Targets (Metabolic Mediators) of Therapeutic Importance in Pancreatic Ductal Adenocarcinoma



Abstract
1. Introduction
2. Tumor Microenvironment
3. Metabolic Pathways
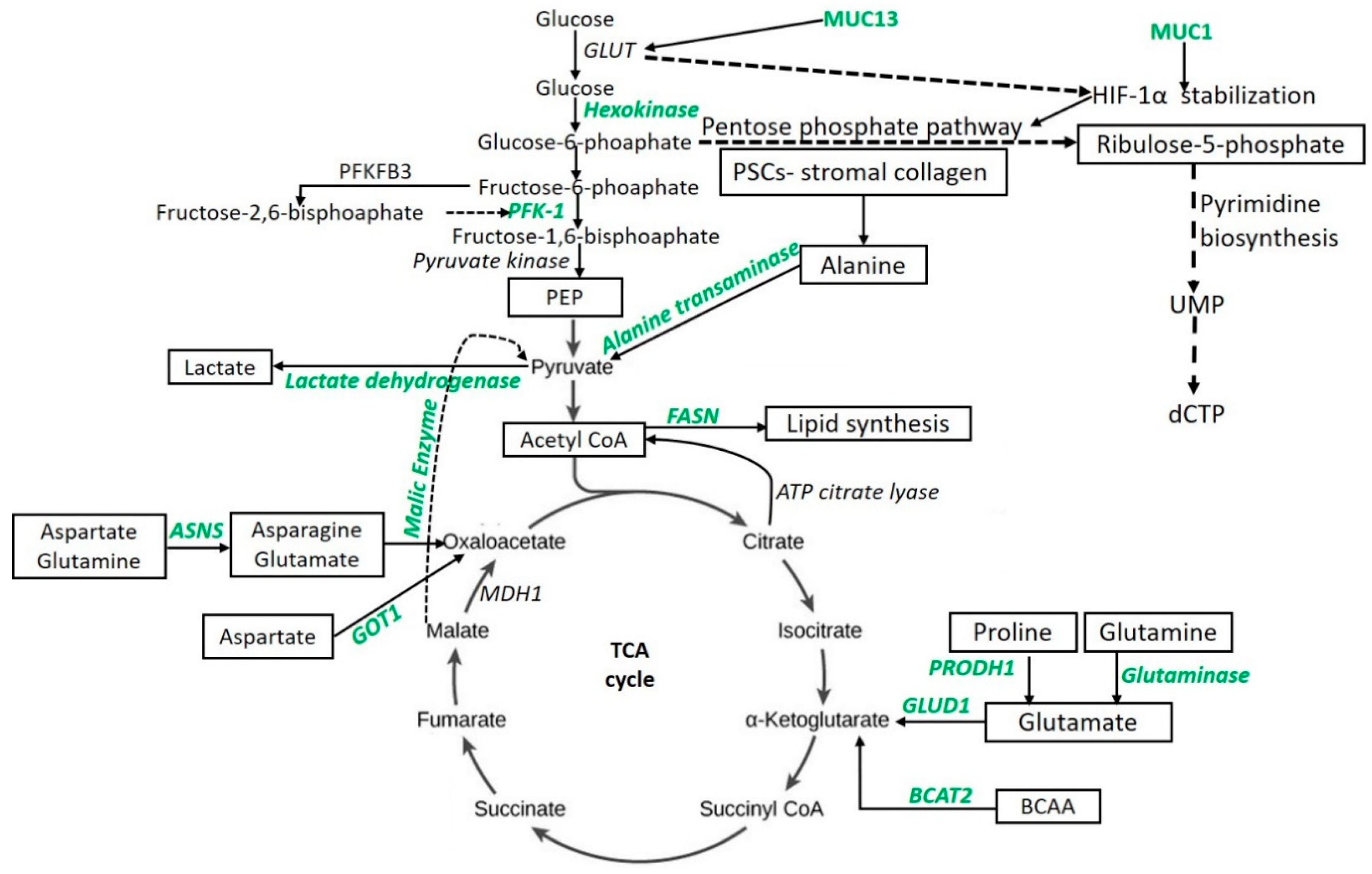
3.1. Glucose Metabolism
3.2. Lipid Metabolism
3.3. Amino-Acid and Nucleotide Metabolism
3.4. Mitochondrial Metabolites and Reactive Oxygen Species
4. Autophagy and Macropinocytosis
5. Aminoacid and Glucose Transporters
6. Metabolic Alterations, Stemness, and Chemo-Resistance
7. Future Directions and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Soloff, E.V.; Zaheer, A.; Meier, J.; Zins, M.; Tamm, E.P. Staging of pancreatic cancer: Resectable, borderline resectable, and unresectable disease. Abdom. Radiol. 2018, 43, 301–313. [Google Scholar] [CrossRef]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef]
- Gunda, V.; Souchek, J.; Abrego, J.; Shukla, S.K.; Goode, G.D.; Vernucci, E.; Dasgupta, A.; Chaika, N.V.; King, R.J.; Li, S.; et al. MUC1-mediated metabolic alterations regulate response to radiotherapy in pancreatic cancer. Clin. Cancer Res. 2017, 23, 5881–5891. [Google Scholar] [CrossRef]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic ductal adenocarcinoma: Current and evolving therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef]
- Manji, G.A.; Olive, K.P.; Saenger, Y.M.; Oberstein, P. Current and Emerging Therapies in Metastatic Pancreatic Cancer; AACR: Philadelphia, PA, USA, 2017. [Google Scholar]
- Perone, J.A.; Riall, T.S.; Olino, K. Palliative care for pancreatic and periampullary cancer. Surg. Clin. 2016, 96, 1415–1430. [Google Scholar] [CrossRef]
- Aung, K.L.; Fischer, S.E.; Denroche, R.E.; Jang, G.-H.; Dodd, A.; Creighton, S.; Southwood, B.; Liang, S.B.; Chadwick, D.; Zhang, A.; et al. Genomics-driven precision medicine for advanced pancreatic cancer: Early results from the COMPASS trial. Clin. Cancer Res. 2018, 24, 1344–1354. [Google Scholar] [CrossRef]
- Ligorio, M.; Sil, S.; Malagon-Lopez, J.; Nieman, L.T.; Misale, S.; Di Pilato, M.; Ebright, R.Y.; Karabacak, M.N.; Kulkarni, A.S.; Liu, A.; et al. Stromal microenvironment shapes the intratumoral architecture of pancreatic cancer. Cell 2019, 178, 160–175.e27. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Katz, M.H.; Pisters, P.W.; Evans, D.B.; Sun, C.C.; Lee, J.E.; Fleming, J.B.; Vauthey, J.N.; Abdalla, E.K.; Crane, C.H.; Wolff, R.A.; et al. Borderline resectable pancreatic cancer: The importance of this emerging stage of disease. J. Am. Coll. Surg. 2008, 206, 833–846. [Google Scholar] [CrossRef]
- Flaveny, C.A.; Griffett, K.; El-Gendy, B.E.-D.M.; Kazantzis, M.; Sengupta, M.; Amelio, A.L.; Chatterjee, A.; Walker, J.; Solt, L.A.; Kamenecka, T.M.; et al. Broad anti-tumor activity of a small molecule that selectively targets the Warburg effect and lipogenesis. Cancer Cell 2015, 28, 42–56. [Google Scholar] [CrossRef]
- Franco, J.; Balaji, U.; Freinkman, E.; Witkiewicz, A.K.; Knudsen, E.S. Metabolic reprogramming of pancreatic cancer mediated by CDK4/6 inhibition elicits unique vulnerabilities. Cell Rep. 2016, 14, 979–990. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Vernucci, E.; Abrego, J.; Gunda, V.; Shukla, S.K.; Dasgupta, A.; Rai, V.; Buettner, K.; Illies, A.; Yu, F.; Lazenby, A.J.; et al. Metabolic Alterations in Pancreatic Cancer Progression. Cancers 2020, 12, 2. [Google Scholar] [CrossRef]
- Abrego, J.; Gunda, V.; Vernucci, E.; Shukla, S.K.; King, R.J.; Dasgupta, A.; Goode, G.; Murthy, D.; Yu, F.; Singh, P.K. GOT1-mediated anaplerotic glutamine metabolism regulates chronic acidosis stress in pancreatic cancer cells. Cancer Lett. 2017, 400, 37–46. [Google Scholar] [CrossRef]
- Nagarajan, A.; Dogra, S.K.; Sun, L.; Gandotra, N.; Ho, T.; Cai, G.; Cline, G.; Kumar, P.; Cowles, R.A.; Wajapeyee, N. Paraoxonase 2 facilitates pancreatic cancer growth and metastasis by stimulating GLUT1-mediated glucose transport. Mol. Cell 2017, 67, 685–701.e6. [Google Scholar] [CrossRef]
- Daemen, A.; Peterson, D.; Sahu, N.; McCord, R.; Du, X.; Liu, B.; Kowanetz, K.; Hong, R.; Moffat, J.; Gao, M.; et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. Proc. Natl. Acad. Sci. USA 2015, 112, E4410–E4417. [Google Scholar] [CrossRef]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic interactions in the tumor microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef]
- Li, J.-T.; Wang, Y.-P.; Yin, M.; Lei, Q.-Y. Metabolism remodeling in pancreatic ductal adenocarcinoma. Cell Stress 2019, 3, 361. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 2016, 5, e10250. [Google Scholar] [CrossRef]
- Qin, C.; Yang, G.; Yang, J.; Ren, B.; Wang, H.; Chen, G.; Zhao, F.; You, L.; Wang, W.; Zhao, Y. Metabolism of pancreatic cancer: Paving the way to better anticancer strategies. Mol. Cancer 2020, 19, 50. [Google Scholar] [CrossRef]
- Yan, L.; Raj, P.; Yao, W.; Ying, H. Glucose Metabolism in Pancreatic Cancer. Cancers 2019, 11, 1460. [Google Scholar] [CrossRef]
- Dey, P.; Baddour, J.; Muller, F.; Wu, C.C.; Wang, H.; Liao, W.-T.; Lan, Z.; Chen, A.; Gutschner, T.; Kang, Y.; et al. Genomic deletion of malic enzyme 2 confers collateral lethality in pancreatic cancer. Nature 2017, 542, 119–123. [Google Scholar] [CrossRef]
- Rajeshkumar, N.; Dutta, P.; Yabuuchi, S.; De Wilde, R.F.; Martinez, G.V.; Le, A.; Kamphorst, J.J.; Rabinowitz, J.D.; Jain, S.K.; Hidalgo, M.; et al. Therapeutic targeting of the Warburg effect in pancreatic cancer relies on an absence of p53 function. Cancer Res. 2015, 75, 3355–3364. [Google Scholar] [CrossRef]
- An, M.-X.; Li, S.; Yao, H.-B.; Li, C.; Wang, J.-M.; Sun, J.; Li, X.Y.; Meng, X.N.; Wang, H.Q. BAG3 directly stabilizes Hexokinase 2 mRNA and promotes aerobic glycolysis in pancreatic cancer cells. J. Cell Biol. 2017, 216, 4091–4105. [Google Scholar] [CrossRef]
- Kumari, S.; Khan, S.; Gupta, S.C.; Kashyap, V.K.; Yallapu, M.M.; Chauhan, S.C.; Jaggi, M. MUC13 contributes to rewiring of glucose metabolism in pancreatic cancer. Oncogenesis 2018, 7, 19. [Google Scholar] [CrossRef]
- Pramanik, K.C.; Makena, M.R.; Bhowmick, K.; Pandey, M.K. Advancement of NF-κB signaling pathway: A novel target in pancreatic Cancer. Int. J. Mol. Sci. 2018, 19, 3890. [Google Scholar] [CrossRef]
- Kabacaoglu, D.; Ruess, D.A.; Ai, J.; Algül, H. NF-κB/Rel Transcription Factors in Pancreatic Cancer: Focusing on RelA, c-Rel, and RelB. Cancers 2019, 11, 937. [Google Scholar] [CrossRef]
- Geismann, C.; Erhart, W.; Grohmann, F.; Schreiber, S.; Schneider, G.; Schäfer, H.; Arlt, A. TRAIL/NF-κB/CX3CL1 mediated onco-immuno crosstalk leading to TRAIL resistance of pancreatic cancer cell lines. Int. J. Mol. Sci. 2018, 19, 1661. [Google Scholar] [CrossRef] [PubMed]
- Nomura, A.; Majumder, K.; Giri, B.; Dauer, P.; Dudeja, V.; Roy, S.; Banerjee, S.; Saluja, A.K. Inhibition of NF-kappa B pathway leads to deregulation of epithelial–mesenchymal transition and neural invasion in pancreatic cancer. Lab. Investig. 2016, 96, 1268–1278. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Chen, C.; Luo, Y.; Liu, M.; Li, Y.; Zheng, S.; Ye, H.; Fu, Z.; Li, M.; Li, Z.; et al. lncRNA-PLACT1 sustains activation of NF-κB pathway through a positive feedback loop with IκBα/E2F1 axis in pancreatic cancer. Mol. Cancer 2020, 19, 35. [Google Scholar] [CrossRef]
- Zubair, H.; Azim, S.; Srivastava, S.K.; Ahmad, A.; Bhardwaj, A.; Khan, M.A.; Patel, G.K.; Arora, S.; Carter, J.E.; Singh, S.; et al. Glucose metabolism reprogrammed by overexpression of IKKε promotes pancreatic tumor growth. Cancer Res. 2016, 76, 7254–7264. [Google Scholar] [CrossRef]
- Sunami, Y.; Rebelo, A.; Kleeff, J. Lipid metabolism and lipid droplets in pancreatic cancer and stellate cells. Cancers 2018, 10, 3. [Google Scholar] [CrossRef]
- Swierczynski, J.; Hebanowska, A.; Sledzinski, T. Role of abnormal lipid metabolism in development, progression, diagnosis and therapy of pancreatic cancer. WJG 2014, 20, 2279. [Google Scholar] [CrossRef]
- Guillaumond, F.; Bidaut, G.; Ouaissi, M.; Servais, S.; Gouirand, V.; Olivares, O.; Lac, S.; Borge, L.; Roques, J.; Gayet, O.; et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, 2473–2478. [Google Scholar] [CrossRef]
- Tadros, S.; Shukla, S.K.; King, R.J.; Gunda, V.; Vernucci, E.; Abrego, J.; Chaika, N.V.; Yu, F.; Lazenby, A.J.; Berim, L.; et al. De novo lipid synthesis facilitates gemcitabine resistance through endoplasmic reticulum stress in pancreatic cancer. Cancer Res. 2017, 77, 5503–5517. [Google Scholar] [CrossRef]
- Bott, A.J.; Shen, J.; Tonelli, C.; Zhan, L.; Sivaram, N.; Jiang, Y.-P.; Yu, X.; Bhatt, V.; Chiles, E.; Zhong, H.; et al. Glutamine Anabolism Plays a Critical Role in Pancreatic Cancer by Coupling Carbon and Nitrogen Metabolism. Cell Rep. 2019, 29, 1287–1298.e6. [Google Scholar] [CrossRef]
- Mayers, J.R.; Torrence, M.E.; Danai, L.V.; Papagiannakopoulos, T.; Davidson, S.M.; Bauer, M.R.; Lau, A.N.; Ji, B.W.; Dixit, P.D.; Hosios, A.M.; et al. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 2016, 353, 1161–1165. [Google Scholar] [CrossRef]
- Gebregiworgis, T.; Purohit, V.; Shukla, S.K.; Tadros, S.; Chaika, N.V.; Abrego, J.; Mulder, S.E.; Gunda, V.; Singh, P.K.; Powers, R. Glucose limitation alters glutamine metabolism in MUC1-overexpressing pancreatic cancer cells. J. Proteome Res. 2017, 16, 3536–3546. [Google Scholar] [CrossRef] [PubMed]
- Biancur, D.E.; Paulo, J.A.; Małachowska, B.; Del Rey, M.Q.; Sousa, C.M.; Wang, X.; Sohn, A.S.W.; Chu, G.C.; Gygi, S.P.; Harper, J.W.; et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat. Commun. 2017, 8, 15965. [Google Scholar] [CrossRef] [PubMed]
- Elgogary, A.; Xu, Q.; Poore, B.; Alt, J.; Zimmermann, S.C.; Zhao, L.; Fu, J.; Chen, B.; Xia, S.; Liu, Y.; et al. Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proc. Natl. Acad. Sci. USA 2016, 113, E5328–E5336. [Google Scholar] [CrossRef] [PubMed]
- Chaiteerakij, R.; Petersen, G.M.; Bamlet, W.R.; Chaffee, K.G.; Zhen, D.B.; Burch, P.A.; Leof, E.R.; Roberts, L.R.; Oberg, A.L. Metformin use and survival of patients with pancreatic cancer: A cautionary lesson. J. Clin. Oncol. 2016, 34, 1898. [Google Scholar] [CrossRef]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine metabolism in cancer: Understanding the heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef]
- Jiang, J.; Srivastava, S.; Zhang, J. Starve cancer cells of glutamine: Break the spell or make a hungry monster? Cancers 2019, 11, 804. [Google Scholar] [CrossRef]
- Shukla, S.K.; Purohit, V.; Mehla, K.; Gunda, V.; Chaika, N.V.; Vernucci, E.; King, R.J.; Abrego, J.; Goode, G.D.; Dasgupta, A.; et al. MUC1 and HIF-1alpha signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell 2017, 32, 71–87.e7. [Google Scholar] [CrossRef]
- Pupo, E.; Avanzato, D.; Middonti, E.; Bussolino, F.; Lanzetti, L. KRAS-driven metabolic rewiring reveals novel actionable targets in cancer. Front. Oncol. 2019, 9, 848. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Roeth, A.A.; Zhang, Y.; Yang, A.; Mashadova, O.; Asara, J.M.; Wang, X.; Bronson, R.T.; Lyssiotis, C.A.; Ying, H.; et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat. Commun. 2018, 9, 4945. [Google Scholar] [CrossRef]
- Ahn, C.S.; Metallo, C.M. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015, 3, 1. [Google Scholar] [CrossRef]
- Lee, J.H.; Cho, Y.-R.; Kim, J.H.; Kim, J.; Nam, H.Y.; Kim, S.W.; Son, J. Branched-chain amino acids sustain pancreatic cancer growth by regulating lipid metabolism. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Elliott, I.A.; Dann, A.M.; Xu, S.; Kim, S.S.; Abt, E.R.; Kim, W.; Poddar, S.; Moore, A.; Zhou, L.; Williams, J.L.; et al. Lysosome inhibition sensitizes pancreatic cancer to replication stress by aspartate depletion. Proc. Natl. Acad. Sci. USA 2019, 116, 6842–6847. [Google Scholar] [CrossRef] [PubMed]
- Olivares, O.; Mayers, J.R.; Gouirand, V.; Torrence, M.E.; Gicquel, T.; Borge, L.; Lac, S.; Roques, J.; Lavaut, M.N.; Berthezène, P.; et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat. Commun. 2017, 8, 16031. [Google Scholar] [CrossRef] [PubMed]
- Sahu, N.; Cruz, D.D.; Gao, M.; Sandoval, W.; Haverty, P.M.; Liu, J.; Stephan, J.P.; Haley, B.; Classon, M.; Hatzivassiliou, G.; et al. Proline starvation induces unresolved ER stress and hinders mTORC1-dependent tumorigenesis. Cell Metab. 2016, 24, 753–761. [Google Scholar] [CrossRef] [PubMed]
- D’aniello, C.; Patriarca, E.J.; Phang, J.M.; Minchiotti, G. Proline Metabolism in Tumor Growth and Metastatic Progression. Front. Oncol. 2020, 10, 776. [Google Scholar] [CrossRef]
- Phang, J.M. Proline metabolism in cell regulation and cancer biology: Recent advances and hypotheses. Antioxid. Redox Signal. 2019, 30, 635–649. [Google Scholar] [CrossRef]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef]
- Parker, S.J.; Amendola, C.R.; Hollinshead, K.E.; Yu, Q.; Yamamoto, K.; Encarnación-Rosado, J.; Rose, R.E.; LaRue, M.M.; Sohn, A.S.W.; Biancur, D.E.; et al. Selective alanine transporter utilization creates a targetable metabolic niche in pancreatic cancer. Cancer Discov. 2020, 10, 1018–1037. [Google Scholar] [CrossRef]
- Dufour, E.; Gay, F.; Aguera, K.; Scoazec, J.-Y.; Horand, F.; Lorenzi, P.L.; Godfrin, Y. Pancreatic tumor sensitivity to plasma L-asparagine starvation. Pancreas 2012, 41, 940–948. [Google Scholar] [CrossRef]
- Cui, H.; Darmanin, S.; Natsuisaka, M.; Kondo, T.; Asaka, M.; Shindoh, M.; Higashino, F.; Hamuro, J.; Okada, F.; Kobayashi, M.; et al. Enhanced expression of asparagine synthetase under glucose-deprived conditions protects pancreatic cancer cells from apoptosis induced by glucose deprivation and cisplatin. Cancer Res. 2007, 67, 3345–3355. [Google Scholar] [CrossRef]
- Pathria, G.; Lee, J.S.; Hasnis, E.; Tandoc, K.; Scott, D.A.; Verma, S.; Feng, Y.; Larue, L.; Sahu, A.D.; Topisirovic, I.; et al. Translational reprogramming marks adaptation to asparagine restriction in cancer. Nat. Cell Biol. 2019, 21, 1590–1603. [Google Scholar] [CrossRef] [PubMed]
- Mayers, J.R.; Wu, C.; Clish, C.B.; Kraft, P.; Torrence, M.E.; Fiske, B.P.; Yuan, C.; Bao, Y.; Townsend, M.K.; Tworoger, S.S.; et al. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat. Med. 2014, 20, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, R.; Goto, A.; Nakagawa, T.; Nishiumi, S.; Kobayashi, T.; Hidaka, A.; Budhathoki, S.; Yamaji, T.; Sawada, N.; Shimazu, T.; et al. Increased Levels of Branched-Chain Amino Acid Associated with Increased Risk of Pancreatic Cancer in a Prospective Case–Control Study of a Large Cohort. Gastroenterology 2018, 155, 1474–1482.e1. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef]
- Yu, M.; Nguyen, N.D.; Huang, Y.; Lin, D.; Fujimoto, T.N.; Molkentine, J.M.; Deorukhkar, A.; Kang, Y.; San Lucas, F.A.; Fernandes, C.J.; et al. Mitochondrial fusion exploits a therapeutic vulnerability of pancreatic cancer. JCI Insight 2019, 4, e126915. [Google Scholar] [CrossRef]
- Martinez-Useros, J.; Li, W.; Cabeza-Morales, M.; Garcia-Foncillas, J. Oxidative stress: A new target for pancreatic cancer prognosis and treatment. J. Clin. Med. 2017, 6, 29. [Google Scholar] [CrossRef]
- Cheung, E.C.; DeNicola, G.M.; Nixon, C.; Blyth, K.; Labuschagne, C.F.; Tuveson, D.A.; Vousden, K.H. Dynamic ROS control by TIGAR regulates the initiation and progression of pancreatic cancer. Cancer Cell 2020, 37, 168–182.e4. [Google Scholar] [CrossRef]
- Chattaragada, M.; Riganti, C.; Sassoe, M.; Principe, M.; Santamorena, M.; Roux, C.; Curcio, C.; Evangelista, A.; Allavena, P.; Salvia, R.; et al. FAM49B, a novel regulator of mitochondrial function and integrity that suppresses tumor metastasis. Oncogene 2018, 37, 697–709. [Google Scholar] [CrossRef]
- Yang, S.; Hwang, S.; Kim, M.; Seo, S.B.; Lee, J.-H.; Jeong, S.M. Mitochondrial glutamine metabolism via GOT2 supports pancreatic cancer growth through senescence inhibition. Cell Death Dis. 2018, 9, 55. [Google Scholar] [CrossRef]
- Zhang, L.; Li, J.; Zong, L.; Chen, X.; Chen, K.; Jiang, Z.; Nan, L.; Li, X.; Li, W.; Shan, T.; et al. Reactive oxygen species and targeted therapy for pancreatic cancer. Oxidative Med. Cell. Longev. 2016, 2016, 1616781. [Google Scholar] [CrossRef]
- Durand, N.; Storz, P. Targeting reactive oxygen species in development and progression of pancreatic cancer. Expert Rev. Anticancer Ther. 2017, 17, 19–31. [Google Scholar] [CrossRef]
- Shao, S.; Qin, T.; Qian, W.; Yue, Y.; Xiao, Y.; Li, X.; Zhang, D.; Wang, Z.; Ma, Q.; Lei, J. Positive feedback in Cav-1-ROS signalling in PSCs mediates metabolic coupling between PSCs and tumour cells. J. Cell. Mol. Med. 2020, 24, 9397–9408. [Google Scholar] [CrossRef]
- Estaras, M.; Martinez-Morcillo, S.; García, A.; Martinez, R.; Estevez, M.; Perez-Lopez, M.; Miguez, M.P.; Fernandez-Bermejo, M.; Mateos, J.M.; Vara, D.; et al. Pancreatic stellate cells exhibit adaptation to oxidative stress evoked by hypoxia. Biol. Cell 2020, 112, 280–299. [Google Scholar] [CrossRef]
- Huang, X.; He, C.; Hua, X.; Kan, A.; Mao, Y.; Sun, S.; Duan, F.; Wang, J.; Huang, P.; Li, S. Oxidative stress induces monocyte-to-myofibroblast transdifferentiation through p38 in pancreatic ductal adenocarcinoma. Clin. Transl. Med. 2020, 10, e41. [Google Scholar] [CrossRef]
- Sim, J.J.; Jeong, K.-Y. Monitoring Epithelial–Mesenchymal Transition of Pancreatic Cancer Cells via Investigation of Mitochondrial Dysfunction. Methods Protoc. 2020, 3, 32. [Google Scholar] [CrossRef]
- Zhao, H.; Duan, Q.; Zhang, Z.; Li, H.; Wu, H.; Shen, Q.; Wang, C.; Yin, T. Up-regulation of glycolysis promotes the stemness and EMT phenotypes in gemcitabine-resistant pancreatic cancer cells. J. Cell. Mol. Med. 2017, 21, 2055–2067. [Google Scholar] [CrossRef]
- Olou, A.A.; King, R.J.; Yu, F.; Singh, P.K. MUC1 oncoprotein mitigates ER stress via CDA-mediated reprogramming of pyrimidine metabolism. Oncogene 2020, 39, 3381–3395. [Google Scholar] [CrossRef]
- Chattopadhyay, M.; Kodela, R.; Santiago, G.; Le, T.T.C.; Nath, N.; Kashfi, K. NOSH-aspirin (NBS-1120) inhibits pancreatic cancer cell growth in a xenograft mouse model: Modulation of FoxM1, p53, NF-κB, iNOS, caspase-3 and ROS. Biochem. Pharmacol. 2020, 176, 113857. [Google Scholar] [CrossRef]
- Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Functions of autophagy in the tumor microenvironment and cancer metastasis. FEBS J. 2018, 285, 1751–1766. [Google Scholar] [CrossRef] [PubMed]
- New, M.; Tooze, S. The Role of Autophagy in Pancreatic Cancer—Recent Advances. Biology 2020, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- New, M.; Van Acker, T.; Long, J.S.; Sakamaki, J.-I.; Ryan, K.M.; Tooze, S.A. Molecular pathways controlling autophagy in pancreatic cancer. Front. Oncol. 2017, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Xu, J.; Meng, Q.; Zhang, B.; Liu, J.; Hua, J.; Zhang, Y.; Shi, S.; Yu, X. TGFB1-induced autophagy affects the pattern of pancreatic cancer progression in distinct ways depending on SMAD4 status. Autophagy 2020, 16, 486–500. [Google Scholar] [CrossRef] [PubMed]
- Sarcar, B.; Li, X.; Fleming, J.B. Hypoxia-Induced Autophagy Degrades Stromal Lumican into Tumor Microenvironment of Pancreatic Ductal Adenocarcinoma: A Mini-Review. J. Cancer Treat. Diagn. 2019, 3, 22. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Herter-Sprie, G.; Zhang, H.; Lin, E.Y.; Biancur, D.; Wang, X.; Deng, J.; Hai, J.; Yang, S.; Wong, K.K.; et al. Autophagy sustains pancreatic cancer growth through both cell-autonomous and nonautonomous mechanisms. Cancer Discov. 2018, 8, 276–287. [Google Scholar] [CrossRef]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef]
- Tsang, Y.H.; Wang, Y.; Kong, K.; Grzeskowiak, C.; Zagorodna, O.; Dogruluk, T.; Lu, H.; Villafane, N.; Bhavana, V.H.; Moreno, D.; et al. Differential expression of MAGEA6 toggles autophagy to promote pancreatic cancer progression. Elife 2020, 9, e48963. [Google Scholar] [CrossRef]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J.; et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020, 581, 100–105. [Google Scholar] [CrossRef]
- Hwang, T.-L.; Lo, C.-H. The Changes of Autophagy in Pancreatic Stem Cells for Patients with Pancreatic Ductal Cancer. Biomed. J. Sci. Tech. Res. 2019, 13, 9939–9947. [Google Scholar] [CrossRef]
- Seo, J.-W.; Choi, J.; Lee, S.-Y.; Sung, S.; Yoo, H.J.; Kang, M.-J.; Cheong, H.; Son, J. Autophagy is required for PDAC glutamine metabolism. Sci. Rep. 2016, 6, 37594. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Dierichs, L.; Gu, J.-N.; Trajkovic-Arsic, M.; Hilger, R.A.; Savvatakis, K.; Vega-Rubin-de-Celis, S.; Liffers, S.T.; Peña-Llopis, S.; Behrens, D.; et al. TFEB-mediated lysosomal biogenesis and lysosomal drug sequestration confer resistance to MEK inhibition in pancreatic cancer. Cell Death Discov. 2020, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF→ MEK→ ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B.; et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013, 497, 633–637. [Google Scholar] [CrossRef]
- Davidson, S.M.; Jonas, O.; Keibler, M.A.; Hou, H.W.; Luengo, A.; Mayers, J.R.; Wyckoff, J.; Del Rosario, A.M.; Whitman, M.; Chin, C.R.; et al. Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nat. Med. 2017, 23, 235. [Google Scholar] [CrossRef]
- Coothankandaswamy, V.; Cao, S.; Xu, Y.; Prasad, P.; Singh, P.K.; Reynolds, C.; Yang, S.; Ogura, J.; Ganapathy, V.; Bhutia, Y.D. Amino acid transporter SLC6A14 is a novel and effective drug target for pancreatic cancer. Br. J. Pharmacol. 2016, 173, 3292–3306. [Google Scholar] [CrossRef]
- Cormerais, Y.; Massard, P.A.; Vucetic, M.; Giuliano, S.; Tambutté, E.; Durivault, J.; Vial, V.; Endou, H.; Wempe, M.F.; Parks, S.K.; et al. The glutamine transporter ASCT2 (SLC1A5) promotes tumor growth independently of the amino acid transporter LAT1 (SLC7A5). J. Biol. Chem. 2018, 293, 2877–2887. [Google Scholar] [CrossRef]
- Altan, B.; Kaira, K.; Watanabe, A.; Kubo, N.; Bao, P.; Dolgormaa, G.; Bilguun, E.O.; Araki, K.; Kanai, Y.; Yokobori, T.; et al. Relationship between LAT1 expression and resistance to chemotherapy in pancreatic ductal adenocarcinoma. Cancer Chemother. Pharmacol. 2018, 81, 141–153. [Google Scholar] [CrossRef]
- Yanagisawa, N.; Ichinoe, M.; Mikami, T.; Nakada, N.; Hana, K.; Koizumi, W.; Endou, H.; Okayasu, I. High expression of L-type amino acid transporter 1 (LAT1) predicts poor prognosis in pancreatic ductal adenocarcinomas. J. Clin. Pathol. 2012, 65, 1019–1023. [Google Scholar] [CrossRef]
- Ikhlef, H.; Phanstiel, O. Development of LAT-1 inhibitors for the treatment of pancreatic cancers. FASEB J. 2019, 33 (Suppl. 1), 652–659. [Google Scholar]
- Kurahara, H.; Maemura, K.; Mataki, Y.; Sakoda, M.; Iino, S.; Kawasaki, Y.; Arigami, T.; Mori, S.; Kijima, Y.; Ueno, S.; et al. Significance of glucose transporter type 1 (GLUT-1) expression in the therapeutic strategy for pancreatic ductal adenocarcinoma. Ann. Surg. Oncol. 2018, 25, 1432–1439. [Google Scholar] [CrossRef]
- Cortese, N.; Capretti, G.; Barbagallo, M.; Rigamonti, A.; Takis, P.G.; Castino, G.F.; Vignali, D.; Maggi, G.; Gavazzi, F.; Ridolfi, C.; et al. Metabolome of pancreatic juice delineates distinct clinical profiles of pancreatic cancer and reveals a link between glucose metabolism and PD-1+ cells. Cancer Immunol. Res. 2020, 8, 493–505. [Google Scholar] [CrossRef]
- Baek, G.; Yan, F.T.; Hu, Z.; Cox, D.; Buboltz, N.; McCue, P.; Yeo, C.J.; White, M.A.; DeBerardinis, R.J.; Knudsen, E.S.; et al. MCT4 defines a glycolytic subtype of pancreatic cancer with poor prognosis and unique metabolic dependencies. Cell Rep. 2014, 9, 2233–2249. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Liu, H. Overview of cancer stem cells and stemness for community oncologists. Target. Oncol. 2017, 12, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Aponte, P.M.; Caicedo, A. Stemness in cancer: Stem cells, cancer stem cells, and their microenvironment. Stem Cells Int. 2017, 2017, 5619472. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Duan, Q.; Zhao, H.; Liu, T.; Wu, H.; Shen, Q.; Wang, C.; Yin, T. Gemcitabine treatment promotes pancreatic cancer stemness through the Nox/ROS/NF-κB/STAT3 signaling cascade. Cancer Lett. 2016, 382, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Srivastava, S.K.; Zubair, H.; Patel, G.K.; Arora, S.; Carter, J.E.; Gorman, G.S.; Singh, S.; Singh, A.P. Co-targeting of CXCR4 and hedgehog pathways disrupts tumor-stromal crosstalk and improves chemotherapeutic efficacy in pancreatic cancer. J. Biol. Chem. 2020, 295, 8413–8424. [Google Scholar] [CrossRef]
- Williet, N.; Saint, A.; Pointet, A.-L.; Tougeron, D.; Pernot, S.; Pozet, A.; Bechade, D.; Trouilloud, I.; Lourenco, N.; Hautefeuille, V.; et al. Folfirinox versus gemcitabine/nab-paclitaxel as first-line therapy in patients with metastatic pancreatic cancer: A comparative propensity score study. Ther. Adv. Gastroenterol. 2019, 12, 1756284819878660. [Google Scholar] [CrossRef]
- Cho, I.R.; Kang, H.; Jo, J.H.; Lee, H.S.; Chung, M.J.; Park, J.Y.; Park, S.W.; Song, S.Y.; An, C.; Park, M.S.; et al. FOLFIRINOX vs gemcitabine/nab-paclitaxel for treatment of metastatic pancreatic cancer: Single-center cohort study. World J. Gastrointest. Oncol. 2020, 12, 182. [Google Scholar] [CrossRef]
- Vogl, U.M.; Andalibi, H.; Klaus, A.; Vormittag, L.; Schima, W.; Heinrich, B.; Kafka, A.; Winkler, T.; Öhler, L. Nab-paclitaxel and gemcitabine or FOLFIRINOX as first-line treatment in patients with unresectable adenocarcinoma of the pancreas: Does sequence matter? BMC Cancer 2019, 19, 28. [Google Scholar] [CrossRef]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef]
- Kuwada, K.; Kagawa, S.; Yoshida, R.; Sakamoto, S.; Ito, A.; Watanabe, M.; Ieda, T.; Kuroda, S.; Kikuchi, S.; Tazawa, H.; et al. The epithelial-to-mesenchymal transition induced by tumor-associated macrophages confers chemoresistance in peritoneally disseminated pancreatic cancer. J. Exp. Clin. Cancer Res. 2018, 37, 307. [Google Scholar] [CrossRef]
- Lankadasari, M.B.; Mukhopadhyay, P.; Mohammed, S.; Harikumar, K.B. TAMing pancreatic cancer: Combat with a double edged sword. Mol. Cancer 2019, 18, 48. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Carmona-Fontaine, C.; Deforet, M.; Akkari, L.; Thompson, C.B.; Joyce, J.A.; Xavier, J.B. Metabolic origins of spatial organization in the tumor microenvironment. Proc. Natl. Acad. Sci. USA 2017, 114, 2934–2939. [Google Scholar] [CrossRef]
- Shukla, S.K.; Markov, S.D.; Attri, K.S.; Vernucci, E.; King, R.J.; Dasgupta, A.; Grandgenett, P.M.; Hollingsworth, M.A.; Singh, P.K.; Yu, F.; et al. Macrophages potentiate STAT3 signaling in skeletal muscles and regulate pancreatic cancer cachexia. Cancer Lett. 2020, 484, 29–39. [Google Scholar] [CrossRef]
- Michaelis, K.A.; Zhu, X.; Burfeind, K.G.; Krasnow, S.M.; Levasseur, P.R.; Morgan, T.K.; Marks, D.L. Establishment and characterization of a novel murine model of pancreatic cancer cachexia. J. Cachexiasarcopenia Muscle 2017, 8, 824–838. [Google Scholar] [CrossRef]
- Penny, H.L.; Sieow, J.L.; Adriani, G.; Yeap, W.H.; See Chi Ee, P.; San Luis, B.; Lee, B.; Lee, T.; Mak, S.Y.; Ho, Y.S.; et al. Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. Oncoimmunology 2016, 5, e1191731. [Google Scholar] [CrossRef]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.-W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity 2017, 47, 323–338.e6. [Google Scholar] [CrossRef]
- Ye, H.; Zhou, Q.; Zheng, S.; Li, G.; Lin, Q.; Wei, L.; Fu, Z.; Zhang, B.; Liu, Y.; Li, Z.; et al. Tumor-associated macrophages promote progression and the Warburg effect via CCL18/NF-kB/VCAM-1 pathway in pancreatic ductal adenocarcinoma. Cell Death Dis. 2018, 9, 453. [Google Scholar] [CrossRef]
- Halbrook, C.J.; Pontious, C.; Kovalenko, I.; Lapienyte, L.; Dreyer, S.; Lee, H.J.; Thurston, G.; Zhang, Y.; Lazarus, J.; Sajjakulnukit, P.; et al. Macrophage-released pyrimidines inhibit gemcitabine therapy in pancreatic cancer. Cell Metab. 2019, 29, 1390–1399.e6. [Google Scholar] [CrossRef]
- Liu, M.; O’Connor, R.S.; Trefely, S.; Graham, K.; Snyder, N.W.; Beatty, G.L. Metabolic rewiring of macrophages by CpG potentiates clearance of cancer cells and overcomes tumor-expressed CD47− mediated ‘don’t-eat-me’ signal. Nat. Immunol. 2019, 20, 265–275. [Google Scholar] [CrossRef]
- Luengo, A.; Gui, D.Y.; Vander Heiden, M.G. Targeting metabolism for cancer therapy. Cell Chem. Biol. 2017, 24, 1161–1180. [Google Scholar] [CrossRef]
- Grasso, C.; Jansen, G.; Giovannetti, E. Drug resistance in pancreatic cancer: Impact of altered energy metabolism. Crit. Rev. Oncol. Hematol. 2017, 114, 139–152. [Google Scholar] [CrossRef]
- Conway, J.R.; Herrmann, D.; Evans, T.J.; Morton, J.P.; Timpson, P. Combating pancreatic cancer with PI3K pathway inhibitors in the era of personalised medicine. Gut 2019, 68, 742–758. [Google Scholar] [CrossRef]
- Punekar, S.; Cho, D.C. Novel Therapeutics Affecting Metabolic Pathways. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, e79–e87. [Google Scholar] [CrossRef]
- Tripathi, S.C.; Fahrmann, J.F.; Vykoukal, J.V.; Dennison, J.B.; Hanash, S.M. Targeting metabolic vulnerabilities of cancer: Small molecule inhibitors in clinic. Cancer Rep. 2019, 2, e1131. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 2014, 19, 637. [Google Scholar] [CrossRef]
- Samaras, P.; Tusup, M.; Nguyen-Kim, T.D.L.; Seifert, B.; Bachmann, H.; von Moos, R.; Knuth, A.; Pascolo, S. Phase I study of a chloroquine–gemcitabine combination in patients with metastatic or unresectable pancreatic cancer. Cancer Chemother. Pharmacol. 2017, 80, 1005–1012. [Google Scholar] [CrossRef]
- Mohammad, G.H.; Olde Damink, S.; Malago, M.; Dhar, D.K.; Pereira, S.P. Pyruvate kinase M2 and lactate dehydrogenase A are overexpressed in pancreatic cancer and correlate with poor outcome. PLoS ONE 2016, 11, e0151635. [Google Scholar] [CrossRef]
- Huang, X.; Wullschleger, S.; Shpiro, N.; McGuire, V.A.; Sakamoto, K.; Woods, Y.L.; McBurnie, W.; Fleming, S.; Alessi, D.R. Important role of the LKB1–AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem. J. 2008, 412, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Stuart, S.D.; Schauble, A.; Gupta, S.; Kennedy, A.D.; Keppler, B.R.; Bingham, P.M.; Zachar, Z. A strategically designed small molecule attacks alpha-ketoglutarate dehydrogenase in tumor cells through a redox process. Cancer Metab. 2014, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Khaira, D.; Rodriguez, R.; Maturo, C.; O’Donnell, K.; Shorr, R. Long-term stable disease of stage IV pancreatic neuroendocrine tumors and without significant adverse effect by CPI-613, an investigational novel anti-cancer agent. Case Study Case Rep. 2011, 1, 137–145. [Google Scholar]
- Detassis, S.; Grasso, M.; Del Vescovo, V.; Denti, M.A. microRNAs make the call in cancer personalized medicine. Front. Cell Dev. Biol. 2017, 5, 86. [Google Scholar] [CrossRef] [PubMed]
- Tesfaye, A.A.; Kamgar, M.; Azmi, A.; Philip, P.A. The evolution into personalized therapies in pancreatic ductal adenocarcinoma: Challenges and opportunities. Expert Rev. Anticancer Ther. 2018, 18, 131–148. [Google Scholar] [CrossRef] [PubMed]
- Leone, P.; Buonavoglia, A.; Fasano, R.; Solimando, A.G.; De Re, V.; Cicco, S.; Vacca, A.; Racanelli, V. Insights into the Regulation of Tumor Angiogenesis by Micro-RNAs. J. Clin. Med. 2019, 8, 2030. [Google Scholar] [CrossRef]
- Wang, S.; Zhou, H.; Wu, D.; Ni, H.; Chen, Z.; Chen, C.; Xiang, Y.; Dai, K.; Chen, X.; Li, X. Micro RNA let-7a regulates angiogenesis by targeting TGFBR 3 mRNA. J. Cell. Mol. Med. 2019, 23, 556–567. [Google Scholar] [CrossRef]
- Sureban, S.M.; May, R.; Qu, D.; Weygant, N.; Chandrakesan, P.; Ali, N.; Lightfoot, S.A.; Pantazis, P.; Rao, C.V.; Postier, R.G.; et al. DCLK1 regulates pluripotency and angiogenic factors via microRNA-dependent mechanisms in pancreatic cancer. PLoS ONE 2013, 8, e73940. [Google Scholar] [CrossRef]
- Golan, T.; Milella, M.; Ackerstein, A.; Berger, R. The changing face of clinical trials in the personalized medicine and immuno-oncology era: Report from the international congress on clinical trials in Oncology & Hemato-Oncology (ICTO 2017). J. Exp. Clin. Cancer Res. 2017, 36, 192. [Google Scholar] [CrossRef]


{kind=link}
| Phase | Trial Id | Target | Compound | Results/Status |
|---|---|---|---|---|
| I | NCT02071862 | GLS | CB-839 | Completed |
| III | NCT03504423 | TCA cycle | FFX Versus CPI-613 with mFFX | Active |
| I | NCT01835041 | TCA cycle | CPI-613 Versus CPI-613 with mFFX | Active |
| I/II | NCT01128296 | autophagy | HCQ + gemcitabine | completed, encouraging results |
| I/II | NCT01506973 | autophagy | HCQ + gemcitabine + abraxane | Active |
| II | NCT03601923 | Poly ADP ribose polymerase | Niraparib | Active |
| II | NCT04409002 | Poly ADP ribose polymerase | niraparib with dostarlimab (antibody attaching to protein called PD-1 on Tcells) and radiation therapy | Active |
| III | NCT03977272 | PD-1 | modified-FOLFIRINOX and Anti-PD-1 antibody in patients with metastatic pancreatic cancer. | Active |
| I | NCT03435289 | PDH and α-KGDH | CPI-613 in combination with gemcitabine and nab-paclitaxel | unknown |
| I | NCT04181645 | PD-1 | SHR-1210 (anti-PD1)/Gemcitabine/Paclitaxel-albumin | Active |
| I | NCT03497819 | mesothelin; tumor associated B cells | CARTmeso; CART19 | Active |
| II/III | NCT03512756 | Reactive oxygen species | SM-88 used with MPS (methoxsalen, phenytoin, sirolimus) | Active |
| II | NCT03509298 | MUC1 | Activated CIK and CD3-MUC1 Bispecific Antibody | Active |
| III | NCT03504423 | Mitochondrial enzymes | FFX versus CPI-613 + mFFX | Active |
| I | NCT01839981 | PDH | 6,8-bis(benzylthio)octanoic acid | Completed, no results posted |
| II | NCT01273805 | autophagy | Hydrooxychloroquine | Completed [129] |
| I | NCT01777477 | autophagy | HCQ + gemcitabine | Completed [130] |
| I/II | NCT00096707 | Hexokinase | 2-DG alone or with docetaxel | No effects [131] |
| I/II | NCT00907166 | PDH and α-KGDH | CPI-613 with gemcitabine | encouraging results [132] |
| II | NCT01167738 | Mitochondrial complex I | metformin with PEXG | negative results [133] |
| II | NCT01210911 | Mitochondrial complex II | metformin with Gemcitabine and erlotinib | negative results [134] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rai, V.; Agrawal, S. Targets (Metabolic Mediators) of Therapeutic Importance in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 8502. https://doi.org/10.3390/ijms21228502
Rai V, Agrawal S. Targets (Metabolic Mediators) of Therapeutic Importance in Pancreatic Ductal Adenocarcinoma. International Journal of Molecular Sciences. 2020; 21(22):8502. https://doi.org/10.3390/ijms21228502
Chicago/Turabian StyleRai, Vikrant, and Swati Agrawal. 2020. "Targets (Metabolic Mediators) of Therapeutic Importance in Pancreatic Ductal Adenocarcinoma" International Journal of Molecular Sciences 21, no. 22: 8502. https://doi.org/10.3390/ijms21228502
APA StyleRai, V., & Agrawal, S. (2020). Targets (Metabolic Mediators) of Therapeutic Importance in Pancreatic Ductal Adenocarcinoma. International Journal of Molecular Sciences, 21(22), 8502. https://doi.org/10.3390/ijms21228502