PNPLA3 I148M Up-Regulates Hedgehog and Yap Signaling in Human Hepatic Stellate Cells

 ,
,  , and
, and
Abstract
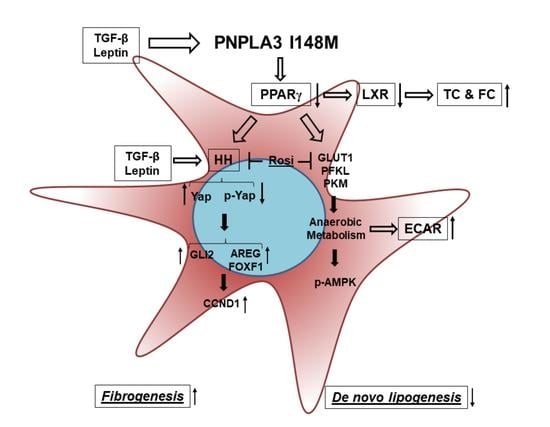
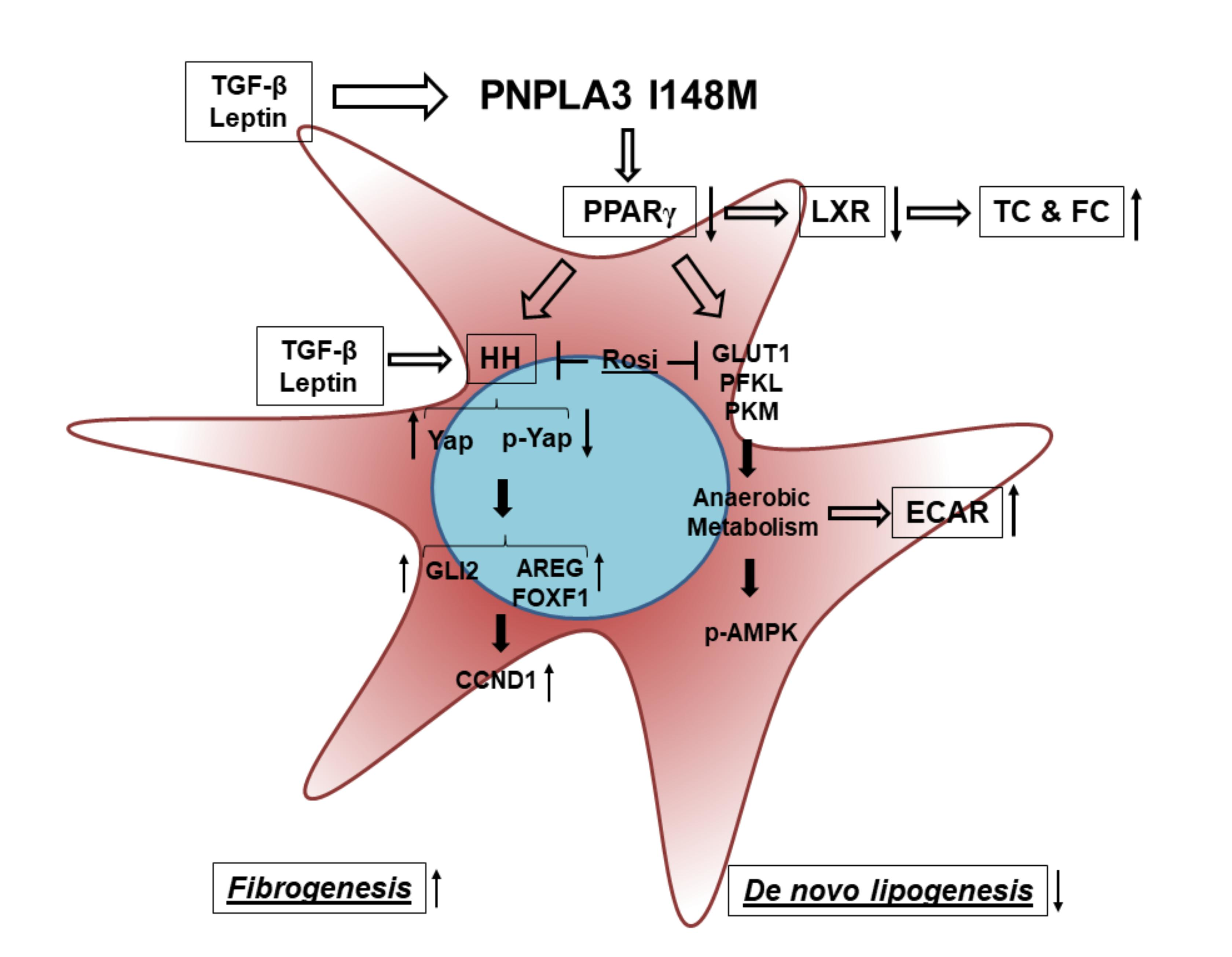{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
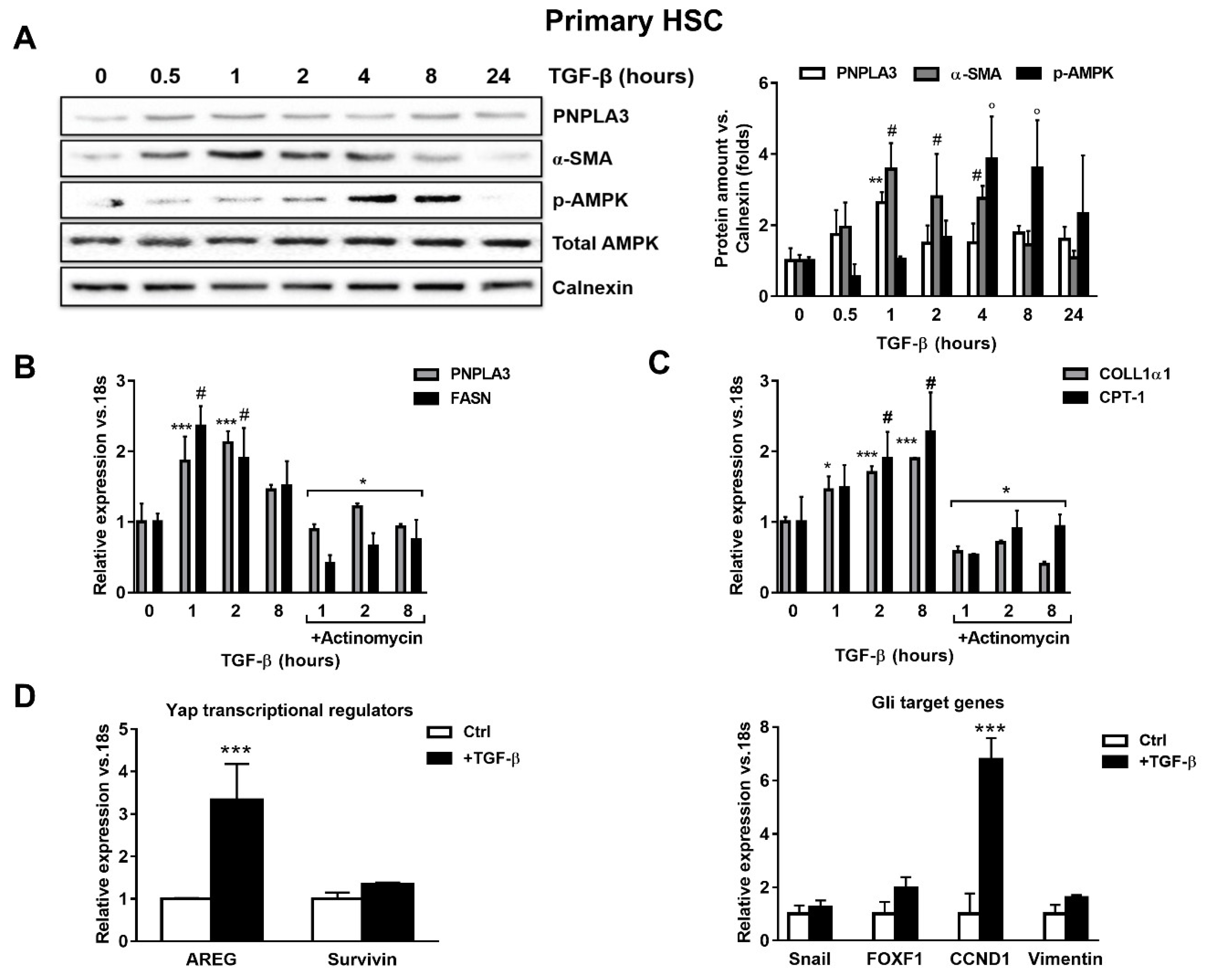
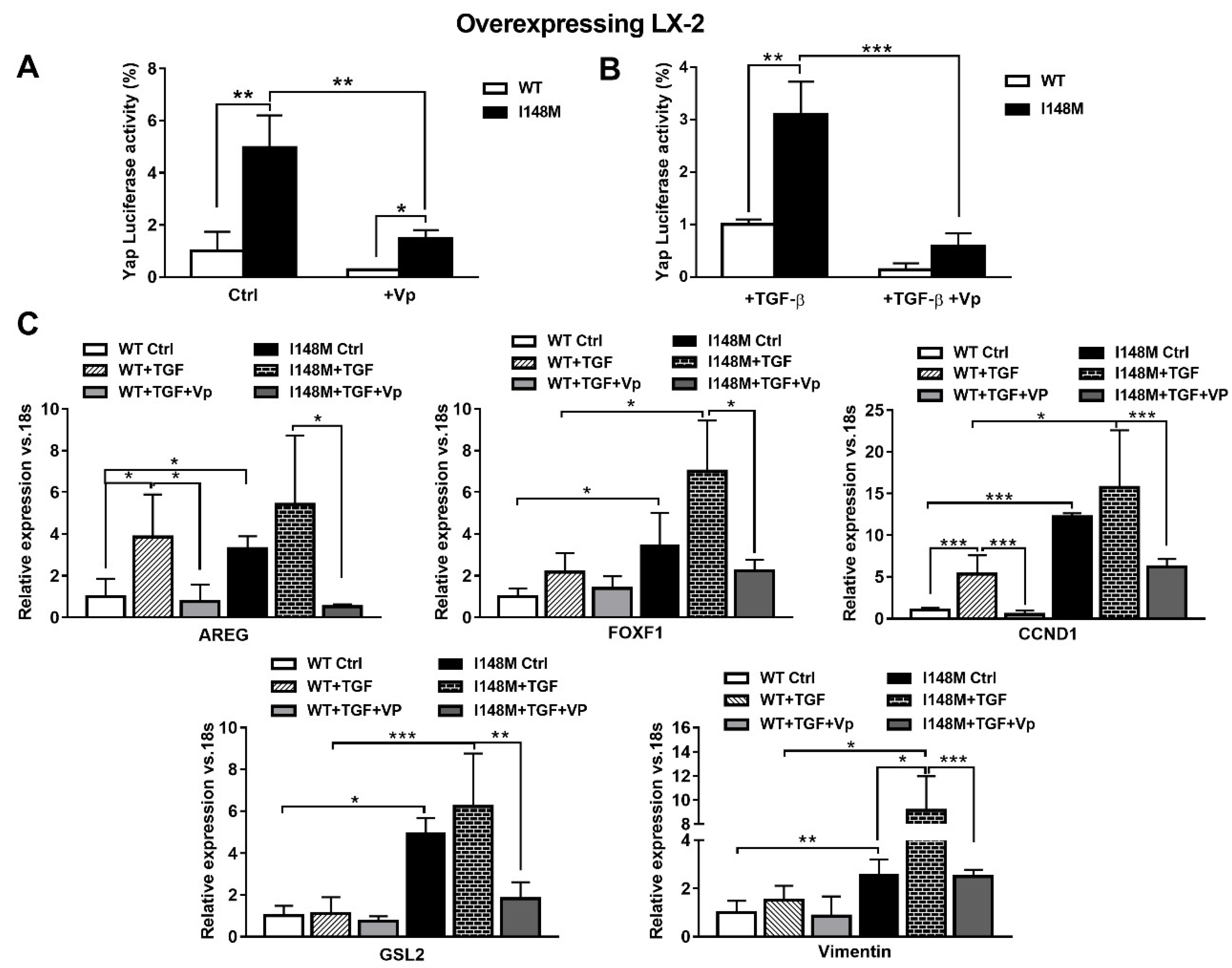
2.1. TGF-β Induces PNPLA3 Protein and Transcript Expression and Up-Regulates Yap/Hedgehog Target Genes in HSCs
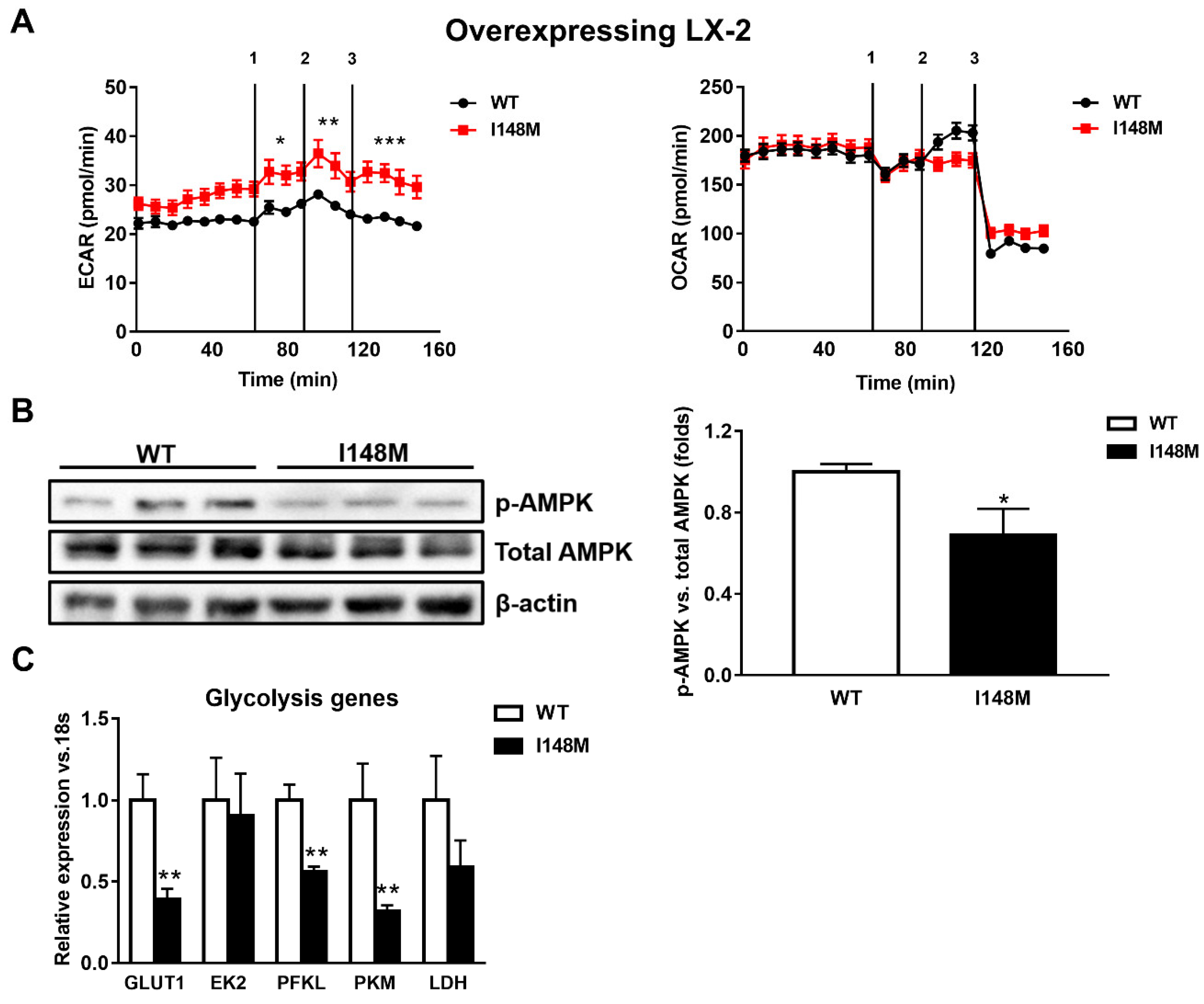
2.2. HSCs Expressing PNPLA3 I148M Have Enhanced Glycolysis Compared to WT Cells
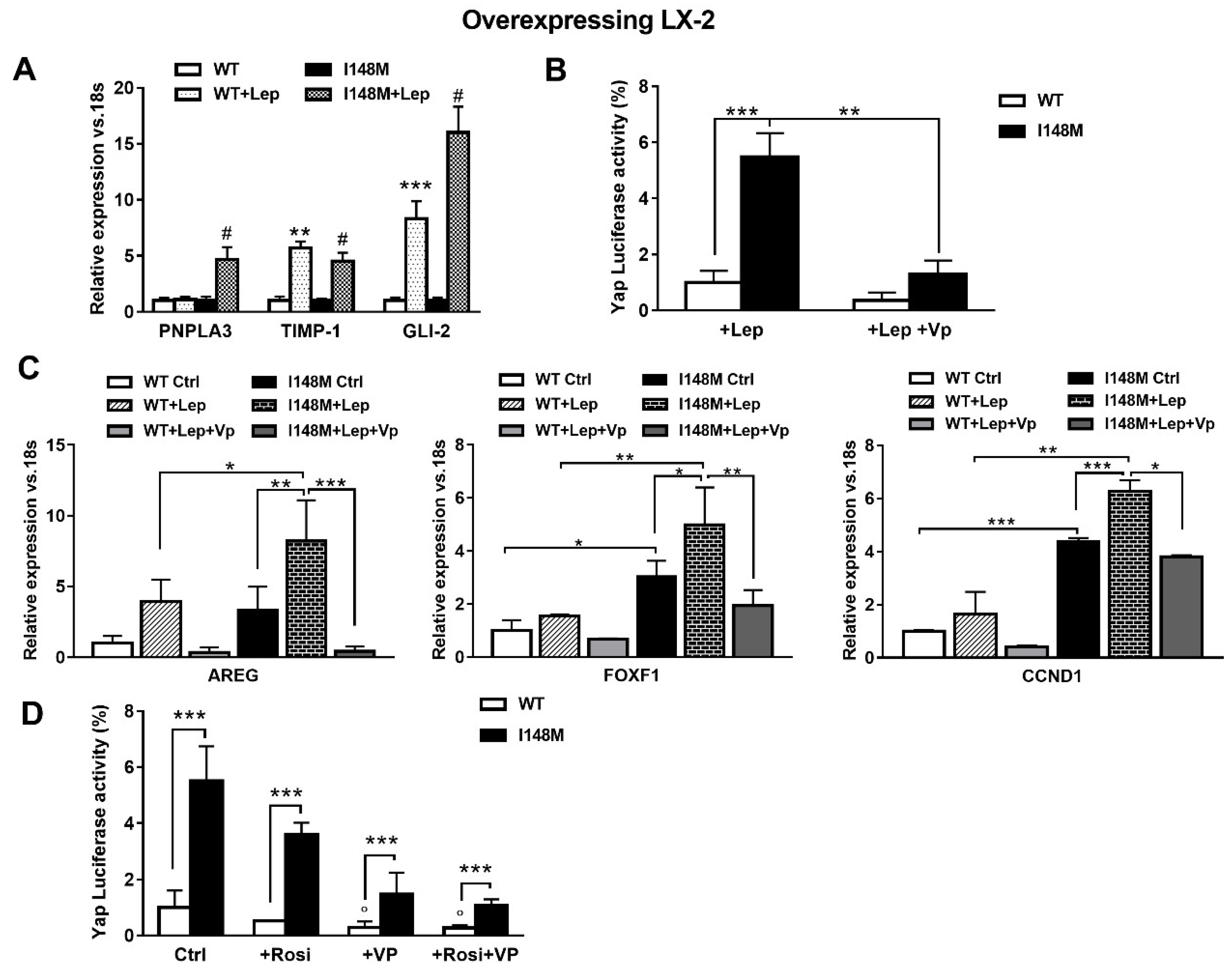
2.3. Hedgehog and Hippo/Yap Signaling are Up-regulated in HSCs with the PNPLA3 I148M Variant
2.4. The Specific Yap Inhibitor Verteporfin Strongly Reduces Yap-Mediated Gene Expression and TGF-β and Leptin Profibrogenic Actions, in Both WT and I148M Overexpressing HSCs
3. Materials and Methods
3.1. Human HSC Isolation and Cell Culture
3.2. Metabolic Characterization
3.3. Luciferase Assay
3.4. Flow Cytometry
3.5. Statistics
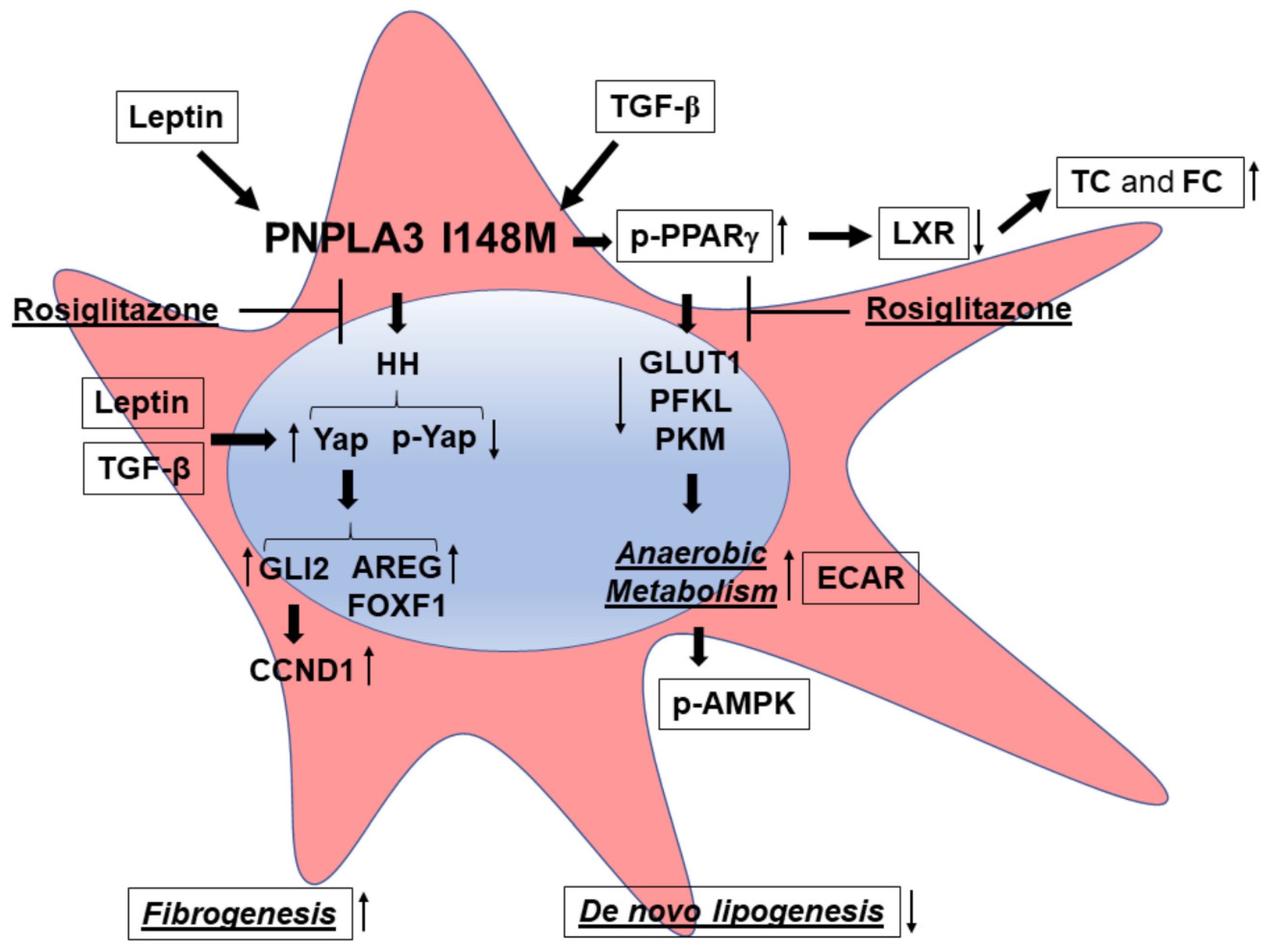
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMPK | 5′ AMP-activated protein kinase |
| AREG | amphiregulin |
| α-SMA | α-smooth muscle actin |
| CCND1 | cyclin D1 |
| COLL1α1 | type I collagen |
| CPT-1 | carnitine palmitoyl-transferase 1 |
| ECM | extracellular matrix |
| FASN | fatty acid synthase |
| FFA | free fatty acid |
| FOXF1 | forkhead box protein F1 |
| GLS-2 | glutaminase 2 |
| GLUT1 | glucose transporter 1 |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| HCC | hepatocellular carcinoma |
| HH | hedgehog |
| HIP | indian hedgehog |
| HHIP | hedgehog-interacting protein |
| HK | hexokinase-2 |
| HSCs | hepatic stellate cells |
| Lep | leptin |
| LPK | liver pyruvate kinase |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| PFKL | phosphofructokinase liver type |
| PKM | pyruvate kinase muscle |
| PNPLA3 | patatin-like phospholipase domain-containing protein 3 (C/C = wild type) |
| PNPLA3 I148M | patatin-like phospholipase domain-containing protein 3 genetic variant I148M (G/G) |
| TG | triglycerides |
| Vp | verteporfin |
| WT | PNPLA3 wild type (C/C) |
| Yap | yes-associated protein 1 |
References
- Alkhouri, N.; Lawitz, E.; Noureddin, M. Looking Into the Crystal Ball: Predicting the Future Challenges of Fibrotic NASH Treatment. Hepatol. Commun. 2019, 3, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P.; Kleiner, D.E.; Dam-Larsen, S.; Adams, L.A.; Bjornsson, E.S.; Charatcharoenwitthaya, P.; Mills, P.R.; Keach, J.C.; Lafferty, H.D.; Stahler, A.; et al. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2015, 149, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Vilar-Gomez, E.; Calzadilla-Bertot, L.; Wong, V.W.-S.; Castellanos, M.; La Fuente, R.A.-D.; Metwally, M.; Eslam, M.; Gonzalez-Fabian, L.; Sanz, M.A.-Q.; Conde-Martin, A.F.; et al. Fibrosis Severity as a Determinant of Cause-Specific Mortality in Patients With Advanced Nonalcoholic Fatty Liver Disease: A Multi-National Cohort Study. Gastroenterology 2018, 155, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Diehl, A.M.; Day, C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef] [PubMed]
- Marcellin, P.; Kutala, B.K. Liver diseases: A major, neglected global public health problem requiring urgent actions and large-scale screening. Liver Int. 2018, 38, 2–6. [Google Scholar] [CrossRef]
- McGlynn, K.A.; Petrick, J.L.; London, W.T. Global Epidemiology of Hepatocellular Carcinoma: An Emphasis on Demographic and Regional Variability. Clin. Liver Dis. 2015, 19, 2232–2238. [Google Scholar] [CrossRef]
- Mederacke, I.; Dapito, D.H.; Affò, S.; Uchinami, H.; Schwabe, R.F. High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat. Protoc. 2015, 10, 305–315. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Patman, G.; Leathart, J.B.S.; Piguet, A.-C.; Burt, A.D.; Dufour, J.-F.; Day, C.; Daly, A.K.; Reeves, H.L.; Anstee, Q.M. Carriage of the PNPLA3 rs738409 C >G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J. Hepatol. 2014, 61, 75–81. [Google Scholar] [CrossRef]
- Hinz, B. The extracellular matrix and transforming growth factor-β1: Tale of a strained relationship. Matrix Biol. 2015, 47, 54–65. [Google Scholar] [CrossRef]
- Javelaud, D.; Pierrat, M.-J.; Mauviel, A. Crosstalk between TGF-β and hedgehog signaling in cancer. FEBS Lett. 2012, 586, 2016–2025. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Choi, S.S.; Michelotti, G.A.; Chan, I.S.; Swiderska-Syn, M.; Karaca, G.F.; Xie, G.; Moylan, C.A.; Garibaldi, F.; Premont, R.; et al. Hedgehog Controls Hepatic Stellate Cell Fate by Regulating Metabolism. Gastroenterology 2012, 143, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef]
- Saxena, N.K.; Ikeda, K.; Rockey, D.C.; Friedman, S.L.; Anania, F.A. Leptin in hepatic fibrosis: Evidence for increased collagen production in stellate cells and lean littermates of ob/ob mice. Hepatology 2002, 35, 762–771. [Google Scholar] [CrossRef]
- Choi, S.S.; Syn, W.-K.; Karaca, G.F.; Omenetti, A.; Moylan, C.A.; Witek, R.P.; Agboola, K.M.; Jung, Y.; Michelotti, G.A.; Diehl, A.M. Leptin Promotes the Myofibroblastic Phenotype in Hepatic Stellate Cells by Activating the Hedgehog Pathway. J. Biol. Chem. 2010, 285, 36551–36560. [Google Scholar] [CrossRef]
- Rotman, Y.; Koh, C.; Zmuda, J.M.; Kleiner, D.E.; Liang, T.J.; Crn, T.N. The association of genetic variability in patatin-like phospholipase domain-containing protein 3 (PNPLA3) with histological severity of nonalcoholic fatty liver disease. Hepatology 2010, 52, 894–903. [Google Scholar] [CrossRef]
- Bruschi, F.V.; Tardelli, M.; Claudel, T.; Trauner, M. PNPLA3 expression and its impact on the liver: Current perspectives. Hepatic Med. Évid. Res. 2017, 9, 55–66. [Google Scholar] [CrossRef]
- Bruschi, F.V.; Tardelli, M.; Herac, M.; Claudel, T.; Trauner, M. Metabolic regulation of hepatic PNPLA3 expression and severity of liver fibrosis in patients with NASH. Liver Int. 2020, 40, 1098–1110. [Google Scholar] [CrossRef]
- Bruschi, F.V.; Claudel, T.; Tardelli, M.; Starlinger, P.; Marra, F.; Trauner, M. PNPLA3 I148M Variant Impairs Liver X Receptor Signaling and Cholesterol Homeostasis in Human Hepatic Stellate Cells. Hepatol. Commun. 2019, 3, 1191–1204. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, F.V.; Claudel, T.; Tardelli, M.; Caligiuri, A.; Stulnig, T.M.; Marra, F.; Trauner, M. The PNPLA3 I148M variant modulates the fibrogenic phenotype of human hepatic stellate cells. Hepatology 2017, 65, 1875–1890. [Google Scholar] [CrossRef] [PubMed]
- She, H.; Xiong, S.; Hazra, S.; Tsukamoto, H. Adipogenic Transcriptional Regulation of Hepatic Stellate Cells. J. Biol. Chem. 2005, 280, 4959–4967. [Google Scholar] [CrossRef]
- Mannaerts, I.; Leite, S.B.; Verhulst, S.; Claerhout, S.; Eysackers, N.; Thoen, L.F.; Hoorens, A.; Reynaert, H.; Halder, G.; Van Grunsven, L.A. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J. Hepatol. 2015, 63, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Swiderska-Syn, M.; Xie, G.; Michelotti, G.A.; Jewell, M.L.; Premont, R.T.; Syn, W.-K.; Diehl, A.M. Hedgehog regulates yes-associated protein 1 in regenerating mouse liver. Hepatology 2016, 64, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Plitzko, B.; Loesgen, S. Measurement of Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR) in Culture Cells for Assessment of the Energy Metabolism. Bio-Protoc. 2018, 8. [Google Scholar] [CrossRef]
- Tardelli, M.; Claudel, T.; Bruschi, F.V.; Moreno-Viedma, V.; Trauner, M. Adiponectin regulates AQP3 via PPARα in human hepatic stellate cells. Biochem. Biophys. Res. Commun. 2017, 490, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cai, B.; Yang, X.; Sonubi, O.O.; Zheng, Z.; Ramakrishnan, R.; Shi, H.; Valenti, L.; Pajvani, U.B.; Sandhu, J.; et al. Cholesterol Stabilizes TAZ in Hepatocytes to Promote Experimental Non-alcoholic Steatohepatitis. Cell Metab. 2020, 31, 969–986. [Google Scholar] [CrossRef]
- Pingitore, P.; Dongiovanni, P.; Motta, B.M.; Meroni, M.; Lepore, S.M.; Mancina, R.M.; Pelusi, S.; Russo, C.; Caddeo, A.; Rossi, G.; et al. PNPLA3 overexpression results in reduction of proteins predisposing to fibrosis. Hum. Mol. Genet. 2016, 25, 5212–5222. [Google Scholar] [CrossRef]
- Viollet, B.; Foretz, M.; Guigas, B.; Horman, S.; Dentin, R.; Bertrand, L.; Hue, L.; Andreelli, F. Activation of AMP-activated protein kinase in the liver: A new strategy for the management of metabolic hepatic disorders. J. Physiol. 2006, 574, 41–53. [Google Scholar] [CrossRef]
- Yu, H.-X.; Yao, Y.; Bu, F.-T.; Chen, Y.; Wu, Y.-T.; Yang, Y.; Chen, X.; Zhu, Y.; Wang, Q.; Pan, X.-Y.; et al. Blockade of YAP alleviates hepatic fibrosis through accelerating apoptosis and reversion of activated hepatic stellate cells. Mol. Immunol. 2019, 107, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xiao, Z.-D.; Li, X.; Aziz, K.E.; Gan, B.; Johnson, R.L.; Chen, J. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat. Cell Biol. 2015, 17, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef]
- Bertero, T.; Cottrill, K.A.; Lu, Y.; Haeger, C.M.; Dieffenbach, P.; Annis, S.; Hale, A.; Bhat, B.; Kaimal, V.; Zhang, Y.-Y.; et al. Matrix Remodeling Promotes Pulmonary Hypertension through Feedback Mechanoactivation of the YAP/TAZ-miR-130/301 Circuit. Cell Rep. 2015, 13, 1016–1032. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Hyun, J.; Premont, R.T.; Choi, S.S.; Michelotti, G.A.; Swiderska-Syn, M.; Dalton, G.D.; Thelen, E.; Rizi, B.S.; Jung, Y.; et al. Hedgehog-YAP Signaling Pathway Regulates Glutaminolysis to Control Activation of Hepatic Stellate Cells. Gastroenterology 2018, 154, 1465–1479. [Google Scholar] [CrossRef]
- Zhou, Y.; Jia, X.; Wang, G.; Wang, X.; Liu, J. PI-3 K/AKT and ERK signaling pathways mediate leptin-induced inhibition of PPARγ gene expression in primary rat hepatic stellate cells. Mol. Cell. Biochem. 2009, 325, 131–139. [Google Scholar] [CrossRef]
- Zhou, Y.; Jia, X.; Qin, J.; Lu, C.; Zhu, H.; Li, X.; Han, X.; Sun, X. Leptin inhibits PPARγ gene expression in hepatic stellate cells in the mouse model of liver damage. Mol. Cell. Endocrinol. 2010, 323, 193–200. [Google Scholar] [CrossRef]
- Boubia, B.; Poupardin, O.; Barth, M.; Binet, J.; Peralba, P.; Mounier, L.; Jacquier, E.; Gauthier, E.; Lepais, V.; Chatar, M.; et al. Design, Synthesis, and Evaluation of a Novel Series of Indole Sulfonamide Peroxisome Proliferator Activated Receptor (PPAR) α/γ/δ Triple Activators: Discovery of Lanifibranor, a New Antifibrotic Clinical Candidate. J. Med. Chem. 2018, 61, 2246–2265. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bruschi, F.V.; Tardelli, M.; Einwallner, E.; Claudel, T.; Trauner, M. PNPLA3 I148M Up-Regulates Hedgehog and Yap Signaling in Human Hepatic Stellate Cells. Int. J. Mol. Sci. 2020, 21, 8711. https://doi.org/10.3390/ijms21228711
Bruschi FV, Tardelli M, Einwallner E, Claudel T, Trauner M. PNPLA3 I148M Up-Regulates Hedgehog and Yap Signaling in Human Hepatic Stellate Cells. International Journal of Molecular Sciences. 2020; 21(22):8711. https://doi.org/10.3390/ijms21228711
Chicago/Turabian StyleBruschi, Francesca Virginia, Matteo Tardelli, Elisa Einwallner, Thierry Claudel, and Michael Trauner. 2020. "PNPLA3 I148M Up-Regulates Hedgehog and Yap Signaling in Human Hepatic Stellate Cells" International Journal of Molecular Sciences 21, no. 22: 8711. https://doi.org/10.3390/ijms21228711
APA StyleBruschi, F. V., Tardelli, M., Einwallner, E., Claudel, T., & Trauner, M. (2020). PNPLA3 I148M Up-Regulates Hedgehog and Yap Signaling in Human Hepatic Stellate Cells. International Journal of Molecular Sciences, 21(22), 8711. https://doi.org/10.3390/ijms21228711