Targeting Adenosine Receptors: A Potential Pharmacological Avenue for Acute and Chronic Pain





Abstract
1. Introduction
2. ARs and Pain
2.1. A1ARs

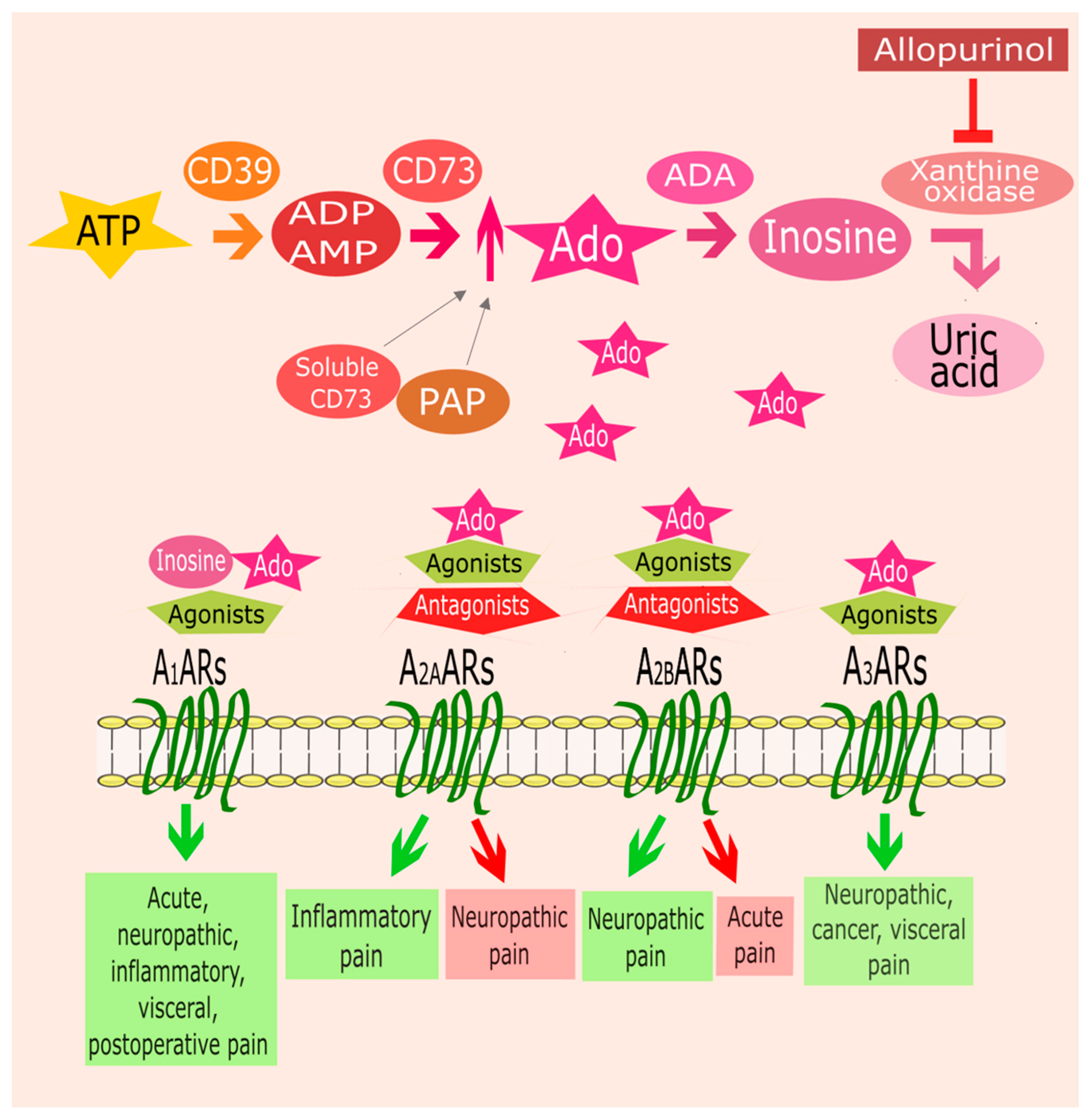{kind=link}
| Ligand | Pharmacological Behavior | Pain Model | Species | Route of Administration |
|---|---|---|---|---|
| 2′-Me-CCPA | agonist | formalin test | rat | intra-PAG, i.p. [20] |
| plantar test | rat | intra-PAG, i.p. [20] | ||
| tail flick test | rat | intra-PAG, i.p. [20] | ||
| CCPA | agonist | formalin test | mouse | i.p. [69] |
| writhing test | mouse | i.p. [69] | ||
| STZ-induced mechanical allodynia | mouse | i.p. [69] | ||
| CFA induced-mechanical allodynia and thermal hyperalgesia | mouse | Zusanli acupoint-injection [28] | ||
| SCNL induced-mechanical allodynia and thermal hyperalgesia | mouse | Zusanli acupoint-injection [28] | ||
| CPA | agonist | formalin test | mouse | i.p. [49]; i.t. [54] |
| CFA-induced-mechanical allodynia and thermal hyperalgesia | mouse | i.m. [30]; i.p. [42]; Weizhong acupoint-injection [46] | ||
| carrageenan-induced mechanical allodynia | rat | i.pl. [18] | ||
| PGE2-induced mechanical allodynia | rat | i.pl. [18] | ||
| SCNL-induced mechanical allodynia and thermal hyperalgesia | rat | i.p. [21] | ||
| colonic distension-induced visceral pain | rat | s.c., i.c. [23,24] | ||
| R-PIA | agonist | plantar incision-induced mechanical allodynia | rat | i.t. [25] |
| photochemical SCI-induced mechanical and thermal allodynia | rat | i.t. [47] | ||
| SCNL-induced mechanical allodynia | rat | i.t. [50] | ||
| photochemical sciatic nerve injury-induced mechanical and thermal allodynia | rat, mouse | i.t. [52] | ||
| carrageenan-induced mechanical and thermal allodynia | rat, mouse | i.t. [52] | ||
| T62 | positive allosteric modulator | SNL-induced mechanical allodynia | rat | i.p. [63]; i.t. [63,64]; p.o. [64] |
| carrageenan-induced thermal hyperalgesia | rat | i.t. [65] | ||
| plantar incision-induced mechanical allodynia | rat | i.t. [66] | ||
| TRR469 | positive allosteric modulator | formalin test | mouse | i.p. [69] |
| writhing test | mouse | i.p. [69] | ||
| STZ-induced mechanical allodynia | mouse | i.p. [69] |
2.2. A2AARs and Pain
| Ligand | Pharmacological Behavior | Pain Model | Species | Route of Administration |
|---|---|---|---|---|
| ATL313 | agonist | CCI-induced mechanical allodynia and thermal hyperalgesia | rat | i.t. [95,96]; peri-sciatic nerve injection [98] |
| SNL-induced mechanical allodynia | rat | i.t. [96] | ||
| SIN-induced mechanical allodynia | rat | i.t. [96] | ||
| SCI-induced mechanical and thermal allodynia | rat | i.t. [97] | ||
| CGS21680 | agonist | formalin test (early phase) | mouse | i.p. [49] |
| CFA-induced-mechanical allodynia and thermal hyperalgesia | rat | i.p. [92] | ||
| CCI-induced mechanical allodynia and thermal hyperalgesia | rat | i.t. [95,96] | ||
| SCI-induced mechanical and thermal allodynia | rat | i.t. [97] | ||
| LASSBio-1359 | agonist | formalin test | mouse | i.p. [94] |
| carrageenan induced-mechanical allodynia and thermal hyperalgesia | mouse | i.p. [94] | ||
| Adonis | agonist-like monoclonal antibody | hot plate test | mouse | i.c.v. [100] |
| tail flick test | mouse | i.c.v. [100] | ||
| TP455 | inverse agonist | writhing test | mouse | i.p. [74] |
| tail immersion test | mouse | i.p. [74] | ||
| ZM241385 | antagonist | writhing test | mouse | i.p. [74] |
| tail immersion test | mouse | i.p. [74] | ||
| carrageenan induced-mechanical allodynia | mouse | s.c. [73] | ||
| sleep deprivation-induced thermal hyperalgesia | rat | i.c.v. [72] | ||
| plantar incision-induced mechanical allodynia and thermal hyperalgesia | rat | i.c.v. [72] |
2.3. A2BARs and Pain
| Ligand | Pharmacological Behavior | Pain Model | Species | Route of Administration |
|---|---|---|---|---|
| BAY606583 | agonist | CCI-induced mechanical allodynia | mouse | i.t. [96] |
| PSB-10 | antagonist | formalin test | mouse | i.p. [105] |
| PSB-36 | antagonist | formalin test | mouse | i.p. [105] |
| PSB-1115 | antagonist | formalin test | mouse | i.p. [105] |
2.4. A3ARs and Pain
| Ligand | Pharmacological Behavior | Pain Model | Species | Route of Administration |
|---|---|---|---|---|
| AR170 | A3AR agonist | colitis-induced visceral hypersensitivity | rat | i.p. [131] |
| Cl-IB-MECA | A3AR agonist | chemotherapy-induced mechanical allodynia | mouse | i.p. [115] |
| CCI-induced mechanical allodynia | mouse | i.p. [115] | ||
| bone cancer-induced mechanical allodynia | rat | i.p. [126] | ||
| colitis-induced visceral hypersensitivity | rat | i.p. [133] | ||
| IB-MECA | A3AR agonist | chemotherapy-induced mechanical allodynia | mouse/rat | i.p. [115,122] |
| CCI-induced mechanical allodynia | mouse | i.p. [115]; i.t. [118] | ||
| STZ-induced mechanical allodynia and thermal hyperalgesia | mouse | i.p. [127] | ||
| opioid-induced thermal hyperalgesia | rat | p.o. [130] | ||
| tibial nerve injury-induced mechanical allodynia | rat | i.p. [120] | ||
| MRS1898 | A3AR agonist | chemotherapy-induced mechanical allodynia | mouse | i.p. [115] |
| CCI-induced mechanical allodynia | mouse | i.p. [115] | ||
| MRS5698 | A3AR agonist | CCI-induced mechanical allodynia | rat | s.c., i.p., i.v. [117]; i.t. [118] |
| spared nerve injury-induced mechanical allodynia | rat | s.c., i.p., i.v. [117] | ||
| SNL-induced mechanical allodynia | rat | s.c., i.p., i.v. [117] | ||
| bone cancer-induced mechanical allodynia | rat | s.c., i.p., i.v. [117] | ||
| chemotherapy-induced mechanical allodynia | rat | s.c., i.p., i.v. [117]; i.t. [124] | ||
| opioid-induced thermal hyperalgesia | rat | p.o. [130] | ||
| MRS5980 | A3AR agonist | colitis-induced visceral hypersensitivity | rat | i.p. [133] |
| MRS7422 | A3AR agonist | CCI-induced mechanical allodynia | mouse | p.o. [121] |
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GPCR | G protein coupled receptors |
| AR | adenosine receptor |
| AC | adenylate cyclase |
| cAMP | cyclic adenosine monophosphate |
| ATP | adenosine triphosphate |
| ADA | adenosine deaminase |
| PKA | protein kinase A |
| PLC | protein lipase C |
| IP3 | inositol triphosphate |
| DAG | diacylglycerol |
| ERK | extracellular signal-regulated kinase |
| NO | nitric oxide |
| cGMP | cyclic guanosine monophosphate |
| PKG | protein kinase G |
| PAG | periaqueductal gray |
| CCI | chronic constriction injury |
| CFA | complete Freund’s adjuvant |
| SCNL | sciatic nerve ligation |
| KO | knockout |
| PAP | prostatic acid phosphatase |
| SCI | spinal cord injury |
| SNL | spinal nerve ligation |
| STZ | streptozotocin |
| ROS | reactive oxygen species |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| TNF-α | tumor necrosis factor-α |
| iNOS | inducible nitric oxide synthase |
| SIN | sciatic inflammatory neuropathy |
| PKC | protein kinase C |
| IL | interleukin |
| PI3K | phosphoinositide 3-kinase |
| MAPK | mitogen-activated protein kinase |
| CREB | cAMP response element-binding protein |
References
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef] [PubMed]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pathological overproduction: The bad side of adenosine. Br. J. Pharmacol. 2017, 174, 1945–1960. [Google Scholar] [CrossRef] [PubMed]
- Borea, P.A.; Gessi, S.; Merighi, S.; Varani, K. Adenosine as a Multi-Signalling Guardian Angel in Human Diseases: When, Where and How Does it Exert its Protective Effects? Trends Pharmacol. Sci. 2016, 37, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Haskó, G.; Linden, J.; Cronstein, B.; Pacher, P. Adenosine receptors: Therapeutic aspects for inflammatory and immune diseases. Nat. Rev. Drug Discov. 2008, 7, 759–770. [Google Scholar] [CrossRef]
- Gompel, J.J.V.; Bower, M.R.; Worrell, G.A.; Stead, M.; Chang, S.-Y.; Goerss, S.J.; Kim, I.; Bennet, K.E.; Meyer, F.B.; Marsh, W.R.; et al. Increased cortical extracellular adenosine correlates with seizure termination. Epilepsia 2014, 55, 233–244. [Google Scholar] [CrossRef]
- During, M.J.; Spencer, D.D. Adenosine: A potential mediator of seizure arrest and postictal refractoriness. Ann. Neurol. 1992, 32, 618–624. [Google Scholar] [CrossRef]
- Ganesana, M.; Venton, B.J. Early changes in transient adenosine during cerebral ischemia and reperfusion injury. PLoS ONE 2018, 13, e0196932. [Google Scholar] [CrossRef]
- Pedata, F.; Dettori, I.; Coppi, E.; Melani, A.; Fusco, I.; Corradetti, R.; Pugliese, A.M. Purinergic signalling in brain ischemia. Neuropharmacology 2016, 104, 105–130. [Google Scholar] [CrossRef]
- Merighi, S.; Battistello, E.; Giacomelli, L.; Varani, K.; Vincenzi, F.; Borea, P.A.; Gessi, S. Targeting A3 and A2A adenosine receptors in the fight against cancer. Expert Opin. Ther. Targets 2019, 23, 669–678. [Google Scholar] [CrossRef]
- Gessi, S.; Merighi, S.; Sacchetto, V.; Simioni, C.; Borea, P.A. Adenosine receptors and cancer. Biochim. Biophys. Acta 2011, 1808, 1400–1412. [Google Scholar] [CrossRef]
- Antonioli, L.; Csóka, B.; Fornai, M.; Colucci, R.; Kókai, E.; Blandizzi, C.; Haskó, G. Adenosine and inflammation: What’s new on the horizon? Drug Discov. Today 2014, 19, 1051–1068. [Google Scholar] [CrossRef] [PubMed]
- Sawynok, J.; Liu, X.J. Adenosine in the spinal cord and periphery: Release and regulation of pain. Prog. Neurobiol. 2003, 69, 313–340. [Google Scholar] [CrossRef]
- Sawynok, J. Adenosine receptor targets for pain. Neuroscience 2016, 338, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B. Adenosine—A physiological or pathophysiological agent? J. Mol. Med. 2014, 92, 201–206. [Google Scholar] [CrossRef]
- Varani, K.; Vincenzi, F.; Merighi, S.; Gessi, S.; Borea, P.A. Biochemical and Pharmacological Role of A1 Adenosine Receptors and Their Modulation as Novel Therapeutic Strategy. Adv. Exp. Med. Biol. 2017, 1051, 193–232. [Google Scholar] [CrossRef]
- Schulte, G.; Robertson, B.; Fredholm, B.B.; DeLander, G.E.; Shortland, P.; Molander, C. Distribution of antinociceptive adenosine a1 receptors in the spinal cord dorsal horn, and relationship to primary afferents and neuronal subpopulations. Neuroscience 2003, 121, 907–916. [Google Scholar] [CrossRef]
- Luongo, L.; Guida, F.; Imperatore, R.; Napolitano, F.; Gatta, L.; Cristino, L.; Giordano, C.; Siniscalco, D.; Di Marzo, V.; Bellini, G.; et al. The A1 adenosine receptor as a new player in microglia physiology. Glia 2014, 62, 122–132. [Google Scholar] [CrossRef]
- Lima, F.O.; Souza, G.R.; Verri, W.A.; Parada, C.A.; Ferreira, S.H.; Cunha, F.Q.; Cunha, T.M. Direct blockade of inflammatory hypernociception by peripheral A1 adenosine receptors: Involvement of the NO/cGMP/PKG/KATP signaling pathway. Pain 2010, 151, 506–515. [Google Scholar] [CrossRef]
- Sawynok, J.; Reid, A.R.; Liu, J. Spinal and peripheral adenosine A1 receptors contribute to antinociception by tramadol in the formalin test in mice. Eur. J. Pharmacol. 2013, 714, 373–378. [Google Scholar] [CrossRef]
- Maione, S.; de Novellis, V.; Cappellacci, L.; Palazzo, E.; Vita, D.; Luongo, L.; Stella, L.; Franchetti, P.; Marabese, I.; Rossi, F.; et al. The antinociceptive effect of 2-chloro-2′-C-methyl-N6-cyclopentyladenosine (2′-Me-CCPA), a highly selective adenosine A1 receptor agonist, in the rat. Pain 2007, 131, 281–292. [Google Scholar] [CrossRef]
- Gong, Q.-J.; Li, Y.-Y.; Xin, W.-J.; Wei, X.-H.; Cui, Y.; Wang, J.; Liu, Y.; Liu, C.-C.; Li, Y.-Y.; Liu, X.-G. Differential effects of adenosine A1 receptor on pain-related behavior in normal and nerve-injured rats. Brain Res. 2010, 1361, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Kan, H.-W.; Chang, C.-H.; Lin, C.-L.; Lee, Y.-C.; Hsieh, S.-T.; Hsieh, Y.-L. Downregulation of adenosine and adenosine A1 receptor contributes to neuropathic pain in resiniferatoxin neuropathy. Pain 2018, 159, 1580–1591. [Google Scholar] [CrossRef] [PubMed]
- Okumura, T.; Nozu, T.; Kumei, S.; Takakusaki, K.; Miyagishi, S.; Ohhira, M. Adenosine A1 receptors mediate the intracisternal injection of orexin-induced antinociceptive action against colonic distension in conscious rats. J. Neurol. Sci. 2016, 362, 106–110. [Google Scholar] [CrossRef]
- Okumura, T.; Nozu, T.; Ishioh, M.; Igarashi, S.; Kumei, S.; Ohhira, M. Adenosine A1 receptor agonist induces visceral antinociception via 5-HT1A, 5-HT2A, dopamine D1 or cannabinoid CB1 receptors, and the opioid system in the central nervous system. Physiol. Behav. 2020, 220, 112881. [Google Scholar] [CrossRef]
- Zahn, P.K.; Straub, H.; Wenk, M.; Pogatzki-Zahn, E.M. Adenosine A1 but not A2a receptor agonist reduces hyperalgesia caused by a surgical incision in rats: A pertussis toxin-sensitive G protein-dependent process. Anesthesiology 2007, 107, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Martins, D.F.; Mazzardo-Martins, L.; Cidral-Filho, F.J.; Stramosk, J.; Santos, A.R.S. Ankle joint mobilization affects postoperative pain through peripheral and central adenosine A1 receptors. Phys. Ther. 2013, 93, 401–412. [Google Scholar] [CrossRef]
- Yamaoka, G.; Horiuchi, H.; Morino, T.; Miura, H.; Ogata, T. Different analgesic effects of adenosine between postoperative and neuropathic pain. J. Orthop. Sci. 2013, 18, 130–136. [Google Scholar] [CrossRef]
- Goldman, N.; Chen, M.; Fujita, T.; Xu, Q.; Peng, W.; Liu, W.; Jensen, T.K.; Pei, Y.; Wang, F.; Han, X.; et al. Adenosine A1 receptors mediate local anti-nociceptive effects of acupuncture. Nat. Neurosci. 2010, 13, 883–888. [Google Scholar] [CrossRef]
- Zhang, M.; Dai, Q.; Liang, D.; Li, D.; Chen, S.; Chen, S.; Han, K.; Huang, L.; Wang, J. Involvement of adenosine A1 receptor in electroacupuncture-mediated inhibition of astrocyte activation during neuropathic pain. Arq. Neuro-Psiquiatr. 2018, 76, 736–742. [Google Scholar] [CrossRef]
- Liao, H.-Y.; Hsieh, C.-L.; Huang, C.-P.; Lin, Y.-W. Electroacupuncture Attenuates CFA-induced Inflammatory Pain by suppressing Nav1.8 through S100B, TRPV1, Opioid, and Adenosine Pathways in Mice. Sci. Rep. 2017, 7, 42531. [Google Scholar] [CrossRef]
- Dai, Q.-X.; Huang, L.-P.; Mo, Y.-C.; Yu, L.-N.; Du, W.-W.; Zhang, A.-Q.; Geng, W.-J.; Wang, J.-L.; Yan, M. Role of spinal adenosine A1 receptors in the analgesic effect of electroacupuncture in a rat model of neuropathic pain. J. Int. Med. Res. 2020, 48, 300060519883748. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Lu, Q.; Chou, G.; Wang, Z.; Pan, R.; Xia, Y.; Hu, H.; Dai, Y. Norisoboldine attenuates inflammatory pain via the adenosine A1 receptor. Eur. J. Pain 2014, 18, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Yin, D.; Liu, Y.-Y.; Wang, T.-X.; Hu, Z.-Z.; Qu, W.-M.; Chen, J.-F.; Cheng, N.-N.; Huang, Z.-L. Paeoniflorin exerts analgesic and hypnotic effects via adenosine A1 receptors in a mouse neuropathic pain model. Psychopharmacology 2016, 233, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Peana, A.T.; Rubattu, P.; Piga, G.G.; Fumagalli, S.; Boatto, G.; Pippia, P.; De Montis, M.G. Involvement of adenosine A1 and A2A receptors in (-)-linalool-induced antinociception. Life Sci. 2006, 78, 2471–2474. [Google Scholar] [CrossRef] [PubMed]
- Valério, D.A.; Ferreira, F.I.; Cunha, T.M.; Alves-Filho, J.C.; Lima, F.O.; De Oliveira, J.R.; Ferreira, S.H.; Cunha, F.Q.; Queiroz, R.H.; Verri, W.A. Fructose-1,6-bisphosphate reduces inflammatory pain-like behaviour in mice: Role of adenosine acting on A1 receptors. Br. J. Pharmacol. 2009, 158, 558–568. [Google Scholar] [CrossRef]
- Nascimento, F.P.; Figueredo, S.M.; Marcon, R.; Martins, D.F.; Macedo, S.J.; Lima, D.A.N.; Almeida, R.C.; Ostroski, R.M.; Rodrigues, A.L.S.; Santos, A.R.S. Inosine reduces pain-related behavior in mice: Involvement of adenosine A1 and A2A receptor subtypes and protein kinase C pathways. J. Pharmacol. Exp. Ther. 2010, 334, 590–598. [Google Scholar] [CrossRef]
- Nascimento, F.P.; Macedo-Júnior, S.J.; Pamplona, F.A.; Luiz-Cerutti, M.; Córdova, M.M.; Constantino, L.; Tasca, C.I.; Dutra, R.C.; Calixto, J.B.; Reid, A.; et al. Adenosine A1 receptor-dependent antinociception induced by inosine in mice: Pharmacological, genetic and biochemical aspects. Mol. Neurobiol. 2015, 51, 1368–1378. [Google Scholar] [CrossRef]
- de Oliveira, E.D.; Schallenberger, C.; Böhmer, A.E.; Hansel, G.; Fagundes, A.C.; Milman, M.; Silva, M.D.P.; Oses, J.P.; Porciúncula, L.O.; Portela, L.V.; et al. Mechanisms involved in the antinociception induced by spinal administration of inosine or guanine in mice. Eur. J. Pharmacol. 2016, 772, 71–82. [Google Scholar] [CrossRef]
- Schmidt, A.P.; Böhmer, A.E.; Antunes, C.; Schallenberger, C.; Porciúncula, L.O.; Elisabetsky, E.; Lara, D.R.; Souza, D.O. Anti-nociceptive properties of the xanthine oxidase inhibitor allopurinol in mice: Role of A1 adenosine receptors. Br. J. Pharmacol. 2009, 156, 163–172. [Google Scholar] [CrossRef]
- Abad, A.S.S.; Falanji, F.; Ghanbarabadi, M.; Rad, A.; Nazemi, S.; Pejhan, A.; Amin, B. Assessment of anti-nociceptive effect of allopurinol in a neuropathic pain model. Brain Res. 2019, 1720, 146238. [Google Scholar] [CrossRef]
- Sowa, N.A.; Voss, M.K.; Zylka, M.J. Recombinant ecto-5’-nucleotidase (CD73) has long lasting antinociceptive effects that are dependent on adenosine A1 receptor activation. Mol. Pain 2010, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Zylka, M.J.; Sowa, N.A.; Taylor-Blake, B.; Twomey, M.A.; Herrala, A.; Voikar, V.; Vihko, P. Prostatic acid phosphatase is an ectonucleotidase and suppresses pain by generating adenosine. Neuron 2008, 60, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Street, S.E.; Walsh, P.L.; Sowa, N.A.; Taylor-Blake, B.; Guillot, T.S.; Vihko, P.; Wightman, R.M.; Zylka, M.J. PAP and NT5E inhibit nociceptive neurotransmission by rapidly hydrolyzing nucleotides to adenosine. Mol. Pain 2011, 7, 80. [Google Scholar] [CrossRef] [PubMed]
- Sowa, N.A.; Street, S.E.; Vihko, P.; Zylka, M.J. Prostatic Acid Phosphatase Reduces Thermal Sensitivity and Chronic Pain Sensitization by Depleting Phosphatidylinositol 4,5-Bisphosphate. J. Neurosci. 2010, 30, 10282–10293. [Google Scholar] [CrossRef] [PubMed]
- Zylka, M.J. Pain-relieving prospects for adenosine receptors and ectonucleotidases. Trends Mol. Med. 2011, 17, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Hurt, J.K.; Zylka, M.J. PAPupuncture has localized and long-lasting antinociceptive effects in mouse models of acute and chronic pain. Mol. Pain 2012, 8, 28. [Google Scholar] [CrossRef]
- von Heijne, M.; Hao, J.X.; Sollevi, A.; Xu, X.J.; Wiesenfeld-Hallin, Z. Marked enhancement of anti-allodynic effect by combined intrathecal administration of the adenosine A1-receptor agonist R-phenylisopropyladenosine and morphine in a rat model of central pain. Acta Anaesthesiol. Scand. 2000, 44, 665–671. [Google Scholar] [CrossRef]
- Ramos-Zepeda, G.A.; Herrero-Zorita, C.; Herrero, J.F. Interaction of the adenosine A1 receptor agonist N6-cyclopentyladenosine and κ-opioid receptors in rat spinal cord nociceptive reflexes. Behav. Pharm. 2014, 25, 741–749. [Google Scholar] [CrossRef]
- Borghi, V.; Przewlocka, B.; Labuz, D.; Maj, M.; Ilona, O.; Pavone, F. Formalin-induced pain and μ-opioid receptor density in brain and spinal cord are modulated by A1 and A2a adenosine agonists in mice. Brain Res. 2002, 956, 339–348. [Google Scholar] [CrossRef]
- Hwang, J.-H.; Hwang, G.-S.; Cho, S.-K.; Han, S.-M. Morphine can enhance the antiallodynic effect of intrathecal R-PIA in rats with nerve ligation injury. Anesth. Analg. 2005, 100, 461–468. [Google Scholar] [CrossRef]
- Zhang, Y.; Conklin, D.R.; Li, X.; Eisenach, J.C. Intrathecal morphine reduces allodynia after peripheral nerve injury in rats via activation of a spinal A1 adenosine receptor. Anesthesiology 2005, 102, 416–420. [Google Scholar] [CrossRef]
- Wu, W.-P.; Hao, J.-X.; Halldner, L.; Lövdahl, C.; DeLander, G.E.; Wiesenfeld-Hallin, Z.; Fredholm, B.B.; Xu, X.-J. Increased nociceptive response in mice lacking the adenosine A1 receptor. Pain 2005, 113, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Reid, A.R.; Sawynok, J. Antinociception by systemically-administered acetaminophen (paracetamol) involves spinal serotonin 5-HT7 and adenosine A1 receptors, as well as peripheral adenosine A1 receptors. Neurosci. Lett. 2013, 536, 64–68. [Google Scholar] [CrossRef]
- Sawynok, J.; Reid, A.R. Caffeine inhibits antinociception by acetaminophen in the formalin test by inhibiting spinal adenosine A1 receptors. Eur. J. Pharmacol. 2012, 674, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Sawynok, J.; Reid, A.R.; Fredholm, B.B. Caffeine reverses antinociception by amitriptyline in wild type mice but not in those lacking adenosine A1 receptors. Neurosci. Lett. 2008, 440, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Reid, A.R.; Sawynok, J. Spinal serotonin 5-HT7 and adenosine A1 receptors, as well as peripheral adenosine A1 receptors, are involved in antinociception by systemically administered amitriptyline. Eur. J. Pharmacol. 2013, 698, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Sawynok, J.; Reid, A.R.; Fredholm, B.B. Caffeine reverses antinociception by oxcarbazepine by inhibition of adenosine A1 receptors: Insights using knockout mice. Neurosci. Lett. 2010, 473, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Stepanović-Petrović, R.M.; Micov, A.M.; Tomić, M.A.; Ugrešić, N.D. The local peripheral antihyperalgesic effect of levetiracetam and its mechanism of action in an inflammatory pain model. Anesth. Analg. 2012, 115, 1457–1466. [Google Scholar] [CrossRef] [PubMed]
- Victor Holanda, A.D.; Asth, L.; Santos, A.R.; Guerrini, R.; Soares-Rachetti, V.d.P.; Caló, G.; André, E.; Gavioli, E.C. Central adenosine A1 and A2A receptors mediate the antinociceptive effects of neuropeptide S in the mouse formalin test. Life Sci. 2015, 120, 8–12. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Tabrizi, M.A.; Gessi, S.; Borea, P.A.; Merighi, S. Allosteric enhancers of A1 adenosine receptors: State of the art and new horizons for drug development. Curr. Med. Chem. 2010, 17, 3488–3502. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Moorman, A.R.; Borea, P.A.; Varani, K. Current status of A1 adenosine receptor allosteric enhancers. Future Med. Chem. 2015, 7, 1247–1259. [Google Scholar] [CrossRef] [PubMed]
- Urwyler, S. Allosteric modulation of family C G-protein-coupled receptors: From molecular insights to therapeutic perspectives. Pharmacol. Rev. 2011, 63, 59–126. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.L.; Xu, Z.; Leung, E.; Eisenach, J.C. Allosteric adenosine modulation to reduce allodynia. Anesthesiology 2001, 95, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Conklin, D.; Ma, W.; Zhu, X.; Eisenach, J.C. Spinal noradrenergic activation mediates allodynia reduction from an allosteric adenosine modulator in a rat model of neuropathic pain. Pain 2002, 97, 117–125. [Google Scholar] [CrossRef]
- Li, X.; Conklin, D.; Pan, H.-L.; Eisenach, J.C. Allosteric adenosine receptor modulation reduces hypersensitivity following peripheral inflammation by a central mechanism. J. Pharmacol. Exp. Ther. 2003, 305, 950–955. [Google Scholar] [CrossRef]
- Obata, H.; Li, X.; Eisenach, J.C. Spinal adenosine receptor activation reduces hypersensitivity after surgery by a different mechanism than after nerve injury. Anesthesiology 2004, 100, 1258–1262. [Google Scholar] [CrossRef]
- Romagnoli, R.; Baraldi, P.G.; Carrion, M.D.; Cara, C.L.; Cruz-Lopez, O.; Salvador, M.K.; Preti, D.; Tabrizi, M.A.; Moorman, A.R.; Vincenzi, F.; et al. Synthesis and biological evaluation of 2-amino-3-(4-chlorobenzoyl)-4-[(4-arylpiperazin-1-yl)methyl]-5-substituted-thiophenes. effect of the 5-modification on allosteric enhancer activity at the A1 adenosine receptor. J. Med. Chem. 2012, 55, 7719–7735. [Google Scholar] [CrossRef]
- Vincenzi, F.; Ravani, A.; Pasquini, S.; Merighi, S.; Gessi, S.; Romagnoli, R.; Baraldi, P.G.; Borea, P.A.; Varani, K. Positive allosteric modulation of A1 adenosine receptors as a novel and promising therapeutic strategy for anxiety. Neuropharmacology 2016, 111, 283–292. [Google Scholar] [CrossRef]
- Vincenzi, F.; Targa, M.; Romagnoli, R.; Merighi, S.; Gessi, S.; Baraldi, P.G.; Borea, P.A.; Varani, K. TRR469, a potent A1 adenosine receptor allosteric modulator, exhibits anti-nociceptive properties in acute and neuropathic pain models in mice. Neuropharmacology 2014, 81, 6–14. [Google Scholar] [CrossRef]
- Gomes, C.; Ferreira, R.; George, J.; Sanches, R.; Rodrigues, D.I.; Gonçalves, N.; Cunha, R.A. Activation of microglial cells triggers a release of brain-derived neurotrophic factor (BDNF) inducing their proliferation in an adenosine A2A receptor-dependent manner: A2A receptor blockade prevents BDNF release and proliferation of microglia. J. Neuroinflammation 2013, 10, 780. [Google Scholar] [CrossRef]
- Hussey, M.J.; Clarke, G.D.; Ledent, C.; Kitchen, I.; Hourani, S.M.O. Genetic deletion of the adenosine A2A receptor in mice reduces the changes in spinal cord NMDA receptor binding and glucose uptake caused by a nociceptive stimulus. Neurosci. Lett. 2010, 479, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Hambrecht-Wiedbusch, V.S.; Gabel, M.; Liu, L.J.; Imperial, J.P.; Colmenero, A.V.; Vanini, G. Preemptive Caffeine Administration Blocks the Increase in Postoperative Pain Caused by Previous Sleep Loss in the Rat: A Potential Role for Preoptic Adenosine A2A Receptors in Sleep–Pain Interactions. Sleep 2017, 40. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Hao, J.X.; Fredholm, B.B.; Schulte, G.; Wiesenfeld-Hallin, Z.; Xu, X.J. Peripheral adenosine A2A receptors are involved in carrageenan-induced mechanical hyperalgesia in mice. Neuroscience 2010, 170, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Varano, F.; Catarzi, D.; Vincenzi, F.; Betti, M.; Falsini, M.; Ravani, A.; Borea, P.A.; Colotta, V.; Varani, K. Design, Synthesis, and Pharmacological Characterization of 2-(2-Furanyl)thiazolo[5,4-d]pyrimidine-5,7-diamine Derivatives: New Highly Potent A2A Adenosine Receptor Inverse Agonists with Antinociceptive Activity. J. Med. Chem. 2016, 59, 10564–10576. [Google Scholar] [CrossRef] [PubMed]
- Varano, F.; Catarzi, D.; Vincenzi, F.; Falsini, M.; Pasquini, S.; Borea, P.A.; Colotta, V.; Varani, K. Structure-activity relationship studies and pharmacological characterization of N5-heteroarylalkyl-substituted-2-(2-furanyl)thiazolo[5,4-d]pyrimidine-5,7-diamine-based derivatives as inverse agonists at human A2A adenosine receptor. Eur. J. Med. Chem. 2018, 155, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Luongo, L.; Salvemini, D. Targeting metabotropic adenosine receptors for neuropathic pain: Focus on A2A. Brain Behav. Immun. 2018, 69, 60–61. [Google Scholar] [CrossRef]
- Bura, A.S.; Nadal, X.; Ledent, C.; Maldonado, R.; Valverde, O. A2A adenosine receptor regulates glia proliferation and pain after peripheral nerve injury. Pain 2008, 140, 95–103. [Google Scholar] [CrossRef]
- Areti, A.; Yerra, V.G.; Naidu, V.; Kumar, A. Oxidative stress and nerve damage: Role in chemotherapy induced peripheral neuropathy. Redox Biol. 2014, 2, 289–295. [Google Scholar] [CrossRef]
- Carrasco, C.; Naziroǧlu, M.; Rodríguez, A.B.; Pariente, J.A. Neuropathic Pain: Delving into the Oxidative Origin and the Possible Implication of Transient Receptor Potential Channels. Front. Physiol. 2018, 9, 95. [Google Scholar] [CrossRef]
- Kim, H.K.; Park, S.K.; Zhou, J.-L.; Taglialatela, G.; Chung, K.; Coggeshall, R.E.; Chung, J.M. Reactive oxygen species (ROS) play an important role in a rat model of neuropathic pain. Pain 2004, 111, 116–124. [Google Scholar] [CrossRef]
- Han, Y.; Smith, M.T. Pathobiology of cancer chemotherapy-induced peripheral neuropathy (CIPN). Front. Pharm. 2013, 4, 156. [Google Scholar] [CrossRef] [PubMed]
- Kerckhove, N.; Collin, A.; Condé, S.; Chaleteix, C.; Pezet, D.; Balayssac, D. Long-Term Effects, Pathophysiological Mechanisms, and Risk Factors of Chemotherapy-Induced Peripheral Neuropathies: A Comprehensive Literature Review. Front. Pharm. 2017, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Naik, A.K.; Tandan, S.K.; Dudhgaonkar, S.P.; Jadhav, S.H.; Kataria, M.; Prakash, V.R.; Kumar, D. Role of oxidative stress in pathophysiology of peripheral neuropathy and modulation by N-acetyl-L-cysteine in rats. Eur. J. Pain 2006, 10, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.S.; Bae, C.; Wang, J.; Lee, K.-H.; Hankerd, K.M.; Kim, H.K.; Chung, J.M.; La, J.-H. Peripheral and central oxidative stress in chemotherapy-induced neuropathic pain. Mol. Pain 2019, 15, 1–11. [Google Scholar] [CrossRef]
- Falsini, M.; Catarzi, D.; Varano, F.; Ceni, C.; Dal Ben, D.; Marucci, G.; Buccioni, M.; Volpini, R.; Di Cesare Mannelli, L.; Lucarini, E.; et al. Antioxidant-Conjugated 1,2,4-Triazolo[4,3-a]pyrazin-3-one Derivatives: Highly Potent and Selective Human A2A Adenosine Receptor Antagonists Possessing Protective Efficacy in Neuropathic Pain. J. Med. Chem. 2019, 62, 8511–8531. [Google Scholar] [CrossRef]
- Betti, M.; Catarzi, D.; Varano, F.; Falsini, M.; Varani, K.; Vincenzi, F.; Pasquini, S.; di Cesare Mannelli, L.; Ghelardini, C.; Lucarini, E.; et al. Modifications on the Amino-3,5-dicyanopyridine Core to Obtain Multifaceted Adenosine Receptor Ligands with Antineuropathic Activity. J. Med. Chem. 2019, 62, 6894–6912. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Giancotti, L.A.; Lauro, F.; Mufti, F.; Salvemini, D. Treatment of chronic neuropathic pain: Purine receptor modulation. Pain 2020, 161, 1425–1441. [Google Scholar] [CrossRef] [PubMed]
- Hussey, M.J.; Clarke, G.D.; Ledent, C.; Kitchen, I.; Hourani, S.M.O. Deletion of the adenosine A2A receptor in mice enhances spinal cord neurochemical responses to an inflammatory nociceptive stimulus. Neurosci. Lett. 2012, 506, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Varani, K.; Padovan, M.; Govoni, M.; Vincenzi, F.; Trotta, F.; Borea, P.A. The role of adenosine receptors in rheumatoid arthritis. Autoimmun. Rev. 2010, 10, 61–64. [Google Scholar] [CrossRef]
- Varani, K.; Padovan, M.; Vincenzi, F.; Targa, M.; Trotta, F.; Govoni, M.; Borea, P.A. A2A and A3 adenosine receptor expression in rheumatoid arthritis: Upregulation, inverse correlation with disease activity score and suppression of inflammatory cytokine and metalloproteinase release. Arthritis Res. Ther. 2011, 13, R197. [Google Scholar] [CrossRef]
- Mazzon, E.; Esposito, E.; Impellizzeri, D.; Di Paola, R.; Melani, A.; Bramanti, P.; Pedata, F.; Cuzzocrea, S. CGS 21680, an Agonist of the Adenosine (A2A) receptor, reduces progression of murine type II collagen-induced arthritis. J. Rheumatol. 2011, 38, 2119–2129. [Google Scholar] [CrossRef] [PubMed]
- Vincenzi, F.; Padovan, M.; Targa, M.; Corciulo, C.; Giacuzzo, S.; Merighi, S.; Gessi, S.; Govoni, M.; Borea, P.A.; Varani, K. A(2A) adenosine receptors are differentially modulated by pharmacological treatments in rheumatoid arthritis patients and their stimulation ameliorates adjuvant-induced arthritis in rats. PLoS ONE 2013, 8, e54195. [Google Scholar] [CrossRef] [PubMed]
- Ravani, A.; Vincenzi, F.; Bortoluzzi, A.; Padovan, M.; Pasquini, S.; Gessi, S.; Merighi, S.; Borea, P.A.; Govoni, M.; Varani, K. Role and Function of A2A and A3 Adenosine Receptors in Patients with Ankylosing Spondylitis, Psoriatic Arthritis and Rheumatoid Arthritis. Int. J. Mol. Sci. 2017, 18, 697. [Google Scholar] [CrossRef] [PubMed]
- Montes, G.C.; Hammes, N.; da Rocha, M.D.; Montagnoli, T.L.; Fraga, C.A.M.; Barreiro, E.J.; Sudo, R.T.; Zapata-Sudo, G. Treatment with Adenosine Receptor Agonist Ameliorates Pain Induced by Acute and Chronic Inflammation. J. Pharmacol. Exp. Ther. 2016, 358, 315–323. [Google Scholar] [CrossRef]
- Loram, L.C.; Harrison, J.A.; Sloane, E.M.; Hutchinson, M.R.; Sholar, P.; Taylor, F.R.; Berkelhammer, D.; Coats, B.D.; Poole, S.; Milligan, E.D.; et al. Enduring Reversal of Neuropathic Pain by a Single Intrathecal Injection of Adenosine 2A Receptor Agonists: A Novel Therapy for Neuropathic Pain. J. Neurosci. 2009, 29, 14015–14025. [Google Scholar] [CrossRef]
- Loram, L.C.; Taylor, F.R.; Strand, K.A.; Harrison, J.A.; RzasaLynn, R.; Sholar, P.; Rieger, J.; Maier, S.F.; Watkins, L.R. Intrathecal injection of adenosine 2A receptor agonists reversed neuropathic allodynia through protein kinase (PK)A/PKC signaling. Brain Behav. Immun. 2013, 33, 112–122. [Google Scholar] [CrossRef]
- Kwilasz, A.J.; Ellis, A.; Wieseler, J.; Loram, L.; Favret, J.; McFadden, A.; Springer, K.; Falci, S.; Rieger, J.; Maier, S.F.; et al. Sustained reversal of central neuropathic pain induced by a single intrathecal injection of adenosine A2A receptor agonists. Brain Behav. Immun. 2018, 69, 470–479. [Google Scholar] [CrossRef]
- Kwilasz, A.J.; Green Fulgham, S.M.; Ellis, A.; Patel, H.P.; Duran-Malle, J.C.; Favret, J.; Harvey, L.O.; Rieger, J.; Maier, S.F.; Watkins, L.R. A single peri-sciatic nerve administration of the adenosine 2A receptor agonist ATL313 produces long-lasting anti-allodynia and anti-inflammatory effects in male rats. Brain Behav. Immun. 2019, 76, 116–125. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Tosh, D.K.; Jain, S.; Gao, Z.-G. Historical and Current Adenosine Receptor Agonists in Preclinical and Clinical Development. Front. Cell Neurosci. 2019, 13, 124. [Google Scholar] [CrossRef]
- By, Y.; Condo, J.; Durand-Gorde, J.-M.; Lejeune, P.-J.; Mallet, B.; Guieu, R.; Ruf, J. Intracerebroventricular injection of an agonist-like monoclonal antibody to adenosine A(2A) receptor has antinociceptive effects in mice. J. Neuroimmunol. 2011, 230, 178–182. [Google Scholar] [CrossRef]
- Feoktistov, I.; Biaggioni, I. Chapter 5—Role of Adenosine A2B Receptors in Inflammation. In Advances in Pharmacology; Jacobson, K.A., Linden, J., Eds.; Pharmacology of Purine and Pyrimidine Receptors; Academic Press: Cambridge, MA, USA, 2011; Volume 61, pp. 115–144. [Google Scholar]
- Popoli, P.; Pepponi, R. Potential Therapeutic Relevance of Adenosine A2B and A2A Receptors in the Central Nervous System. CNS Neurol. Disord.-Drug Targets (Former. Curr. Drug Targets CNS Neurol. Disord.) 2012, 11, 664–674. [Google Scholar] [CrossRef]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Müller, C.E. International union of basic and clinical pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—An update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Abo-Salem, O.M.; Hayallah, A.M.; Bilkei-Gorzo, A.; Filipek, B.; Zimmer, A.; Müller, C.E. Antinociceptive effects of novel A2B adenosine receptor antagonists. J. Pharmacol. Exp. Ther. 2004, 308, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Bilkei-Gorzo, A.; Abo-Salem, O.M.; Hayallah, A.M.; Michel, K.; Müller, C.E.; Zimmer, A. Adenosine receptor subtype-selective antagonists in inflammation and hyperalgesia. Naunyn. Schmiedebergs. Arch. Pharmacol. 2008, 377, 65–76. [Google Scholar] [CrossRef]
- Yoon, M.H.; Bae, H.B.; Choi, J.I.; Kim, S.J.; Chung, S.T.; Kim, C.M. Roles of Adenosine Receptor Subtypes in the Antinociceptive Effect of Intrathecal Adenosine in a Rat Formalin Test. PHA 2006, 78, 21–26. [Google Scholar] [CrossRef]
- Asano, T.; Tanaka, K.-I.; Tada, A.; Shimamura, H.; Tanaka, R.; Maruoka, H.; Takenaga, M.; Mizushima, T. Aminophylline suppresses stress-induced visceral hypersensitivity and defecation in irritable bowel syndrome. Sci. Rep. 2017, 7, 40214. [Google Scholar] [CrossRef] [PubMed]
- Asano, T.; Takenaga, M. Adenosine A2B Receptors: An Optional Target for the Management of Irritable Bowel Syndrome with Diarrhea? J. Clin. Med. 2017, 6, 104. [Google Scholar] [CrossRef]
- Savegnago, L.; Jesse, C.R.; Nogueira, C.W. Caffeine and a selective adenosine A(2B) receptor antagonist but not imidazoline receptor antagonists modulate antinociception induced by diphenyl diselenide in mice. Neurosci. Lett. 2008, 436, 120–123. [Google Scholar] [CrossRef]
- Hu, X.; Adebiyi, M.G.; Luo, J.; Sun, K.; Le, T.-T.T.; Zhang, Y.; Wu, H.; Zhao, S.; Karmouty-Quintana, H.; Liu, H.; et al. Sustained Elevated Adenosine via ADORA2B Promotes Chronic Pain through Neuro-immune Interaction. Cell Rep. 2016, 16, 106–119. [Google Scholar] [CrossRef]
- Chen, J.-F.; Lee, C.; Chern, Y. Chapter One—Adenosine Receptor Neurobiology: Overview. In International Review of Neurobiology; Mori, A., Ed.; Adenosine Receptors in Neurology and Psychiatry; Academic Press: Cambridge, MA, USA, 2014; Volume 119, pp. 1–49. [Google Scholar]
- Borea, P.A.; Varani, K.; Vincenzi, F.; Baraldi, P.G.; Tabrizi, M.A.; Merighi, S.; Gessi, S. The A3 Adenosine Receptor: History and Perspectives. Pharm. Rev. 2015, 67, 74–102. [Google Scholar] [CrossRef]
- Fishman, P.; Bar-Yehuda, S.; Liang, B.T.; Jacobson, K.A. Pharmacological and therapeutic effects of A3 adenosine receptor agonists. Drug Discov. Today 2012, 17, 359–366. [Google Scholar] [CrossRef]
- Wu, W.-P.; Hao, J.-X.; Halldner-Henriksson, L.; Xu, X.-J.; Jacobson, M.A.; Wiesenfeld-Hallin, Z.; Fredholm, B.B. Decreased inflammatory pain due to reduced carrageenan-induced inflammation in mice lacking adenosine A3 receptors. Neuroscience 2002, 114, 523–527. [Google Scholar] [CrossRef]
- Chen, Z.; Janes, K.; Chen, C.; Doyle, T.; Bryant, L.; Tosh, D.K.; Jacobson, K.A.; Salvemini, D. Controlling murine and rat chronic pain through A3 adenosine receptor activation. FASEB J. 2012, 26, 1855–1865. [Google Scholar] [CrossRef] [PubMed]
- Janes, K.; Symons-Liguori, A.; Jacobson, K.A.; Salvemini, D. Identification of A3 adenosine receptor agonists as novel non-narcotic analgesics. Br. J. Pharm. 2016, 173, 1253–1267. [Google Scholar] [CrossRef]
- Little, J.W.; Ford, A.; Symons-Liguori, A.M.; Chen, Z.; Janes, K.; Doyle, T.; Xie, J.; Luongo, L.; Tosh, D.K.; Maione, S.; et al. Endogenous adenosine A3 receptor activation selectively alleviates persistent pain states. Brain 2015, 138, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Ford, A.; Castonguay, A.; Cottet, M.; Little, J.W.; Chen, Z.; Symons-Liguori, A.M.; Doyle, T.; Egan, T.M.; Vanderah, T.W.; Koninck, Y.D.; et al. Engagement of the GABA to KCC2 Signaling Pathway Contributes to the Analgesic Effects of A3AR Agonists in Neuropathic Pain. J. Neurosci. 2015, 35, 6057–6067. [Google Scholar] [CrossRef] [PubMed]
- Coppi, E.; Cherchi, F.; Fusco, I.; Failli, P.; Vona, A.; Dettori, I.; Gaviano, L.; Lucarini, E.; Jacobson, K.A.; Tosh, D.K.; et al. Adenosine A3 receptor activation inhibits pronociceptive N-type Ca2+ currents and cell excitability in dorsal root ganglion neurons. Pain 2019, 160, 1103–1118. [Google Scholar] [CrossRef]
- Terayama, R.; Tabata, M.; Maruhama, K.; Iida, S. A3 adenosine receptor agonist attenuates neuropathic pain by suppressing activation of microglia and convergence of nociceptive inputs in the spinal dorsal horn. Exp. Brain Res. 2018, 236, 3203–3213. [Google Scholar] [CrossRef]
- Suresh, R.R.; Jain, S.; Chen, Z.; Tosh, D.K.; Ma, Y.; Podszun, M.C.; Rotman, Y.; Salvemini, D.; Jacobson, K.A. Design and in vivo activity of A3 adenosine receptor agonist prodrugs. Purinergic Signal. 2020, 16, 367–377. [Google Scholar] [CrossRef]
- Janes, K.; Esposito, E.; Doyle, T.; Cuzzocrea, S.; Tosh, D.K.; Jacobson, K.A.; Salvemini, D. A3 adenosine receptor agonist prevents the development of paclitaxel-induced neuropathic pain by modulating spinal glial-restricted redox-dependent signaling pathways. Pain 2014, 155, 2560–2567. [Google Scholar] [CrossRef]
- Kim, Y.; Kwon, S.Y.; Jung, H.S.; Park, Y.J.; Kim, Y.S.; In, J.H.; Choi, J.W.; Kim, J.A.; Joo, J.D. Amitriptyline inhibits the MAPK/ERK and CREB pathways and proinflammatory cytokines through A3AR activation in rat neuropathic pain models. Korean J. Anesth. 2019, 72, 60–67. [Google Scholar] [CrossRef]
- Wahlman, C.; Doyle, T.M.; Little, J.W.; Luongo, L.; Janes, K.; Chen, Z.; Esposito, E.; Tosh, D.K.; Cuzzocrea, S.; Jacobson, K.A.; et al. Chemotherapy-induced pain is promoted by enhanced spinal adenosine kinase levels through astrocyte-dependent mechanisms. Pain 2018, 159, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Haskó, G. Immunity, inflammation and cancer: A leading role for adenosine. Nat. Rev. Cancer 2013, 13, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Varani, K.; Vincenzi, F.; Targa, M.; Paradiso, B.; Parrilli, A.; Fini, M.; Lanza, G.; Borea, P.A. The stimulation of A3 adenosine receptors reduces bone-residing breast cancer in a rat preclinical model. Eur. J. Cancer 2013, 49, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhang, E.; Feng, C.; Zhao, X. Role of A3 adenosine receptor in diabetic neuropathy. J. Neurosci. Res. 2016, 94, 936–946. [Google Scholar] [CrossRef]
- Hayhurst, C.J.; Durieux, M.E. Differential Opioid Tolerance and Opioid-induced HyperalgesiaA Clinical Reality. Anesthesiology 2016, 124, 483–488. [Google Scholar] [CrossRef]
- Rivat, C.; Ballantyne, J. The dark side of opioids in pain management: Basic science explains clinical observation. Pain Rep. 2016, 1, e570. [Google Scholar] [CrossRef]
- Doyle, T.M.; Largent-Milnes, T.M.; Chen, Z.; Staikopoulos, V.; Esposito, E.; Dalgarno, R.; Fan, C.; Tosh, D.K.; Cuzzocrea, S.; Jacobson, K.A.; et al. Chronic Morphine-Induced Changes in Signaling at the A3 Adenosine Receptor Contribute to Morphine-Induced Hyperalgesia, Tolerance, and Withdrawal. J. Pharmacol. Exp. Ther. 2020, 374, 331–341. [Google Scholar] [CrossRef]
- Antonioli, L.; Lucarini, E.; Lambertucci, C.; Fornai, M.; Pellegrini, C.; Benvenuti, L.; Di Cesare Mannelli, L.; Spinaci, A.; Marucci, G.; Blandizzi, C.; et al. The Anti-Inflammatory and Pain-Relieving Effects of AR170, an Adenosine A3 Receptor Agonist, in a Rat Model of Colitis. Cells 2020, 9, 1509. [Google Scholar] [CrossRef]
- Lucarini, E.; Coppi, E.; Micheli, L.; Parisio, C.; Vona, A.; Cherchi, F.; Pugliese, A.M.; Pedata, F.; Failli, P.; Palomino, S.; et al. Acute visceral pain relief mediated by A3AR agonists in rats: Involvement of N-type voltage-gated calcium channels. Pain 2020. [Google Scholar] [CrossRef]
- Grundy, L.; Harrington, A.M.; Castro, J.; Garcia-Caraballo, S.; Deiteren, A.; Maddern, J.; Rychkov, G.Y.; Ge, P.; Peters, S.; Feil, R.; et al. Chronic linaclotide treatment reduces colitis-induced neuroplasticity and reverses persistent bladder dysfunction. JCI Insight 2018, 3, e121841. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Barer, F.; Bar-Yehuda, S.; IJzerman, A.P.; Jacobson, K.A.; Fishman, P. A₃ adenosine receptor allosteric modulator induces an anti-inflammatory effect: In vivo studies and molecular mechanism of action. Mediat. Inflamm. 2014, 2014, 708746. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vincenzi, F.; Pasquini, S.; Borea, P.A.; Varani, K. Targeting Adenosine Receptors: A Potential Pharmacological Avenue for Acute and Chronic Pain. Int. J. Mol. Sci. 2020, 21, 8710. https://doi.org/10.3390/ijms21228710
Vincenzi F, Pasquini S, Borea PA, Varani K. Targeting Adenosine Receptors: A Potential Pharmacological Avenue for Acute and Chronic Pain. International Journal of Molecular Sciences. 2020; 21(22):8710. https://doi.org/10.3390/ijms21228710
Chicago/Turabian StyleVincenzi, Fabrizio, Silvia Pasquini, Pier Andrea Borea, and Katia Varani. 2020. "Targeting Adenosine Receptors: A Potential Pharmacological Avenue for Acute and Chronic Pain" International Journal of Molecular Sciences 21, no. 22: 8710. https://doi.org/10.3390/ijms21228710
APA StyleVincenzi, F., Pasquini, S., Borea, P. A., & Varani, K. (2020). Targeting Adenosine Receptors: A Potential Pharmacological Avenue for Acute and Chronic Pain. International Journal of Molecular Sciences, 21(22), 8710. https://doi.org/10.3390/ijms21228710