased Alternaria-induced release of TSLP, whereas the induction of TSLP was prevented by the type 1 (T1) cytokine interferon (IFN)-γ [78]. These findings suggest that the local microenvironment in the airways is likely to dictate the outcome of allergen-epithelial cell interactions, and may partly explain why allergens do not cause inflammation in healthy individuals, despite their capacity for direct activation of airway epithelial cells. Another important question is whether there is differential regulation of allergen-induced innate immune responses in asthmatic and healthy airway epithelium. Although few reports of altered epithelial innate immune responses to allergens in asthma are available to date, an in vitro study in HBECs demonstrated that cells from asthmatic individuals released more CCL20 compared with cells from healthy controls in response to stimulation with the HDM allergen Der p1 [87], indicating that such dysregulation may exist.
5.2. Virus-Airway Epithelial Interactions
In healthy individuals, upper respiratory tract viral infections are usually self-limiting and manifest as a common cold with relatively mild symptoms. In individuals with asthma, however, respiratory viruses, particularly RV, are able to subvert host immune defense systems and act as major triggers of exacerbations in both children and adults [88,89,90]. These acute, disease-worsening events impair quality of life, are a major cause of hospitalization, and can, in their most severe form, be fatal [91]. In addition to the causative role of viruses in asthma exacerbations, there is considerable evidence that virus-induced wheezing illnesses early in life are a significant risk factor for later asthma development, especially in genetically susceptible children [92,93,94]. Again, this association is particularly strong with RV [92,93,94,95,96], but RSV has also been suggested to be a risk factor [97,98].
Although important progress has been made over the past decade, the precise pathogenic mechanisms by which respiratory viruses may drive asthma inception and exacerbations are not completely understood. Bronchial epithelial cells are the primary targets of respiratory viruses and the main site of viral replication in the lower airways [99,100]. Hence, dysregulated epithelial production of mediators that influence the immune response has been suggested as one explanation to why respiratory infections trigger asthmatic and allergic reactions in susceptible individuals. Even though RSV, influenza virus, and some additional respiratory viruses have been detected in airway samples from asthma patients with exacerbating disease, RV infections are by far the most frequent cause of viral-induced asthma exacerbations, accounting for up to 70–80% of all cases [101]. As a consequence, experimental studies on epithelial responses to RV infection in asthma currently dominate the research field.
Early host recognition of respiratory viruses by the airway epithelium mainly occurs through a number of PRRs that sense viral RNA. Replication of single-stranded RNA viruses such as RV leads to the production of double-stranded RNA (dsRNA), which is recognized as a potent stimulus for antiviral innate immune responses [102]. Airway epithelial cells constitutively express TLR3, which in a coordinated manner with the IFN-inducible RLRs melanoma differentiation-associated gene (MDA)-5 and RIG-I, interacts with dsRNA, leading to activation of downstream signaling pathways involving NF-κB and IRF3/IRF7 [102,103]. Ultimately, this results in the production of type I (IFN-α/β) and III (IFN-λ1, 2, and 3) IFNs [104]. In addition, a wide range of proinflammatory cytokines (IL-1β, IL-6), chemokines (CXCL8, CXCL5, CXCL10, CCL5/RANTES), and growth factors (G-CSF, GM-CSF) are produced, which can contribute to the activation and recruitment of various immune cells to the airways [104].
Epithelial generation of IFNs is essential for effective antiviral responses and viral clearance. IFNs are able to induce hundreds of IFN-stimulated genes, which cooperate to limit viral replication and invasion by a number of mechanisms [105]. Despite some controversial reports, there is evidence that RV-induced epithelial production of IFNs is reduced in some individuals with asthma, providing one plausible explanation to the increased susceptibility to viral infections in at least a subgroup of asthmatic individuals. Wark et al. were the first to demonstrate impaired IFN production in the asthmatic epithelium. In their study they found that HBECs from subjects with asthma exhibited increased RV replication in vitro compared with healthy individuals, and that this was reflected by delayed and deficient IFN-β induction [106]. Contoli et al. later made similar observations of deficient IFN-λ induction in RV-infected HBECs from atopic individuals with asthma [107]. By using a human experimental model of RV exacerbation, the authors further showed that exacerbation severity was inversely proportional to IFN-λ generation. Though there have been contradictory results [108,109,110], these initial findings have since been confirmed in several reports [111,112,113,114,115,116], and different factors have been proposed to negatively regulate RV-induced epithelial IFN production, including suppressor of cytokine signaling (SOCS)1, the T2 cytokines IL-4 and IL-13, HDM, and oxidative stress [117,118,119,120]. Despite extensive research, however, mechanistic insight into the impaired IFN responses in asthmatic individuals is still lacking and further studies are warranted.
Viral infections are classically associated with the induction of a T1 immune response. Yet, emerging evidence suggests the involvement of the T2-promoting cytokines IL-25, IL-33, and TSLP in the response to respiratory viruses and associated exacerbations in asthma. By using a human experimental model of RV exacerbation, Jackson et al. found that subjects with asthma had increased levels of IL-33 and that this correlated with the T2 cytokines IL-5 and IL-13 in the airway lining fluid as well as exacerbation severity after virus inoculation [121]. They also showed that supernatants from RV-infected HBECs triggered IL-33-dependent induction of IL-4, IL-5, and IL-13 in human T cells and ILC2s in vitro. In a similar model, Beale et al. showed that IL-25 was induced by experimental RV infection, and that IL-25 expression both at baseline and during infection was higher in asthmatic individuals [122]. They further demonstrated in vitro that RV-infected HBECs from asthmatic subjects had greater IL-25 induction compared with cells from healthy individuals. Moreover, several studies have found overproduction of TSLP in response to dsRNA or RSV infection ex vivo in HBECs from asthmatic individuals compared with healthy controls [112,123,124]. Taken together, these data suggest that airway epithelial cells from asthmatic individuals may have an increased capacity for IL-33, IL-25, and TSLP production in response to virus infections, and that these cytokines may be important mediators in exaggerated T2 inflammatory responses in viral-induced asthma exacerbations. The biological functions of IL-33, IL-25, and TSLP, as well as their proposed roles in T2 immunity and asthma, will be further discussed below.
6. Epithelial-Derived Cytokines as Master Regulators of T2 Immunity
Overwhelming evidence supports a central role for the three epithelial-derived cytokines, TSLP, IL-33, and IL-25, in asthma. These three cytokines, commonly referred to as alarmins, act as master regulators that mediate both innate and adaptive immune responses, leading to sustained T2-skewed inflammation (Figure 2). Although distinct in their mode of action, crosstalk within this triad of alarmins is likely to exist and is underpinned by the findings that some of the triggers for release are shared by all three cytokines. In light of the strong indication that they act as upstream drivers of T2-mediated disease, TSLP, IL-33, and IL-25 have attracted a lot of interest as potential therapeutic targets in asthma, and monoclonal antibodies targeting TSLP and IL-33 are currently under clinical evaluation.
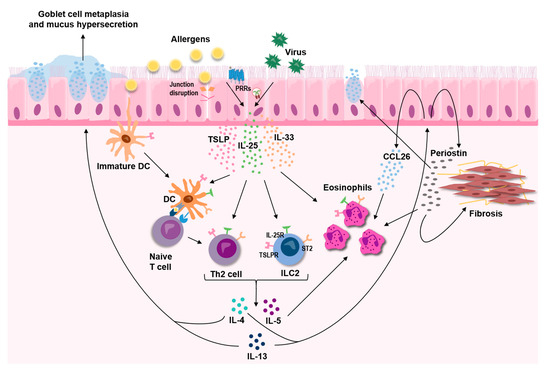
Figure 2.
The epithelial-derived cytokines TSLP, IL-33, and IL-25 are released in response to various insults, including allergens and respiratory viruses, and act as key upstream drivers of type 2 inflammation in the airways through effects on both innate and adaptive immune cells.
6.1. TSLP
TSLP is a member of the IL-2 cytokine family and considered a pivotal upstream cytokine driving a pronounced T2 immune response [125,126,127]. The airway epithelium is a major source of TSLP under both homeostatic and inflammatory conditions [128,129,130]. A range of stimuli involved in asthma pathogenesis, including respiratory viruses, proinflammatory (TNF-α and IL-1β) and T2 (IL-4 and IL-13) cytokines, and proteolytic allergens have been shown to cause increased expression and release of TSLP from airway epithelial cells through activation of different PRRs and cytokine receptors, supporting its function as an alarmin signaling a compromised airway epithelium [78,123,126,131,132,133,134].
TSLP binds to a heterodimeric receptor complex composed of the TSLP receptor (TSLPR) and the IL-7Rα chain. The broad effect of TSLP on the immune response in the airways is reflected by the various cell types that express the TSLPR, including many cells of the hematopoietic system, but also structural cells like airway smooth muscle cells [135,136]. The role of TSLP in asthma and allergic inflammation has been extensively investigated. Both in vitro and in vivo studies have demonstrated a strong link between TSLP expression and the production of IL-4, IL-5, and IL-13, which are central in the development of a T2 phenotype in asthma [125,133,137,138,139,140]. It is believed that a major T2 promoting effect of TSLP is its ability to induce OX40 ligand on DCs, priming them to drive differentiation of naive CD4+ T cells into functional Th2 cells, which produce IL-4, IL-5, and IL-13 [125,139,140]. In addition, TSLP can interact directly with other cells of the immune system, such as mast cells, eosinophils, Th2 cells, and ILC2s to promote a T2-biased inflammatory response [126,133,138,141,142,143].
As previously mentioned, several single-nucleotide polymorphisms (SNPs) in the TSLP gene associated with increased asthma risk have been identified through GWA studies, indicating a role of TSLP in asthma pathogenesis [39,48,144]. In support of these findings, a number of studies have shown that TSLP expression is elevated in the airway epithelium and bronchoalveolar lavage (BAL) fluid of individuals with asthma and that it correlates with disease severity and loss of lung function [128,129,130]. Furthermore, clinical trials have now provided strong evidence for a central role of TSLP as an important modulator in asthma. A humanized monoclonal anti-TSLP antibody, tezepelumab, which blocks the interaction of TSLP with TSLPR recently completed a phase 2 clinical trial for uncontrolled, severe asthma where tezepelumab-treated individuals displayed significant reduction in asthma exacerbation rate together with improved lung function and asthma control [145]. In addition, an earlier clinical trial demonstrated that anti-TSLP reduced bronchoconstriction and airway inflammation in mild asthmatic individuals before and after allergen challenge [146].
6.2. IL-33
IL-33 is a member of the IL-1 superfamily and has been forwarded as a multifactorial alarmin cytokine with critical roles in T2 immunity and asthma pathophysiology [147]. Airway epithelial cells are the primary cell type in the human airways that express IL-33 under basal conditions, where it is predominantly localized to the nucleus in a full-length precursor form [148,149]. Cellular release of immunologically active full-length IL-33 (IL-33FL) occurs rapidly following epithelial injury or exposure to environmental stressors such as airborne allergens and viruses [80,81,121]. Although IL-33 has primarily been considered to be passively released due to cell necrosis, findings also indicate active mechanisms of IL-33 secretion in response to allergens, mediated via purinergic receptor-dependent signaling or dual oxidase 1 (DUOX1)-dependent activation of EGFR-signaling [80,81].
Similar to other IL-1 family cytokines, the activity of IL-33 is regulated by both its cellular localization and by proteolytic cleavage. Recent studies have shed new light on how IL-33 activity can be regulated by direct sensing of proteolytic activities, as well as oxidative changes. Cayrol et al. demonstrated that various allergens with protease activity, including HDM, Alternaria, Aspergillus fumigatus and pollens, can induce IL-33FL release and subsequent cleavage in a central sensor domain into a shorter form with considerably enhanced bioactivity that potently stimulates ILC2s [150]. In another study, Scott et al. demonstrated that IL-33 activity can be enhanced by proteolytic mechanisms involving allergen proteases as well as endogenous proteases from damaged airway epithelial cells [151]. In addition, they showed that allergen proteases degraded mature oxidized forms of released IL-33, suggesting a regulatory mechanism for rapid inactivation of IL-33 in an oxidative milieu, such as during tissue injury.
IL-33 binds to a heterodimeric receptor formed by IL-1 receptor-like 1 (IL1RL1, also known as ST2) and the IL-1 receptor accessory protein, which leads to activation of NF-kB and MAPK signaling pathways [147,152]. In addition to membrane-bound ST2, there is also a soluble form (sST2), which can act as a decoy receptor to sequester free IL-33, preventing IL-33/ST2 signaling [152]. The most established function of IL-33 is activation of ST2 expressing immune cells involved in T2 immunity, such as ILC2s, Th2 cells, mast cells, eosinophils, basophils, and DCs [153,154,155,156,157,158,159]. The functional role of IL-33 in T2 immunity-associated allergic responses and asthma has been extensively investigated in vivo and numerous studies have shown that inhibition of IL-33/ST2 signaling attenuates T2 inflammation in murine models of allergic asthma [155,157,160,161,162].
Significant associations between genetic variants of IL33 and IL1RL1 and human asthma have consistently been identified in several genetic studies, suggesting that the IL-33/ST2-axis is likely to play a role in the disease [39,144,163]. In support of this, IL-33 has been shown to be upregulated in the airway epithelium and BAL fluid from individuals with moderate to severe asthma, and release of IL-33 is increased during experimental RV-induced asthma exacerbation [121,149]. Clinical phase 2 trials with monoclonal antibodies targeting either IL-33 or ST2 are currently ongoing and will evaluate the potential of IL-33 as a therapeutic target in asthma.
6.3. IL-25
IL-25, also known as IL-17E, is a member of the IL-17 cytokine family, consisting of six structurally related but functionally distinct proteins [164]. Whereas other IL-17 cytokine members such as IL-17A and IL-17F seem to have important roles in neutrophilic inflammation, proinflammatory cytokine induction, and T1 immunity, IL-25 is unique in that it promotes T2 immune responses, including eosinophilic inflammation and overproduction of IL-4, IL-5, and IL-13 [165]. A specialized group of epithelial cells called solitary chemosensory cells were recently identified to be the main epithelial source of IL-25 in the upper airways [166]. Airway epithelial cells have been demonstrated to contain preformed IL-25, which is stored or sequestered in the cytoplasm [79]. When exposed to protease-containing allergens such as HDM, epithelial cells rapidly release IL-25, implying a role in allergic disease [79]. Other proteases, such as papain and trypsin, or breakdown of cell-cell adhesion molecules may also trigger epithelial release of IL-25 [79,167]. The exact mechanisms of IL-25 release and regulation, however, have yet to be defined.
IL-25 binds to a heterodimeric receptor, IL-17RA/IL-17RB (IL-25R), which is expressed on several cell types such as ILC2s, activated memory Th2 cells, TSLP-activated DCs, mast cells, eosinophils, and endothelial cells [164,168,169,170]. Thus, IL-25 is able to mediate both innate and adaptive immune responses to induce a sustained T2-biased mucosal inflammation. For example, IL-25 may amplify Th2-cell dependent pathways leading to enhanced allergic inflammation [171]. Although IL-33 seems to be superior in driving the development and activation of ILC2s, IL-25 also functions as an ILC2-inducing cytokine [159].
Studies in both mice and humans suggest a role for IL-25 in asthma. Different experimental murine models have shown that allergic inflammation can be attenuated by blocking IL-25 signaling [122,167,172]. In a study by Cheng et al., the role of IL-25 in the lower airways was investigated in steroid naive, newly diagnosed asthmatic individuals and healthy control subjects [173]. By analyzing bronchial brushings and biopsies, BAL, sputum, and blood, the authors could reveal an “IL-25-high” subgroup among asthmatic individuals who exhibited increased airway epithelial expression of IL-25 associated with severe airway eosinophilia, marked subepithelial fibrosis, higher expression of MUC5AC and elevated IgE levels. Further, plasma IL-25 levels correlated with epithelial IL-25 expression, suggesting that IL-25 may have potential as a systemic biomarker for stratifying patients for treatment. As previously described, a role for IL-25 in viral-induced asthma exacerbations has also been indicated [122]. Although the collected data supports an association between IL-25 and asthma, clinical trials using anti-IL-25 antibodies, as for TSLP and IL-33, have not yet been conducted.
7. The Effect of T2 Inflammation on the Airway Epithelium
Airway epithelial cells are able to respond to many cytokines, both pro- and anti-inflammatory, including the key T2 cytokines IL-4, IL-5, and IL-13 [174,175,176]. Several studies by Woodruff and colleagues have demonstrated the ability of the airway epithelium to respond with unique gene and protein signatures in response to T2 cytokines, and that these signatures could be used as biomarkers for T2 asthma [177,178,179]. One of the proteins from this T2 signature is periostin, which is increased in epithelial cells in response to IL-13 +/− IL-4 [177,180,181], and which may have potential as a systemic biomarker of eosinophilic airway inflammation [182,183]. Periostin, a matricellular protein, has been implicated in processes related to airway remodeling such as cell proliferation, collagen production and epithelial-to-mesenchymal transition [184], as well as subepithelial fibrosis [185], mainly driven by TGF-β activation. Periostin could also increase the expression of mucin genes in airway epithelial cells [186], as well as act as a binding partner for eosinophils and thereby promote their migration [187], both processes involved in T2 airway inflammation and asthma (Figure 2).
A widely studied effect from T2 cytokines on the epithelium is the induction of mucus production [11,188], an important clinical feature in asthma (Figure 2). The effect is mediated by altered expression and secretion of the mucins MUC5AC and MUC5B, which have different viscoelastic properties and are therefore believed to have different roles in the human airways. MUC5AC is commonly found to be increased in asthmatic individuals and in airway epithelia exposed to IL-13, whereas there are conflicting results around levels of MUC5B, meaning that the ratio between the two mucins may be altered in asthma, leading to altered properties of the mucin gel layer [189,190]. Linked to increased mucus production are SPDEF (SAM pointed domain-containing Ets transcription factor) and CLCA1, both also induced by T2 cytokines in the airway epithelium and the latter, as mentioned previously, is also linked genetically to asthma and part of the epithelial T2 signature introduced above [47,177,191,192]. Out of these, SPDEF, a transcription factor, appears to be most critical for goblet cell hyperplasia and increased mucus release in human [191,193], which when silenced or knocked out abolishes goblet cell differentiation and mucus release, by decreasing both CLCA1 and MUC5AC [192,194]. How CLCA1 modulates mucus production and secretion is, however, not yet fully elucidated and points towards possible differences between mice and man [195,196], but there are indications that CLCA1 could activate MAPK13 which in turn increases MUC5AC expression [197] whereas blocking chloride channels by niflumic acid lowers MUC5AC expression [198].
The increase in mucus production can lead to mucus plugging and impaired mucociliary clearance, the latter possibly due to IL-13-induced tethering of the mucus gel to the epithelial cells rather than impaired ciliary function [199]. A recent single-cell sequencing study identified a novel mucous ciliated cell state in asthma, induced by T2 cytokines, which could contribute to mucous cell metaplasia [200]. Mechanistic studies are being employed to further understand this effect and thus aid in how to possibly modulate mucus hypersecretion in asthma. Studies have highlighted the importance of autophagy in IL-13-induced mucus production [201], as well as the involvement of NOTCH3 signaling [202], both providing possible pathways for therapeutic intervention. The airway epithelium also responds to T2 cytokines by secreting proteins that could aid in counteracting impaired mucociliary clearance. An example of this being gelsolin, which has been found to be increased in epithelial cell cultures treated with IL-4, where it may improve fluidity of the airway surface liquid through the breakdown of released filamentous actin [203].
Another important clinical feature in asthma is increased nitric oxide, a gas involved in airway reactivity and inflammation, which is detectable in exhaled breath and used as a biomarker for T2 asthma [204,205,206,207]. Higher levels of nitric oxide are usually linked to increased levels of inducible nitric oxide synthase (NOS2) in the airway epithelium, which can be normalized through the administration of corticosteroids [208,209,210]. Several cytokines can increase the levels of NOS2 on both the gene and protein level [208,210], though the effect is suggested to be stronger from T2 cytokines including IL-4 and IL-13 [181,211].
A key inflammatory cell in T2 asthma is the eosinophil, of which there are increased numbers both locally and systemically in individuals with asthma. Eosinophils are recruited by eotaxins, a group of chemokines with three members in humans, comprised of CCL11 (eotaxin-1), CCL24 (eotaxin-2), and CCL26 (eotaxin-3). Of these, CCL26 has been shown to be the strongest inducer of eosinophil migration in asthmatic individuals [212]. Several studies have shown that this chemokine is strongly induced by IL-13 in airway epithelial cells (Figure 3) [181,207,213,214] and further that it is elevated in serum and bronchial biopsies from asthmatic individuals [183,207] and correlates with sputum eosinophil counts [213]. Recently, interest has grown when it comes to the involvement of extracellular vesicles, or exosomes, in asthma pathology and their potential as biomarkers. Airway epithelial cells have been shown to secrete extracellular vesicles that could be involved in cell-cell communication both during homeostasis and disease. These epithelium-derived vesicles tend to be coated with mucins, and the number of vesicles increase upon stimulation with T2 cytokines [179,215,216,217,218]. Additionally, in T2 conditions, epithelium-derived exosomes can induce chemotaxis of macrophages [206] and their cargo, such as proteins and microRNA (miRNA)s, are altered [181,219,220], all possibly contributing to pathological pathways observed in asthma.
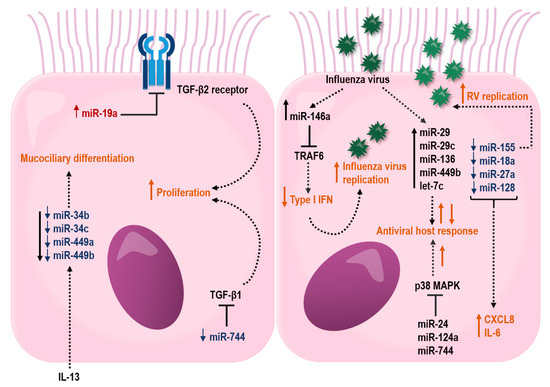
Figure 3.
Proposed effects of miRNAs in the airway epithelium that may influence barrier functions and host response to respiratory virus infections. miRNAs that are up- or downregulated in bronchial epithelial cells from asthmatic individuals are depicted in red color with upward arrow and blue color with downward arrow, respectively. Black lines ending with a perpendicular line indicate an inhibitory effect. Dotted lines illustrate regulated miRNAs or processes (orange color), where up- and downward arrows indicate stimulation and suppression, respectively.
8. Epigenetic Regulation of the Airway Epithelium in Asthma
The common feature of epigenetic mechanisms is that they regulate gene expression without affecting the nucleotide sequence of the genomic DNA [221]. The classical epigenetic modifications are DNA methylation and histone modifications, both of which are highly relevant to asthma pathophysiology. A recent study by Stefanowicz et al. showed six histone modifiers to be differently expressed in airway epithelial cells derived from individuals with asthma as compared to healthy individuals [222,223]. In addition to human studies, beneficial effects by targeting histone deacetylases have been carried out in animal models of asthma (can be reviewed in [224]). However, DNA methylation is probably the most studied epigenetic modification in general, but also in asthma. Indeed, a recent study demonstrated that SNPs identified by GWA studies affect asthma risk through DNA methylation and expression of cis-genes in airway epithelium [225]. In another study, bronchial mucous tissues obtained from atopic or non-atopic individuals with asthma revealed similar DNA methylation levels as in healthy control. Importantly, a set of loci was identified with significant differences in DNA methylation between the asthma groups [226]. Several studies have demonstrated DNA methylation changes in asthma genes, induced by environmental factors [227,228,229]. RV infection was shown to induce DNA methylation changes in nasal epithelial cells derived from both children and adults with asthma. Furthermore, diesel exhaust particle exposure and allergen challenge induced DNA methylation patterns in human airway epithelial cells 48 h post exposure. In addition to DNA methylation changes, airway epithelial gene expression can be modulated by miRNAs. Thus, miRNAs have shown to be differentially expressed in bronchial epithelial cells following tobacco smoke exposure, diesel exhaust particle exposure, and virus exposure (reviewed in [230]). These data highlight the role of environmental exposures in epigenetic regulation of gene expression in airway epithelium in asthma.
8.1. Epigenetic Regulation of the Airway Epithelium through miRNAs
Although the classical epigenetic modifications mentioned above are the most studied mechanisms to date, noncoding RNAs such as miRNAs are involved in the epigenetic regulation of epithelial gene expression. miRNAs are gene-regulatory small noncoding RNAs that bind to target mRNAs leading to mRNA degradation and in some cases translational repression [231,232,233]. miRNAs are crucial in most biological and pathological processes, including immune responses, cell proliferation, cell differentiation, and apoptosis. To date there are a limited number of studies examining miRNA expression in the airway epithelium of asthmatic individuals compared to healthy individuals. However, one of the first studies using bronchial brushing samples of airway epithelial cells from asthmatic individuals, cultured at ALI, demonstrated higher expression of let-7f, miR-181c, -487b and lower expression of miR-203 compared with healthy control samples. Network analysis has suggested the aquaporin gene AQP4 as a target gene of miR-203 and this gene was also shown to be highly increased in cells from asthma patients [234]. However, recent studies have identified Abelson tyrosine kinase (Abl) as a target for miR-203, which may have functional impact on epithelial cell proliferation, adhesion, growth, and development [235]. Therefore, a reduced level of this miRNA in airway epithelium in asthma may lead to cell proliferation and goblet cell hyperplasia [234].
In another study, bronchial brushings from asthma patients have shown a reduced expression of miR-181b, which was inversely correlated with sputum eosinophilia. Interestingly, overexpression of miR-181b reduced levels of IL-13 induced secretion of IL-1B and the eosinophil chemoattractant, eotaxin-1 (CCL11) by targeting SSP1 in a bronchial epithelial cell line. Furthermore, dexamethasone restored IL-13-induced miR-181b downregulation and inhaled corticosteroid treatment increased miR-181b in plasma from asthma patients. This study highlights a possible role for miR-181b as a biomarker for eosinophilic asthma [236]. In addition, the miR-34/449 family (miR-34b, miR-34c, miR-449a, and miR-449b) were shown to be suppressed in bronchial brushings from individuals with asthma, which was associated with an increased IL-13 expression (Figure 3). Interestingly, IL-13-induced inhibition of miR-34/449 family members resulted in an altered mucociliary differentiation towards a reduced number of ciliated cells and increased number of mucous cells, suggesting a role for this miRNA family in asthma pathogenesis [237].
A recent study has suggested that reduced levels of miR-146a in HBECs from asthma patients may contribute to neutrophilic asthma. However, reduced levels of miR-146a were found irrespective of asthma phenotype, but the neutrophil chemoattractant CXCL8 and CXCL1 were only increased in the neutrophilic asthma phenotype [238]. Both stimulated and unstimulated HBECs transfected with miR-146a mimics revealed reduced levels of both CXCL8 and CXCL1 mRNA. Even though the exact target for miR-146a was not identified, this study suggests that reduced levels of miR-146a may contribute to the development of a neutrophilic asthma phenotype [238].
8.2. miRNAs and the Airway Epithelial Barrier in Asthma
Little is known about how miRNAs influence the epithelial barrier function in asthma. However, studies have suggested that the impact of miRNAs on the airway epithelium may differ with severity of disease. Notably, several differentially expressed miRNAs in airway epithelial cells target genes involved in airway epithelial barrier functions (Figure 3). In severe asthma, miR-19, -221, and -744 were shown to be differently expressed in HBECs, specifically from individuals with an eosinophilic allergic asthma [239,240,241]. miR-19a was shown to enhance proliferation of HBECs, specifically in severe asthmatics by targeting the TGF-β2 receptor in HBECs [240]. In contrast to miR-19 and -221, the expression miR-744 was reduced in HBECs from severe asthma. This miRNA inhibits proliferation of HBECs by regulating the Smad3 pathway via targeting TGF-β1, a major proinflammatory mediator involved in fibrotic tissue remodeling within the asthmatic lung, highlighting a possible role for this miRNA in asthma pathogenesis [241].
8.3. miRNAs and Airway Epithelial Cell Responses to Virus Infection
Common viruses that affect the respiratory system are human RVs, RSV, and influenza viruses. As previously described, these viruses are known to cause illness and exacerbations in individuals with asthma. One of the most studied miRNAs is miR-155, which, in addition to regulating T2 inflammation in animal models of asthma [242,243], has been shown to be involved in RV replication in HBECs [244]. Inhibition of miR-155 in HBECs resulted in an increased viral replication of RV-1B [244]. An additional study demonstrated decreased expression of miR-18a, -27a, -128 and -155 in HBECs derived from individuals with asthma, and further knockdown of these miRNAs led to increased expression of the proinflammatory cytokines IL-6 and CXCL8. Indeed, IL-6 and CXCL8 are known to be increased in HBECs from asthmatics both under baseline conditions and after stimulation, suggesting a regulatory role for miRNAs in the control of IL-6 and CXCL8 expression in asthma [245].
In addition, miRNAs have also been shown to take part in essential pro-viral host factors through their regulation of the downstream p38 MAPK kinases, MK2 and Myc. Thus, miR-24, -124a, and -744 were shown to have antiviral effects on influenza A virus in the human lung epithelial cell line A549, whereas miR-124a and -744 had antiviral effects in RSV infection. These antiviral effects were through the suppression of the p38 MAPK pathway [246]. Influenza A viruses also increase the expression of miR-29, -29c, -136, 449b, and let-7c in A549 cells [247,248,249,250,251,252]. All of these miRNAs affect the influenza A viral response by targeting genes involved in antiviral host defense. In addition, miR-146a is induced in influenza virus A infection and downregulation of miR-146a was shown to inhibit influenza A virus replication by enhancing IFN type 1 responses by directly targeting the tumor necrosis factor receptor association factor 6 (TRAF6) [253].
Together, these data suggest a role for miRNA regulation of immune responses to respiratory viruses (Figure 3), and it is tempting to speculate that the miRNAs that affect virus replication therefore play a pivotal role in virus-induced exacerbations in asthma.
9. Conclusions
Today it is evident that the airway epithelium is not merely a passive barrier, but an essential part of the local immune response in the airways, bridging innate and adaptive immunity against various environmental insults. Overwhelming evidence indicates that the airway epithelium is dysfunctional in asthma, and plays a critical role in the development, progression, and exacerbation of the disease. This impairment includes both structural and immunological components, which collectively may influence the outcome of environmental challenges and contribute to asthma pathology. A role of the airway epithelium in asthma is further supported by findings from GWA studies, where many genes associated with asthma development are expressed in the airway epithelium.
Furthermore, epigenetic regulatory mechanisms may also contribute to abnormalities in asthma. miRNAs have recently been recognized as important modulators of airway epithelial functions. Although several studies have described differential expression of miRNAs in the asthmatic airway epithelium, the functional consequences of such dysregulation have not been investigated to the same extent. Hence, more mechanistic studies delineating the role of miRNAs in various aspects of airway epithelial dysfunction, including barrier impairment and deficient antiviral responses, are warranted, and could potentially pave the way for new treatment strategies in asthma.
About half of all individuals with asthma exhibit active T2 inflammation. It is now recognized that factors beyond IgE-mediated allergen sensitization, such as respiratory viruses, can trigger T2 immune responses. In addition, various allergens, including HDM, have the capacity to stimulate T2-skewed innate immune responses through epithelial PRR activation. The airway epithelium is a major source of TSLP, IL-33, and IL-25, which are rapidly released in response to both viruses and allergens. These alarmin cytokines activate various cells of both the innate and adaptive immune system, importantly DCs and ILC2s, thus acting as key upstream drivers of the T2 inflammatory cascade in asthma. This notion has led to an immense interest in targeting these epithelial-derived cytokines as a potential treatment strategy in asthma, and recent clinical trials blocking TSLP have shown promising results.
When activated by the alarmins, immune cells secrete T2 cytokines, which further affects the airway epithelium. This includes increased production of mucus and immune cell recruiting chemokines, as well as effects on the epithelial barrier such as remodeling and fibrosis. These changes thus lead to sustained inflammation and a persisting disease. Additionally, the epithelium is a potential source of both local and systemic biomarkers in the form of for example proteins or miRNAs, as the large surface area of the epithelium would enable production of high enough levels to be detected.
Taken together, it is clear that the airway epithelium is a central player in asthma, and further insight into the regulatory mechanisms underlying airway epithelial dysfunction could identify novel targets for future asthma intervention.
Author Contributions
J.C., E.A. and M.R. wrote the manuscript, J.C. prepared the figures and J.C., E.A. and M.R. revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the Swedish Research Council (2017-02302), the Swedish Heart and Lung Foundation (20170392), the VBG Group Herman Krefting Foundation for Asthma and Allergy Research and the Swedish Foundation for Strategic Research (ID16-0083).
Acknowledgments
We would like to thank Reghan Borer for constructive comments on the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AMP | Antimicrobial peptide |
| DC | Dendritic cell |
| ILC2 | Type 2 innate lymphoid cell |
| GWA | Genome-wide association |
| ALI | Air-liquid interface |
| T2 | Type 2 |
| PAR-2 | Protease-activated receptor 2 |
| HDM | House dust mite |
| Th2 | T helper 2 |
| EGFR | Epidermal growth factor receptor |
| RV | Rhinovirus |
| RSV | Respiratory syncytial virus |
| PRR | Pattern recognition receptors |
| TLR | Toll-like receptor |
| HBEC | Human bronchial epithelial cell |
| TSLP | Thymic stromal lymphopoietin |
| IFN | Interferon |
| dsRNA | Double-stranded RNA |
| SNP | Single nucleotide polymorphism |
| BAL | Bronchoalveolar lavage |
| miRNA | microRNA |
References
- Global Burden of Disease Study Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet (Lond. Engl.) 2015, 386, 743–800. [CrossRef]
- Fanta, C.H. Asthma. N. Engl. J. Med. 2009, 360, 1002–1014. [Google Scholar] [CrossRef]
- Holgate, S.T.; Wenzel, S.; Postma, D.S.; Weiss, S.T.; Renz, H.; Sly, P.D. Asthma. Nat. Rev. Dis Primers 2015, 1, 15025. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, B.N.; Hammad, H.; Fahy, J.V. The Cytokines of Asthma. Immunity 2019, 50, 975–991. [Google Scholar] [CrossRef] [PubMed]
- Haldar, P.; Pavord, I.D.; Shaw, D.E.; Berry, M.A.; Thomas, M.; Brightling, C.E.; Wardlaw, A.J.; Green, R.H. Cluster analysis and clinical asthma phenotypes. Am. J. Respir. Crit. Care Med. 2008, 178, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Fahy, J.V. Type 2 inflammation in asthma—Present in most, absent in many. Nat. Rev. Immunol. 2015, 15, 57–65. [Google Scholar] [CrossRef]
- Lotvall, J.; Akdis, C.A.; Bacharier, L.B.; Bjermer, L.; Casale, T.B.; Custovic, A.; Lemanske, R.F., Jr.; Wardlaw, A.J.; Wenzel, S.E.; Greenberger, P.A. Asthma endotypes: A new approach to classification of disease entities within the asthma syndrome. J. Allergy Clin. Immunol. 2011, 127, 355–360. [Google Scholar] [CrossRef]
- Gohy, S.; Hupin, C.; Ladjemi, M.Z.; Hox, V.; Pilette, C. Key role of the epithelium in chronic upper airways diseases. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2020, 50, 135–146. [Google Scholar] [CrossRef]
- Holgate, S.T. The sentinel role of the airway epithelium in asthma pathogenesis. Immunol Rev. 2011, 242, 205–219. [Google Scholar] [CrossRef]
- Matsui, H.; Randell, S.H.; Peretti, S.W.; Davis, C.W.; Boucher, R.C. Coordinated clearance of periciliary liquid and mucus from airway surfaces. J. Clin. Investig. 1998, 102, 1125–1131. [Google Scholar] [CrossRef]
- Fahy, J.V.; Dickey, B.F. Airway mucus function and dysfunction. N. Engl. J. Med. 2010, 363, 2233–2247. [Google Scholar] [CrossRef] [PubMed]
- Boers, J.E.; Ambergen, A.W.; Thunnissen, F.B. Number and proliferation of basal and parabasal cells in normal human airway epithelium. Am. J. Respir. Crit. Care Med. 1998, 157 Pt 1, 2000–2006. [Google Scholar] [CrossRef] [PubMed]
- Rock, J.R.; Onaitis, M.W.; Rawlins, E.L.; Lu, Y.; Clark, C.P.; Xue, Y.; Randell, S.H.; Hogan, B.L. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 12771–12775. [Google Scholar] [CrossRef] [PubMed]
- Pardo-Saganta, A.; Law, B.M.; Tata, P.R.; Villoria, J.; Saez, B.; Mou, H.; Zhao, R.; Rajagopal, J. Injury induces direct lineage segregation of functionally distinct airway basal stem/progenitor cell subpopulations. Cell Stem Cell 2015, 16, 184–197. [Google Scholar] [CrossRef]
- Ruiz Garcia, S.; Deprez, M.; Lebrigand, K.; Cavard, A.; Paquet, A.; Arguel, M.J.; Magnone, V.; Truchi, M.; Caballero, I.; Leroy, S.; et al. Novel dynamics of human mucociliary differentiation revealed by single-cell RNA sequencing of nasal epithelial cultures. Development 2019, 146, dev177428. [Google Scholar] [CrossRef]
- Linden, S.K.; Sutton, P.; Karlsson, N.G.; Korolik, V.; McGuckin, M.A. Mucins in the mucosal barrier to infection. Mucosal Immunol. 2008, 1, 183–197. [Google Scholar] [CrossRef]
- Bals, R.; Hiemstra, P.S. Innate immunity in the lung: How epithelial cells fight against respiratory pathogens. Eur. Respir. J. 2004, 23, 327–333. [Google Scholar] [CrossRef]
- Singh, P.K.; Tack, B.F.; McCray, P.B., Jr.; Welsh, M.J. Synergistic and additive killing by antimicrobial factors found in human airway surface liquid. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L799–L805. [Google Scholar] [CrossRef]
- Bartlett, J.A.; Fischer, A.J.; McCray, P.B.J. Innate immune functions of the airway epithelium. Contrib. Microbiol. 2008, 15, 147–163. [Google Scholar]
- Cookson, W. The immunogenetics of asthma and eczema: A new focus on the epithelium. Nat. Rev. Immunol. 2004, 4, 978–988. [Google Scholar] [CrossRef]
- Beisswenger, C.; Kandler, K.; Hess, C.; Garn, H.; Felgentreff, K.; Wegmann, M.; Renz, H.; Vogelmeier, C.; Bals, R. Allergic airway inflammation inhibits pulmonary antibacterial host defense. J. Immunol. 2006, 177, 1833–1837. [Google Scholar] [CrossRef] [PubMed]
- Lasser, C.; O’Neil, S.E.; Shelke, G.V.; Sihlbom, C.; Hansson, S.F.; Gho, Y.S.; Lundback, B.; Lotvall, J. Exosomes in the nose induce immune cell trafficking and harbour an altered protein cargo in chronic airway inflammation. J. Transl. Med. 2016, 14, 181. [Google Scholar] [CrossRef] [PubMed]
- Steelant, B.; Seys, S.F.; Boeckxstaens, G.; Akdis, C.A.; Ceuppens, J.L.; Hellings, P.W. Restoring airway epithelial barrier dysfunction: A new therapeutic challenge in allergic airway disease. Rhinology 2016, 54, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.; Fogg, V.C.; Margolis, B. Tight junctions and cell polarity. Annu. Rev. Cell Dev. Biol. 2006, 22, 207–235. [Google Scholar] [CrossRef]
- Hartsock, A.; Nelson, W.J. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta 2008, 1778, 660–669. [Google Scholar] [CrossRef]
- de Boer, W.I.; Sharma, H.S.; Baelemans, S.M.; Hoogsteden, H.C.; Lambrecht, B.N.; Braunstahl, G.J. Altered expression of epithelial junctional proteins in atopic asthma: Possible role in inflammation. Can. J. Physiol. Pharm. 2008, 86, 105–112. [Google Scholar] [CrossRef]
- Xiao, C.; Puddicombe, S.M.; Field, S.; Haywood, J.; Broughton-Head, V.; Puxeddu, I.; Haitchi, H.M.; Vernon-Wilson, E.; Sammut, D.; Bedke, N.; et al. Defective epithelial barrier function in asthma. J. Allergy Clin. Immunol. 2011, 128, 549–556. [Google Scholar] [CrossRef]
- Knight, D.A.; Holgate, S.T. The airway epithelium: Structural and functional properties in health and disease. Respirology 2003, 8, 432–446. [Google Scholar] [CrossRef]
- Green, K.J.; Jones, J.C. Desmosomes and hemidesmosomes: Structure and function of molecular components. FASEB J.: Off. Publ. Fed. Am. Soc. Exp. Biol. 1996, 10, 871–881. [Google Scholar] [CrossRef]
- Zepp, J.A.; Morrisey, E.E. Cellular crosstalk in the development and regeneration of the respiratory system. Nat. Rev. Mol. Cell Biol. 2019, 20, 551–566. [Google Scholar] [CrossRef]
- Loxham, M.; Davies, D.E.; Blume, C. Epithelial function and dysfunction in asthma. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2014, 44, 1299–1313. [Google Scholar] [CrossRef] [PubMed]
- Moheimani, F.; Hsu, A.C.-Y.; Reid, A.T.; Williams, T.; Kicic, A.; Stick, S.; Hansbro, P.M.; Wark, P.A.; Knight, D. The genetic and epigenetic landscapes of the epithelium in asthma. Respir. Res. 2016, 17, 119. [Google Scholar] [CrossRef] [PubMed]
- Heijink, I.H.; Kuchibhotla, V.N.S.; Roffel, M.P.; Maes, T.; Knight, D.A.; Sayers, I.; Nawijn, M.C. Epithelial cell dysfunction, a major driver of asthma development. Allergy 2020, 75, 1902–1917. [Google Scholar] [CrossRef] [PubMed]
- Koppelman, G.H.; Meyers, D.A.; Howard, T.D.; Zheng, S.L.; Hawkins, G.A.; Ampleford, E.J.; Xu, J.; Koning, H.; Bruinenberg, M.; Nolte, I.M.; et al. Identification of PCDH1 as a novel susceptibility gene for bronchial hyperresponsiveness. Am. J. Respir. Crit. Care Med. 2009, 180, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Bonnelykke, K.; Sleiman, P.; Nielsen, K.; Kreiner-Moller, E.; Mercader, J.M.; Belgrave, D.; den Dekker, H.T.; Husby, A.; Sevelsted, A.; Faura-Tellez, G.; et al. A genome-wide association study identifies CDHR3 as a susceptibility locus for early childhood asthma with severe exacerbations. Nat. Genet. 2014, 46, 51–55. [Google Scholar] [CrossRef]
- Li, X.; Hastie, A.T.; Hawkins, G.A.; Moore, W.C.; Ampleford, E.J.; Milosevic, J.; Li, H.; Busse, W.W.; Erzurum, S.C.; Kaminski, N.; et al. eQTL of bronchial epithelial cells and bronchial alveolar lavage deciphers GWAS-identified asthma genes. Allergy 2015, 70, 1309–1318. [Google Scholar] [CrossRef]
- Basnet, S.; Bochkov, Y.A.; Brockman-Schneider, R.A.; Kuipers, I.; Aesif, S.W.; Jackson, D.J.; Lemanske, R.F., Jr.; Ober, C.; Palmenberg, A.C.; Gern, J.E. CDHR3 Asthma-Risk Genotype Affects Susceptibility of Airway Epithelium to Rhinovirus C Infections. Am. J. Respir. Cell Mol. Biol. 2019, 61, 450–458. [Google Scholar] [CrossRef]
- Moffatt, M.F.; Kabesch, M.; Liang, L.; Dixon, A.L.; Strachan, D.; Heath, S.; Depner, M.; von Berg, A.; Bufe, A.; Rietschel, E.; et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature 2007, 448, 470–473. [Google Scholar] [CrossRef]
- Moffatt, M.F.; Gut, I.G.; Demenais, F.; Strachan, D.P.; Bouzigon, E.; Heath, S.; von Mutius, E.; Farrall, M.; Lathrop, M.; Cookson, W.; et al. A large-scale, consortium-based genomewide association study of asthma. N. Engl. J. Med. 2010, 363, 1211–1221. [Google Scholar] [CrossRef]
- Ferreira, M.A.; McRae, A.F.; Medland, S.E.; Nyholt, D.R.; Gordon, S.D.; Wright, M.J.; Henders, A.K.; Madden, P.A.; Visscher, P.M.; Wray, N.R.; et al. Association between ORMDL3, IL1RL1 and a deletion on chromosome 17q21 with asthma risk in Australia. Eur. J. Hum. Genet. 2011, 19, 458–464. [Google Scholar] [CrossRef]
- Miller, M.; Rosenthal, P.; Beppu, A.; Mueller, J.L.; Hoffman, H.M.; Tam, A.B.; Doherty, T.A.; McGeough, M.D.; Pena, C.A.; Suzukawa, M.; et al. ORMDL3 transgenic mice have increased airway remodeling and airway responsiveness characteristic of asthma. J. Immunol. 2014, 192, 3475–3487. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.; Tam, A.B.; Mueller, J.L.; Rosenthal, P.; Beppu, A.; Gordillo, R.; McGeough, M.D.; Vuong, C.; Doherty, T.A.; Hoffman, H.M.; et al. Cutting Edge: Targeting Epithelial ORMDL3 Increases, Rather than Reduces, Airway Responsiveness and Is Associated with Increased Sphingosine-1-Phosphate. J. Immunol. 2017, 198, 3017–3022. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.; Heinzmann, A.; Noguchi, E.; Abecasis, G.; Broxholme, J.; Ponting, C.P.; Bhattacharyya, S.; Tinsley, J.; Zhang, Y.; Holt, R.; et al. Positional cloning of a novel gene influencing asthma from chromosome 2q14. Nat. Genet. 2003, 35, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, T.; Polvi, A.; Rydman, P.; Vendelin, J.; Pulkkinen, V.; Salmikangas, P.; Makela, S.; Rehn, M.; Pirskanen, A.; Rautanen, A.; et al. Characterization of a common susceptibility locus for asthma-related traits. Science 2004, 304, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, K.; Byrne, C.; Griesinger, G.; Leung, A.; Chung, A.; Hill, A.S.; Swallow, D.M. Allelic association and recombination hotspots in the mucin gene (MUC) complex on chromosome 11p15.5. Ann. Hum. Genet. 2007, 71 Pt 5, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Shrine, N.; Portelli, M.A.; John, C.; Soler Artigas, M.; Bennett, N.; Hall, R.; Lewis, J.; Henry, A.P.; Billington, C.K.; Ahmad, A.; et al. Moderate-to-severe asthma in individuals of European ancestry: A genome-wide association study. Lancet Respir. Med. 2019, 7, 20–34. [Google Scholar] [CrossRef]
- Kamada, F.; Suzuki, Y.; Shao, C.; Tamari, M.; Hasegawa, K.; Hirota, T.; Shimizu, M.; Takahashi, N.; Mao, X.Q.; Doi, S.; et al. Association of the hCLCA1 gene with childhood and adult asthma. Genes Immun. 2004, 5, 540–547. [Google Scholar] [CrossRef]
- He, J.Q.; Hallstrand, T.S.; Knight, D.; Chan-Yeung, M.; Sandford, A.; Tripp, B.; Zamar, D.; Bosse, Y.; Kozyrskyj, A.L.; James, A.; et al. A thymic stromal lymphopoietin gene variant is associated with asthma and airway hyperresponsiveness. J. Allergy Clin. Immunol. 2009, 124, 222–229. [Google Scholar] [CrossRef]
- Harada, M.; Hirota, T.; Jodo, A.I.; Hitomi, Y.; Sakashita, M.; Tsunoda, T.; Miyagawa, T.; Doi, S.; Kameda, M.; Fujita, K.; et al. Thymic stromal lymphopoietin gene promoter polymorphisms are associated with susceptibility to bronchial asthma. Am. J. Respir. Cell Mol. Biol. 2011, 44, 787–793. [Google Scholar] [CrossRef]
- Ferreira, M.A.; Matheson, M.C.; Tang, C.S.; Granell, R.; Ang, W.; Hui, J.; Kiefer, A.K.; Duffy, D.L.; Baltic, S.; Danoy, P.; et al. Genome-wide association analysis identifies 11 risk variants associated with the asthma with hay fever phenotype. J. Allergy Clin. Immunol. 2014, 133, 1564–1571. [Google Scholar] [CrossRef]
- Gudbjartsson, D.F.; Bjornsdottir, U.S.; Halapi, E.; Helgadottir, A.; Sulem, P.; Jonsdottir, G.M.; Thorleifsson, G.; Helgadottir, H.; Steinthorsdottir, V.; Stefansson, H.; et al. Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nat. Genet. 2009, 41, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Hackett, T.L.; Singhera, G.K.; Shaheen, F.; Hayden, P.; Jackson, G.R.; Hegele, R.G.; Van Eeden, S.; Bai, T.R.; Dorscheid, D.R.; Knight, D.A. Intrinsic phenotypic differences of asthmatic epithelium and its inflammatory responses to respiratory syncytial virus and air pollution. Am. J. Respir. Cell Mol. Biol. 2011, 45, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Hackett, T.L.; de Bruin, H.G.; Shaheen, F.; van den Berge, M.; van Oosterhout, A.J.; Postma, D.S.; Heijink, I.H. Caveolin-1 controls airway epithelial barrier function. Implications for asthma. Am. J. Respir. Cell Mol. Biol. 2013, 49, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Leino, M.S.; Loxham, M.; Blume, C.; Swindle, E.J.; Jayasekera, N.P.; Dennison, P.W.; Shamji, B.W.; Edwards, M.J.; Holgate, S.T.; Howarth, P.H.; et al. Barrier disrupting effects of alternaria alternata extract on bronchial epithelium from asthmatic donors. PLoS ONE 2013, 8, e71278. [Google Scholar] [CrossRef]
- Post, S.; Nawijn, M.C.; Jonker, M.R.; Kliphuis, N.; van den Berge, M.; van Oosterhout, A.J.; Heijink, I.H. House dust mite-induced calcium signaling instigates epithelial barrier dysfunction and CCL20 production. Allergy 2013, 68, 1117–1125. [Google Scholar] [CrossRef]
- Jacquet, A. Interactions of airway epithelium with protease allergens in the allergic response. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2011, 41, 305–311. [Google Scholar] [CrossRef]
- Heijink, I.H.; van Oosterhout, A.; Kapus, A. Epidermal growth factor receptor signalling contributes to house dust mite-induced epithelial barrier dysfunction. Eur. Respir. J. 2010, 36, 1016–1026. [Google Scholar] [CrossRef]
- Wan, H.; Winton, H.L.; Soeller, C.; Gruenert, D.C.; Thompson, P.J.; Cannell, M.B.; Stewart, G.A.; Garrod, D.R.; Robinson, C. Quantitative structural and biochemical analyses of tight junction dynamics following exposure of epithelial cells to house dust mite allergen Der p 1. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2000, 30, 685–698. [Google Scholar] [CrossRef]
- Wan, H.; Winton, H.L.; Soeller, C.; Tovey, E.R.; Gruenert, D.C.; Thompson, P.J.; Stewart, G.A.; Taylor, G.W.; Garrod, D.R.; Cannell, M.B.; et al. Der p 1 facilitates transepithelial allergen delivery by disruption of tight junctions. J. Clin. Investig. 1999, 104, 123–133. [Google Scholar] [CrossRef]
- Lee, S.I.; Pham le, D.; Shin, Y.S.; Suh, D.H.; Park, H.S. Environmental changes could enhance the biological effect of Hop J pollens on human airway epithelial cells. J. Allergy Clin. Immunol. 2014, 134, 470–472. [Google Scholar] [CrossRef]
- Runswick, S.; Mitchell, T.; Davies, P.; Robinson, C.; Garrod, D.R. Pollen proteolytic enzymes degrade tight junctions. Respirology 2007, 12, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Vinhas, R.; Cortes, L.; Cardoso, I.; Mendes, V.M.; Manadas, B.; Todo-Bom, A.; Pires, E.; Veríssimo, P. Pollen proteases compromise the airway epithelial barrier through degradation of transmembrane adhesion proteins and lung bioactive peptides. Allergy 2011, 66, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Ahdieh, M.; Vandenbos, T.; Youakim, A. Lung epithelial barrier function and wound healing are decreased by IL-4 and IL-13 and enhanced by IFN-gamma. Am. J. Physiol. Cell Physiol. 2001, 281, C2029–C2038. [Google Scholar] [CrossRef] [PubMed]
- Saatian, B.; Rezaee, F.; Desando, S.; Emo, J.; Chapman, T.; Knowlden, S.; Georas, S.N. Interleukin-4 and interleukin-13 cause barrier dysfunction in human airway epithelial cells. Tissue Barriers 2013, 1, e24333. [Google Scholar] [CrossRef] [PubMed]
- Sugita, K.; Steer, C.A.; Martinez-Gonzalez, I.; Altunbulakli, C.; Morita, H.; Castro-Giner, F.; Kubo, T.; Wawrzyniak, P.; Rückert, B.; Sudo, K.; et al. Type 2 innate lymphoid cells disrupt bronchial epithelial barrier integrity by targeting tight junctions through IL-13 in asthmatic patients. J. Allergy Clin. Immunol. 2018, 141, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Sweerus, K.; Lachowicz-Scroggins, M.; Gordon, E.; LaFemina, M.; Huang, X.; Parikh, M.; Kanegai, C.; Fahy, J.V.; Frank, J.A. Claudin-18 deficiency is associated with airway epithelial barrier dysfunction and asthma. J. Allergy Clin. Immunol. 2017, 139, 72–81. [Google Scholar] [CrossRef]
- Comstock, A.T.; Ganesan, S.; Chattoraj, A.; Faris, A.N.; Margolis, B.L.; Hershenson, M.B.; Sajjan, U.S. Rhinovirus-induced barrier dysfunction in polarized airway epithelial cells is mediated by NADPH oxidase 1. J. Virol. 2011, 85, 6795–6808. [Google Scholar] [CrossRef] [PubMed]
- Looi, K.; Buckley, A.G.; Rigby, P.J.; Garratt, L.W.; Iosifidis, T.; Zosky, G.R.; Larcombe, A.N.; Lannigan, F.J.; Ling, K.M.; Martinovich, K.M.; et al. Effects of human rhinovirus on epithelial barrier integrity and function in children with asthma. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2018, 48, 513–524. [Google Scholar] [CrossRef]
- Rezaee, F.; DeSando, S.A.; Ivanov, A.I.; Chapman, T.J.; Knowlden, S.A.; Beck, L.A.; Georas, S.N. Sustained protein kinase D activation mediates respiratory syncytial virus-induced airway barrier disruption. J. Virol. 2013, 87, 11088–11095. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Hammad, H. The airway epithelium in asthma. Nat. Med. 2012, 18, 684–692. [Google Scholar] [CrossRef]
- Matsuwaki, Y.; Wada, K.; White, T.; Moriyama, H.; Kita, H. Alternaria fungus induces the production of GM-CSF, interleukin-6 and interleukin-8 and calcium signaling in human airway epithelium through protease-activated receptor 2. Int. Arch. Allergy Immunol. 2012, 158 (Suppl. 1), 19–29. [Google Scholar] [CrossRef] [PubMed]
- Day, S.B.; Ledford, J.R.; Zhou, P.; Lewkowich, I.P.; Page, K. German cockroach proteases and protease-activated receptor-2 regulate chemokine production and dendritic cell recruitment. J. Innate Immun. 2012, 4, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.E.; Kim, J.W.; Jeong, K.Y.; Kim, K.E.; Yong, T.S.; Sohn, M.H. Regulation of German cockroach extract-induced IL-8 expression in human airway epithelial cells. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2007, 37, 1364–1373. [Google Scholar] [CrossRef] [PubMed]
- Nathan, A.T.; Peterson, E.A.; Chakir, J.; Wills-Karp, M. Innate immune responses of airway epithelium to house dust mite are mediated through beta-glucan-dependent pathways. J. Allergy Clin. Immunol. 2009, 123, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Osterlund, C.; Grönlund, H.; Polovic, N.; Sundström, S.; Gafvelin, G.; Bucht, A. The non-proteolytic house dust mite allergen Der p 2 induce NF-kappaB and MAPK dependent activation of bronchial epithelial cells. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2009, 39, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Basnet, S.; Palmenberg, A.C.; Gern, J.E. Rhinoviruses and Their Receptors. Chest 2019, 155, 1018–1025. [Google Scholar] [CrossRef]
- Trompette, A.; Divanovic, S.; Visintin, A.; Blanchard, C.; Hegde, R.S.; Madan, R.; Thorne, P.S.; Wills-Karp, M.; Gioannini, T.L.; Weiss, J.P.; et al. Allergenicity resulting from functional mimicry of a Toll-like receptor complex protein. Nature 2009, 457, 585–588. [Google Scholar] [CrossRef]
- Kouzaki, H.; O’Grady, S.M.; Lawrence, C.B.; Kita, H. Proteases induce production of thymic stromal lymphopoietin by airway epithelial cells through protease-activated receptor-2. J. Immunol. 2009, 183, 1427–1434. [Google Scholar] [CrossRef]
- Kouzaki, H.; Tojima, I.; Kita, H.; Shimizu, T. Transcription of interleukin-25 and extracellular release of the protein is regulated by allergen proteases in airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2013, 49, 741–750. [Google Scholar] [CrossRef]
- Kouzaki, H.; Iijima, K.; Kobayashi, T.; O’Grady, S.M.; Kita, H. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL-33 release and innate Th2-type responses. J. Immunol. 2011, 186, 4375–4387. [Google Scholar] [CrossRef]
- Hristova, M.; Habibovic, A.; Veith, C.; Janssen-Heininger, Y.M.; Dixon, A.E.; Geiszt, M.; van der Vliet, A. Airway epithelial dual oxidase 1 mediates allergen-induced IL-33 secretion and activation of type 2 immune responses. J. Allergy Clin. Immunol. 2016, 137, 1545–1556. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, Y.; Lv, Z.; Chen, Y.; Li, Y.; Huang, K.; Corrigan, C.J.; Ying, S. Bronchial Allergen Challenge of Patients with Atopic Asthma Triggers an Alarmin (IL-33, TSLP, and IL-25) Response in the Airways Epithelium and Submucosa. J. Immunol. 2018, 201, 2221–2231. [Google Scholar] [CrossRef] [PubMed]
- Ramu, S.; Menzel, M.; Bjermer, L.; Andersson, C.; Akbarshahi, H.; Uller, L. Allergens produce serine proteases-dependent distinct release of metabolite DAMPs in human bronchial epithelial cells. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2018, 48, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.; Bauer, W.; Braun, R.K.; Menz, G.; Braun, P.; Schwarz, F.; Hansel, T.T.; Villiger, B. Activated T cells and cytokines in bronchoalveolar lavages from patients with various lung diseases associated with eosinophilia. Am. J. Respir. Crit. Care Med. 1994, 150, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Redington, A.E.; Madden, J.; Frew, A.J.; Djukanovic, R.; Roche, W.R.; Holgate, S.T.; Howarth, P.H. Transforming growth factor-beta 1 in asthma. Measurement in bronchoalveolar lavage fluid. Am. J. Respir. Crit. Care Med. 1997, 156 Pt 1, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Heijink, I.H.; Marcel Kies, P.; van Oosterhout, A.J.; Postma, D.S.; Kauffman, H.F.; Vellenga, E. Der p, IL-4, and TGF-beta cooperatively induce EGFR-dependent TARC expression in airway epithelium. Am. J. Respir. Cell Mol. Biol. 2007, 36, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Pichavant, M.; Charbonnier, A.S.; Taront, S.; Brichet, A.; Wallaert, B.; Pestel, J.; Tonnel, A.B.; Gosset, P. Asthmatic bronchial epithelium activated by the proteolytic allergen Der p 1 increases selective dendritic cell recruitment. J. Allergy Clin. Immunol. 2005, 115, 771–778. [Google Scholar] [CrossRef]
- Johnston, N.W.; Johnston, S.L.; Duncan, J.M.; Greene, J.M.; Kebadze, T.; Keith, P.K.; Roy, M.; Waserman, S.; Sears, M.R. The September epidemic of asthma exacerbations in children: A search for etiology. J. Allergy Clin. Immunol. 2005, 115, 132–138. [Google Scholar] [CrossRef]
- Nicholson, K.G.; Kent, J.; Ireland, D.C. Respiratory viruses and exacerbations of asthma in adults. BMJ (Clin. Res. Ed.) 1993, 307, 982–986. [Google Scholar] [CrossRef]
- Johnston, S.L.; Pattemore, P.K.; Sanderson, G.; Smith, S.; Lampe, F.; Josephs, L.; Symington, P.; O’Toole, S.; Myint, S.H.; Tyrrell, D.A.; et al. Community study of role of viral infections in exacerbations of asthma in 9-11 year old children. BMJ (Clin. Res. Ed.) 1995, 310, 1225–1229. [Google Scholar] [CrossRef]
- O’Byrne, P.M.; Pedersen, S.; Lamm, C.J.; Tan, W.C.; Busse, W.W. Severe exacerbations and decline in lung function in asthma. Am. J. Respir. Crit. Care Med. 2009, 179, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.J.; Evans, M.D.; Gangnon, R.E.; Tisler, C.J.; Pappas, T.E.; Lee, W.M.; Gern, J.E.; Lemanske, R.F., Jr. Evidence for a causal relationship between allergic sensitization and rhinovirus wheezing in early life. Am. J. Respir. Crit. Care Med. 2012, 185, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.J.; Gangnon, R.E.; Evans, M.D.; Roberg, K.A.; Anderson, E.L.; Pappas, T.E.; Printz, M.C.; Lee, W.M.; Shult, P.A.; Reisdorf, E.; et al. Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am. J. Respir. Crit. Care Med. 2008, 178, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Kusel, M.M.; de Klerk, N.H.; Kebadze, T.; Vohma, V.; Holt, P.G.; Johnston, S.L.; Sly, P.D. Early-life respiratory viral infections, atopic sensitization, and risk of subsequent development of persistent asthma. J. Allergy Clin. Immunol. 2007, 119, 1105–1110. [Google Scholar] [CrossRef]
- Kotaniemi-Syrjänen, A.; Vainionpää, R.; Reijonen, T.M.; Waris, M.; Korhonen, K.; Korppi, M. Rhinovirus-induced wheezing in infancy--the first sign of childhood asthma? J. Allergy Clin. Immunol. 2003, 111, 66–71. [Google Scholar] [CrossRef]
- Rubner, F.J.; Jackson, D.J.; Evans, M.D.; Gangnon, R.E.; Tisler, C.J.; Pappas, T.E.; Gern, J.E.; Lemanske, R.F., Jr. Early life rhinovirus wheezing, allergic sensitization, and asthma risk at adolescence. J. Allergy Clin. Immunol. 2017, 139, 501–507. [Google Scholar] [CrossRef]
- Wu, P.; Hartert, T.V. Evidence for a causal relationship between respiratory syncytial virus infection and asthma. Expert Rev. Anti-Infect. Ther. 2011, 9, 731–745. [Google Scholar] [CrossRef]
- Sigurs, N.; Aljassim, F.; Kjellman, B.; Robinson, P.D.; Sigurbergsson, F.; Bjarnason, R.; Gustafsson, P.M. Asthma and allergy patterns over 18 years after severe RSV bronchiolitis in the first year of life. Thorax 2010, 65, 1045–1052. [Google Scholar] [CrossRef]
- Papadopoulos, N.G.; Bates, P.J.; Bardin, P.G.; Papi, A.; Leir, S.H.; Fraenkel, D.J.; Meyer, J.; Lackie, P.M.; Sanderson, G.; Holgate, S.T.; et al. Rhinoviruses infect the lower airways. J. Infect. Dis. 2000, 181, 1875–1884. [Google Scholar] [CrossRef]
- Wos, M.; Sanak, M.; Soja, J.; Olechnowicz, H.; Busse, W.W.; Szczeklik, A. The presence of rhinovirus in lower airways of patients with bronchial asthma. Am. J. Respir. Crit. Care Med. 2008, 177, 1082–1089. [Google Scholar] [CrossRef]
- Papadopoulos, N.G.; Christodoulou, I.; Rohde, G.; Agache, I.; Almqvist, C.; Bruno, A.; Bonini, S.; Bont, L.; Bossios, A.; Bousquet, J.; et al. Viruses and bacteria in acute asthma exacerbations—A GA2 LEN-DARE systematic review. Allergy 2011, 66, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Nagarkar, D.R.; Bowman, E.R.; Schneider, D.; Gosangi, B.; Lei, J.; Zhao, Y.; McHenry, C.L.; Burgens, R.V.; Miller, D.J.; et al. Role of double-stranded RNA pattern recognition receptors in rhinovirus-induced airway epithelial cell responses. J. Immunol. 2009, 183, 6989–6997. [Google Scholar] [CrossRef] [PubMed]
- Slater, L.; Bartlett, N.W.; Haas, J.J.; Zhu, J.; Message, S.D.; Walton, R.P.; Sykes, A.; Dahdaleh, S.; Clarke, D.L.; Belvisi, M.G.; et al. Co-ordinated role of TLR3, RIG-I and MDA5 in the innate response to rhinovirus in bronchial epithelium. PLoS Pathog. 2010, 6, e1001178. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hamati, E.; Lee, P.K.; Lee, W.M.; Wachi, S.; Schnurr, D.; Yagi, S.; Dolganov, G.; Boushey, H.; Avila, P.; et al. Rhinovirus induces airway epithelial gene expression through double-stranded RNA and IFN-dependent pathways. Am. J. Respir. Cell Mol. Biol. 2006, 34, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Wark, P.A.; Johnston, S.L.; Bucchieri, F.; Powell, R.; Puddicombe, S.; Laza-Stanca, V.; Holgate, S.T.; Davies, D.E. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J. Exp. Med. 2005, 201, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Contoli, M.; Message, S.D.; Laza-Stanca, V.; Edwards, M.R.; Wark, P.A.; Bartlett, N.W.; Kebadze, T.; Mallia, P.; Stanciu, L.A.; Parker, H.L.; et al. Role of deficient type III interferon-lambda production in asthma exacerbations. Nat. Med. 2006, 12, 1023–1026. [Google Scholar] [CrossRef]
- Bochkov, Y.A.; Hanson, K.M.; Keles, S.; Brockman-Schneider, R.A.; Jarjour, N.N.; Gern, J.E. Rhinovirus-induced modulation of gene expression in bronchial epithelial cells from subjects with asthma. Mucosal Immunol. 2010, 3, 69–80. [Google Scholar] [CrossRef]
- Sykes, A.; Macintyre, J.; Edwards, M.R.; Del Rosario, A.; Haas, J.; Gielen, V.; Kon, O.M.; McHale, M.; Johnston, S.L. Rhinovirus-induced interferon production is not deficient in well controlled asthma. Thorax 2014, 69, 240–246. [Google Scholar] [CrossRef]
- Lopez-Souza, N.; Favoreto, S.; Wong, H.; Ward, T.; Yagi, S.; Schnurr, D.; Finkbeiner, W.E.; Dolganov, G.M.; Widdicombe, J.H.; Boushey, H.A.; et al. In vitro susceptibility to rhinovirus infection is greater for bronchial than for nasal airway epithelial cells in human subjects. J. Allergy Clin. Immunol. 2009, 123, 1384–1390. [Google Scholar] [CrossRef]
- Baraldo, S.; Contoli, M.; Bazzan, E.; Turato, G.; Padovani, A.; Marku, B.; Calabrese, F.; Caramori, G.; Ballarin, A.; Snijders, D.; et al. Deficient antiviral immune responses in childhood: Distinct roles of atopy and asthma. J. Allergy Clin. Immunol. 2012, 130, 1307–1314. [Google Scholar] [CrossRef] [PubMed]
- Uller, L.; Leino, M.; Bedke, N.; Sammut, D.; Green, B.; Lau, L.; Howarth, P.H.; Holgate, S.T.; Davies, D.E. Double-stranded RNA induces disproportionate expression of thymic stromal lymphopoietin versus interferon-beta in bronchial epithelial cells from donors with asthma. Thorax 2010, 65, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.R.; Regamey, N.; Vareille, M.; Kieninger, E.; Gupta, A.; Shoemark, A.; Saglani, S.; Sykes, A.; Macintyre, J.; Davies, J.; et al. Impaired innate interferon induction in severe therapy resistant atopic asthmatic children. Mucosal Immunol. 2013, 6, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Kicic, A.; Stevens, P.T.; Sutanto, E.N.; Kicic-Starcevich, E.; Ling, K.M.; Looi, K.; Martinovich, K.M.; Garratt, L.W.; Iosifidis, T.; Shaw, N.C.; et al. Impaired airway epithelial cell responses from children with asthma to rhinoviral infection. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2016, 46, 1441–1455. [Google Scholar] [CrossRef]
- Wark, P.A.; Grissell, T.; Davies, B.; See, H.; Gibson, P.G. Diversity in the bronchial epithelial cell response to infection with different rhinovirus strains. Respirology 2009, 14, 180–186. [Google Scholar] [CrossRef]
- Collison, A.; Hatchwell, L.; Verrills, N.; Wark, P.A.; de Siqueira, A.P.; Tooze, M.; Carpenter, H.; Don, A.S.; Morris, J.C.; Zimmermann, N.; et al. The E3 ubiquitin ligase midline 1 promotes allergen and rhinovirus-induced asthma by inhibiting protein phosphatase 2A activity. Nat. Med. 2013, 19, 232–237. [Google Scholar] [CrossRef]
- Gielen, V.; Sykes, A.; Zhu, J.; Chan, B.; Macintyre, J.; Regamey, N.; Kieninger, E.; Gupta, A.; Shoemark, A.; Bossley, C.; et al. Increased nuclear suppressor of cytokine signaling 1 in asthmatic bronchial epithelium suppresses rhinovirus induction of innate interferons. J. Allergy Clin. Immunol. 2015, 136, 177–188. [Google Scholar] [CrossRef]
- Contoli, M.; Ito, K.; Padovani, A.; Poletti, D.; Marku, B.; Edwards, M.R.; Stanciu, L.A.; Gnesini, G.; Pastore, A.; Spanevello, A.; et al. Th2 cytokines impair innate immune responses to rhinovirus in respiratory epithelial cells. Allergy 2015, 70, 910–920. [Google Scholar] [CrossRef]
- Akbarshahi, H.; Menzel, M.; Ramu, S.; Mahmutovic Persson, I.; Bjermer, L.; Uller, L. House dust mite impairs antiviral response in asthma exacerbation models through its effects on TLR3. Allergy 2018, 73, 1053–1063. [Google Scholar] [CrossRef]
- Menzel, M.; Ramu, S.; Calvén, J.; Olejnicka, B.; Sverrild, A.; Porsbjerg, C.; Tufvesson, E.; Bjermer, L.; Akbarshahi, H.; Uller, L. Oxidative Stress Attenuates TLR3 Responsiveness and Impairs Anti-viral Mechanisms in Bronchial Epithelial Cells From COPD and Asthma Patients. Front. Immunol. 2019, 10, 2765. [Google Scholar] [CrossRef]
- Jackson, D.J.; Makrinioti, H.; Rana, B.M.; Shamji, B.W.; Trujillo-Torralbo, M.B.; Footitt, J.; Jerico, D.-R.; Telcian, A.G.; Nikonova, A.; Zhu, J.; et al. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am. J. Respir. Crit. Care Med. 2014, 190, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Beale, J.; Jayaraman, A.; Jackson, D.J.; Macintyre, J.D.R.; Edwards, M.R.; Walton, R.P.; Zhu, J.; Man Ching, Y.; Shamji, B.; Edwards, M.; et al. Rhinovirus-induced IL-25 in asthma exacerbation drives type 2 immunity and allergic pulmonary inflammation. Sci. Transl. Med. 2014, 6, 256ra134. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Headley, M.B.; Loo, Y.M.; Berlin, A.; Gale, M., Jr.; Debley, J.S.; Lukacs, N.W.; Ziegler, S.F. Thymic stromal lymphopoietin is induced by respiratory syncytial virus-infected airway epithelial cells and promotes a type 2 response to infection. J. Allergy Clin. Immunol. 2012, 130, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Brandelius, A.; Yudina, Y.; Calvén, J.; Bjermer, L.; Andersson, M.; Persson, C.; Uller, L. dsRNA-induced expression of thymic stromal lymphopoietin (TSLP) in asthmatic epithelial cells is inhibited by a small airway relaxant. Pulm. Pharmacol. Ther. 2011, 24, 59–66. [Google Scholar] [CrossRef]
- Soumelis, V.; Reche, P.A.; Kanzler, H.; Yuan, W.; Edward, G.; Homey, B.; Gilliet, M.; Ho, S.; Antonenko, S.; Lauerma, A.; et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat. Immunol. 2002, 3, 673–680. [Google Scholar] [CrossRef]
- Allakhverdi, Z.; Comeau, M.R.; Jessup, H.K.; Yoon, B.R.; Brewer, A.; Chartier, S.; Paquette, N.; Ziegler, S.F.; Sarfati, M.; Delespesse, G. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J. Exp. Med. 2007, 204, 253–258. [Google Scholar] [CrossRef]
- Kitajima, M.; Lee, H.C.; Nakayama, T.; Ziegler, S.F. TSLP enhances the function of helper type 2 cells. Eur. J. Immunol. 2011, 41, 1862–1871. [Google Scholar] [CrossRef]
- Ying, S.; O’Connor, B.; Ratoff, J.; Meng, Q.; Fang, C.; Cousins, D.; Zhang, G.; Gu, S.; Gao, Z.; Shamji, B.; et al. Expression and cellular provenance of thymic stromal lymphopoietin and chemokines in patients with severe asthma and chronic obstructive pulmonary disease. J. Immunol. 2008, 181, 2790–2798. [Google Scholar] [CrossRef]
- Ying, S.; O’Connor, B.; Ratoff, J.; Meng, Q.; Mallett, K.; Cousins, D.; Robinson, D.; Zhang, G.; Zhao, J.; Lee, T.H.; et al. Thymic stromal lymphopoietin expression is increased in asthmatic airways and correlates with expression of Th2-attracting chemokines and disease severity. J. Immunol. 2005, 174, 8183–8190. [Google Scholar] [CrossRef]
- Shikotra, A.; Choy, D.F.; Ohri, C.M.; Doran, E.; Butler, C.; Hargadon, B.; Shelley, M.; Abbas, A.R.; Austin, C.D.; Jackman, J.; et al. Increased expression of immunoreactive thymic stromal lymphopoietin in patients with severe asthma. J. Allergy Clin. Immunol. 2012, 129, 104–111. [Google Scholar] [CrossRef]
- Calvén, J.; Yudina, Y.; Hallgren, O.; Westergren-Thorsson, G.; Davies, D.E.; Brandelius, A.; Uller, L. Viral stimuli trigger exaggerated thymic stromal lymphopoietin expression by chronic obstructive pulmonary disease epithelium: Role of endosomal TLR3 and cytosolic RIG-I-like helicases. J. Innate Immun. 2012, 4, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Favoreto, S., Jr.; Avila, P.C.; Schleimer, R.P. TLR3- and Th2 cytokine-dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J. Immunol. 2007, 179, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Nagarkar, D.R.; Poposki, J.A.; Comeau, M.R.; Biyasheva, A.; Avila, P.C.; Schleimer, R.P.; Kato, A. Airway epithelial cells activate TH2 cytokine production in mast cells through IL-1 and thymic stromal lymphopoietin. J. Allergy Clin. Immunol. 2012, 130, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Ziegler, S.F. Inducible expression of the proallergic cytokine thymic stromal lymphopoietin in airway epithelial cells is controlled by NFkappaB. Proc. Natl. Acad. Sci. USA 2007, 104, 914–919. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, B. Functions of thymic stromal lymphopoietin in immunity and disease. Immunol. Res. 2012, 52, 211–223. [Google Scholar] [CrossRef]
- Ziegler, S.F.; Roan, F.; Bell, B.D.; Stoklasek, T.A.; Kitajima, M.; Han, H. The biology of thymic stromal lymphopoietin (TSLP). Adv. Pharmacol. (San Diego Calif.) 2013, 66, 129–155. [Google Scholar]
- Zhou, B.; Comeau, M.R.; De Smedt, T.; Liggitt, H.D.; Dahl, M.E.; Lewis, D.B.; Gyarmati, D.; Aye, T.; Campbell, D.J.; Ziegler, S.F. Thymic stromal lymphopoietin as a key initiator of allergic airway inflammation in mice. Nat. Immunol. 2005, 6, 1047–1053. [Google Scholar] [CrossRef]
- Halim, T.Y.; Krauss, R.H.; Sun, A.C.; Takei, F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity 2012, 36, 451–463. [Google Scholar] [CrossRef]
- Omori, M.; Ziegler, S. Induction of IL-4 expression in CD4(+) T cells by thymic stromal lymphopoietin. J. Immunol. 2007, 178, 1396–1404. [Google Scholar] [CrossRef]
- Ito, T.; Wang, Y.H.; Duramad, O.; Hori, T.; Delespesse, G.J.; Watanabe, N.; Qin, F.X.; Yao, Z.; Cao, W.; Liu, Y.J. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J. Exp. Med. 2005, 202, 1213–1223. [Google Scholar] [CrossRef]
- Wong, C.K.; Hu, S.; Cheung, P.F.; Lam, C.W. Thymic stromal lymphopoietin induces chemotactic and prosurvival effects in eosinophils: Implications in allergic inflammation. Am. J. Respir. Cell Mol. Biol. 2010, 43, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, S.; Jagot, F.; Kyle, R.L.; Hyde, E.; White, R.F.; Prout, M.; Schmidt, A.J.; Yamane, H.; Lamiable, O.; Le Gros, G.; et al. Thymic stromal lymphopoietin drives the development of IL-13(+) Th2 cells. Proc. Natl. Acad. Sci. USA 2018, 115, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Verma, M.; Michalec, L.; Liu, W.; Sripada, A.; Rollins, D.; Good, J.; Ito, Y.; Chu, H.; Gorska, M.M.; et al. Steroid resistance of airway type 2 innate lymphoid cells from patients with severe asthma: The role of thymic stromal lymphopoietin. J. Allergy Clin. Immunol. 2018, 141, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Torgerson, D.G.; Ampleford, E.J.; Chiu, G.Y.; Gauderman, W.J.; Gignoux, C.R.; Graves, P.E.; Himes, B.E.; Levin, A.M.; Mathias, R.A.; Hancock, D.B.; et al. Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nat. Genet. 2011, 43, 887–892. [Google Scholar]
- Corren, J.; Parnes, J.R.; Wang, L.; Mo, M.; Roseti, S.L.; Griffiths, J.M.; van der Merwe, R. Tezepelumab in Adults with Uncontrolled Asthma. N. Engl. J. Med. 2017, 377, 936–946. [Google Scholar] [CrossRef]
- Gauvreau, G.M.; O’Byrne, P.M.; Boulet, L.P.; Wang, Y.; Cockcroft, D.; Bigler, J.; FitzGerald, J.M.; Boedigheimer, M.; Davis, B.E.; Dias, C.; et al. Effects of an anti-TSLP antibody on allergen-induced asthmatic responses. N. Engl. J. Med. 2014, 370, 2102–2110. [Google Scholar] [CrossRef]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef]
- Haenuki, Y.; Matsushita, K.; Futatsugi-Yumikura, S.; Ishii, K.J.; Kawagoe, T.; Imoto, Y.; Fujieda, S.; Yasuda, M.; Hisa, Y.; Akira, S.; et al. A critical role of IL-33 in experimental allergic rhinitis. J. Allergy Clin. Immunol. 2012, 130, 184–194. [Google Scholar] [CrossRef]
- Préfontaine, D.; Nadigel, J.; Chouiali, F.; Audusseau, S.; Semlali, A.; Chakir, J.; Martin, J.G.; Hamid, Q. Increased IL-33 expression by epithelial cells in bronchial asthma. J. Allergy Clin. Immunol. 2010, 125, 752–754. [Google Scholar] [CrossRef]
- Cayrol, C.; Duval, A.; Schmitt, P.; Roga, S.; Camus, M.; Stella, A.; Burlet-Schiltz, O.; Gonzalez-de-Peredo, A.; Girard, J.P. Environmental allergens induce allergic inflammation through proteolytic maturation of IL-33. Nat. Immunol. 2018, 19, 375–385. [Google Scholar] [CrossRef]
- Cohen, E.S.; Scott, I.C.; Majithiya, J.B.; Rapley, L.; Kemp, B.P.; England, E.; Rees, D.G.; Overed-Sayer, C.L.; Woods, J.; Bond, N.J.; et al. Oxidation of the alarmin IL-33 regulates ST2-dependent inflammation. Nat. Commun. 2015, 6, 8327. [Google Scholar] [CrossRef] [PubMed]
- Palmer, G.; Lipsky, B.P.; Smithgall, M.D.; Meininger, D.; Siu, S.; Talabot-Ayer, D.; Gabay, C.; Smith, D.E. The IL-1 receptor accessory protein (AcP) is required for IL-33 signaling and soluble AcP enhances the ability of soluble ST2 to inhibit IL-33. Cytokine 2008, 42, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Blom, L.; Poulsen, L.K. IL-1 family members IL-18 and IL-33 upregulate the inflammatory potential of differentiated human Th1 and Th2 cultures. J. Immunol. 2012, 189, 4331–4337. [Google Scholar] [CrossRef] [PubMed]
- Allakhverdi, Z.; Smith, D.E.; Comeau, M.R.; Delespesse, G. Cutting edge: The ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J. Immunol. 2007, 179, 2051–2054. [Google Scholar] [CrossRef]
- Kurowska-Stolarska, M.; Kewin, P.; Murphy, G.; Russo, R.C.; Stolarski, B.; Garcia, C.C.; Komai-Koma, M.; Pitman, N.; Li, Y.; Niedbala, W.; et al. IL-33 induces antigen-specific IL-5+ T cells and promotes allergic-induced airway inflammation independent of IL-4. J. Immunol. 2008, 181, 4780–4790. [Google Scholar] [CrossRef]
- Rank, M.A.; Kobayashi, T.; Kozaki, H.; Bartemes, K.R.; Squillace, D.L.; Kita, H. IL-33-activated dendritic cells induce an atypical TH2-type response. J. Allergy Clin. Immunol. 2009, 123, 1047–1054. [Google Scholar] [CrossRef]
- Besnard, A.G.; Togbe, D.; Guillou, N.; Erard, F.; Quesniaux, V.; Ryffel, B. IL-33-activated dendritic cells are critical for allergic airway inflammation. Eur. J. Immunol. 2011, 41, 1675–1686. [Google Scholar] [CrossRef]
- Pecaric-Petkovic, T.; Didichenko, S.A.; Kaempfer, S.; Spiegl, N.; Dahinden, C.A. Human basophils and eosinophils are the direct target leukocytes of the novel IL-1 family member IL-33. Blood 2009, 113, 1526–1534. [Google Scholar] [CrossRef]
- Barlow, J.L.; Peel, S.; Fox, J.; Panova, V.; Hardman, C.S.; Camelo, A.; Bucks, C.; Wu, X.; Kane, C.M.; Neill, D.R.; et al. IL-33 is more potent than IL-25 in provoking IL-13-producing nuocytes (type 2 innate lymphoid cells) and airway contraction. J. Allergy Clin. Immunol. 2013, 132, 933–941. [Google Scholar] [CrossRef]
- Tjota, M.Y.; Williams, J.W.; Lu, T.; Clay, B.S.; Byrd, T.; Hrusch, C.L.; Decker, D.C.; de Araujo, C.A.; Bryce, P.J.; Sperling, A.I. IL-33-dependent induction of allergic lung inflammation by FcγRIII signaling. J. Clin. Investig. 2013, 123, 2287–2297. [Google Scholar] [CrossRef]
- Kamijo, S.; Takeda, H.; Tokura, T.; Suzuki, M.; Inui, K.; Hara, M.; Matsuda, H.; Matsuda, A.; Oboki, K.; Ohno, T.; et al. IL-33-mediated innate response and adaptive immune cells contribute to maximum responses of protease allergen-induced allergic airway inflammation. J. Immunol. 2013, 190, 4489–4499. [Google Scholar] [CrossRef] [PubMed]
- Halim, T.Y.; Steer, C.A.; Mathä, L.; Gold, M.J.; Martinez-Gonzalez, I.; McNagny, K.M.; McKenzie, A.N.; Takei, F. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 2014, 40, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Savenije, O.E.; Mahachie John, J.M.; Granell, R.; Kerkhof, M.; Dijk, F.N.; de Jongste, J.C.; Smit, H.A.; Brunekreef, B.; Postma, D.S.; Van Steen, K.; et al. Association of IL33-IL-1 receptor-like 1 (IL1RL1) pathway polymorphisms with wheezing phenotypes and asthma in childhood. J. Allergy Clin. Immunol. 2014, 134, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Liu, Y.J. The IL-17 cytokine family and their role in allergic inflammation. Curr. Opin. Immunol. 2008, 20, 697–702. [Google Scholar] [CrossRef]
- Hurst, S.D.; Muchamuel, T.; Gorman, D.M.; Gilbert, J.M.; Clifford, T.; Kwan, S.; Menon, S.; Seymour, B.; Jackson, C.; Kung, T.T.; et al. New IL-17 family members promote Th1 or Th2 responses in the lung: In vivo function of the novel cytokine IL-25. J. Immunol. 2002, 169, 443–453. [Google Scholar] [CrossRef]
- Kohanski, M.A.; Workman, A.D.; Patel, N.N.; Hung, L.Y.; Shtraks, J.P.; Chen, B.; Blasetti, M.; Doghramji, L.; Kennedy, D.W.; Adappa, N.D.; et al. Solitary chemosensory cells are a primary epithelial source of IL-25 in patients with chronic rhinosinusitis with nasal polyps. J. Allergy Clin. Immunol. 2018, 142, 460–469. [Google Scholar] [CrossRef]
- Gregory, L.G.; Jones, C.P.; Walker, S.A.; Sawant, D.; Gowers, K.H.; Campbell, G.A.; McKenzie, A.N.; Lloyd, C.M. IL-25 drives remodelling in allergic airways disease induced by house dust mite. Thorax 2013, 68, 82–90. [Google Scholar] [CrossRef]
- Wang, Y.H.; Angkasekwinai, P.; Lu, N.; Voo, K.S.; Arima, K.; Hanabuchi, S.; Hippe, A.; Corrigan, C.J.; Dong, C.; Homey, B.; et al. IL-25 augments type 2 immune responses by enhancing the expansion and functions of TSLP-DC-activated Th2 memory cells. J. Exp. Med. 2007, 204, 1837–1847. [Google Scholar] [CrossRef]
- Wang, Y.H.; Ito, T.; Wang, Y.H.; Homey, B.; Watanabe, N.; Martin, R.; Barnes, C.J.; McIntyre, B.W.; Gilliet, M.; Kumar, R.; et al. Maintenance and polarization of human TH2 central memory T cells by thymic stromal lymphopoietin-activated dendritic cells. Immunity 2006, 24, 827–838. [Google Scholar] [CrossRef]
- Corrigan, C.J.; Wang, W.; Meng, Q.; Fang, C.; Eid, G.; Caballero, M.R.; Lv, Z.; An, Y.; Wang, Y.H.; Liu, Y.J.; et al. Allergen-induced expression of IL-25 and IL-25 receptor in atopic asthmatic airways and late-phase cutaneous responses. J. Allergy Clin. Immunol. 2011, 128, 116–124. [Google Scholar] [CrossRef]
- Tamachi, T.; Maezawa, Y.; Ikeda, K.; Kagami, S.; Hatano, M.; Seto, Y.; Suto, A.; Suzuki, K.; Watanabe, N.; Saito, Y.; et al. IL-25 enhances allergic airway inflammation by amplifying a TH2 cell-dependent pathway in mice. J. Allergy Clin. Immunol. 2006, 118, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Ballantyne, S.J.; Barlow, J.L.; Jolin, H.E.; Nath, P.; Williams, A.S.; Chung, K.F.; Sturton, G.; Wong, S.H.; McKenzie, A.N. Blocking IL-25 prevents airway hyperresponsiveness in allergic asthma. J. Allergy Clin. Immunol. 2007, 120, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Xue, Z.; Yi, L.; Shi, H.; Zhang, K.; Huo, X.; Bonser, L.R.; Zhao, J.; Xu, Y.; Erle, D.J.; et al. Epithelial interleukin-25 is a key mediator in Th2-high, corticosteroid-responsive asthma. Am. J. Respir. Crit. Care Med. 2014, 190, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Adler, K.B.; Fischer, B.M.; Wright, D.T.; Cohn, L.A.; Becker, S. Interactions between respiratory epithelial cells and cytokines: Relationships to lung inflammation. Ann. N. Y. Acad. Sci. 1994, 725, 128–145. [Google Scholar] [CrossRef]
- Kato, A.; Schleimer, R.P. Beyond inflammation: Airway epithelial cells are at the interface of innate and adaptive immunity. Curr. Opin. Immunol. 2007, 19, 711–720. [Google Scholar] [CrossRef]
- Gras, D.; Chanez, P.; Vachier, I.; Petit, A.; Bourdin, A. Bronchial epithelium as a target for innovative treatments in asthma. Pharmacol. Ther. 2013, 140, 290–305. [Google Scholar] [CrossRef]
- Woodruff, P.G.; Boushey, H.A.; Dolganov, G.M.; Barker, C.S.; Yang, Y.H.; Donnelly, S.; Ellwanger, A.; Sidhu, S.S.; Dao-Pick, T.P.; Pantoja, C.; et al. Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc. Natl. Acad. Sci. USA 2007, 104, 15858–15863. [Google Scholar] [CrossRef]
- Woodruff, P.G.; Modrek, B.; Choy, D.F.; Jia, G.; Abbas, A.R.; Ellwanger, A.; Koth, L.L.; Arron, J.R.; Fahy, J.V. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am. J. Respir. Crit. Care Med. 2009, 180, 388–395. [Google Scholar] [CrossRef]
- Bhakta, N.R.; Solberg, O.D.; Nguyen, C.P.; Nguyen, C.N.; Arron, J.R.; Fahy, J.V.; Woodruff, P.G. A qPCR-based metric of Th2 airway inflammation in asthma. Clin. Transl. Allergy 2013, 3, 24. [Google Scholar] [CrossRef]
- Choy, D.F.; Hart, K.M.; Borthwick, L.A.; Shikotra, A.; Nagarkar, D.R.; Siddiqui, S.; Jia, G.; Ohri, C.M.; Doran, E.; Vannella, K.M.; et al. TH2 and TH17 inflammatory pathways are reciprocally regulated in asthma. Sci. Transl. Med. 2015, 7, 301ra129. [Google Scholar] [CrossRef]
- Ax, E.; Jevnikar, Z.; Cvjetkovic, A.; Malmhall, C.; Olsson, H.; Radinger, M.; Lasser, C. T2 and T17 cytokines alter the cargo and function of airway epithelium-derived extracellular vesicles. Respir. Res. 2020, 21, 155. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Erickson, R.W.; Choy, D.F.; Mosesova, S.; Wu, L.C.; Solberg, O.D.; Shikotra, A.; Carter, R.; Audusseau, S.; Hamid, Q.; et al. Periostin is a systemic biomarker of eosinophilic airway inflammation in asthmatic patients. J. Allergy Clin. Immunol. 2012, 130, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Pavlidis, S.; Takahashi, K.; Ng Kee Kwong, F.; Xie, J.; Hoda, U.; Sun, K.; Elyasigomari, V.; Agapow, P.; Loza, M.; Baribaud, F.; et al. “T2-high” in severe asthma related to blood eosinophil, exhaled nitric oxide and serum periostin. Eur. Respir. J. 2019, 53, 1800938. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, D.N.; Moore, B.B. The role of periostin in lung fibrosis and airway remodeling. Cell. Mol. Life Sci. CMLS 2017, 74, 4305–4314. [Google Scholar]
- Sidhu, S.S.; Yuan, S.; Innes, A.L.; Kerr, S.; Woodruff, P.G.; Hou, L.; Muller, S.J.; Fahy, J.V. Roles of epithelial cell-derived periostin in TGF-beta activation, collagen production, and collagen gel elasticity in asthma. Proc. Natl. Acad. Sci. USA 2010, 107, 14170–14175. [Google Scholar] [CrossRef]
- Burgess, J.K.; Jonker, M.R.; Berg, M.; Ten Hacken, N.T.H.; Meyer, K.B.; van den Berge, M.; Nawijn, M.C.; Heijink, I.H. Periostin: Contributor to abnormal airway epithelial function in asthma? Eur. Respir. J. 2020. [Google Scholar] [CrossRef]
- Johansson, M.W.; Khanna, M.; Bortnov, V.; Annis, D.S.; Nguyen, C.L.; Mosher, D.F. IL-5-stimulated eosinophils adherent to periostin undergo stereotypic morphological changes and ADAM8-dependent migration. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2017, 47, 1263–1274. [Google Scholar] [CrossRef]
- Wang, Y.; Bai, C.; Li, K.; Adler, K.B.; Wang, X. Role of airway epithelial cells in development of asthma and allergic rhinitis. Respir. Med. 2008, 102, 949–955. [Google Scholar] [CrossRef]
- Bonser, L.R.; Erle, D.J. Airway Mucus and Asthma: The Role of MUC5AC and MUC5B. J. Clin. Med. 2017, 6, 112. [Google Scholar] [CrossRef]
- Lachowicz-Scroggins, M.E.; Yuan, S.; Kerr, S.C.; Dunican, E.M.; Yu, M.; Carrington, S.D.; Fahy, J.V. Abnormalities in MUC5AC and MUC5B Protein in Airway Mucus in Asthma. Am. J. Respir. Crit. Care Med. 2016, 194, 1296–1299. [Google Scholar] [CrossRef]
- Park, K.S.; Korfhagen, T.R.; Bruno, M.D.; Kitzmiller, J.A.; Wan, H.; Wert, S.E.; Khurana Hershey, G.K.; Chen, G.; Whitsett, J.A. SPDEF regulates goblet cell hyperplasia in the airway epithelium. J. Clin. Investig. 2007, 117, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Li, Q.; Kolosov, V.P.; Perelman, J.M.; Zhou, X. Interleukin-13 induces mucin 5AC production involving STAT6/SPDEF in human airway epithelial cells. Cell Commun. Adhes. 2010, 17, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Rajavelu, P.; Chen, G.; Xu, Y.; Kitzmiller, J.A.; Korfhagen, T.R.; Whitsett, J.A. Airway epithelial SPDEF integrates goblet cell differentiation and pulmonary Th2 inflammation. J. Clin. Investig. 2015, 125, 2021–2031. [Google Scholar] [CrossRef] [PubMed]
- Koh, K.D.; Siddiqui, S.; Cheng, D.; Bonser, L.R.; Sun, D.I.; Zlock, L.T.; Finkbeiner, W.E.; Woodruff, P.G.; Erle, D.J. Efficient RNP-directed Human Gene Targeting Reveals SPDEF Is Required for IL-13-induced Mucostasis. Am. J. Respir. Cell Mol. Biol. 2020, 62, 373–381. [Google Scholar] [CrossRef]
- Liu, C.L.; Shi, G.P. Calcium-activated chloride channel regulator 1 (CLCA1): More than a regulator of chloride transport and mucus production. World Allergy Organ. J. 2019, 12, 100077. [Google Scholar] [CrossRef]
- Ha, E.V.; Rogers, D.F. Novel Therapies to Inhibit Mucus Synthesis and Secretion in Airway Hypersecretory Diseases. Pharmacology 2016, 97, 84–100. [Google Scholar] [CrossRef]
- Alevy, Y.G.; Patel, A.C.; Romero, A.G.; Patel, D.A.; Tucker, J.; Roswit, W.T.; Miller, C.A.; Heier, R.F.; Byers, D.E.; Brett, T.J.; et al. IL-13-induced Sairway mucus production is attenuated by MAPK13 inhibition. J. Clin. Investig. 2012, 122, 4555–4568. [Google Scholar] [CrossRef]
- Kim, H.K.; Kook, J.H.; Kang, K.R.; Oh, D.J.; Kim, T.H.; Lee, S.H. Increased expression of hCLCA1 in chronic rhinosinusitis and its contribution to produce MUC5AC. Laryngoscope 2016, 126, E347–E355. [Google Scholar] [CrossRef]
- Bonser, L.R.; Zlock, L.; Finkbeiner, W.; Erle, D.J. Epithelial tethering of MUC5AC-rich mucus impairs mucociliary transport in asthma. J. Clin. Investig. 2016, 126, 2367–2371. [Google Scholar] [CrossRef]
- Vieira Braga, F.A.; Kar, G.; Berg, M.; Carpaij, O.A.; Polanski, K.; Simon, L.M.; Brouwer, S.; Gomes, T.; Hesse, L.; Jiang, J.; et al. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat. Med. 2019, 25, 1153–1163. [Google Scholar] [CrossRef]
- Dickinson, J.D.; Alevy, Y.; Malvin, N.P.; Patel, K.K.; Gunsten, S.P.; Holtzman, M.J.; Stappenbeck, T.S.; Brody, S.L. IL13 activates autophagy to regulate secretion in airway epithelial cells. Autophagy 2016, 12, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Reid, A.T.; Nichol, K.S.; Chander Veerati, P.; Moheimani, F.; Kicic, A.; Stick, S.M.; Bartlett, N.W.; Grainge, C.L.; Wark, P.A.B.; Hansbro, P.M.; et al. Blocking Notch3 Signaling Abolishes MUC5AC Production in Airway Epithelial Cells from Individuals with Asthma. Am. J. Respir. Cell Mol. Biol. 2020, 62, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Candiano, G.; Bruschi, M.; Pedemonte, N.; Caci, E.; Liberatori, S.; Bini, L.; Pellegrini, C.; Vigano, M.; O’Connor, B.J.; Lee, T.H.; et al. Gelsolin secretion in interleukin-4-treated bronchial epithelia and in asthmatic airways. Am. J. Respir. Crit. Care Med. 2005, 172, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Hamid, Q.; Springall, D.R.; Riveros-Moreno, V.; Chanez, P.; Howarth, P.; Redington, A.; Bousquet, J.; Godard, P.; Holgate, S.; Polak, J.M. Induction of nitric oxide synthase in asthma. Lancet (Lond. Engl.) 1993, 342, 1510–1513. [Google Scholar] [CrossRef]
- Massaro, A.F.; Gaston, B.; Kita, D.; Fanta, C.; Stamler, J.S.; Drazen, J.M. Expired nitric oxide levels during treatment of acute asthma. Am. J. Respir. Crit. Care Med. 1995, 152, 800–803. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, R.; Lahti, A.; Kankaanranta, H.; Moilanen, E. Nitric oxide production and signaling in inflammation. Curr Drug Targets Inflamm Allergy 2005, 4, 471–479. [Google Scholar] [CrossRef]
- Silkoff, P.E.; Laviolette, M.; Singh, D.; FitzGerald, J.M.; Kelsen, S.; Backer, V.; Porsbjerg, C.M.; Girodet, P.O.; Berger, P.; Kline, J.N.; et al. Identification of airway mucosal type 2 inflammation by using clinical biomarkers in asthmatic patients. J. Allergy Clin. Immunol. 2017, 140, 710–719. [Google Scholar] [CrossRef]
- Asano, K.; Chee, C.B.; Gaston, B.; Lilly, C.M.; Gerard, C.; Drazen, J.M.; Stamler, J.S. Constitutive and inducible nitric oxide synthase gene expression, regulation, and activity in human lung epithelial cells. Proc. Natl. Acad. Sci. USA 1994, 91, 10089–10093. [Google Scholar] [CrossRef]
- Lane, C.; Knight, D.; Burgess, S.; Franklin, P.; Horak, F.; Legg, J.; Moeller, A.; Stick, S. Epithelial inducible nitric oxide synthase activity is the major determinant of nitric oxide concentration in exhaled breath. Thorax 2004, 59, 757–760. [Google Scholar] [CrossRef]
- Guo, F.H.; Comhair, S.A.; Zheng, S.; Dweik, R.A.; Eissa, N.T.; Thomassen, M.J.; Calhoun, W.; Erzurum, S.C. Molecular mechanisms of increased nitric oxide (NO) in asthma: Evidence for transcriptional and post-translational regulation of NO synthesis. J. Immunol. 2000, 164, 5970–5980. [Google Scholar] [CrossRef]
- Chibana, K.; Trudeau, J.B.; Mustovich, A.T.; Hu, H.; Zhao, J.; Balzar, S.; Chu, H.W.; Wenzel, S.E. IL-13 induced increases in nitrite levels are primarily driven by increases in inducible nitric oxide synthase as compared with effects on arginases in human primary bronchial epithelial cells. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2008, 38, 936–946. [Google Scholar] [CrossRef] [PubMed]
- Provost, V.; Larose, M.C.; Langlois, A.; Rola-Pleszczynski, M.; Flamand, N.; Laviolette, M. CCL26/eotaxin-3 is more effective to induce the migration of eosinophils of asthmatics than CCL11/eotaxin-1 and CCL24/eotaxin-2. J. Leukoc. Biol. 2013, 94, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Larose, M.C.; Chakir, J.; Archambault, A.S.; Joubert, P.; Provost, V.; Laviolette, M.; Flamand, N. Correlation between CCL26 production by human bronchial epithelial cells and airway eosinophils: Involvement in patients with severe eosinophilic asthma. J. Allergy Clin. Immunol. 2015, 136, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Al Mubarak, R.; Roberts, N.; Correll, K.; Janssen, W.; Finigan, J.; Mishra, R.; Chu, H.W. IL-13 induces periostin and eotaxin expression in human primary alveolar epithelial cells: Comparison with paired airway epithelial cells. PLoS ONE 2018, 13, e0196256. [Google Scholar] [CrossRef]
- Gupta, R.; Radicioni, G.; Abdelwahab, S.; Dang, H.; Carpenter, J.; Chua, M.; Mieczkowski, P.A.; Sheridan, J.T.; Randell, S.H.; Kesimer, M. Intercellular Communication between Airway Epithelial Cells Is Mediated by Exosome-Like Vesicles. Am. J. Respir. Cell Mol. Biol. 2019, 60, 209–220. [Google Scholar] [CrossRef]
- Kesimer, M.; Scull, M.; Brighton, B.; DeMaria, G.; Burns, K.; O’Neal, W.; Pickles, R.J.; Sheehan, J.K. Characterization of exosome-like vesicles released from human tracheobronchial ciliated epithelium: A possible role in innate defense. FASEB J.: Off. Publ. Fed. Am. Soc. Exp. Biol. 2009, 23, 1858–1868. [Google Scholar] [CrossRef]
- Kesimer, M.; Gupta, R. Physical characterization and profiling of airway epithelial derived exosomes using light scattering. Methods (San Diego, Calif.) 2015, 87, 59–63. [Google Scholar] [CrossRef]
- Kulshreshtha, A.; Ahmad, T.; Agrawal, A.; Ghosh, B. Proinflammatory role of epithelial cell-derived exosomes in allergic airway inflammation. J. Allergy Clin. Immunol. 2013, 131, 1194–1203. [Google Scholar] [CrossRef]
- Bartel, S.; La Grutta, S.; Cilluffo, G.; Perconti, G.; Bongiovanni, A.; Giallongo, A.; Behrends, J.; Kruppa, J.; Hermann, S.; Chiang, D.; et al. Human airway epithelial extracellular vesicle miRNA signature is altered upon asthma development. Allergy 2019, 75, 346–356. [Google Scholar] [CrossRef]
- Mitchel, J.A.; Antoniak, S.; Lee, J.H.; Kim, S.H.; McGill, M.; Kasahara, D.I.; Randell, S.H.; Israel, E.; Shore, S.A.; Mackman, N.; et al. IL-13 Augments Compressive Stress-Induced Tissue Factor Expression in Human Airway Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2016, 54, 524–531. [Google Scholar] [CrossRef]
- Lacal, I.; Ventura, R. Epigenetic Inheritance: Concepts, Mechanisms and Perspectives. Front. Mol. Neurosci. 2018, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Stefanowicz, D.; Lee, J.Y.; Lee, K.; Shaheen, F.; Koo, H.K.; Booth, S.; Knight, D.A.; Hackett, T.L. Elevated H3K18 acetylation in airway epithelial cells of asthmatic subjects. Respir. Res. 2015, 16, 95. [Google Scholar] [CrossRef] [PubMed]
- Stefanowicz, D.; Ullah, J.; Lee, K.; Shaheen, F.; Olumese, E.; Fishbane, N.; Koo, H.K.; Hallstrand, T.S.; Knight, D.A.; Hackett, T.L. Epigenetic modifying enzyme expression in asthmatic airway epithelial cells and fibroblasts. BMC Pulm. Med. 2017, 17, 24. [Google Scholar] [CrossRef] [PubMed]
- Alashkar Alhamwe, B.; Miethe, S.; Pogge von Strandmann, E.; Potaczek, D.P.; Garn, H. Epigenetic Regulation of Airway Epithelium Immune Functions in Asthma. Front. Immunol. 2020, 11, 1747. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Forno, E.; Yan, Q.; Jiang, Y.; Zhang, R.; Boutaoui, N.; Acosta-Perez, E.; Canino, G.; Chen, W.; Celedon, J.C. SNPs identified by GWAS affect asthma risk through DNA methylation and expression of cis-genes in airway epithelium. Eur. Respir. J. 2020, 55, 1902079. [Google Scholar] [CrossRef]
- Kim, Y.J.; Park, S.W.; Kim, T.H.; Park, J.S.; Cheong, H.S.; Shin, H.D.; Park, C.S. Genome-wide methylation profiling of the bronchial mucosa of asthmatics: Relationship to atopy. BMC Med. Genet. 2013, 14, 39. [Google Scholar] [CrossRef]
- Pech, M.; Weckmann, M.; Konig, I.R.; Franke, A.; Heinsen, F.A.; Oliver, B.; Ricklefs, I.; Fuchs, O.; Rabe, K.; Hansen, G.; et al. Rhinovirus infections change DNA methylation and mRNA expression in children with asthma. PLoS ONE 2018, 13, e0205275. [Google Scholar] [CrossRef]
- Lund, R.J.; Osmala, M.; Malonzo, M.; Lukkarinen, M.; Leino, A.; Salmi, J.; Vuorikoski, S.; Turunen, R.; Vuorinen, T.; Akdis, C.; et al. Atopic asthma after rhinovirus-induced wheezing is associated with DNA methylation change in the SMAD3 gene promoter. Allergy 2018, 73, 1735–1740. [Google Scholar] [CrossRef]
- McErlean, P.; Favoreto, S., Jr.; Costa, F.F.; Shen, J.; Quraishi, J.; Biyasheva, A.; Cooper, J.J.; Scholtens, D.M.; Vanin, E.F.; de Bonaldo, M.F.; et al. Human rhinovirus infection causes different DNA methylation changes in nasal epithelial cells from healthy and asthmatic subjects. BMC Med. Genom. 2014, 7, 37. [Google Scholar] [CrossRef]
- Weidner, J.; Bartel, S.; Kilic, A.; Zissler, U.M.; Renz, H.; Schwarze, J.; Schmidt-Weber, C.B.; Maes, T.; Rebane, A.; Krauss-Etschmann, S.; et al. Spotlight on microRNAs in allergy and asthma. Allergy 2020. [Google Scholar] [CrossRef]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Baltimore, D. MicroRNAs as regulatory elements in immune system logic. Nat. Rev. Immunol. 2016, 16, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Johansson, K.; Weidner, J.; Radinger, M. MicroRNAs in type 2 immunity. Cancer Lett 2018, 425, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Jardim, M.J.; Dailey, L.; Silbajoris, R.; Diaz-Sanchez, D. Distinct microRNA expression in human airway cells of asthmatic donors identifies a novel asthma-associated gene. Am. J. Respir. Cell Mol. Biol. 2012, 47, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Liao, G.; Panettieri, R.A.; Tang, D.D. MicroRNA-203 negatively regulates c-Abl, ERK1/2 phosphorylation, and proliferation in smooth muscle cells. Physiol. Rep. 2015, 3, e12541. [Google Scholar] [CrossRef]
- Huo, X.; Zhang, K.; Yi, L.; Mo, Y.; Liang, Y.; Zhao, J.; Zhang, Z.; Xu, Y.; Zhen, G. Decreased epithelial and plasma miR-181b-5p expression associates with airway eosinophilic inflammation in asthma. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2016, 46, 1281–1290. [Google Scholar] [CrossRef]
- Solberg, O.D.; Ostrin, E.J.; Love, M.I.; Peng, J.C.; Bhakta, N.R.; Hou, L.; Nguyen, C.; Solon, M.; Nguyen, C.; Barczak, A.J.; et al. Airway epithelial miRNA expression is altered in asthma. Am. J. Respir. Crit. Care Med. 2012, 186, 965–974. [Google Scholar] [CrossRef]
- Kivihall, A.; Aab, A.; Soja, J.; Sladek, K.; Sanak, M.; Altraja, A.; Jakiela, B.; Bochenek, G.; Rebane, A. Reduced expression of miR-146a in human bronchial epithelial cells alters neutrophil migration. Clin. Transl. Allergy 2019, 9, 62. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, Y.; Rong, W.; Fan, L.; Cai, Y.; Qu, Q.; Gao, Y.; Zhao, H. miR-221 participates in the airway epithelial cells injury in asthma via targeting SIRT1. Exp. Lung Res. 2018, 44, 272–279. [Google Scholar] [CrossRef]
- Haj-Salem, I.; Fakhfakh, R.; Bérubé, J.C.; Jacques, E.; Plante, S.; Simard, M.J.; Bossé, Y.; Chakir, J. MicroRNA-19a enhances proliferation of bronchial epithelial cells by targetingTGFβR2gene in severe asthma. Allergy 2015, 70, 212–219. [Google Scholar] [CrossRef]
- Huang, H.; Lu, H.; Liang, L.; Zhi, Y.; Huo, B.; Wu, L.; Xu, L.; Shen, Z. MicroRNA-744 Inhibits Proliferation of Bronchial Epithelial Cells by Regulating Smad3 Pathway via Targeting Transforming Growth Factor-beta1 (TGF-beta1) in Severe Asthma. Med. Sci. Monit. 2019, 25, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Malmhall, C.; Alawieh, S.; Lu, Y.; Sjostrand, M.; Bossios, A.; Eldh, M.; Radinger, M. MicroRNA-155 is essential for T(H)2-mediated allergen-induced eosinophilic inflammation in the lung. J. Allergy Clin. Immunol. 2014, 133, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Johansson, K.; Malmhall, C.; Ramos-Ramirez, P.; Radinger, M. MicroRNA-155 is a critical regulator of type 2 innate lymphoid cells and IL-33 signaling in experimental models of allergic airway inflammation. J. Allergy Clin. Immunol. 2017, 139, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Bondanese, V.P.; Francisco-Garcia, A.; Bedke, N.; Davies, D.E.; Sanchez-Elsner, T. Identification of host miRNAs that may limit human rhinovirus replication. World J. Biol. Chem. 2014, 5, 437–456. [Google Scholar] [CrossRef]
- Martinez-Nunez, R.T.; Bondanese, V.P.; Louafi, F.; Francisco-Garcia, A.S.; Rupani, H.; Bedke, N.; Holgate, S.; Howarth, P.H.; Davies, D.E.; Sanchez-Elsner, T. A microRNA network dysregulated in asthma controls IL-6 production in bronchial epithelial cells. PLoS ONE 2014, 9, e111659. [Google Scholar] [CrossRef]
- McCaskill, J.L.; Ressel, S.; Alber, A.; Redford, J.; Power, U.F.; Schwarze, J.; Dutia, B.M.; Buck, A.H. Broad-Spectrum Inhibition of Respiratory Virus Infection by MicroRNA Mimics Targeting p38 MAPK Signaling. Mol. Nucleic Acids 2017, 7, 256–266. [Google Scholar] [CrossRef]
- Fang, J.; Hao, Q.; Liu, L.; Li, Y.; Wu, J.; Huo, X.; Zhu, Y. Epigenetic changes mediated by microRNA miR29 activate cyclooxygenase 2 and lambda-1 interferon production during viral infection. J. Virol. 2012, 86, 1010–1020. [Google Scholar] [CrossRef]
- Guan, Z.; Shi, N.; Song, Y.; Zhang, X.; Zhang, M.; Duan, M. Induction of the cellular microRNA-29c by influenza virus contributes to virus-mediated apoptosis through repression of antiapoptotic factors BCL2L2. Biochem. Biophys. Res. Commun. 2012, 425, 662–667. [Google Scholar] [CrossRef]
- Zhang, X.; Dong, C.; Sun, X.; Li, Z.; Zhang, M.; Guan, Z.; Duan, M. Induction of the cellular miR-29c by influenza virus inhibits the innate immune response through protection of A20 mRNA. Biochem. Biophys. Res. Commun. 2014, 450, 755–761. [Google Scholar] [CrossRef]
- Zhao, L.; Zhu, J.; Zhou, H.; Zhao, Z.; Zou, Z.; Liu, X.; Lin, X.; Zhang, X.; Deng, X.; Wang, R.; et al. Identification of cellular microRNA-136 as a dual regulator of RIG-I-mediated innate immunity that antagonizes H5N1 IAV replication in A549 cells. Sci. Rep. 2015, 5, 14991. [Google Scholar] [CrossRef]
- Buggele, W.A.; Krause, K.E.; Horvath, C.M. Small RNA profiling of influenza A virus-infected cells identifies miR-449b as a regulator of histone deacetylase 1 and interferon beta. PLoS ONE 2013, 8, e76560. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.J.; Yang, J.; Fan, X.L.; Zhao, H.B.; Hu, W.; Li, Z.P.; Yu, G.C.; Ding, X.R.; Wang, J.Z.; Bo, X.C.; et al. Cellular microRNA let-7c inhibits M1 protein expression of the H1N1 influenza A virus in infected human lung epithelial cells. J. Cell Mol. Med. 2012, 16, 2539–2546. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Sun, X.; Zhu, Y.; Qin, W. Downregulation of miR-146a inhibits influenza A virus replication by enhancing the type I interferon response in vitro and in vivo. Biomed. Pharm. 2019, 111, 740–750. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).