New Aspects of the Kidney in the Regulation of Fibroblast Growth Factor 23 (FGF23) and Mineral Homeostasis

Abstract
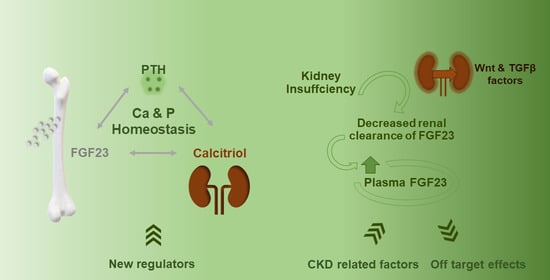
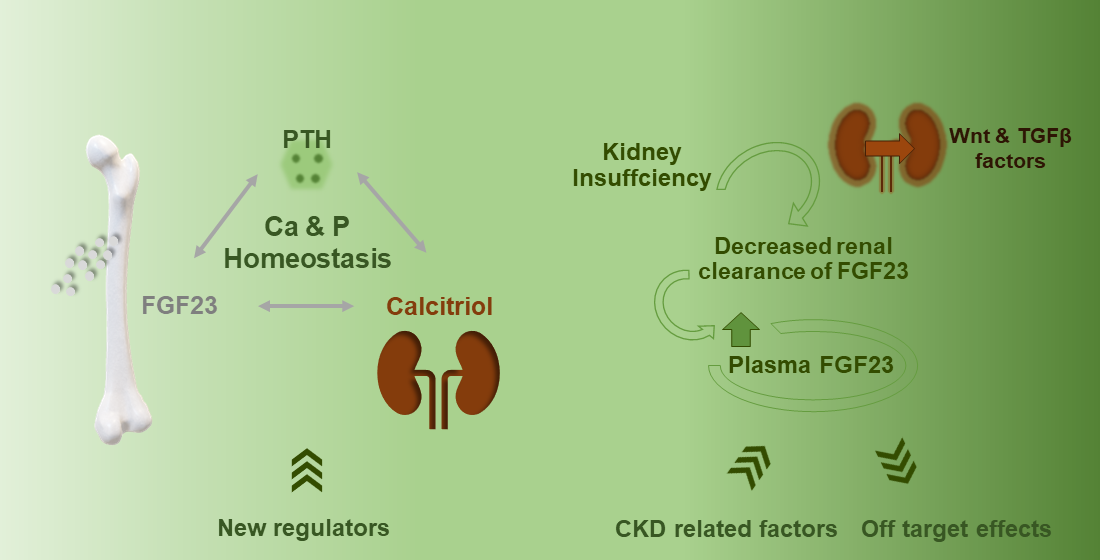{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. FGF23 in Normal Physiology
2.1. Secretion and Metabolism of FGF23
2.2. Circadian Rhythm of Plasma FGF23
2.3. FGF23, Klotho and Kidney
2.4. FGF23 Regulates PTH Production
3. FGF23 and the Disturbed Mineral Balance in Kidney Disease
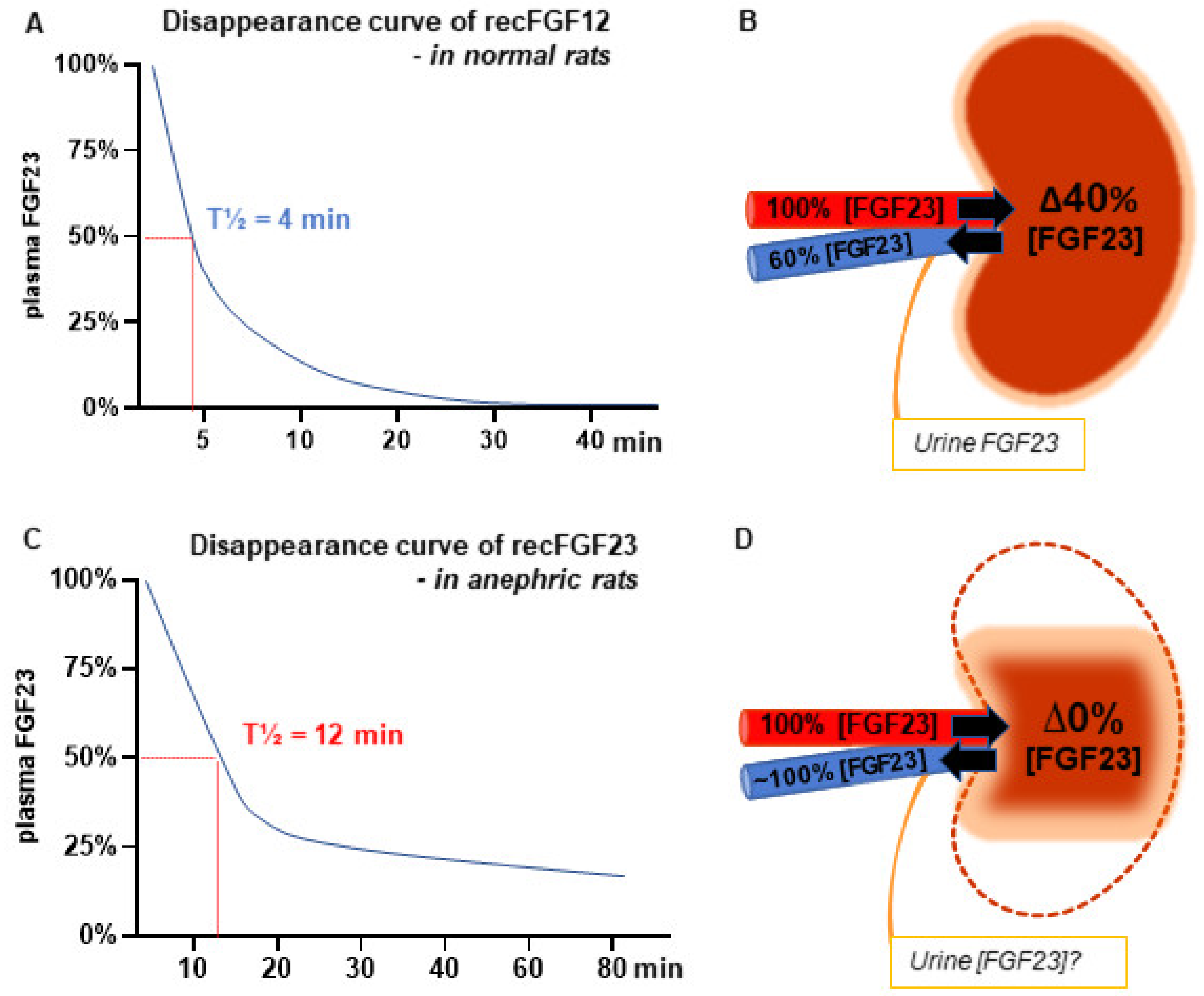
3.1. Role of the Kidney in Secretion and Metabolism of FGF23
3.2. FGF23 Regulation in Kidney Disease
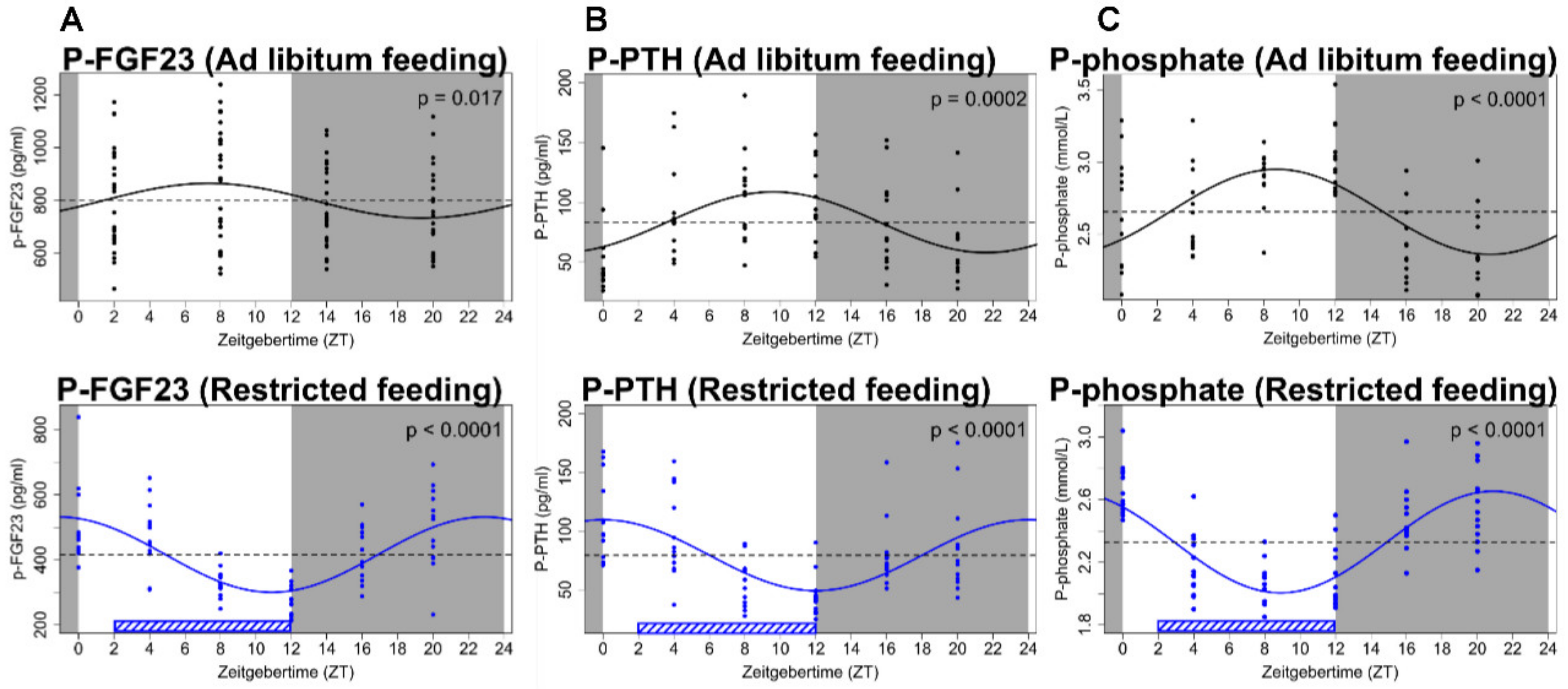
3.3. Kidney Insufficiency Causes Disturbed Circadian Rhythm of Plasma FGF23, Phosphate and Hormones Involved in the Mineral Homeostasis
3.4. FGF23/Klotho System in Kidney Failure
3.5. The Complex Interplay between FGF23 and PTH in Uremia
4. New Factors Involved in CKD-MBD
5. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yamashita, T.; Yoshioka, M.; Itoh, N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem. Biophys. Res. Commun. 2000, 277, 494–498. [Google Scholar] [CrossRef]
- Larsson, T.; Nisbeth, U.; Ljunggren, O.; Juppner, H.; Jonsson, K.B. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int. 2003, 64, 2272–2279. [Google Scholar] [CrossRef]
- Martin, A.; David, V.; Quarles, L.D. Regulation and function of the FGF23/klotho endocrine pathways. Physiol. Rev. 2012, 92, 131–155. [Google Scholar] [CrossRef]
- Fukumoto, S. Post-translational modification of Fibroblast Growth Factor 23. Ther. Apher. Dial. 2005, 9, 319–322. [Google Scholar] [CrossRef]
- Goetz, R.; Nakada, Y.; Hu, M.C.; Kurosu, H.; Wang, L.; Nakatani, T.; Shi, M.; Eliseenkova, A.V.; Razzaque, M.S.; Moe, O.W.; et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc. Natl. Acad. Sci. USA 2010, 107, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, H.; Ogawa, Y.; Miyoshi, M.; Yamamoto, M.; Nandi, A.; Rosenblatt, K.P.; Baum, M.G.; Schiavi, S.; Hu, M.C.; Moe, O.W.; et al. Regulation of fibroblast growth factor-23 signaling by klotho. J. Biol. Chem. 2006, 281, 6120–6123. [Google Scholar] [CrossRef] [PubMed]
- Urakawa, I.; Yamazaki, Y.; Shimada, T.; Iijima, K.; Hasegawa, H.; Okawa, K.; Fujita, T.; Fukumoto, S.; Yamashita, T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 2006, 444, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Ben-Dov, I.Z.; Galitzer, H.; Lavi-Moshayoff, V.; Goetz, R.; Kuro-O, M.; Mohammadi, M.; Sirkis, R.; Naveh-Many, T.; Silver, J. The parathyroid is a target organ for FGF23 in rats. J. Clin. Investig. 2007, 117, 4003–4008. [Google Scholar] [CrossRef] [PubMed]
- Perwad, F.; Azam, N.; Zhang, M.Y.; Yamashita, T.; Tenenhouse, H.S.; Portale, A.A. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology 2005, 146, 5358–5364. [Google Scholar] [CrossRef]
- Scanni, R.; von Rotz, M.; Jehle, S.; Hulter, H.N.; Krapf, R. The human response to acute enteral and parenteral phosphate loads. J. Am. Soc. Nephrol. 2014, 25, 2730–2739. [Google Scholar] [CrossRef] [PubMed]
- Mace, M.L.; Gravesen, E.; Hofman-Bang, J.; Olgaard, K.; Lewin, E. Key role of the kidney in the regulation of fibroblast growth factor 23. Kidney Int. 2015, 88, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Bon, N.; Frangi, G.; Sourice, S.; Guicheux, J.; Beck-Cormier, S.; Beck, L. Phosphate-dependent FGF23 secretion is modulated by PiT2/Slc20a2. Mol. Metab. 2018, 11, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Takashi, Y.; Kosako, H.; Sawatsubashi, S.; Kinoshita, Y.; Ito, N.; Tsoumpra, M.K.; Nangaku, M.; Abe, M.; Matsuhisa, M.; Kato, S.; et al. Activation of unliganded FGF receptor by extracellular phosphate potentiates proteolytic protection of FGF23 by its O-glycosylation. Proc. Natl. Acad. Sci. USA 2019, 116, 11418–11427. [Google Scholar] [CrossRef] [PubMed]
- White, K.E.; Cabral, J.M.; Davis, S.I.; Fishburn, T.; Evans, W.E.; Ichikawa, S.; Fields, J.; Yu, X.; Shaw, N.J.; McLellan, N.J.; et al. Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. Am. J. Hum. Genet. 2005, 76, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.L.; Feng, B.; Chen, M.Z.; Kolumam, G.; Zavala-Solorio, J.; Wyatt, S.K.; Gandham, V.D.; Carano, R.A.; Sonoda, J. Antibody-mediated activation of FGFR1 induces FGF23 production and hypophosphatemia. PLoS ONE 2013, 8, e57322. [Google Scholar] [CrossRef]
- Xiao, Z.; Huang, J.; Cao, L.; Liang, Y.; Han, X.; Quarles, L.D. Osteocyte-specific deletion of Fgfr1 suppresses FGF23. PLoS ONE 2014, 9, e104154. [Google Scholar] [CrossRef]
- Wohrle, S.; Bonny, O.; Beluch, N.; Gaulis, S.; Stamm, C.; Scheibler, M.; Muller, M.; Kinzel, B.; Thuery, A.; Brueggen, J.; et al. FGF receptors control vitamin D and phosphate homeostasis by mediating renal FGF-23 signaling and regulating FGF-23 expression in bone. J. Bone Miner. Res. 2011, 26, 2486–2497. [Google Scholar] [CrossRef]
- Mace, M.L.; Gravesen, E.; Nordholm, A.; Hofman-Bang, J.; Secher, T.; Olgaard, K.; Lewin, E. Kidney fibroblast growth factor 23 does not contribute to elevation of its circulating levels in uremia. Kidney Int. 2017, 92, 165–178. [Google Scholar] [CrossRef]
- Mace, M.L.; Gravesen, E.; Nordholm, A.; Olgaard, K.; Lewin, E. Fibroblast Growth Factor (FGF) 23 Regulates the Plasma Levels of Parathyroid Hormone In Vivo Through the FGF Receptor in Normocalcemia, But Not in Hypocalcemia. Calcif. Tissue Int. 2018, 102, 85–92. [Google Scholar] [CrossRef]
- Hori, M.; Kinoshita, Y.; Taguchi, M.; Fukumoto, S. Phosphate enhances Fgf23 expression through reactive oxygen species in UMR-106 cells. J. Bone Miner. Metab. 2016, 34, 132–139. [Google Scholar] [CrossRef]
- Meir, T.; Durlacher, K.; Pan, Z.; Amir, G.; Richards, W.G.; Silver, J.; Naveh-Many, T. Parathyroid hormone activates the orphan nuclear receptor Nurr1 to induce FGF23 transcription. Kidney Int. 2014, 86, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Rhee, Y.; Bivi, N.; Farrow, E.; Lezcano, V.; Plotkin, L.I.; White, K.E.; Bellido, T. Parathyroid hormone receptor signaling in osteocytes increases the expression of fibroblast growth factor-23 in vitro and in vivo. Bone 2011, 49, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Barthel, T.K.; Mathern, D.R.; Whitfield, G.K.; Haussler, C.A.; Hopper, H.A.; Hsieh, J.C.; Slater, S.A.; Hsieh, G.; Kaczmarska, M.; Jurutka, P.W.; et al. 1,25-Dihydroxyvitamin D3/VDR-mediated induction of FGF23 as well as transcriptional control of other bone anabolic and catabolic genes that orchestrate the regulation of phosphate and calcium mineral metabolism. J. Steroid Biochem. Mol. Biol. 2007, 103, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Yamamoto, L.; Karaplis, A.C.; St-Arnaud, R.; Goltzman, D. Fibroblast Growth Factor 23 Regulation by Systemic and Local Osteoblast-Synthesized 1,25-Dihydroxyvitamin D. J. Am. Soc. Nephrol. 2017, 28, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Edmonston, D.; Wolf, M. FGF23 at the crossroads of phosphate, iron economy and erythropoiesis. Nat. Rev. Nephrol. 2020, 16, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Leaf, D.E.; Jacob, K.A.; Srivastava, A.; Chen, M.E.; Christov, M.; Juppner, H.; Sabbisetti, V.S.; Martin, A.; Wolf, M.; Waikar, S.S. Fibroblast Growth Factor 23 Levels Associate with AKI and Death in Critical Illness. J. Am. Soc. Nephrol. 2017, 28, 1877–1885. [Google Scholar] [CrossRef]
- Wesseling-Perry, K.; Pereira, R.C.; Tsai, E.; Ettenger, R.; Juppner, H.; Salusky, I.B. FGF23 and mineral metabolism in the early post-renal transplantation period. Pediatr. Nephrol. 2013, 28, 2207–2215. [Google Scholar] [CrossRef]
- Zajac, M.; Rybi-Szuminska, A.; Wasilewska, A. Urine fibroblast growth factor 23 levels in hypertensive children and adolescents. Croat. Med. J. 2015, 56, 344–350. [Google Scholar] [CrossRef]
- Van Ballegooijen, A.J.; Rhee, E.P.; Elmariah, S.; de Boer, I.H.; Kestenbaum, B. Renal Clearance of Mineral Metabolism Biomarkers. J. Am. Soc. Nephrol. 2016, 27, 392–397. [Google Scholar] [CrossRef]
- Nordholm, A.; Egstrand, S.; Gravesen, E.; Mace, M.L.; Morevati, M.; Olgaard, K.; Lewin, E. Circadian rhythm of activin A and related parameters of mineral metabolism in normal and uremic rats. Pflugers Arch. 2019, 471, 1079–1094. [Google Scholar] [CrossRef] [PubMed]
- Egstrand, S.; Nordholm, A.; Morevati, M.; Mace, M.L.; Hassan, A.; Naveh-Many, T.; Rukov, J.L.; Gravesen, E.; Olgaard, K.; Lewin, E. A molecular circadian clock operates in the parathyroid gland and is disturbed in chronic kidney disease associated bone and mineral disorder. Kidney Int. 2020. [Google Scholar] [CrossRef] [PubMed]
- Reppert, S.M.; Weaver, D.R. Coordination of circadian timing in mammals. Nature 2002, 418, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Egstrand, S.; Olgaard, K.; Lewin, E. Circadian rhythms of mineral metabolism in chronic kidney disease-mineral bone disorder. Curr. Opin. Nephrol. Hypertens. 2020, 29, 367–377. [Google Scholar] [CrossRef]
- Zuber, A.M.; Centeno, G.; Pradervand, S.; Nikolaeva, S.; Maquelin, L.; Cardinaux, L.; Bonny, O.; Firsov, D. Molecular clock is involved in predictive circadian adjustment of renal function. Proc. Natl. Acad. Sci. USA 2009, 106, 16523–16528. [Google Scholar] [CrossRef]
- Ansermet, C.; Centeno, G.; Nikolaeva, S.; Maillard, M.P.; Pradervand, S.; Firsov, D. The intrinsic circadian clock in podocytes controls glomerular filtration rate. Sci. Rep. 2019, 9, 16089. [Google Scholar] [CrossRef]
- Maré, A.; D’Haese, P.C.; Verhulst, A. The role of sclerostin in bone and ectopic calcifications. Int. J. Mol. Sci. 2020, 21, 3199. [Google Scholar] [CrossRef]
- Swanson, C.M.; Kohrt, W.M.; Buxton, O.M.; Everson, C.A.; Wright, K.P., Jr.; Orwoll, E.S.; Shea, S.A. The importance of the circadian system & sleep for bone health. Metabolism 2018, 84, 28–43. [Google Scholar]
- Zaidi, M.; New, M.I.; Blair, H.C.; Zallone, A.; Baliram, R.; Davies, T.F.; Cardozo, C.; Iqbal, J.; Sun, L.; Rosen, C.J.; et al. Actions of pituitary hormones beyond traditional targets. J. Endocrinol. 2018, 237, R83–R98. [Google Scholar] [CrossRef]
- Sze, L.; Bernays, R.L.; Zwimpfer, C.; Wiesli, P.; Brandle, M.; Schmid, C. Excessively high soluble Klotho in patients with acromegaly. J. Intern. Med. 2012, 272, 93–97. [Google Scholar] [CrossRef]
- Hu, M.C.; Shi, M.; Zhang, J.; Pastor, J.; Nakatani, T.; Lanske, B.; Razzaque, M.S.; Rosenblatt, K.P.; Baum, M.G.; Kuro-o, M.; et al. Klotho: A novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 2010, 24, 3438–3450. [Google Scholar] [CrossRef] [PubMed]
- Andrukhova, O.; Zeitz, U.; Goetz, R.; Mohammadi, M.; Lanske, B.; Erben, R.G. FGF23 acts directly on renal proximal tubules to induce phosphaturia through activation of the ERK1/2-SGK1 signaling pathway. Bone 2012, 51, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Gattineni, J.; Alphonse, P.; Zhang, Q.; Mathews, N.; Bates, C.M.; Baum, M. Regulation of renal phosphate transport by FGF23 is mediated by FGFR1 and FGFR4. Am. J. Physiol. Ren. Physiol. 2014, 306, F351–F358. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yang, J.; Li, L.; Huang, J.; King, G.; Quarles, L.D. Conditional Deletion of Fgfr1 in the Proximal and Distal Tubule Identifies Distinct Roles in Phosphate and Calcium Transport. PLoS ONE 2016, 11, e0147845. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Liu, Y.; Goetz, R.; Fu, L.; Jayaraman, S.; Hu, M.C.; Moe, O.W.; Liang, G.; Li, X.; Mohammadi, M. Alpha-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signalling. Nature 2018, 553, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Imura, A.; Iwano, A.; Tohyama, O.; Tsuji, Y.; Nozaki, K.; Hashimoto, N.; Fujimori, T.; Nabeshima, Y. Secreted Klotho protein in sera and CSF: Implication for post-translational cleavage in release of Klotho protein from cell membrane. FEBS Lett. 2004, 565, 143–147. [Google Scholar] [CrossRef]
- Lewin, E.; Olgaard, K. The vascular secret of Klotho. Kidney Int. 2015, 87, 1089–1091. [Google Scholar] [CrossRef]
- Andrukhova, O.; Smorodchenko, A.; Egerbacher, M.; Streicher, C.; Zeitz, U.; Goetz, R.; Shalhoub, V.; Mohammadi, M.; Pohl, E.E.; Lanske, B.; et al. FGF23 promotes renal calcium reabsorption through the TRPV5 channel. EMBO J. 2014, 33, 229–246. [Google Scholar] [CrossRef]
- Shimada, T.; Kakitani, M.; Yamazaki, Y.; Hasegawa, H.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Tomizuka, K.; Yamashita, T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Investig. 2004, 113, 561–568. [Google Scholar] [CrossRef]
- Perwad, F.; Zhang, M.Y.; Tenenhouse, H.S.; Portale, A.A. Fibroblast growth factor 23 impairs phosphorus and vitamin D metabolism in vivo and suppresses 25-hydroxyvitamin D-1alpha-hydroxylase expression in vitro. Am. J. Physiol. Ren. Physiol. 2007, 293, F1577–F1583. [Google Scholar] [CrossRef]
- Liu, S.; Tang, W.; Zhou, J.; Stubbs, J.R.; Luo, Q.; Pi, M.; Quarles, L.D. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J. Am. Soc. Nephrol. 2006, 17, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Canalejo, R.; Canalejo, A.; Martinez-Moreno, J.M.; Rodriguez-Ortiz, M.E.; Estepa, J.C.; Mendoza, F.J.; Munoz-Castaneda, J.R.; Shalhoub, V.; Almaden, Y.; Rodriguez, M. FGF23 fails to inhibit uremic parathyroid glands. J. Am. Soc. Nephrol. 2010, 21, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K.; Takeshita, A.; Furushima, K.; Miyajima, M.; Hatamura, I.; Kuro, O.; Furuta, Y.; Sakaguchi, K. Persistent fibroblast growth factor 23 signalling in the parathyroid glands for secondary hyperparathyroidism in mice with chronic kidney disease. Sci. Rep. 2017, 7, 40534. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J.; Zhong, M.; Ritter, C.; Brown, E.M.; Slatopolsky, E. Loss of calcium responsiveness in cultured bovine parathyroid cells is associated with decreased calcium receptor expression. Biochem. Biophys. Res. Commun. 1995, 212, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Imura, A.; Tsuji, Y.; Murata, M.; Maeda, R.; Kubota, K.; Iwano, A.; Obuse, C.; Togashi, K.; Tominaga, M.; Kita, N.; et al. Alpha-Klotho as a regulator of calcium homeostasis. Science 2007, 316, 1615–1618. [Google Scholar] [CrossRef]
- Martuseviciene, G.; Hofman-Bang, J.; Clausen, T.; Olgaard, K.; Lewin, E. The secretory response of parathyroid hormone to acute hypocalcemia in vivo is independent of parathyroid glandular sodium/potassium-ATPase activity. Kidney Int. 2011, 79, 742–748. [Google Scholar] [CrossRef]
- Olauson, H.; Lindberg, K.; Amin, R.; Sato, T.; Jia, T.; Goetz, R.; Mohammadi, M.; Andersson, G.; Lanske, B.; Larsson, T.E. Parathyroid-specific deletion of Klotho unravels a novel calcineurin-dependent FGF23 signaling pathway that regulates PTH secretion. PLoS Genet. 2013, 9, e1003975. [Google Scholar] [CrossRef]
- Moe, S.; Drueke, T.; Cunningham, J.; Goodman, W.; Martin, K.; Olgaard, K.; Ott, S.; Sprague, S.; Lameire, N.; Eknoyan, G. Definition, evaluation, and classification of renal osteodystrophy: A position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2006, 69, 1945–1953. [Google Scholar] [CrossRef]
- Isakova, T.; Wahl, P.; Vargas, G.S.; Gutierrez, O.M.; Scialla, J.; Xie, H.; Appleby, D.; Nessel, L.; Bellovich, K.; Chen, J.; et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011, 79, 1370–1378. [Google Scholar] [CrossRef]
- Naveh-Many, T.; Volovelsky, O. Parathyroid Cell Proliferation in Secondary Hyperparathyroidism of Chronic Kidney Disease. Int. J. Mol. Sci. 2020, 21, 4332. [Google Scholar] [CrossRef]
- Kendrick, J.; Cheung, A.K.; Kaufman, J.S.; Greene, T.; Roberts, W.L.; Smits, G.; Chonchol, M. FGF-23 associates with death, cardiovascular events, and initiation of chronic dialysis. J. Am. Soc. Nephrol. 2011, 22, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, O.M.; Mannstadt, M.; Isakova, T.; Rauh-Hain, J.A.; Tamez, H.; Shah, A.; Smith, K.; Lee, H.; Thadhani, R.; Juppner, H.; et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N. Engl. J. Med. 2008, 359, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Liu, X.; Song, M.; Zhou, Q.; Yu, J.; Zhou, B.; Wu, Y.; He, Y.; Huang, L. Fibroblast growth factor 23 as a predictor of cardiovascular and all-cause mortality in prospective studies. Atherosclerosis 2017, 261, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.; Molnar, M.Z.; Amaral, A.P.; Czira, M.E.; Rudas, A.; Ujszaszi, A.; Kiss, I.; Rosivall, L.; Kosa, J.; Lakatos, P.; et al. Elevated fibroblast growth factor 23 is a risk factor for kidney transplant loss and mortality. J. Am. Soc. Nephrol. 2011, 22, 956–966. [Google Scholar] [CrossRef]
- Isakova, T.; Xie, H.; Yang, W.; Xie, D.; Anderson, A.H.; Scialla, J.; Wahl, P.; Gutierrez, O.M.; Steigerwalt, S.; He, J.; et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA 2011, 305, 2432–2439. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; Cai, X.; Lee, J.; Xie, D.; Wang, X.; Mehta, R.; Allen, N.B.; Scialla, J.J.; Pencina, M.J.; Anderson, A.H.; et al. Longitudinal FGF23 Trajectories and Mortality in Patients with CKD. J. Am. Soc. Nephrol. 2018, 29, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M. Forging forward with 10 burning questions on FGF23 in kidney disease. J. Am. Soc. Nephrol. 2010, 21, 1427–1435. [Google Scholar] [CrossRef]
- Musgrove, J.; Wolf, M. Regulation and Effects of FGF23 in Chronic Kidney Disease. Annu. Rev. Physiol. 2020, 82, 365–390. [Google Scholar] [CrossRef]
- Gutierrez, O.; Isakova, T.; Rhee, E.; Shah, A.; Holmes, J.; Collerone, G.; Juppner, H.; Wolf, M. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J. Am. Soc. Nephrol. 2005, 16, 2205–2215. [Google Scholar] [CrossRef]
- Marsell, R.; Grundberg, E.; Krajisnik, T.; Mallmin, H.; Karlsson, M.; Mellstrom, D.; Orwoll, E.; Ohlsson, C.; Jonsson, K.B.; Ljunggren, O.; et al. Fibroblast growth factor-23 is associated with parathyroid hormone and renal function in a population-based cohort of elderly men. Eur. J. Endocrinol. 2008, 158, 125–129. [Google Scholar] [CrossRef]
- Wan, M.; Smith, C.; Shah, V.; Gullet, A.; Wells, D.; Rees, L.; Shroff, R. Fibroblast growth factor 23 and soluble klotho in children with chronic kidney disease. Nephrol. Dial. Transpl. 2013, 28, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Hsu, R.; Hsu, C.Y.; Kordesch, K.; Nicasio, E.; Cortez, A.; McAlpine, I.; Brady, S.; Zhuo, H.; Kangelaris, K.N.; et al. FGF-23 and PTH levels in patients with acute kidney injury: A cross-sectional case series study. Ann. Intensive Care 2011, 1, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Leaf, D.E.; Wolf, M.; Waikar, S.S.; Chase, H.; Christov, M.; Cremers, S.; Stern, L. FGF-23 levels in patients with AKI and risk of adverse outcomes. Clin. J. Am. Soc. Nephrol. 2012, 7, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Christov, M.; Waikar, S.S.; Pereira, R.C.; Havasi, A.; Leaf, D.E.; Goltzman, D.; Pajevic, P.D.; Wolf, M.; Juppner, H. Plasma FGF23 levels increase rapidly after acute kidney injury. Kidney Int. 2013, 84, 776–785. [Google Scholar] [CrossRef]
- Shimada, T.; Urakawa, I.; Isakova, T.; Yamazaki, Y.; Epstein, M.; Wesseling-Perry, K.; Wolf, M.; Salusky, I.B.; Jüppner, H. Circulating fibroblast growth factor 23 in patients with end-stage renal disease treated by peritoneal dialysis is intact and biologically active. J. Clin. Endocrinol. Metab. 2010, 95, 578–585. [Google Scholar] [CrossRef]
- Hofman-Bang, J.; Martuseviciene, G.; Santini, M.A.; Olgaard, K.; Lewin, E. Increased parathyroid expression of klotho in uremic rats. Kidney Int. 2010, 78, 1119–1127. [Google Scholar] [CrossRef][Green Version]
- Rukov, J.L.; Gravesen, E.; Mace, M.L.; Hofman-Bang, J.; Vinther, J.; Andersen, C.B.; Lewin, E.; Olgaard, K. Effect of chronic uremia on the transcriptional profile of the calcified aorta analysed by RNA-sequencing. Am. J. Physiol. Ren. Physiol. 2016, 310, F477–F491. [Google Scholar] [CrossRef]
- Gravesen, E.; Lerche Mace, M.; Nordholm, A.; Hofman-Bang, J.; Hruska, K.; Haagen Nielsen, C.; Kjaer, A.; Olgaard, K.; Lewin, E. Exogenous BMP7 in aortae of rats with chronic uremia ameliorates expression of profibrotic genes, but does not reverse established vascular calcification. PLoS ONE 2018, 13, e0190820. [Google Scholar] [CrossRef]
- Gravesen, E.; Nordholm, A.; Mace, M.; Morevati, M.; Hogdall, E.; Nielsen, C.; Kjaer, A.; Olgaard, K.; Lewin, E. Effect of inhibition of CBP-coactivated beta-catenin-mediated Wnt signalling in uremic rats with vascular calcifications. PLoS ONE 2018, 13, e0201936. [Google Scholar] [CrossRef]
- Bohnert, B.N.; Daniel, C.; Amann, K.; Voelkl, J.; Alesutan, I.; Lang, F.; Heyne, N.; Haring, H.U.; Artunc, F. Impact of phosphorus restriction and vitamin D-substitution on secondary hyperparathyroidism in a proteinuric mouse model. Kidney Blood Press. Res. 2015, 40, 153–165. [Google Scholar] [CrossRef]
- Finch, J.L.; Lee, D.H.; Liapis, H.; Ritter, C.; Zhang, S.; Suarez, E.; Ferder, L.; Slatopolsky, E. Phosphate restriction significantly reduces mortality in uremic rats with established vascular calcification. Kidney Int. 2013, 84, 1145–1153. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; Barchi-Chung, A.; Enfield, G.; Smith, K.; Vargas, G.; Houston, J.; Xie, H.; Wahl, P.; Schiavenato, E.; Dosch, A.; et al. Effects of dietary phosphate restriction and phosphate binders on FGF23 levels in CKD. Clin. J. Am. Soc. Nephrol. 2013, 8, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Covic, A.; Passlick-Deetjen, J.; Kroczak, M.; Buschges-Seraphin, B.; Ghenu, A.; Ponce, P.; Marzell, B.; de Francisco, A.L. A comparison of calcium acetate/magnesium carbonate and sevelamer-hydrochloride effects on fibroblast growth factor-23 and bone markers: Post hoc evaluation from a controlled, randomized study. Nephrol. Dial. Transplant. 2013, 28, 2383–2392. [Google Scholar] [CrossRef]
- Goto, S.; Nakai, K.; Kono, K.; Yonekura, Y.; Ito, J.; Fujii, H.; Nishi, S. Dietary phosphorus restriction by a standard low-protein diet decreased serum fibroblast growth factor 23 levels in patients with early and advanced stage chronic kidney disease. Clin. Exp. Nephrol. 2014, 18, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Ix, J.H.; Isakova, T.; Larive, B.; Raphael, K.L.; Raj, D.S.; Cheung, A.K.; Sprague, S.M.; Fried, L.F.; Gassman, J.J.; Middleton, J.P.; et al. Effects of Nicotinamide and Lanthanum Carbonate on Serum Phosphate and Fibroblast Growth Factor-23 in CKD: The COMBINE Trial. J. Am. Soc. Nephrol. 2019, 30, 1096–1108. [Google Scholar] [CrossRef]
- Cozzolino, M.; Ketteler, M.; Martin, K.J.; Sharma, A.; Goldsmith, D.; Khan, S. Paricalcitol- or cinacalcet-centred therapy affects markers of bone mineral disease in patients with secondary hyperparathyroidism receiving haemodialysis: Results of the IMPACT-SHPT study. Nephrol. Dial. Transplant. 2014, 29, 899–905. [Google Scholar] [CrossRef]
- McKnight, Q.; Jenkins, S.; Li, X.; Nelson, T.; Marlier, A.; Cantley, L.G.; Finberg, K.E.; Fretz, J.A. IL-1beta Drives Production of FGF-23 at the Onset of Chronic Kidney Disease in Mice. J. Bone Miner. Res. 2020, 35, 1352–1362. [Google Scholar] [CrossRef]
- David, V.; Martin, A.; Isakova, T.; Spaulding, C.; Qi, L.; Ramirez, V.; Zumbrennen-Bullough, K.B.; Sun, C.C.; Lin, H.Y.; Babitt, J.L.; et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2016, 89, 135–146. [Google Scholar] [CrossRef]
- Toro, L.; Barrientos, V.; Leon, P.; Rojas, M.; Gonzalez, M.; Gonzalez-Ibanez, A.; Illanes, S.; Sugikawa, K.; Abarzua, N.; Bascunan, C.; et al. Erythropoietin induces bone marrow and plasma fibroblast growth factor 23 during acute kidney injury. Kidney Int. 2018, 93, 1131–1141. [Google Scholar] [CrossRef]
- Gravesen, E.; Hofman-Bang, J.; Mace, M.L.; Lewin, E.; Olgaard, K. High dose intravenous iron, mineral homeostasis and intact FGF23 in normal and uremic rats. BMC Nephrol. 2013, 14, 281. [Google Scholar] [CrossRef]
- David, V.; Francis, C.; Babitt, J.L. Ironing out the cross talk between FGF23 and inflammation. Am. J. Physiol. Ren. Physiol. 2017, 312, F1–F8. [Google Scholar] [CrossRef] [PubMed]
- Parfit, A.M. Calcium Homeostasis. In Handbook of Experimental Pharmacology; Mundy, G.R., Martin, T.J., Eds.; Springer: Berlin/Heidelberg, Germany, 1993; Volume 107, pp. 1–65. [Google Scholar]
- Huan, J.; Martuseviciene, G.; Olgaard, K.; Lewin, E. Calcium-sensing receptor and recovery from hypocalcaemia in thyroparathyroidectomized rats. Eur. J. Clin. Investig. 2007, 37, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Mosyak, L.; Kurinov, I.; Zuo, H.; Sturchler, E.; Cheng, T.C.; Subramanyam, P.; Brown, A.P.; Brennan, S.C.; Mun, H.C.; et al. Structural mechanism of ligand activation in human calcium-sensing receptor. Elife 2016, 5, e13662. [Google Scholar] [CrossRef] [PubMed]
- Centeno, P.P.; Herberger, A.; Mun, H.C.; Tu, C.; Nemeth, E.F.; Chang, W.; Conigrave, A.D.; Ward, D.T. Phosphate acts directly on the calcium-sensing receptor to stimulate parathyroid hormone secretion. Nat. Commun. 2019, 10, 4693. [Google Scholar] [CrossRef] [PubMed]
- Nordholm, A.; Mace, M.L.; Gravesen, E.; Olgaard, K.; Lewin, E. A potential kidney-bone axis involved in the rapid minute-to-minute regulation of plasma Ca2+. BMC Nephrol. 2015, 16, 29. [Google Scholar] [CrossRef]
- Lewin, E.; Wang, W.; Olgaard, K. Rapid recovery of plasma ionized calcium after acute induction of hypocalcaemia in parathyroidectomized and nephrectomized rats. Nephrol. Dial. Transplant. 1999, 14, 604–609. [Google Scholar] [CrossRef][Green Version]
- Wang, W.; Lewin, E.; Olgaard, K. Parathyroid hormone is not a key hormone in the rapid minute-to-minute regulation of plasma Ca2+ homeostasis in rats. Eur. J. Clin. Investig. 1999, 29, 309–320. [Google Scholar] [CrossRef]
- Wang, W.; Lewin, E.; Olgaard, K. 1,25(OH)2D3 only affects long-term levels of plasma Ca2+ but not the rapid minute-to-minute plasma Ca2+ homeostasis in the rat. Steroids 1999, 64, 726–734. [Google Scholar] [CrossRef]
- Wang, W.; Lewin, E.; Olgaard, K. Role of calcitonin in the rapid minute-to-minute regulation of plasma Ca2+ homeostasis in the rat. Eur. J. Clin. Investig. 2002, 32, 674–681. [Google Scholar] [CrossRef]
- Crnko, S.; Du Pre, B.C.; Sluijter, J.P.G.; Van Laake, L.W. Circadian rhythms and the molecular clock in cardiovascular biology and disease. Nat. Rev. Cardiol. 2019, 16, 437–447. [Google Scholar] [CrossRef]
- Sulli, G.; Lam, M.T.Y.; Panda, S. Interplay between Circadian Clock and Cancer: New Frontiers for Cancer Treatment. Trends Cancer 2019, 5, 475–494. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, S.M.; Pusztaszeri, M.; Brulhart-Meynet, M.C.; Petrenko, V.; De Vito, C.; Sobel, J.; Delucinge-Vivier, C.; Kebebew, E.; Regazzi, R.; Philippe, J.; et al. Identification of Differential Transcriptional Patterns in Primary and Secondary Hyperparathyroidism. J. Clin. Endocrinol. Metab. 2018, 103, 2189–2198. [Google Scholar] [CrossRef] [PubMed]
- Nordholm, A.; Mace, M.L.; Gravesen, E.; Hofman-Bang, J.; Morevati, M.; Olgaard, K.; Lewin, E. Klotho and activin A in kidney injury: Plasma Klotho is maintained in unilateral obstruction despite no upregulation of Klotho biosynthesis in the contralateral kidney. Am. J. Physiol. Ren. Physiol. 2018, 314, F753–F762. [Google Scholar] [CrossRef] [PubMed]
- Shalhoub, V.; Shatzen, E.M.; Ward, S.C.; Davis, J.; Stevens, J.; Bi, V.; Renshaw, L.; Hawkins, N.; Wang, W.; Chen, C.; et al. FGF23 neutralization improves chronic kidney disease-associated hyperparathyroidism yet increases mortality. J. Clin. Investig. 2012, 122, 2543–2553. [Google Scholar] [CrossRef] [PubMed]
- Spichtig, D.; Zhang, H.; Mohebbi, N.; Pavik, I.; Petzold, K.; Stange, G.; Saleh, L.; Edenhofer, I.; Segerer, S.; Biber, J.; et al. Renal expression of FGF23 and peripheral resistance to elevated FGF23 in rodent models of polycystic kidney disease. Kidney Int. 2014, 85, 1340–1350. [Google Scholar] [CrossRef] [PubMed]
- Zanchi, C.; Locatelli, M.; Benigni, A.; Corna, D.; Tomasoni, S.; Rottoli, D.; Gaspari, F.; Remuzzi, G.; Zoja, C. Renal expression of FGF23 in progressive renal disease of diabetes and the effect of ACE inhibitor. PLoS ONE 2013, 8, e70775. [Google Scholar] [CrossRef]
- Smith, E.R.; Tan, S.J.; Holt, S.G.; Hewitson, T.D. FGF23 is synthesised locally by renal tubules and activates injury-primed fibroblasts. Sci. Rep. 2017, 7, 3345. [Google Scholar] [CrossRef]
- Prie, D.; Forand, A.; Francoz, C.; Elie, C.; Cohen, I.; Courbebaisse, M.; Eladari, D.; Lebrec, D.; Durand, F.; Friedlander, G. Plasma fibroblast growth factor 23 concentration is increased and predicts mortality in patients on the liver-transplant waiting list. PLoS ONE 2013, 8, e66182. [Google Scholar] [CrossRef]
- Zhang, Q.; Doucet, M.; Tomlinson, R.E.; Han, X.; Quarles, L.D.; Collins, M.T.; Clemens, T.L. The hypoxia-inducible factor-1alpha activates ectopic production of fibroblast growth factor 23 in tumor-induced osteomalacia. Bone Res. 2016, 4, 16011. [Google Scholar] [CrossRef]
- Andrukhova, O.; Slavic, S.; Odorfer, K.I.; Erben, R.G. Experimental Myocardial Infarction Upregulates Circulating Fibroblast Growth Factor-23. J. Bone Miner. Res. 2015, 30, 1831–1839. [Google Scholar] [CrossRef]
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.C.; Sloan, A.; Isakova, T.; Gutierrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 induces left ventricular hypertrophy. J. Clin. Investig. 2011, 121, 4393–4408. [Google Scholar] [CrossRef]
- Grabner, A.; Amaral, A.P.; Schramm, K.; Singh, S.; Sloan, A.; Yanucil, C.; Li, J.; Shehadeh, L.A.; Hare, J.M.; David, V.; et al. Activation of Cardiac Fibroblast Growth Factor Receptor 4 Causes Left Ventricular Hypertrophy. Cell Metab. 2015, 22, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Frias, O.; Gil-Pena, H.; Perez-Roldan, J.M.; Gonzalez-Sanchez, S.; Ariceta, G.; Chocron, S.; Loza, R.; de la Cerda Ojeda, F.; Madariaga, L.; Vergara, I.; et al. Risk of cardiovascular involvement in pediatric patients with X-linked hypophosphatemia. Pediatr. Nephrol. 2019, 34, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Pastor-Arroyo, E.M.; Gehring, N.; Krudewig, C.; Costantino, S.; Bettoni, C.; Knopfel, T.; Sabrautzki, S.; Lorenz-Depiereux, B.; Pastor, J.; Strom, T.M.; et al. The elevation of circulating fibroblast growth factor 23 without kidney disease does not increase cardiovascular disease risk. Kidney Int. 2018, 94, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Andrukhova, O.; Slavic, S.; Smorodchenko, A.; Zeitz, U.; Shalhoub, V.; Lanske, B.; Pohl, E.E.; Erben, R.G. FGF23 regulates renal sodium handling and blood pressure. EMBO Mol. Med. 2014, 6, 744–759. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Grabner, A.; Yanucil, C.; Schramm, K.; Czaya, B.; Krick, S.; Czaja, M.J.; Bartz, R.; Abraham, R.; Di Marco, G.S.; et al. Fibroblast growth factor 23 directly targets hepatocytes to promote inflammation in chronic kidney disease. Kidney Int. 2016, 90, 985–996. [Google Scholar] [CrossRef]
- Rossaint, J.; Oehmichen, J.; Van, A.H.; Reuter, S.; Pavenstadt, H.J.; Meersch, M.; Unruh, M.; Zarbock, A. FGF23 signaling impairs neutrophil recruitment and host defense during CKD. J. Clin. Investig. 2016, 126, 962–974. [Google Scholar] [CrossRef]
- Flythe, J.E.; Assimon, M.M.; Tugman, M.J.; Chang, E.H.; Gupta, S.; Shah, J.; Sosa, M.A.; De Mauro Renaghan, A.; Melamed, M.L.; Wilson, F.P.; et al. Characteristics and Outcomes of Individuals with Pre-existing Kidney Disease and COVID-19 Admitted to Intensive Care Units in the United States. Am. J. Kidney Dis. 2020. [Google Scholar] [CrossRef]
- Jager, K.J.; Kramer, A.; Chesnaye, N.C.; Couchoud, C.; Sánchez-Álvarez, J.E.; Garneata, L.; Collart, F.; Hemmelder, M.H.; Ambühl, P.; Kerschbaum, J.; et al. Results from the ERA-EDTA Registry indicate a high mortality due to COVID-19 in dialysis patients and kidney transplant recipients across Europe. Kidney Int. 2020. S0085-2538(20)31081-4. [Google Scholar] [CrossRef]
- Komaba, H.; Fukagawa, M. FGF23-parathyroid interaction: Implications in chronic kidney disease. Kidney Int. 2010, 77, 292–298. [Google Scholar] [CrossRef]
- Galitzer, H.; Ben-Dov, I.Z.; Silver, J.; Naveh-Many, T. Parathyroid cell resistance to fibroblast growth factor 23 in secondary hyperparathyroidism of chronic kidney disease. Kidney Int. 2010, 77, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Kumata, C.; Mizobuchi, M.; Ogata, H.; Koiwa, F.; Nakazawa, A.; Kondo, F.; Kadokura, Y.; Kinugasa, E.; Akizawa, T. Involvement of alpha-klotho and fibroblast growth factor receptor in the development of secondary hyperparathyroidism. Am. J. Nephrol. 2010, 31, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Ohkido, I.; Yokoyama, K.; Imura, A.; Utsunomiya, Y.; Hosoya, T.; Nabeshima, Y. Persistent alpha-Klotho (a-Kl) expression in the parathyroid glands of patients with secondary hyperparathyroidism. Nephrol. Dial. Transplant. 2010, 25, 1007–1008. [Google Scholar] [CrossRef] [PubMed]
- Lavi-Moshayoff, V.; Wasserman, G.; Meir, T.; Silver, J.; Naveh-Many, T. PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: A bone parathyroid feedback loop. Am. J. Physiol. Ren. Physiol. 2010, 299, F882–F889. [Google Scholar] [CrossRef]
- Yu, M.; Malik Tyagi, A.; Li, J.Y.; Adams, J.; Denning, T.L.; Weitzmann, M.N.; Jones, R.M.; Pacifici, R. PTH induces bone loss via microbial-dependent expansion of intestinal TNF(+) T cells and Th17 cells. Nat. Commun. 2020, 11, 468. [Google Scholar] [CrossRef]
- Liao, S.C.; Moi, S.H.; Chou, F.F.; Yang, C.H.; Chen, J.B. Changes in Serum Concentrations of Fibroblast Growth Factor 23 and Soluble Klotho in Hemodialysis Patients after Total Parathyroidectomy. BioMed Res. Int. 2016, 2016, 6453803. [Google Scholar] [CrossRef]
- Sato, T.; Tominaga, Y.; Ueki, T.; Goto, N.; Matsuoka, S.; Katayama, A.; Haba, T.; Uchida, K.; Nakanishi, S.; Kazama, J.J.; et al. Total parathyroidectomy reduces elevated circulating fibroblast growth factor 23 in advanced secondary hyperparathyroidism. Am. J. Kidney Dis. 2004, 44, 481–487. [Google Scholar] [CrossRef]
- Susantitaphong, P.; Vadcharavivad, S.; Susomboon, T.; Singhan, W.; Dumrongpisutikul, N.; Jakchairoongruang, K.; Eiam-Ong, S.; Praditpornsilpa, K. The effectiveness of cinacalcet: A randomized, open label study in chronic hemodialysis patients with severe secondary hyperparathyroidism. Ren. Fail. 2019, 41, 326–333. [Google Scholar] [CrossRef]
- Hruska, K.A.; Sugatani, T.; Agapova, O.; Fang, Y. The chronic kidney disease—Mineral bone disorder (CKD-MBD): Advances in pathophysiology. Bone 2017, 100, 80–86. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, C.; Hong, X.; Miao, J.; Liao, Y.; Hou, F.F.; Zhou, L.; Liu, Y. Wnt/β-catenin signaling mediates both heart and kidney injury in type 2 cardiorenal syndrome. Kidney Int. 2019, 95, 815–829. [Google Scholar] [CrossRef]
- Kuro-o, M. Klotho and endocrine fibroblast growth factors: Markers of chronic kidney disease progression and cardiovascular complications? Nephrol. Dial. Transplant. 2019, 34, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Castañeda, J.R.; Herencia, C.; de Pendón-Ruiz Mier, M.V.; Rodriguez-Ortiz, M.E.; Diaz-Tocados, J.M.; Vergara, N.; Martínez-Moreno, J.M.; Salmerón, M.D.; Richards, W.G.; Felsenfeld, A.; et al. Differential regulation of renal Klotho and FGFR1 in normal and uremic rats. FASEB J. 2017, 31, 3858–3867. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Castañeda, J.R.; Rodelo-Haad, C.; de Pendon-Ruiz Mier, M.V.; Martin-Malo, A.; Santamaria, R.; Rodriguez, M. Klotho/FGF23 and Wnt signaling as important players in the comorbidities associated with chronic kidney disease. Toxins (Basel) 2020, 12, 185. [Google Scholar] [CrossRef] [PubMed]
- Sugatani, T.; Alvarez, U.M.; Hruska, K.A. Activin A stimulates IkappaB-alpha/NFkappaB and RANK expression for osteoclast differentiation, but not AKT survival pathway in osteoclast precursors. J. Cell Biochem. 2003, 90, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Agapova, O.A.; Fang, Y.; Sugatani, T.; Seifert, M.E.; Hruska, K.A. Ligand trap for the activin type IIA receptor protects against vascular disease and renal fibrosis in mice with chronic kidney disease. Kidney Int. 2016, 89, 1231–1243. [Google Scholar] [CrossRef]
- Sugatani, T.; Agapova, O.A.; Fang, Y.; Berman, A.G.; Wallace, J.M.; Malluche, H.H.; Faugere, M.C.; Smith, W.; Sung, V.; Hruska, K.A. Ligand trap of the activin receptor type IIA inhibits osteoclast stimulation of bone remodeling in diabetic mice with chronic kidney disease. Kidney Int. 2017, 91, 86–95. [Google Scholar] [CrossRef]
- Williams, M.J.; Sugatani, T.; Agapova, O.A.; Fang, Y.; Gaut, J.P.; Faugere, M.C.; Malluche, H.H.; Hruska, K.A. The activin receptor is stimulated in the skeleton, vasculature, heart, and kidney during chronic kidney disease. Kidney Int. 2018, 93, 147–158. [Google Scholar] [CrossRef]
- Fang, Y.; Ginsberg, C.; Seifert, M.; Agapova, O.; Sugatani, T.; Register, T.C.; Freedman, B.I.; Monier-Faugere, M.C.; Malluche, H.; Hruska, K.A. CKD-induced wingless/integration1 inhibitors and phosphorus cause the CKD-mineral and bone disorder. J. Am. Soc. Nephrol. 2014, 25, 1760–1773. [Google Scholar] [CrossRef]
- Moe, S.M.; Chen, N.X.; Newman, C.L.; Organ, J.M.; Kneissel, M.; Kramer, I.; Gattone, V.H., 2nd; Allen, M.R. Anti-sclerostin antibody treatment in a rat model of progressive renal osteodystrophy. J. Bone Miner. Res. 2015, 30, 499–509. [Google Scholar] [CrossRef]
- Simic, P.; Kim, W.; Zhou, W.; Pierce, K.A.; Chang, W.; Sykes, D.B.; Aziz, N.B.; Elmariah, S.; Ngo, D.; Pajevic, P.D.; et al. Glycerol-3-phosphate is an FGF23 regulator derived from the injured kidney. J. Clin. Investig. 2020, 130, 1513–1526. [Google Scholar] [CrossRef]
- Mace, M.L.; Gravesen, E.; Nordholm, A.; Egstrand, S.; Morevati, M.; Nielsen, C.; Kjaer, A.; Behets, G.; D’Haese, P.; Olgaard, K.; et al. Chronic kidney disease induced vascular calcification impairs bone metabolism. J. Bone Min. Res. 2020, in press. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mace, M.L.; Olgaard, K.; Lewin, E. New Aspects of the Kidney in the Regulation of Fibroblast Growth Factor 23 (FGF23) and Mineral Homeostasis. Int. J. Mol. Sci. 2020, 21, 8810. https://doi.org/10.3390/ijms21228810
Mace ML, Olgaard K, Lewin E. New Aspects of the Kidney in the Regulation of Fibroblast Growth Factor 23 (FGF23) and Mineral Homeostasis. International Journal of Molecular Sciences. 2020; 21(22):8810. https://doi.org/10.3390/ijms21228810
Chicago/Turabian StyleMace, Maria L., Klaus Olgaard, and Ewa Lewin. 2020. "New Aspects of the Kidney in the Regulation of Fibroblast Growth Factor 23 (FGF23) and Mineral Homeostasis" International Journal of Molecular Sciences 21, no. 22: 8810. https://doi.org/10.3390/ijms21228810
APA StyleMace, M. L., Olgaard, K., & Lewin, E. (2020). New Aspects of the Kidney in the Regulation of Fibroblast Growth Factor 23 (FGF23) and Mineral Homeostasis. International Journal of Molecular Sciences, 21(22), 8810. https://doi.org/10.3390/ijms21228810