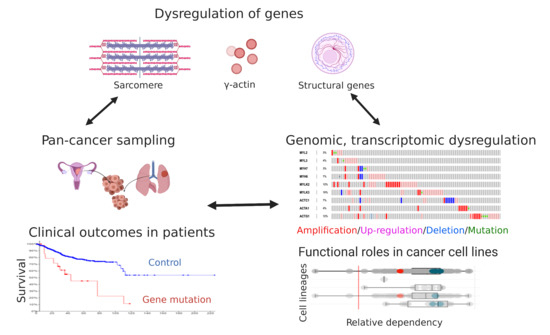
Genomic Amplification and Functional Dependency of the Gamma Actin Gene ACTG1 in Uterine Cancer

 , , and
, , and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
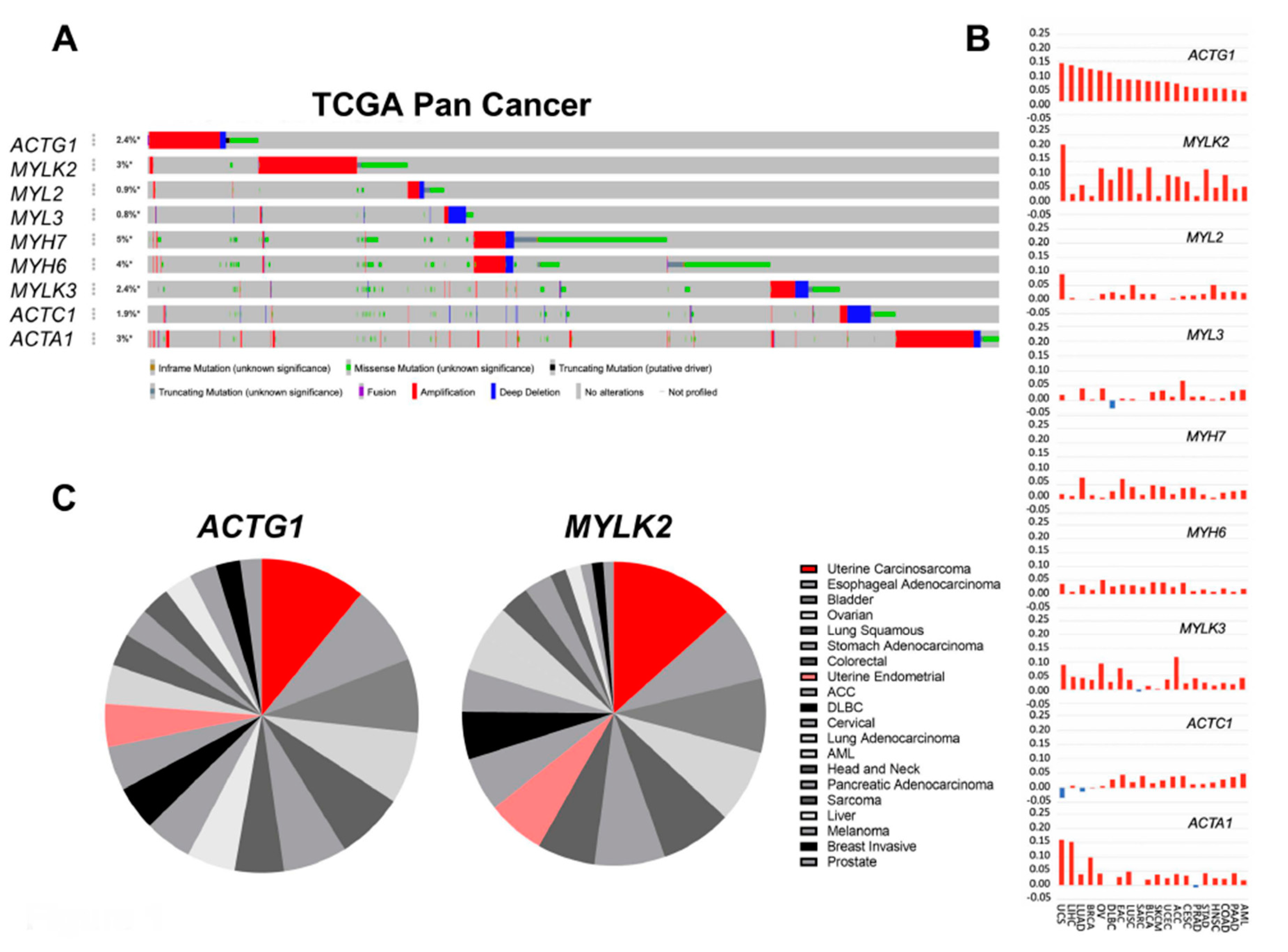
2.1. Actomyosin Genes Are Dysregulated in Cancer
2.2. Uterine Cancer Subtypes Harbor Significant Gains of ACTG1 with Poor Prognosis in Patients
2.3. Clinical, Histological and Molecular Features of ACTG1 Gains
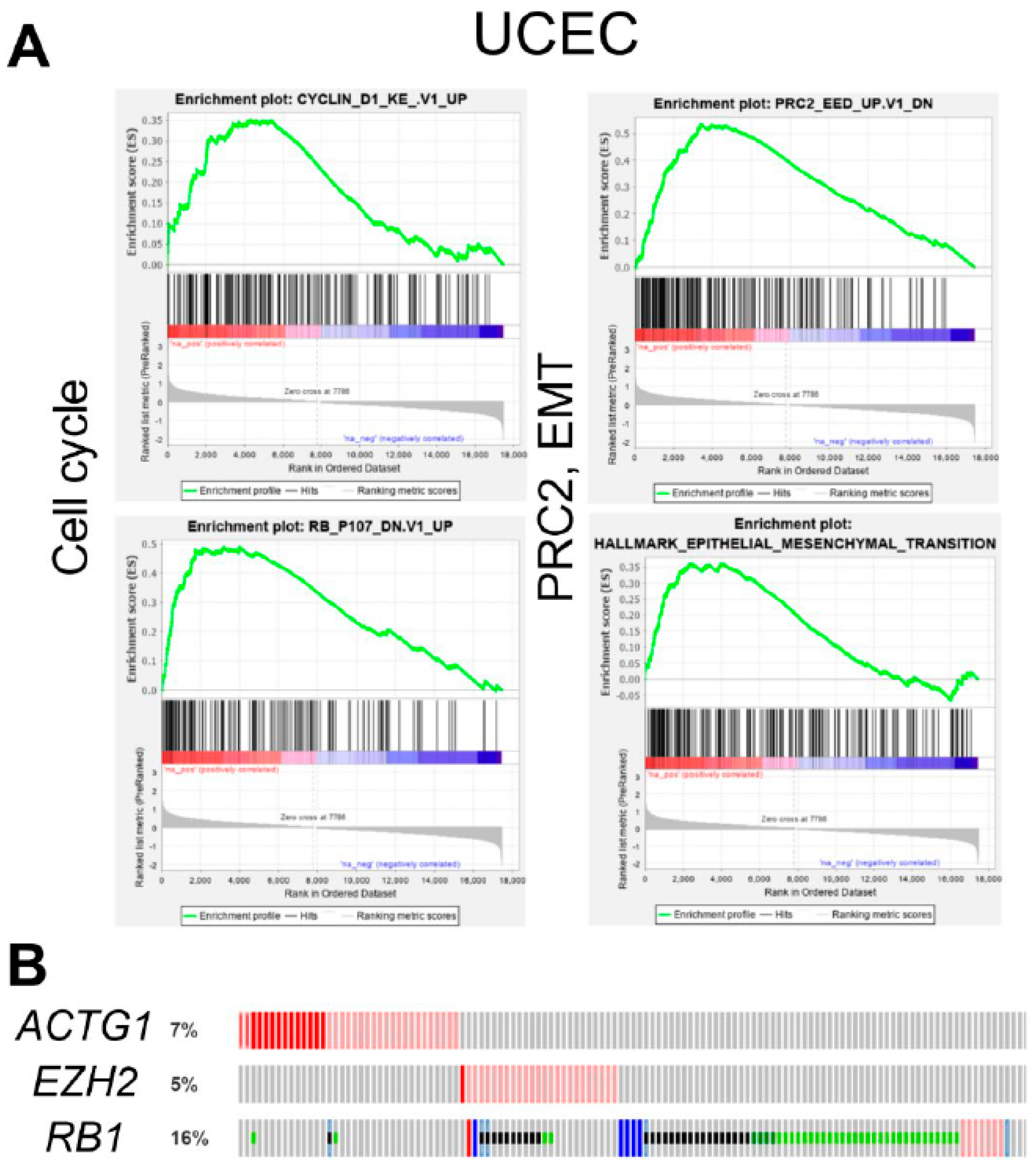
2.4. ACTG1 Gains in UCEC are Associated with Cell Cycle, PRC2 Activity, and Epithelial to Mesenchymal Transition
2.5. ACTG1 Gains Correspond to Repressed Immune Signaling Response and Lymphocyte Infiltration
2.6. ACTG1 is a Dependency Gene in Uterine Cancer
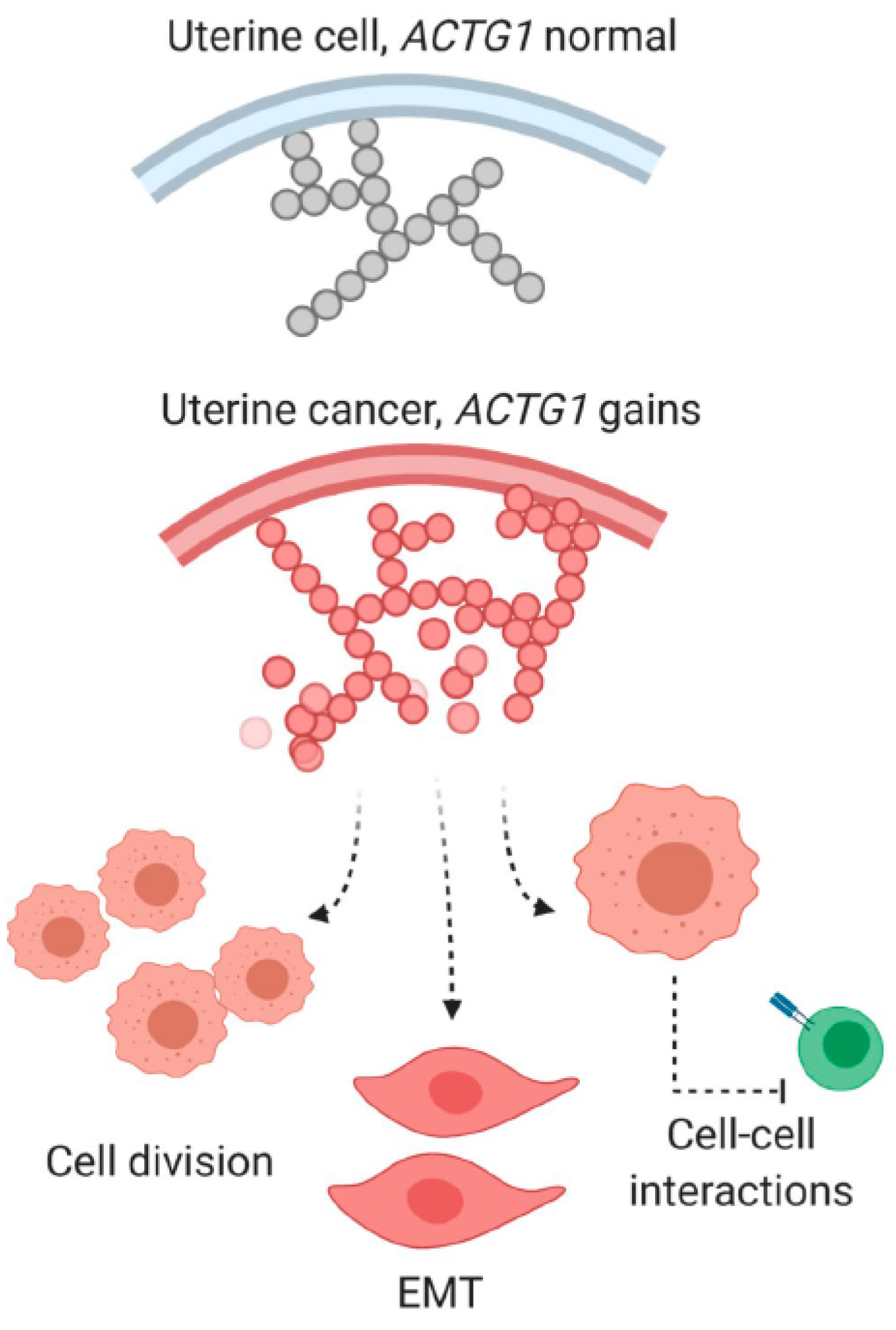
3. Discussion
3.1. ACTG1 is an Amplified Gene That Predicts Poor Prognosis in Uterine Cancer Subtypes
3.2. Intrinsic Functions of ACTG1 in Uterine Cancer Cells
3.3. Immune Regulatory Functions of Uterine Cancers with ACTG1 Gains
3.4. Clinical Utility of ACTG1 Gains in Uterine Cancer Patients
4. Methods
4.1. Gene Dysregulation Status Analysis
4.2. Survival Curves
4.3. Dependency Analysis
4.4. Gene Set Enrichment Analysis (GSEA) of Uterine Cancers
4.5. TCIA Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACTA1 | Skeletal α-actin |
| ACTC1 | Cardiac α-actin |
| ACTG1 | Cytoplasmic γ-actin |
| MYH6 | α-Myosin Heavy Chain (MHC) |
| MYH7 | β-MHC |
| MYL2 | Myosin Regulatory Light Chain (RLC) |
| MYL3 | Myosin Essential Light Chain (ELC) |
| MYLK2 | Skeletal Myosin Light Chain Kinase (MLCK) |
| MYLK3 | Cardiac MLCK |
| TCGA | The Cancer Genome Atlas |
| TCIA | The Cancer Immunome Atlas |
| GSEA | Gene Set Enrichment Analysis |
| UCEC | Uterine Corpus Endometrial Carcinoma |
| UCS | Uterine Carcinosarcoma |
| UEC | Uterine Endometroid Carcinoma |
| UMEC | Uterine Mixed Endometrial Carcinoma |
| USC | Uterine Serous Carcinoma |
References
- Olson, T.M.; Michels, V.V.; Thibodeau, S.N.; Tai, Y.S.; Keating, M.T. Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science 1998, 280, 750–752. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.J.; Moore, J.R. Actin in action and inaction: The differential effects of hypertrophic and dilated cardiomyopathy actin mutations on thin filament regulation. J. Mol. Cell. Cardiol. 2010, 48, 277–278. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Trivedi, D.V.; Sarkar, S.S.; Adhikari, A.S.; Sunitha, M.S.; Sutton, S.; Ruppel, K.M.; Spudich, J.A. The myosin mesa and the basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Nat. Struct. Mol. Biol. 2017, 24, 525. [Google Scholar] [CrossRef] [PubMed]
- Zaleta-Rivera, K.; Dainis, A.; Ribeiro, A.J.S.; Cordero, P.; Rubio, G.; Shang, C.; Liu, J.; Finsterbach, T.; Parikh, V.N.; Sutton, S.; et al. Allele-Specific Silencing Ameliorates Restrictive Cardiomyopathy Attributable to a Human Myosin Regulatory Light Chain Mutation. Circulation 2019, 140, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.C.; Kazmierczak, K.; Liang, J.; Zhou, Z.; Yadav, S.; Gomes, A.V.; Irving, T.C.; Szczesna-Cordary, D. Sarcomeric perturbations of myosin motors lead to dilated cardiomyopathy in genetically modified MYL2 mice. Proc. Natl. Acad. Sci. USA 2018, 115, E2338–E2347. [Google Scholar] [CrossRef]
- Lu, X.; Kazmierczak, K.; Jiang, X.; Jones, M.; Watt, J.; Helfman, D.M.; Moore, J.R.; Szczesna-Cordary, D.; Lossos, I.S. Germinal center-specific protein human germinal center associated lymphoma directly interacts with both myosin and actin and increases the binding of myosin to actin. FEBS J. 2011, 278, 1922–1931. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, Y.R.; Yang, W.X. Myosins as fundamental components during tumorigenesis: Diverse and indispensable. Oncotarget 2016, 7, 46785–46812. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pavia, P.; Kim, Y.; Restrepo-Cordoba, M.A.; Lunde, I.G.; Wakimoto, H.; Smith, A.M.; Toepfer, C.N.; Getz, K.; Gorham, J.; Patel, P.; et al. Genetic Variants Associated With Cancer Therapy-Induced Cardiomyopathy. Circulation 2019, 140, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Surcel, A.; Schiffhauer, E.S.; Thomas, D.G.; Zhu, Q.; DiNapoli, K.T.; Herbig, M.; Otto, O.; West-Foyle, H.; Jacobi, A.; Krater, M.; et al. Targeting Mechanoresponsive Proteins in Pancreatic Cancer: 4-Hydroxyacetophenone Blocks Dissemination and Invasion by Activating MYH14. Cancer Res. 2019, 79, 4665–4678. [Google Scholar] [CrossRef] [PubMed]
- Broders-Bondon, F.; Nguyen Ho-Bouldoires, T.H.; Fernandez-Sanchez, M.E.; Farge, E. Mechanotransduction in tumor progression: The dark side of the force. J. Cell Biol. 2018, 217, 1571–1587. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.; Xia, Y.; Discher, D.E.; Janmey, P.A. Mechanotransduction in cancer. Curr. Opin. Chem. Eng. 2016, 11, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Northcott, J.M.; Dean, I.S.; Mouw, J.K.; Weaver, V.M. Feeling Stress: The Mechanics of Cancer Progression and Aggression. Front. Cell Dev. Biol. 2018, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Wicki, A.; Lehembre, F.; Wick, N.; Hantusch, B.; Kerjaschki, D.; Christofori, G. Tumor invasion in the absence of epithelial-mesenchymal transition: Podoplanin-mediated remodeling of the actin cytoskeleton. Cancer Cell 2006, 9, 261–272. [Google Scholar] [CrossRef]
- McGrail, D.J.; Mezencev, R.; Kieu, Q.M.N.; McDonald, J.F.; Dawson, M.R. SNAIL-induced epithelial-to-mesenchymal transition produces concerted biophysical changes from altered cytoskeletal gene expression. FASEB J. 2015, 29, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Cowley, G.S.; Weir, B.A.; Vazquez, F.; Tamayo, P.; Scott, J.A.; Rusin, S.; East-Seletsky, A.; Ali, L.D.; Gerath, W.F.; Pantel, S.E.; et al. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci. Data 2014, 1, 140035. [Google Scholar] [CrossRef]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef]
- Kawasaki, T.; Yokoi, S.; Tsuda, H.; Izumi, H.; Kozaki, K.; Aida, S.; Ozeki, Y.; Yoshizawa, Y.; Imoto, I.; Inazawa, J. BCL2L2 is a probable target for novel 14q11.2 amplification detected in a non-small cell lung cancer cell line. Cancer Sci. 2007, 98, 1070–1077. [Google Scholar] [CrossRef]
- Levine, D.; The Cancer Genome Atlas Research Network. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Soslow, R.A.; Tornos, C.; Park, K.J.; Malpica, A.; Matias-Guiu, X.; Oliva, E.; Parkash, V.; Carlson, J.; McCluggage, W.G.; Gilks, C.B. Endometrial Carcinoma Diagnosis: Use of FIGO Grading and Genomic Subcategories in Clinical Practice: Recommendations of the International Society of Gynecological Pathologists. Int. J. Gynecol. Pathol. 2019, 38, S64–S74. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Hung, M.C. The role of EZH2 in tumour progression. Br. J. Cancer 2012, 106, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Li, C.H.; Chen, Y. Targeting EZH2 for cancer therapy: Progress and perspective. Curr. Protein Pept. Sci. 2015, 16, 559–570. [Google Scholar]
- He, L.R.; Liu, M.Z.; Li, B.K.; Jia, W.H.; Zhang, Y.; Liao, Y.J.; Chen, Y.C.; Zhang, L.J.; Guan, X.Y.; Zeng, Y.X.; et al. High expression of EZH2 is associated with tumor aggressiveness and poor prognosis in patients with esophageal squamous cell carcinoma treated with definitive chemoradiotherapy. Int. J. Cancer 2010, 127, 138–147. [Google Scholar] [CrossRef]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Cherniack, A.D.; Shen, H.; Walter, V.; Stewart, C.; Murray, B.A.; Bowlby, R.; Hu, X.; Ling, S.; Soslow, R.A.; Broaddus, R.R.; et al. Integrated Molecular Characterization of Uterine Carcinosarcoma. Cancer Cell 2017, 31, 411–423. [Google Scholar] [CrossRef]
- McConechy, M.K.; Hoang, L.N.; Chui, M.H.; Senz, J.; Yang, W.; Rozenberg, N.; Mackenzie, R.; McAlpine, J.N.; Huntsman, D.G.; Clarke, B.A.; et al. In-depth molecular profiling of the biphasic components of uterine carcinosarcomas. J. Pathol. Clin. Res. 2015, 1, 173–185. [Google Scholar] [CrossRef]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef]
- Herman, I.M. Actin isoforms. Curr. Opin. Cell Biol. 1993, 5, 48–55. [Google Scholar] [CrossRef]
- Pollock, C.B.; Shirasawa, S.; Sasazuki, T.; Kolch, W.; Dhillon, A.S. Oncogenic K-RAS is required to maintain changes in cytoskeletal organization, adhesion, and motility in colon cancer cells. Cancer Res. 2005, 65, 1244–1250. [Google Scholar] [CrossRef] [PubMed]
- Spaans, V.M.; Trietsch, M.D.; Crobach, S.; Stelloo, E.; Kremer, D.; Osse, E.M.; Haar, N.T.; van Eijk, R.; Muller, S.; van Wezel, T.; et al. Designing a high-throughput somatic mutation profiling panel specifically for gynaecological cancers. PLoS ONE 2014, 9, e93451. [Google Scholar] [CrossRef] [PubMed]
- Drosten, M.; Sum, E.Y.; Lechuga, C.G.; Simón-Carrasco, L.; Jacob, H.K.; García-Medina, R.; Huang, S.; Beijersbergen, R.L.; Bernards, R.; Barbacid, M. Loss of p53 induces cell proliferation via Ras-independent activation of the Raf/Mek/Erk signaling pathway. Proc. Natl. Acad. Sci. USA 2014, 111, 15155–15160. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Goebeler, M.; Posern, G.; Feller, S.M.; Seitz, C.S.; Brocker, E.B.; Rapp, U.R.; Ludwig, S. Ras-independent activation of the Raf/MEK/ERK pathway upon calcium-induced differentiation of keratinocytes. J. Biol. Chem. 2000, 275, 41011–41017. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Xiao, Y.; Freeman, G.J. The microsatellite instable subset of colorectal cancer is a particularly good candidate for checkpoint blockade immunotherapy. Cancer Discov. 2015, 5, 16–18. [Google Scholar] [CrossRef]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef]
- Di Tucci, C.; Capone, C.; Galati, G.; Iacobelli, V.; Schiavi, M.C.; Di Donato, V.; Muzii, L.; Panici, P.B. Immunotherapy in endometrial cancer: New scenarios on the horizon. J. Gynecol. Oncol. 2019, 30, e46. [Google Scholar] [CrossRef]
- Akalay, I.; Janji, B.; Hasmim, M.; Noman, M.Z.; Andre, F.; De Cremoux, P.; Bertheau, P.; Badoual, C.; Vielh, P.; Larsen, A.K.; et al. Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis. Cancer Res. 2013, 73, 2418–2427. [Google Scholar] [CrossRef]
- Chouaib, S.; Janji, B.; Tittarelli, A.; Eggermont, A.; Thiery, J.P. Tumor plasticity interferes with anti-tumor immunity. Crit. Rev. Immunol. 2014, 34, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; Savagner, P.; Ortiz-Cuaran, S.; Mahjoubi, L.; Saintigny, P.; Thiery, J.P.; Chouaib, S. New insights into the role of EMT in tumor immune escape. Mol. Oncol. 2017, 11, 824–846. [Google Scholar] [CrossRef] [PubMed]
- Datar, I.; Schalper, K.A. Epithelial-Mesenchymal Transition and Immune Evasion during Lung Cancer Progression: The Chicken or the Egg? Clin. Cancer Res. 2016, 22, 3422–3424. [Google Scholar] [CrossRef] [PubMed]
- Foerster, F.; Braig, S.; Moser, C.; Kubisch, R.; Busse, J.; Wagner, E.; Schmoeckel, E.; Mayr, D.; Schmitt, S.; Huettel, S.; et al. Targeting the actin cytoskeleton: Selective antitumor action via trapping PKCvarepsilon. Cell Death Dis. 2014, 5, e1398. [Google Scholar] [CrossRef]
- Stehn, J.R.; Haass, N.K.; Bonello, T.; Desouza, M.; Kottyan, G.; Treutlein, H.; Zeng, J.; Nascimento, P.R.; Sequeira, V.B.; Butler, T.L.; et al. A novel class of anticancer compounds targets the actin cytoskeleton in tumor cells. Cancer Res. 2013, 73, 5169–5182. [Google Scholar] [CrossRef]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richter, C.; Mayhew, D.; Rennhack, J.P.; So, J.; Stover, E.H.; Hwang, J.H.; Szczesna-Cordary, D. Genomic Amplification and Functional Dependency of the Gamma Actin Gene ACTG1 in Uterine Cancer. Int. J. Mol. Sci. 2020, 21, 8690. https://doi.org/10.3390/ijms21228690
Richter C, Mayhew D, Rennhack JP, So J, Stover EH, Hwang JH, Szczesna-Cordary D. Genomic Amplification and Functional Dependency of the Gamma Actin Gene ACTG1 in Uterine Cancer. International Journal of Molecular Sciences. 2020; 21(22):8690. https://doi.org/10.3390/ijms21228690
Chicago/Turabian StyleRichter, Camden, David Mayhew, Jonathan P. Rennhack, Jonathan So, Elizabeth H. Stover, Justin H. Hwang, and Danuta Szczesna-Cordary. 2020. "Genomic Amplification and Functional Dependency of the Gamma Actin Gene ACTG1 in Uterine Cancer" International Journal of Molecular Sciences 21, no. 22: 8690. https://doi.org/10.3390/ijms21228690
APA StyleRichter, C., Mayhew, D., Rennhack, J. P., So, J., Stover, E. H., Hwang, J. H., & Szczesna-Cordary, D. (2020). Genomic Amplification and Functional Dependency of the Gamma Actin Gene ACTG1 in Uterine Cancer. International Journal of Molecular Sciences, 21(22), 8690. https://doi.org/10.3390/ijms21228690