Recombinant Chromosome 7 Driven by Maternal Chromosome 7 Pericentric Inversion in a Girl with Features of Silver-Russell Syndrome

 , ,
, ,  and
and
Abstract
1. Introduction
2. Results
2.1. Clinical Report
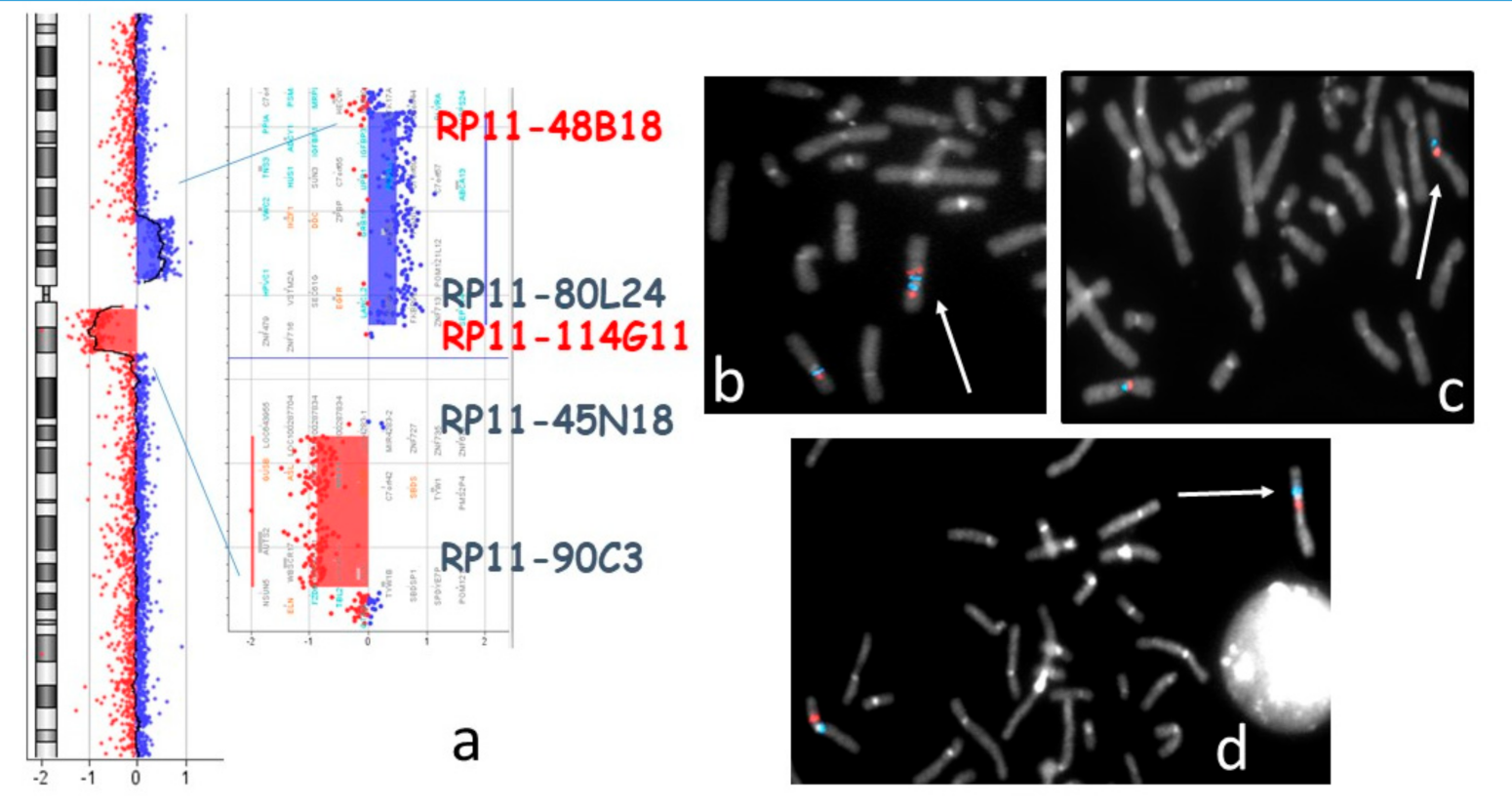
2.2. Cytogenetics Analysis
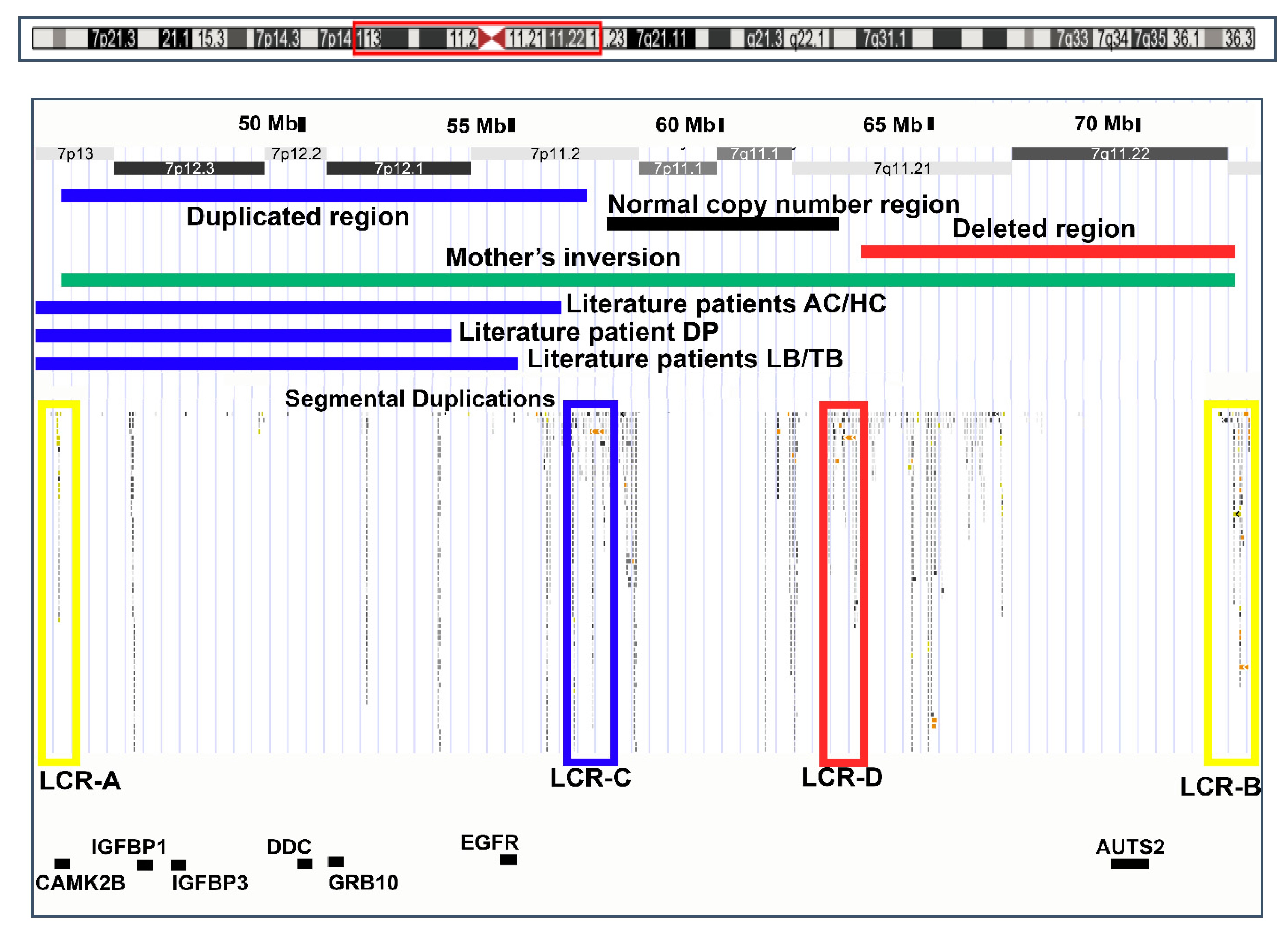
2.3. Molecular Genetic Analyses
3. Discussion
3.1. Genotype–Phenotype Correlation
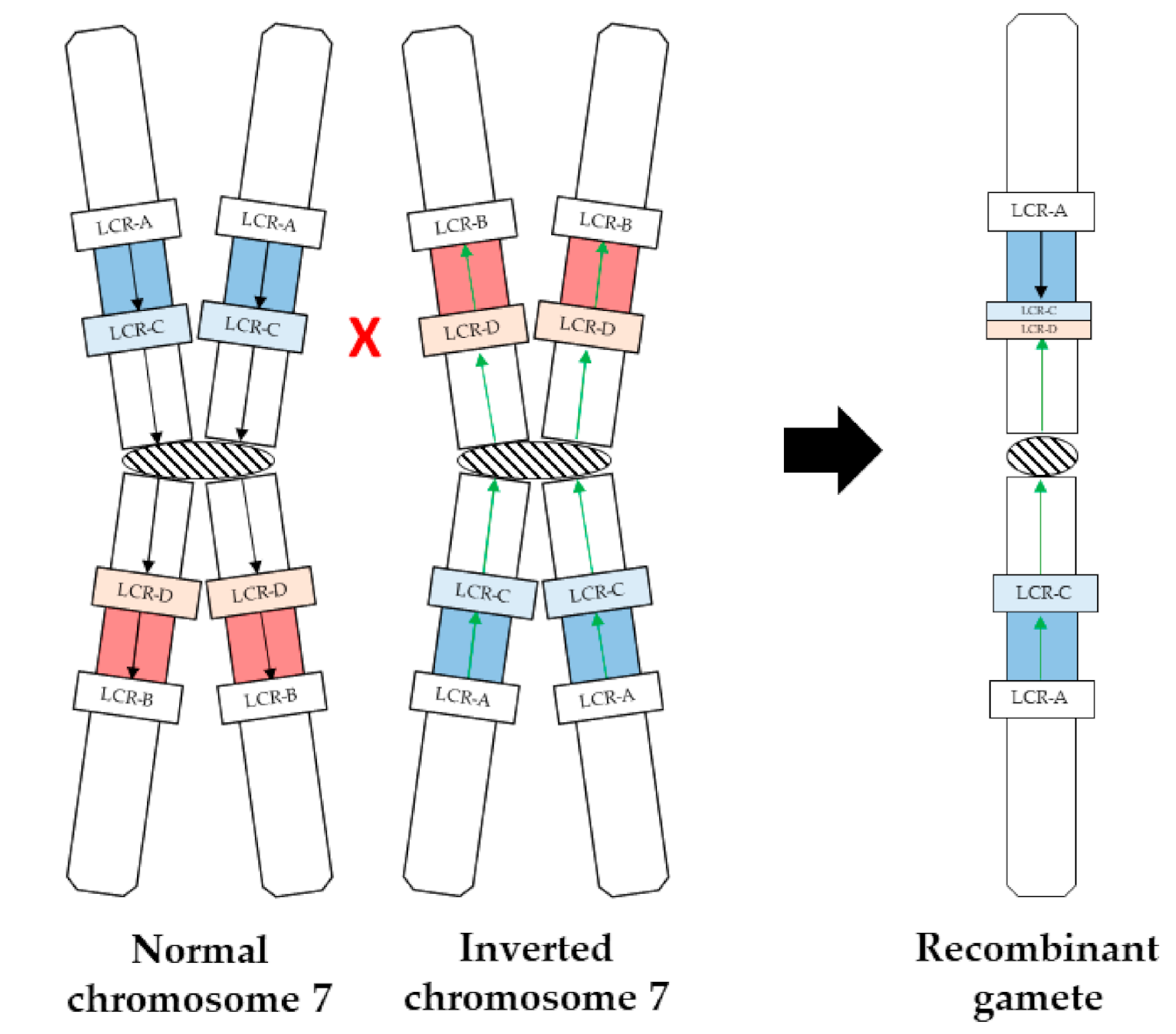
3.2. Rearrangement Mechanism
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wakeling, E.L.; Brioude, F.; Lokulo-Sodipe, O.; O’Connell, S.M.; Salem, J.; Bliek, J.; Canton, A.P.M.; Chrzanowska, K.H.; Davies, J.H.; Dias, R.P.; et al. Diagnosis and management of Silver–Russell syndrome: First international consensus statement. Nat. Rev. Endocrinol. 2017, 13, 105–124. [Google Scholar] [CrossRef] [PubMed]
- Monk, D.; Wakeling, E.L.; Proud, V.; Hitchins, M.; Abu-Amero, S.N.; Stanier, P.; Preece, M.A.; Moore, G.E. Duplication of 7p11.2-p13, Including GRB10, in Silver-Russell Syndrome. Am. J. Hum. Genet. 2000, 66, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Monk, D.; Bentley, L.; Hitchins, M.; Myler, R.A.; Clayton-Smith, J.; Ismail, S.; Price, S.M.; Preece, M.A.; Stanier, P.; Moore, G.E. Chromosome 7p disruptions in Silver Russell syndrome: Delineating an imprinted candidate gene region. Qual. Life Res. 2002, 111, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Joyce, C.; Sharp, A.; Walker, J.; Bullman, H.; Temple, I. Duplication of 7p12.1-p13, including GRB10 and IGFBP1, in a mother and daughter with features of Silver-Russell syndrome. Qual. Life Res. 1999, 105, 273–280. [Google Scholar] [CrossRef]
- Eggermann, T.; Begemann, M.; Gogiel, M.; Palomares, M.; Vallespin, E.; Fernandez, L.; Cazorla, R.; Spengler, S.; García-Miñaur, S. Heterogeneous growth patterns in carriers of chromosome 7p12.2 imbalances affecting GRB10. Am. J. Med. Genet. Part A 2012, 158A, 2815–2819. [Google Scholar] [CrossRef] [PubMed]
- DECIPHER Database (DatabasE of genomiC varIation and Phenotype in Humans Using Ensembl Resources). Available online: https://decipher.sanger.ac.uk/ (accessed on 31 October 2020).
- Online Mendelian Inheritance in Men (OMIM). Available online: omim.org (accessed on 22 September 2020).
- Eggermann, T.; Begemann, M.; Kurth, I.; Elbracht, M. Contribution of GRB10 to the prenatal phenotype in Silver-Russell syndrome? Lessons from 7p12 copy number variations. Eur. J. Med. Genet. 2019, 62, 103671. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, P.; Monk, D.; Hitchins, M.; Gordon, E.; Dean, W.; Beechey, C.V.; Peters, J.; Craigen, W.; Preece, M.; Stanier, P.; et al. Conserved methylation imprints in the human and mouse GRB10 genes with divergent allelic expression suggests differential reading of the same mark. Hum. Mol. Genet. 2003, 12, 1005–1019. [Google Scholar] [CrossRef] [PubMed]
- Beunders, G.; Van De Kamp, J.; Vasudevan, P.; Morton, J.; Smets, K.; Kleefstra, T.; A De Munnik, S.; Schuurs-Hoeijmakers, J.; Ceulemans, B.; Zollino, M.; et al. A detailed clinical analysis of 13 patients with AUTS2 syndrome further delineates the phenotypic spectrum and underscores the behavioural phenotype. J. Med. Genet. 2016, 53, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Sanders, A.D.; Hills, M.; Porubský, D.; Guryev, V.; Falconer, E.; Lansdorp, P.M. Characterizing polymorphic inversions in human genomes by single-cell sequencing. Genome Res. 2016, 26, 1575–1587. [Google Scholar] [CrossRef] [PubMed]
- Catacchio, C.R.; Maggiolini, F.A.M.; D′addabbo, P.; Bitonto, M.; Capozzi, O.; Signorile, M.L.; Miroballo, M.; Archidiacono, N.; Eichler, E.E.; Ventura, M.; et al. Inversion variants in human and primate genomes. Genome Res. 2018, 28, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Anton, E.; Blanco, J.; Egozcue, J.; Vidal, F. Sperm studies in heterozygote inversion carriers: A review. Cytogenet. Genome Res. 2005, 111, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Anton, E.; Vidal, F.; Egozcue, J.; Blanco, J. Genetic reproductive risk in inversion carriers. Fertil. Steril. 2006, 85, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Morel, F.; Laudier, B.; Guérif, F.; Couet, M.; Royère, D.; Roux, C.; Bresson, J.; Amice, V.; De Braekeleer, M.; Douet-Guilbert, N. Meiotic segregation analysis in spermatozoa of pericentric inversion carriers using fluorescence in-situ hybridization. Hum. Reprod. 2007, 22, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Dong, M.; Sun, Y.; Wang, L.; Chen, S.; Jin, F. Different segregation patterns in five carriers due to a pericentric inversion of chromosome 1. Syst. Biol. Reprod. Med. 2014, 60, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Navarro, J.; Benet, J.; Martorell, M.R.; Templado, C.; Egozcue, J. Segregation analysis in a man heterozygous for a pericentric inversion of chromosome 7 (p13;q36) by sperm chromosome studies. Am. J. Hum. Genet. 1993, 53, 214–219. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Region on chr.7 | SNP Probes | Genotype | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SNP_ID | Probe Name | Cytoband | Father | Proband | Mother | |||||
| Deleted region | rs6945241 | A_20_P00145608 | 7q11.22 | C | T | C | T | T | ||
| rs2103132 | A_20_P00247516 | 7q11.22 | C | G | C | G | G | |||
| rs6460543 | A_20_P00145611 | 7q11.22 | A | G | A | G | G | |||
| rs7793970 | A_20_P00247563 | 7q11.22 | A | G | A | G | G | |||
| rs6979389 | A_20_P00145689 | 7q11.22 | C | T | C | T | T | |||
| Region on chr.7 | Microsatellites | Number of Repeats | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Marker | Locus | Cytoband | Range | Father | Proband | Mother | |||||
| Duplicated region | 12 | D7S519 | 7p13 | 257–285 | 269 | 263 | 269 | 261 | 257 | 257 | 261 |
| 21 | D7S2422 | 7p12.1 | 195–227 | 208 | 194 | 208 | 192 | 211 | 211 | 192 | |
| 22 | D7S2467 | 7p12.1 | 240–248 | 239 | 239 | 239 | 241 | 241 | 241 | 241 | |
| 33 | D7S506 | 7p12.1 | 117–146 | 128 | 112 | 128 | 128 | 128 | 128 | 128 | |
| 24 | D7S2552 | 7p11.2 | 232–282 | 270 | 256 | 270 | 270 | 276 | 276 | 270 | |
| Patient | Sex | Age | SRS Diagnostic Criteria | PSS (SDS) | Additional SRS Features | DDs | Duplication 7p | Other CNV (hg19) | References | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | Breakpoints on chr.7 (hg19) | Size (Mb) | Inheritance | N. of RefSeq Genes | ||||||||
| Present case | F | 17 y 6 m | − | + | + | − | − | − | + (−2.81) | micrognatia, hypoglycaemia | motor and speech delay, severe ID, head stereotypy | 44,114,508-56,786,860 | 12.67 | de novo, mat origin | 64 | 7q11.21q11.23(63,374,309-72,365,957)x1 | Present study |
| AC § | M | 4 y | + | + | Na | − | − | na | + (−2.00) | 5th finger clinodactyly | not reported | 39,747,723-56,160,689 | 16.4 | mat | 83 | − | Monk, 2002 [3] |
| HC § # (AC’s mother) | F | 48 y | na | + | Na | − | − | na | + (−2.00) | 5th finger clinodactyly | not reported | Idem | idem | na | idem | − | |
| DP | F | 5 y | − | − | Na | + | − | + | + (−2.90) | triangular face, micrognatia, down-turned mouth, 5th finger clinodactyly, hypoglycaemia, excessive sweating | mild DD | 39,668,287-53,521,622 | 13.85 | de novo, mat origin | 69 | 7p12.1p11.2(52,885,014-54,748,619)x1 | Monk 2000; 2002 [2,3] |
| LB | F | 6.3 y | − | na | − | − | − | na | + (−3.56) | micrognatia, 5th finger clinodactyly | LD | 42,000,548-55,129,179 | 13.12 | mat | 65 | − | Joyce, 1999; Monk, 2002 [3,4] |
| TB (LB’s mother) | F | adult | na | na | Na | − | − | − | + (−2.00) | micrognatia, 5th finger clinodactyly | LD | Idem | idem | de novo, pat origin | idem | − | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catusi, I.; Bonati, M.T.; Mainini, E.; Russo, S.; Orlandini, E.; Larizza, L.; Recalcati, M.P. Recombinant Chromosome 7 Driven by Maternal Chromosome 7 Pericentric Inversion in a Girl with Features of Silver-Russell Syndrome. Int. J. Mol. Sci. 2020, 21, 8487. https://doi.org/10.3390/ijms21228487
Catusi I, Bonati MT, Mainini E, Russo S, Orlandini E, Larizza L, Recalcati MP. Recombinant Chromosome 7 Driven by Maternal Chromosome 7 Pericentric Inversion in a Girl with Features of Silver-Russell Syndrome. International Journal of Molecular Sciences. 2020; 21(22):8487. https://doi.org/10.3390/ijms21228487
Chicago/Turabian StyleCatusi, Ilaria, Maria Teresa Bonati, Ester Mainini, Silvia Russo, Eleonora Orlandini, Lidia Larizza, and Maria Paola Recalcati. 2020. "Recombinant Chromosome 7 Driven by Maternal Chromosome 7 Pericentric Inversion in a Girl with Features of Silver-Russell Syndrome" International Journal of Molecular Sciences 21, no. 22: 8487. https://doi.org/10.3390/ijms21228487
APA StyleCatusi, I., Bonati, M. T., Mainini, E., Russo, S., Orlandini, E., Larizza, L., & Recalcati, M. P. (2020). Recombinant Chromosome 7 Driven by Maternal Chromosome 7 Pericentric Inversion in a Girl with Features of Silver-Russell Syndrome. International Journal of Molecular Sciences, 21(22), 8487. https://doi.org/10.3390/ijms21228487