Trans-Ancestral Fine-Mapping and Epigenetic Annotation as Tools to Delineate Functionally Relevant Risk Alleles at IKZF1 and IKZF3 in Systemic Lupus Erythematosus


Abstract
:1. Introduction
2. Results
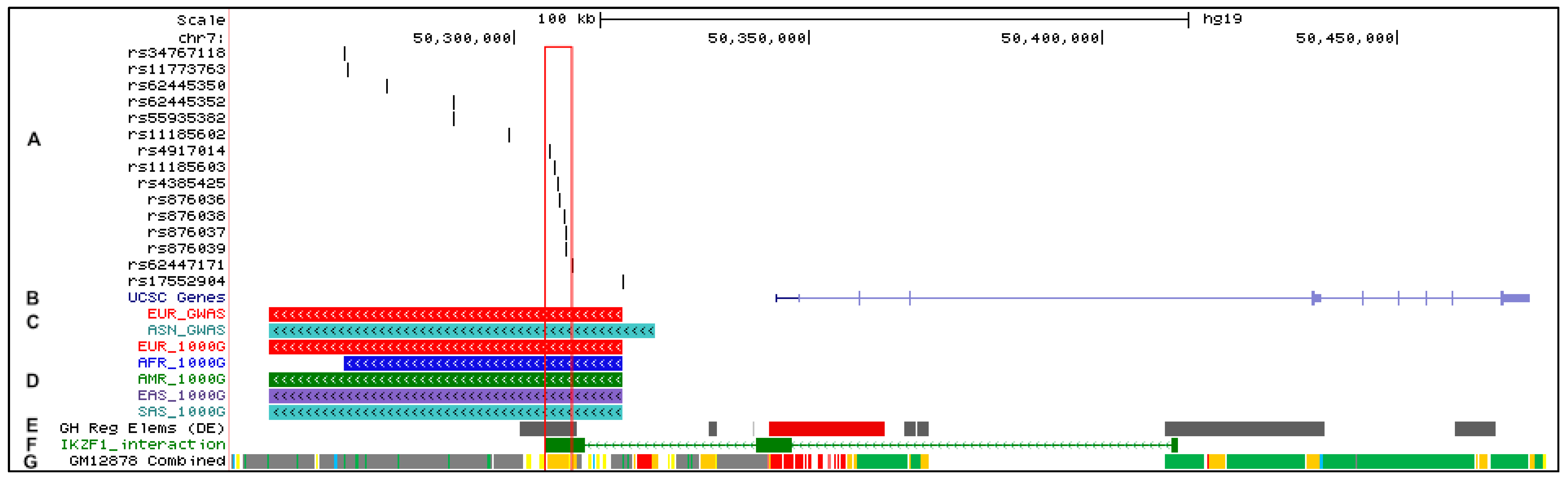
2.1. Defining the Risk Haplotype at IKZF1 in SLE
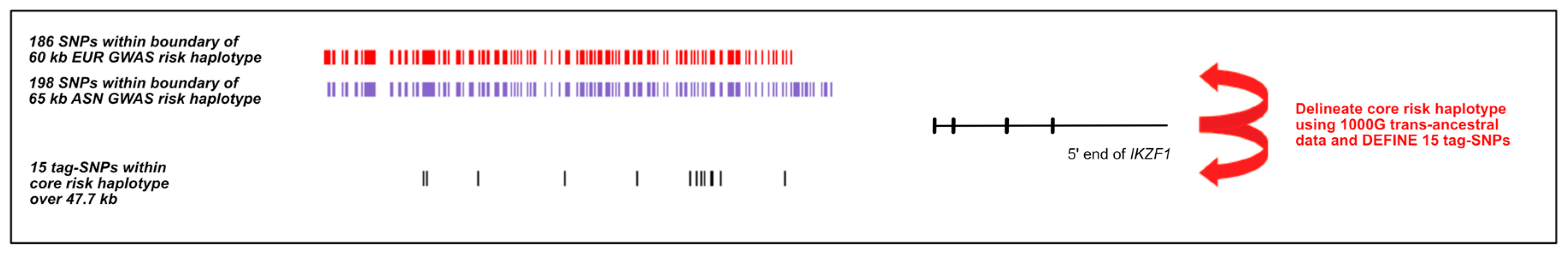
2.2. Refining the IKZF1 Risk Haplotype Using the 1000 Genomes Super-Populations
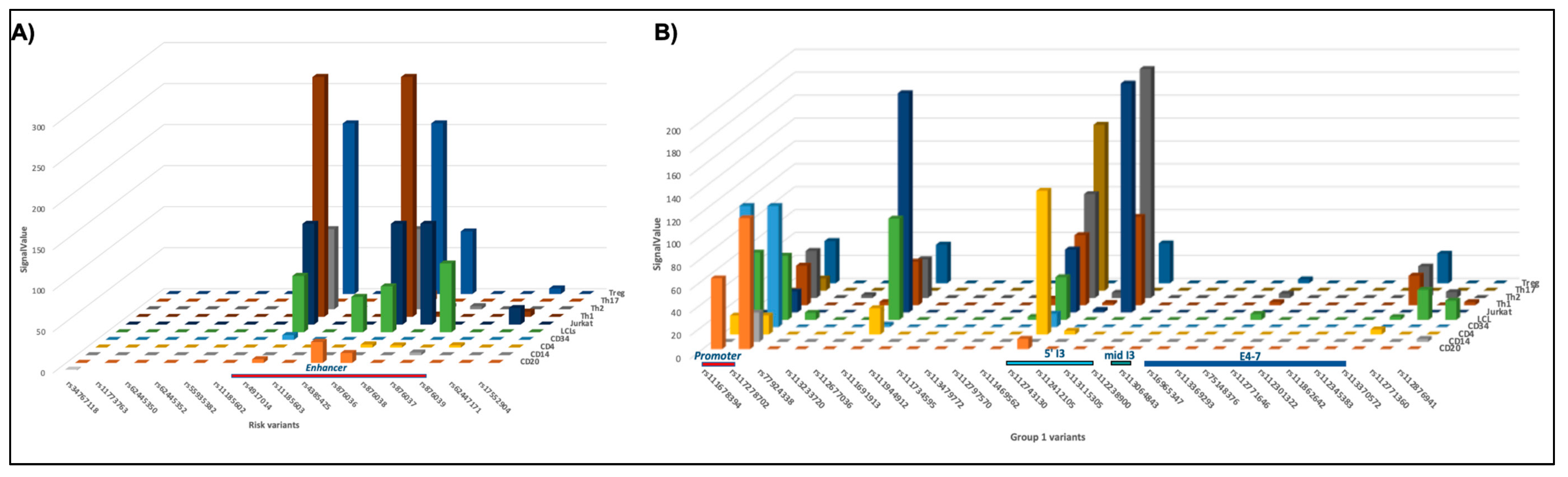
2.3. Functional Annotation of IKZF1 Risk Alleles
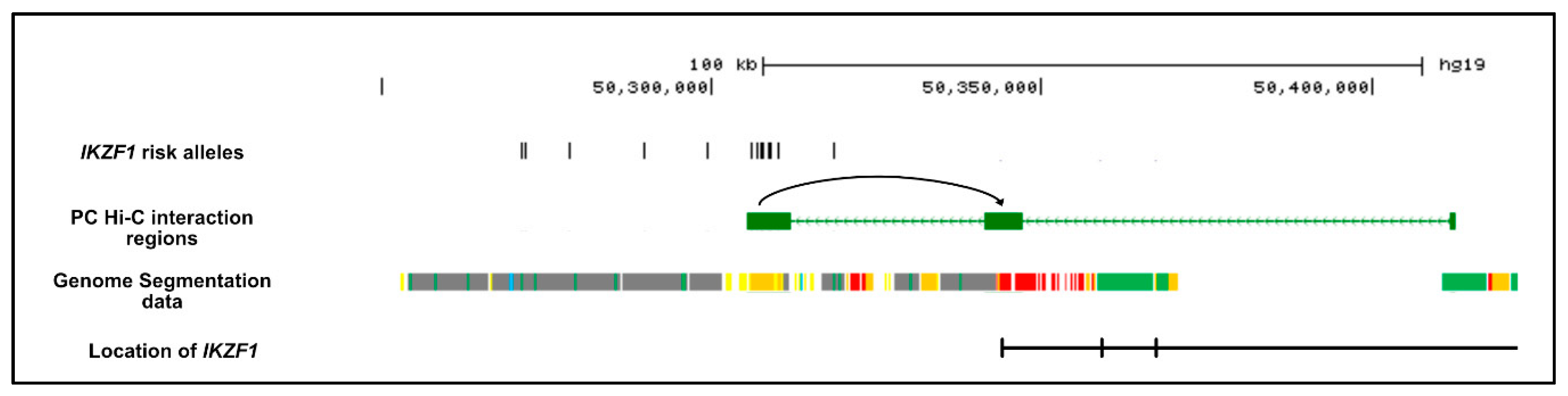
2.3.1. Determination of Chromatin Status
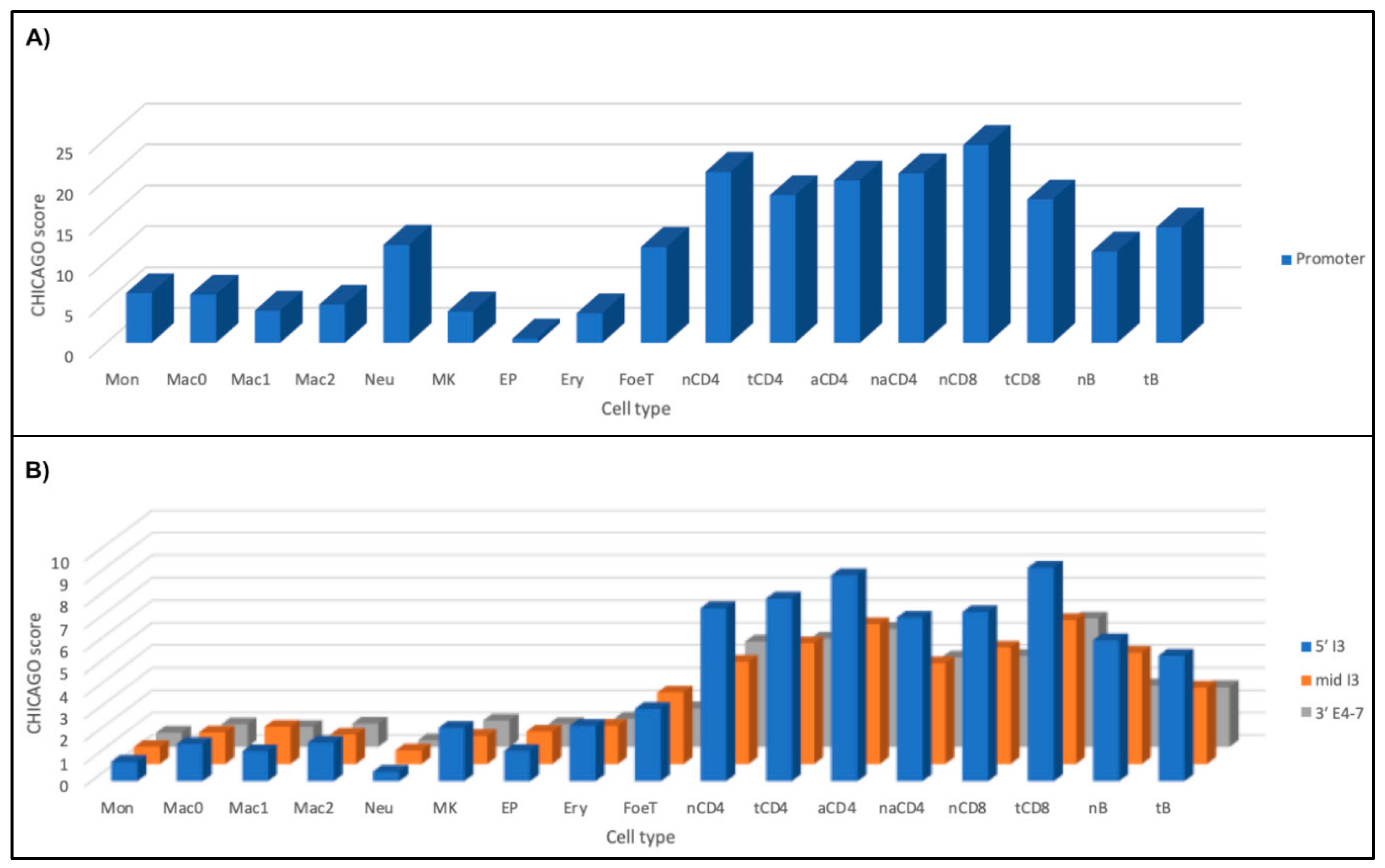
2.3.2. Chromatin Looping with Risk Alleles
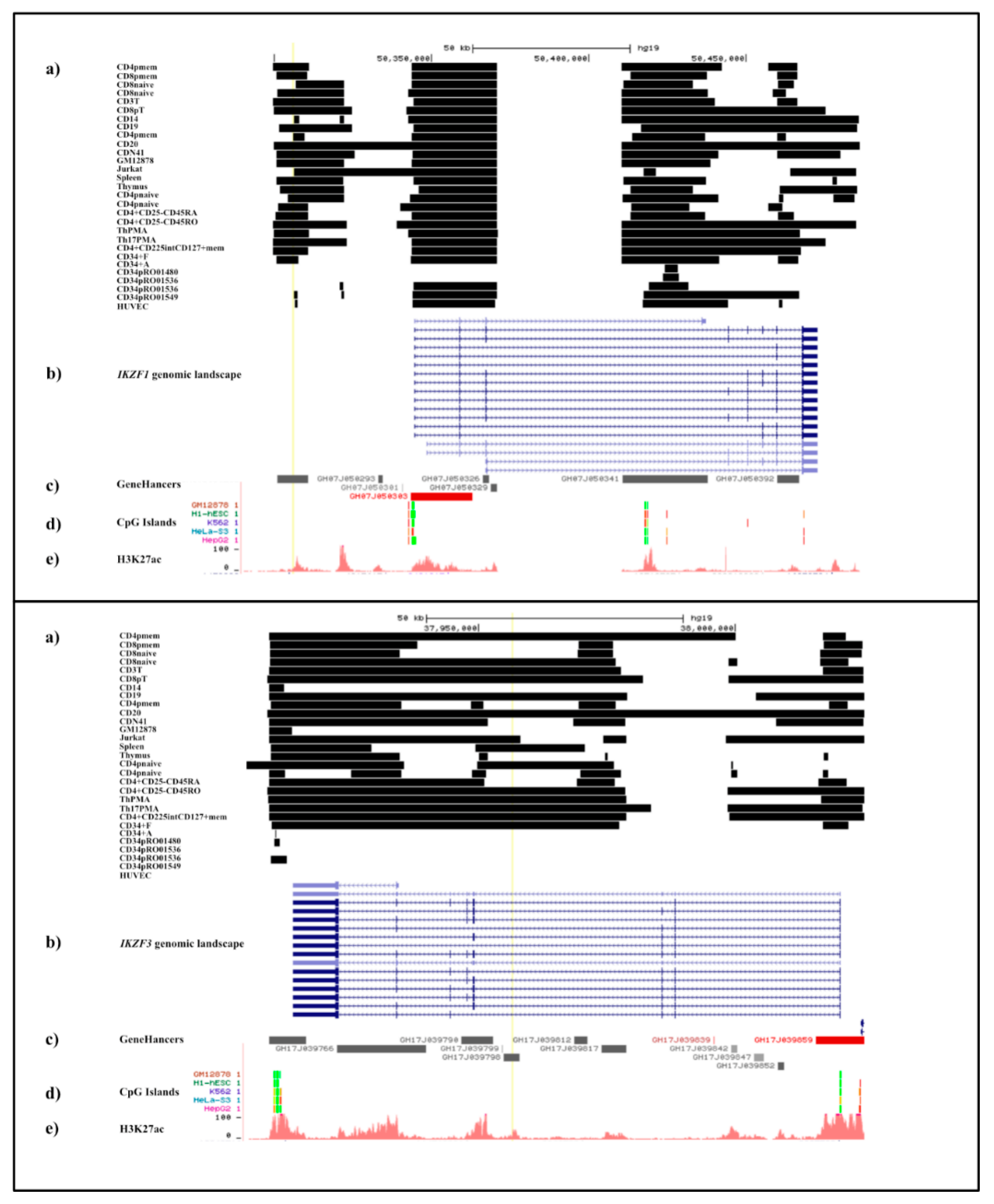
2.3.3. Cell-Type Specificity in DNAse Sensitivity in the IKZF1 Enhancer Region
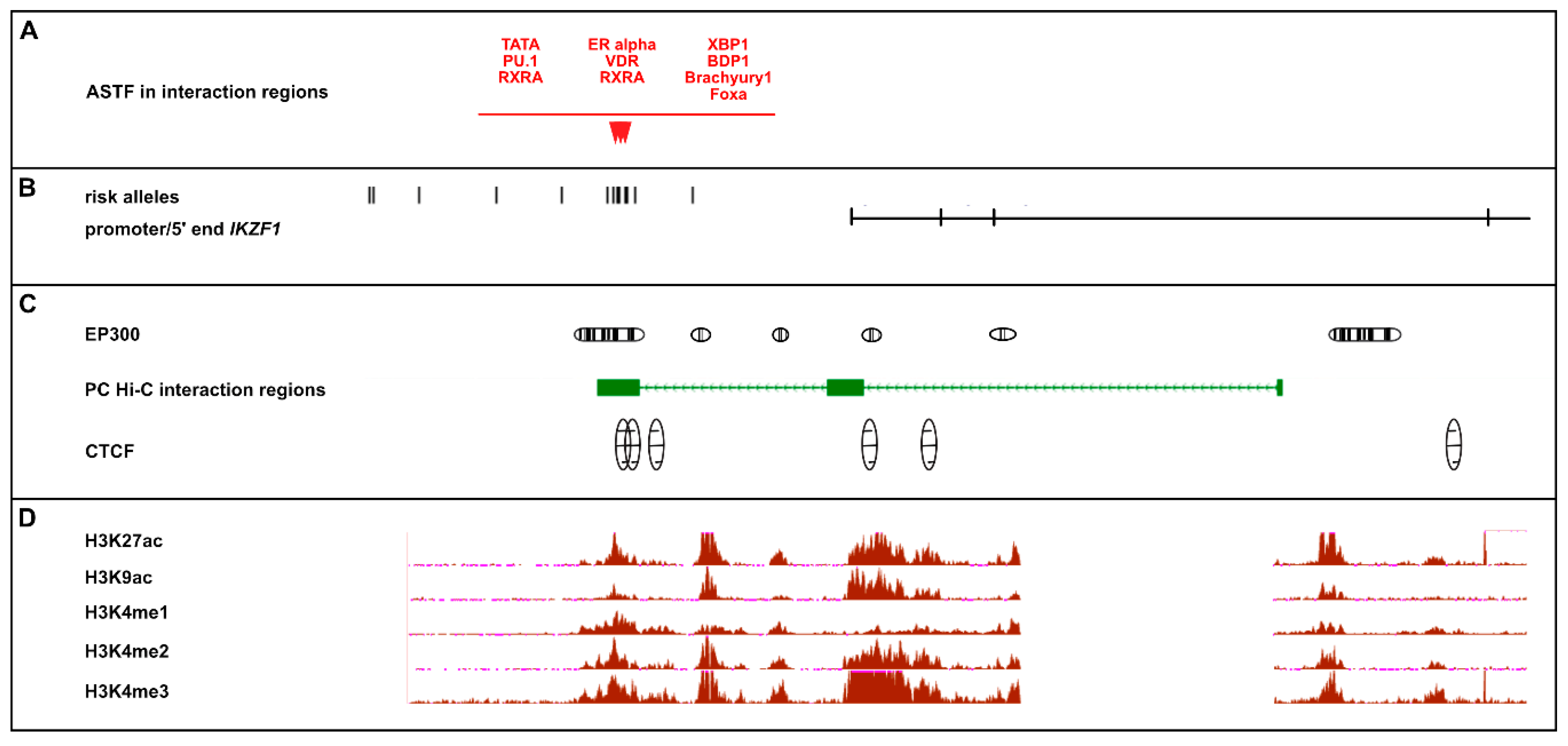
2.3.4. Discovery of Allele-Specific Transcription Factor Binding Sites
2.3.5. Identification of cis-eQTLs at IKZF1
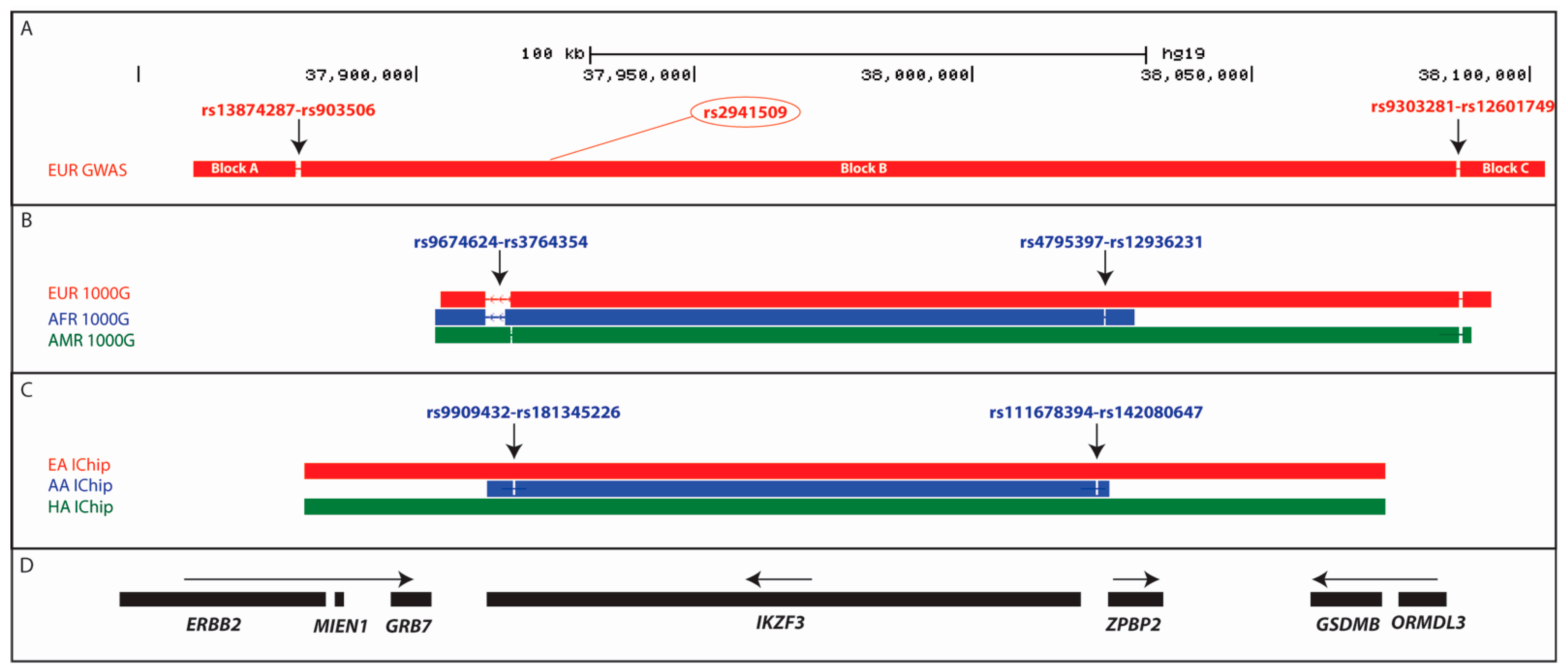
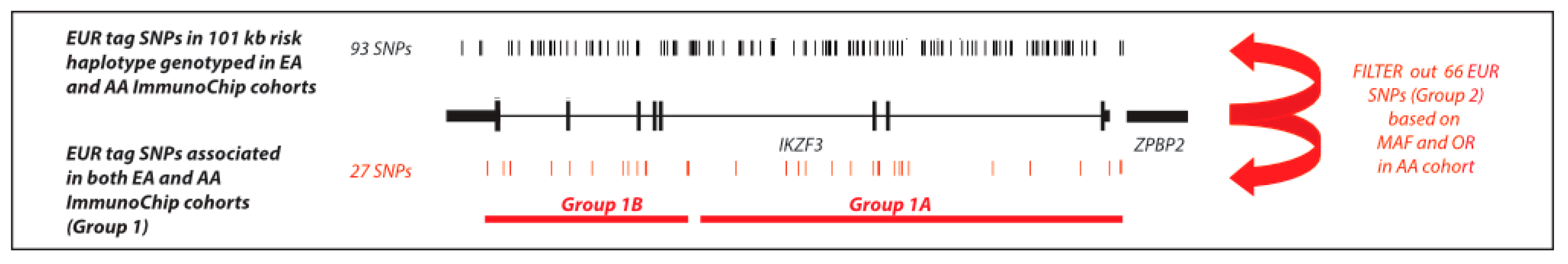
2.4. Extended IKZF3 Haplotype across Multiple Genes in European SLE GWAS Study
2.5. Fine-Mapping the IKZF3 Risk Haplotype Using the 1000 Genomes Super-Populations
2.6. Trans-Ancestral Exclusion Mapping of IKZF3 using the SLE ImmunoChip Data
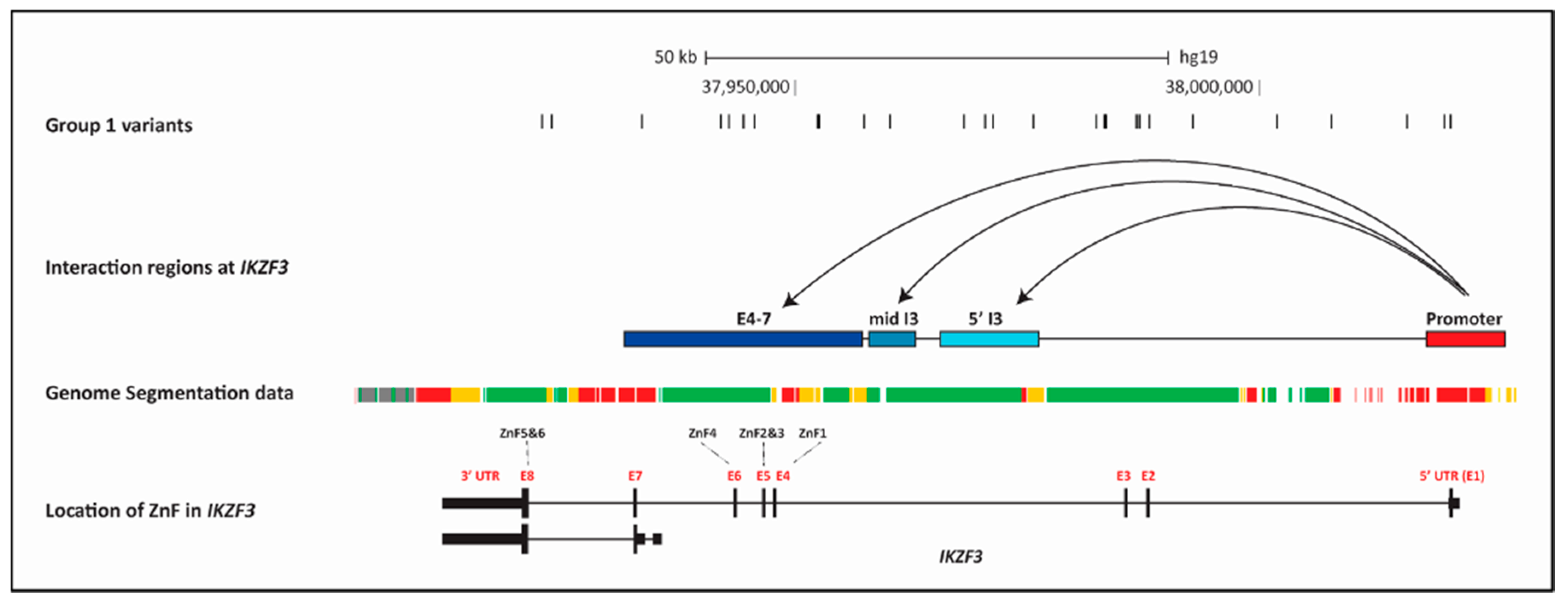
2.7. Functional Annotation of Risk Alleles at IKZF3
2.7.1. Analysis of Expression Levels
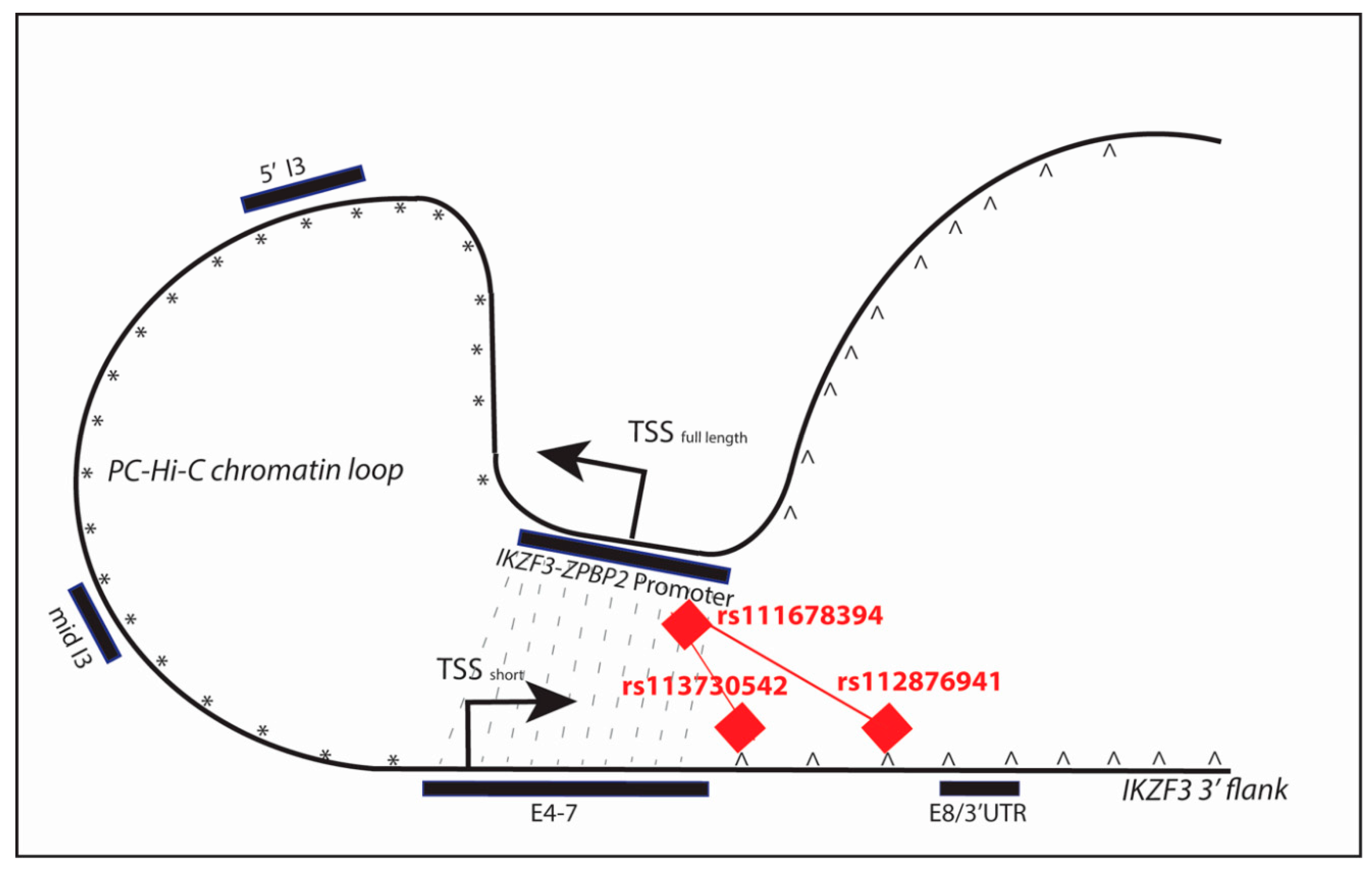
2.7.2. Determination of Chromatin Looping at IKZF3
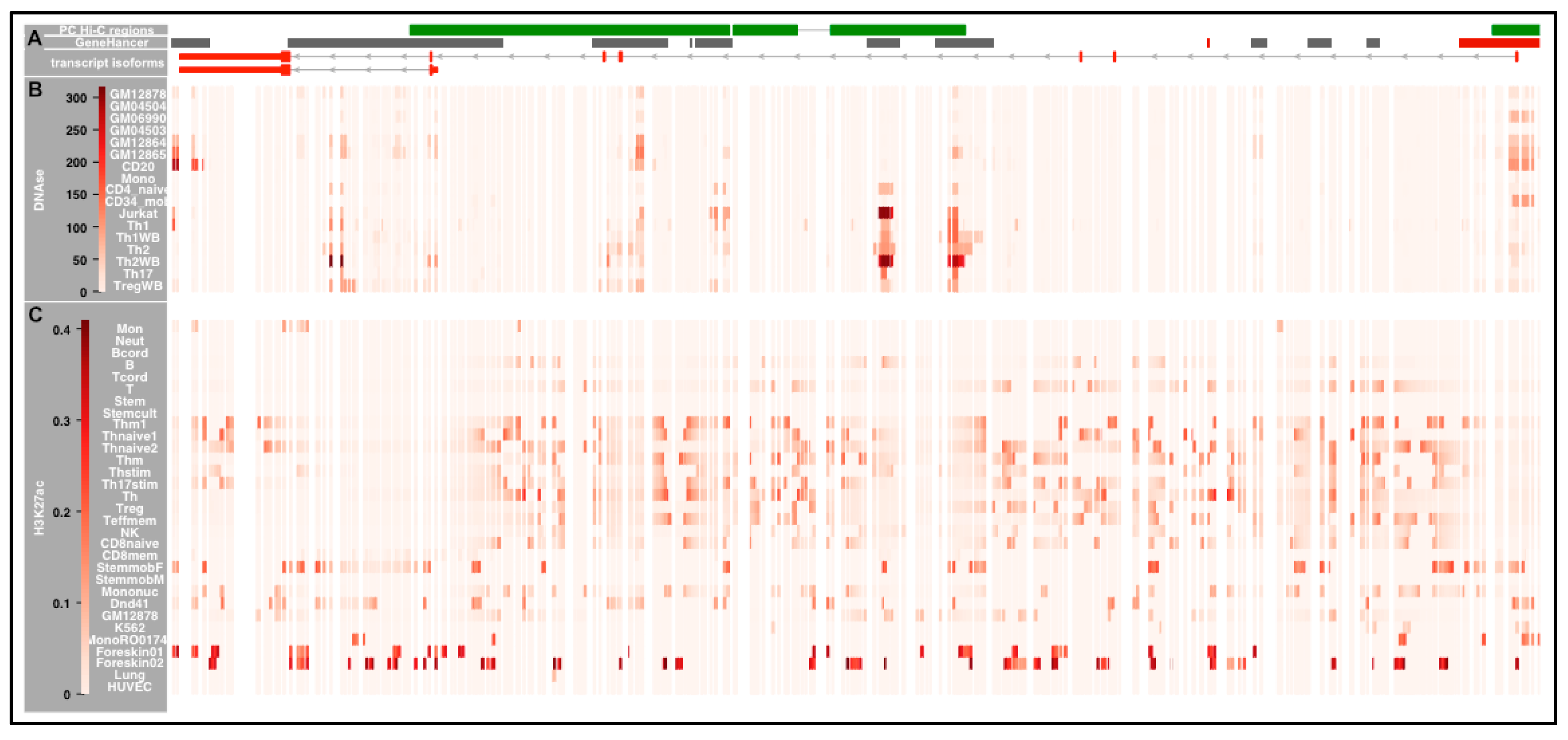
2.7.3. Accessibility of the Chromatin across IKZF3
2.7.4. Cell-Type Specificity in DNAse Sensitivity in the IKZF3 Interaction Regions
2.7.5. Discovery of Allele-Specific Transcription Factor Binding Sites
2.7.6. IKZF3 Risk Alleles Lie within a SuperEnhancer in B Cells
3. Discussion
4. Materials and Methods
4.1. Datasets
4.2. Haplotype Analysis of the Genetic Datasets
4.3. Trans-Ancestral Meta-Analysis
4.4. Trans-Ancestral Exclusion Mapping
4.5. Functional Annotation of Risk Alleles
4.6. Allele-Specific Transcription Factor Binding
4.7. Visualisation of Genomic Data
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Tag-SNPs | Single nucleotide polymorphism tagging a haplotype |
| SLE GWAS | Genome-Wide association study in Systemic Lupus Erythematosus |
| ASTF | Allele-Specific transcription factor binding site |
| eQTL | Expression quantitative trait locus |
| GWAS | Genome-Wide association study |
| MAF | Minor allele frequency |
| PC Hi-C | Promoter Capture Hi-C |
| SLE | Systemic Lupus Erythematosus |
| TF | Transcription factor |
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNP | Pos (hg19) | African American 2970 Cases, 2452 Controls | European 6748 Cases, 11,516 Controls | Hispanic 1872 Cases and 2016 Controls | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| p Value | ORAA (CI) | MAFAA | p Value | OREA (CI) | MAFEA | p Value | ORHisp (CI) | MAFHisp | ||
| rs4917014 | 7:50305863 | 1.48 × 10−5 | 0.728 (0.631–0.841) | 0.09 (G) | 3.67 × 10−9 | 0.866 (0.826–0.909) | 0.32 (G) | 0.021 | 0.897 (0.818–0.984) | 0.48 (T) |
| rs11185603 | 7:50306810 | 4.29 × 10−5 | 0.742 (0.643–0.856) | 0.09 (G) | 8.99 × 10−9 | 0.870 (0.829–0.912) | 0.32 (G) | 0.021 | 0.898 (0.819–0.984) | 0.48 (C) |
| rs4385425 | 7:50307334 | 1.83 × 10−5 | 0.831 (0.771–0.897) | 0.49 (G) | 1.51 × 10−9 | 0.872 (0.832–0.914) | 0.32 (G) | 0.148 | 0.934 (0.852–1.026) | 0.50 (A) |
| rs876036 | 7:50307710 | 9.52 × 10−3 | 0.890 (0.815–0.972) | 0.25 (C) | 7.49 × 10−9 | 0.869 (0.829–0.912) | 0.32 (C) | 0.053 | 0.913 (0.833–1.001) | 0.49 (T) |
| rs876037 | 7:50308692 | 1.87 × 10−5 | 0.731 (0.633–0.844) | 0.09 (A) | 2.23 × 10−8 | 0.873 (0.832–0.915) | 0.31 (A) | 0.020 | 0.897 (0.818–0.983) | 0.48 (T) |
| Locus | Element | Name | Position (hg19) |
|---|---|---|---|
| IKZF1 | PC Hi-C interaction regions | Enhancer (Enh) | chr7:50305428-50311993 |
| Transcriptional Start Site/Promoter (TSS) | chr7:50341186-50347256 | ||
| intron 3 (I3) | chr7:50411807-50412756 | ||
| GeneHancer regions | GH07J050261 | chr7:50300992-50310765 | |
| GH07J050293 | chr7:50333047-50334464 | ||
| GH07J050301 | chr7:50340632-50340761 | ||
| GH07J050303 | chr7:50343395-50362927 | ||
| GH07J050326 | chr7:50366368-50368325 | ||
| GH07J050329 | chr7:50368690-50370631 | ||
| GH07J050341 | chr7:50410631-50437890 | ||
| GH07J050392 | chr7:50459865-50466852 | ||
| IKZF3 | PC Hi-C interaction regions | IKZF3-ZPBP2 bi-directional promoter | chr17:38018444-38027003 |
| 5′ I3 | chr17:37965773-37976506 | ||
| mid I3 | chr17:37958027-37963133 | ||
| 3′ E4-7 | chr17:37932293-37957717 | ||
| GeneHancer regions | GH17J039753 | chr17:37909296-37916397 | |
| GH17J039766 | chr17:37922530-37939749 | ||
| GH17J039790 | chr17:37946728-37952847 | ||
| GH17J039799 | chr17:37954622-37954701 | ||
| GH17J039798 | chr17:37954998-37957986 | ||
| GH17J039812 | chr17:37968642-37971311 | ||
| GH17J039817 | chr17:37974070-37978821 | ||
| GH17J039839 | chr17:37995815-37995875 | ||
| GH17J039842 | chr17:37999223-38000547 | ||
| GH17J039847 | chr17:38003768-38005630 |
| Order | Risk SNP | Pos (hg19) | TF Showing Allele-Specific Binding (ASTF) | Strand | Ref | Alt | Alt-Ref Enrichment |
|---|---|---|---|---|---|---|---|
| 1 | rs34767118 | 50271064 | Sox_5 | + | 12.5 | 11.4 | −1.1 |
| VDR_1 | + | −8.1 | 3.9 | 12 | |||
| Zbtb12 | + | 11.8 | 14.4 | 2.6 | |||
| 2 | rs11773763 | 50271499 | CDP_4 | - | 12.6 | 13.2 | 0.6 |
| Fox | - | 13.3 | 2.5 | −10.8 | |||
| Foxd1_1 | - | 4.5 | 2.5 | −2 | |||
| Foxi1 | - | 13.1 | 11.9 | −1.2 | |||
| Foxj1_2 | - | 14.2 | 13.9 | −0.3 | |||
| Foxj2_1 | - | 12.1 | 12 | −0.1 | |||
| Gm397 | - | 6.6 | 10.7 | 4.1 | |||
| Pou3f2_2 | + | −9.4 | 2.6 | 12 | |||
| Zfp105 | + | 10.8 | 11 | 0.2 | |||
| p53_1 | + | −25.8 | −27.5 | −1.7 | |||
| 3 | rs62445350 | 50278187 | none | 0 | |||
| 4 | rs62445352 | 50289504 | Arid3a_2 | - | 8.4 | 10.9 | 2.5 |
| Barx2 | - | 10.5 | 11.9 | 1.4 | |||
| Cdx2_2 | - | 10.6 | 11.2 | 0.6 | |||
| Dbx1 | - | 8.7 | 10.6 | 1.9 | |||
| Dbx2 | + | 8.8 | 11.5 | 2.7 | |||
| Dlx3 | - | 12.1 | 10 | −2.1 | |||
| Evi-1_4 | + | 4 | 15.5 | 11.5 | |||
| HNF1_1 | - | 12.7 | 10.7 | −2 | |||
| HNF1_6 | - | 13.8 | 11.2 | −2.6 | |||
| HNF1_7 | + | 11.7 | 10.2 | −1.5 | |||
| Hoxa10 | - | 11 | 12.8 | 1.8 | |||
| Hoxa3_2 | - | 13.9 | 13.1 | −0.8 | |||
| Hoxa5_3 | - | 11.6 | 10.2 | −1.4 | |||
| Hoxa7_2 | - | 11 | 12.5 | 1.5 | |||
| Hoxb4 | - | 11.2 | 12.4 | 1.2 | |||
| Hoxc6 | - | 12.1 | 13 | 0.9 | |||
| Hoxc9 | - | 12.2 | 12.7 | 0.5 | |||
| Hoxd8 | + | 12.9 | 16.1 | 3.2 | |||
| Msx-1_2 | - | 10.7 | 13.2 | 2.5 | |||
| Ncx_2 | - | 11.4 | 15.1 | 3.7 | |||
| Nkx6-1_2 | - | 9.9 | 14.8 | 4.9 | |||
| Nkx6-1_3 | - | 9.7 | 14.9 | 5.2 | |||
| Nkx6-2 | - | 11.6 | 12.6 | 1 | |||
| Pax-4_2 | - | 11.2 | 8.1 | −3.1 | |||
| Pou2f2_known4 | + | 12.8 | 13.3 | 0.5 | |||
| Pou3f4 | - | 6 | 11.7 | 5.7 | |||
| Pou4f3 | - | 9.1 | 15.1 | 6 | |||
| Pou5f1_known1 | + | 11.6 | 4.7 | −6.9 | |||
| Prrx1 | + | 11 | 10.4 | −0.6 | |||
| 5 | rs55935382 | 50289669 | SRF_known5 | + | −0.8 | 11 | 11.8 |
| 6 | rs11185602 | 50299077 | Cart1 | + | 15.2 | 11.7 | −3.5 |
| Cdx | + | 9.6 | 12.1 | 2.5 | |||
| HNF1_2 | - | 6.2 | 11.3 | 5.1 | |||
| Lhx3_2 | + | 10.7 | 3 | −7.7 | |||
| PLZF | + | 13.2 | 13 | −0.2 | |||
| Pou2f2_known2 | + | 12.8 | 8.4 | −4.4 | |||
| Pou2f2_known9 | + | 7.4 | −4.5 | −11.9 | |||
| Pou6f1_1 | - | 10.2 | 13.9 | 3.7 | |||
| 7 | rs4917014 * | 50305863 | Nkx2_2 | + | 10.9 | 12 | 1.1 |
| 8 | rs11185603 * | 50306810 | CCNT2_disc2 | + | 12.5 | 7.1 | −5.4 |
| ELF1_known1 | - | 13 | 2 | −11 | |||
| Nkx2_2 | - | 11.9 | 10.3 | −1.6 | |||
| PU.1_disc3 | - | 12.3 | 0.4 | −11.9 | |||
| RXRA_disc4 | + | 12.8 | 1.7 | −11.1 | |||
| TATA_disc7 | - | 13.6 | 7.3 | −6.3 | |||
| 9 | rs4385425 * | 50307334 | none | 0 | |||
| 10 | rs876036 * | 50307710 | ERalpha-a_disc4 | + | 0.2 | 10.7 | 10.5 |
| LXR_3 | - | 11.3 | 7.4 | −3.9 | |||
| RXRA_known4 | + | 10.4 | −0.2 | −10.6 | |||
| VDR_2 | + | 12.4 | 4.6 | −7.8 | |||
| VDR_3 | + | 12.2 | 8.3 | −3.9 | |||
| 11 | rs876038 * | 50308527 | BDP1_disc1 | - | 2.7 | 2.1 | −0.6 |
| Brachyury_1 | - | −2.4 | −5.6 | −3.2 | |||
| XBP-1_1 | + | 12.2 | 0.2 | −12 | |||
| 12 | rs876037 * | 50308527 | none | 0 | |||
| 13 | rs876039 * | 50308811 | Foxa_known2 | - | 11.5 | 12.6 | 1.1 |
| Foxa_known3 | - | 12.7 | 13.3 | 0.6 |
| # | Group | rs | Chr | Pos (hg19) | A1/A2 | ImmunoChip Association Data | Meta-Analysis | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MAFEA | PEA | OREA | MAFEA | PAA | ORAA | s | P(R) | OR | OR(R) | Q | I | ||||||
| 1 | 1 | rs111678394 | 17 | 38021116 | C/G | 0.035 | 2.50 × 10−6 | 1.29 (1.16–1.44) | 0.005 | 0.042 | 1.656 (1.01–2.71) | 5.29 × 10−7 | 5.29 × 10−7 | 1.31 | 1.31 | 0.335 | 0 |
| 2 | 1 | rs117278702 | 17 | 38020420 | A/G | 0.032 | 1.13 × 10−5 | 1.28 (1.15–1.44) | 0.004 | 0.136 | 1.50 (0.877–2.55) | 4.38 × 10−6 | 4.38 × 10−6 | 1.30 | 1.30 | 0.588 | 0 |
| 3 | 2 | rs9905881 | 17 | 38018954 | A/G | 0.036 | 4.44 × 10−6 | 1.28 (1.15–1.43) | 0.256 | 0.004 | 1.13 (1.04–1.24) | 3.16 × 10−7 | 0.003 | 1.19 | 1.20 | 0.079 | 67.8 |
| 4 | 2 | rs9899336 | 17 | 38017779 | T/C | 0.036 | 3.60 × 10−6 | 1.28 (1.16–1.43) | 0.256 | 0.005 | 1.13 (1.04–1.23) | 3.50 × 10−7 | 0.004 | 1.19 | 1.20 | 0.066 | 70.5 |
| 5 | 2 | rs9899006 | 17 | 38017064 | A/T | 0.042 | 1.28 × 10−5 | 1.25 (1.13–1.38) | 0.257 | 0.005 | 1.13 (1.04–1.23) | 7.23 × 10−7 | 0.001 | 1.18 | 1.18 | 0.137 | 54.8 |
| 6 | 1 | rs77924338 | 17 | 38016356 | T/C | 0.035 | 2.50 × 10−6 | 1.29 (1.16–1.44) | 0.005 | 0.042 | 1.66 (1.01–2.71) | 5.29 × 10−7 | 5.29 × 10−7 | 1.31 | 1.31 | 0.335 | 0 |
| 7 | 2 | rs9915797 | 17 | 38014867 | A/G | 0.036 | 2.75 × 10−6 | 1.29 (1.16–1.44) | 0.256 | 0.005 | 1.13 (1.04–1.23) | 3.40 × 10−7 | 0.005 | 1.19 | 1.20 | 0.056 | 72.6 |
| 8 | 2 | rs16965367 | 17 | 38014315 | C/T | 0.036 | 3.99 × 10−6 | 1.28 (1.15–1.43) | 0.256 | 0.005 | 1.13 (1.04–1.23) | 3.68 × 10−7 | 0.004 | 1.19 | 1.20 | 0.069 | 69.9 |
| 9 | 2 | rs113466546 | 17 | 38012586 | A/G | 0.036 | 2.10 × 10−6 | 1.29 (1.16–1.44) | 0.130 | 0.026 | 1.13 (1.02–1.27) | 7.90 × 10−7 | 0.004 | 1.21 | 1.21 | 0.090 | 65.2 |
| 10 | 2 | rs9907291 | 17 | 38010036 | G/A | 0.036 | 2.75 × 10−6 | 1.29 (1.16–1.44) | 0.257 | 0.003 | 1.14 (1.05–1.24) | 1.57 × 10−7 | 0.003 | 1.20 | 1.21 | 0.072 | 69.1 |
| 11 | 2 | rs8069531 | 17 | 38009343 | T/A | 0.036 | 3.24 × 10−6 | 1.29 (1.16–1.43) | 0.256 | 0.005 | 1.13 (1.04–1.23) | 3.36 × 10−7 | 0.004 | 1.19 | 1.21 | 0.064 | 70.9 |
| 12 | 2 | rs8068894 | 17 | 38008999 | G/A | 0.036 | 2.75 × 10−6 | 1.29 (1.16–1.44) | 0.256 | 0.005 | 1.13 (1.04–1.23) | 2.87 × 10−7 | 0.005 | 1.19 | 1.20 | 0.060 | 71.9 |
| 13 | 1 | rs113233720 | 17 | 38008190 | T/C | 0.035 | 2.50 × 10−6 | 1.29 (1.16–1.44) | 0.005 | 0.042 | 1.66 (1.01–2.71) | 5.29 × 10−7 | 5.29 × 10−7 | 1.31 | 1.31 | 0.335 | 0 |
| 14 | 1 | rs112677036 | 17 | 38002152 | A/G | 0.035 | 2.50 × 10−6 | 1.29 (1.16–1.44) | 0.005 | 0.042 | 1.66 (1.01–2.71) | 5.29 × 10−7 | 5.29 × 10−7 | 1.31 | 1.31 | 0.335 | 0 |
| 15 | 2 | rs67600807 | 17 | 38001558 | G/A | 0.036 | 2.75 × 10−6 | 1.29 (1.16–1.44) | 0.262 | 0.007 | 1.13 (1.03–1.22) | 4.54 × 10−7 | 0.008 | 1.19 | 1.20 | 0.049 | 74.2 |
| 16 | 2 | rs9908694 | 17 | 37997771 | T/C | 0.036 | 2.90 × 10−6 | 1.29 (1.16–1.43) | 0.256 | 0.005 | 1.13 (1.04–1.23) | 2.95 × 10−7 | 0.004 | 1.19 | 1.20 | 0.061 | 71.6 |
| 17 | 2 | rs9900541 | 17 | 37996070 | C/T | 0.036 | 2.75 × 10−6 | 1.29 (1.16–1.44) | 0.256 | 0.005 | 1.13 (1.04–1.23) | 3.12 × 10−7 | 0.005 | 1.19 | 1.20 | 0.058 | 72.3 |
| 18 | 1 | rs111691913 | 17 | 37993238 | T/C | 0.035 | 2.24 × 10−6 | 1.30 (1.16–1.44) | 0.005 | 0.042 | 1.66 (1.01–2.71) | 4.60 × 10−7 | 4.60 × 10−7 | 1.31 | 1.31 | 0.338 | 0 |
| 19 | 2 | rs28449671 | 17 | 37991630 | C/T | 0.036 | 2.47 × 10−6 | 1.29 (1.16–1.44) | 0.256 | 0.005 | 1.13 (1.036–1.23) | 3.27 × 10−7 | 0.006 | 1.19 | 1.20 | 0.055 | 73.0 |
| 20 | 1 | rs111944912 | 17 | 37988476 | C/T | 0.035 | 3.64 × 10−6 | 1.29 (1.16–1.43) | 0.005 | 0.042 | 1.66 (1.01–2.71) | 7.80 × 10−7 | 7.80 × 10−7 | 1.30 | 1.30 | 0.326 | 0 |
| 21 | 2 | rs73304123 | 17 | 37987588 | T/C | 0.036 | 3.07 × 10−6 | 1.29 (1.16–1.43) | 0.128 | 0.025 | 1.14 (1.02–1.27) | 9.54 × 10−7 | 0.003 | 1.21 | 1.21 | 0.108 | 61.4 |
| 22 | 2 | rs112141468 | 17 | 37987464 | T/C | 0.036 | 3.99 × 10−6 | 1.28 (1.15–1.43) | 0.259 | 0.006 | 1.13 (1.04–1.23) | 4.64 × 10−7 | 0.005 | 1.19 | 1.20 | 0.063 | 71.2 |
| 23 | 1 | rs111734595 | 17 | 37987399 | T/C | 0.035 | 3.64 × 10−6 | 1.29 (1.16–1.43) | 0.005 | 0.042 | 1.66 (1.01–2.71) | 7.80 × 10−7 | 7.80 × 10−7 | 1.30 | 1.30 | 0.326 | 0 |
| 24 | 1 | rs113479772 | 17 | 37987042 | A/G | 0.035 | 3.64 × 10−6 | 1.29 (1.16–1.43) | 0.005 | 0.042 | 1.66 (1.01–2.71) | 7.80 × 10−7 | 7.80 × 10−7 | 1.30 | 1.30 | 0.326 | 0 |
| 25 | 1 | rs112797570 | 17 | 37983751 | A/G | 0.035 | 3.64 × 10−6 | 1.29 (1.16–1.43) | 0.005 | 0.042 | 1.66 (1.01–2.71) | 7.80 × 10−7 | 7.80 × 10−7 | 1.30 | 1.30 | 0.326 | 0 |
| 26 | 1 | rs112437508 | 17 | 37983512 | A/G | 0.035 | 3.09 × 10−6 | 1.29 (1.16–1.44) | 0.023 | 0.564 | 1.07 (0.840–1.38) | 6.90 × 10−6 | 0.016 | 1.25 | 1.22 | 0.190 | 41.7 |
| 27 | 2 | rs35130019 | 17 | 37983141 | G/A | 0.037 | 6.29 × 10−6 | 1.28 (1.15–1.42) | 0.255 | 0.007 | 1.13 (1.03–1.23) | 7.56 × 10−7 | 0.004 | 1.18 | 1.19 | 0.073 | 68.8 |
| 28 | 1 | rs111469562 | 17 | 37982696 | C/T | 0.036 | 4.51 − 06 | 1.28 (1.15–1.43) | 0.005 | 0.042 | 1.66 (1.01–2.71) | 9.58 × 10−7 | 9.58 × 10−7 | 1.30 | 1.30 | 0.321 | 0 |
| 29 | 2 | rs12942660 | 17 | 37982037 | T/C | 0.036 | 4.44 × 10−6 | 1.28 (1.15–1.43) | 0.252 | 0.003 | 1.14 (1.04–1.24) | 2.54 × 10−7 | 0.002 | 1.19 | 1.20 | 0.085 | 66.2 |
| 30 | 2 | rs8076347 | 17 | 37977540 | T/G | 0.036 | 3.78 × 10−6 | 1.28 (1.16–1.42) | 0.252 | 0.003 | 1.14 (1.04–1.24) | 2.29 × 10−7 | 0.002 | 1.19 | 1.20 | 0.081 | 67.1 |
| 31 | 2 | rs9908983 | 17 | 37976926 | A/G | 0.036 | 3.42 × 10−6 | 1.29 (1.16–1.43) | 0.124 | 0.023 | 1.14 (1.02–1.27) | 8.39 × 10−7 | 0.002 | 1.21 | 1.21 | 0.122 | 58.2 |
| 32 | 2 | rs9911069 | 17 | 37976601 | C/T | 0.036 | 3.07 × 10−6 | 1.29 (1.16–1.43) | 0.124 | 0.023 | 1.14 (1.02–1.27) | 7.99 × 10−7 | 0.002 | 1.22 | 1.21 | 0.120 | 58.7 |
| 33 | 2 | rs9901917 | 17 | 37976205 | C/G | 0.036 | 3.42 × 10−6 | 1.29 (1.16–1.43) | 0.124 | 0.023 | 1.14 (1.02–1.27) | 8.39 × 10−7 | 0.002 | 1.21 | 1.21 | 0.122 | 58.2 |
| 34 | 1 | rs112743130 | 17 | 37975855 | C/G | 0.035 | 3.46 × 10−6 | 1.29 (1.16–1.43) | 0.005 | 0.059 | 1.59 (0.979–2.58) | 8.55 × 10−7 | 8.55 × 10−7 | 1.30 | 1.30 | 0.406 | 0 |
| 35 | 2 | rs34053394 | 17 | 37975660 | G/A | 0.036 | 3.42 × 10−6 | 1.29 (1.16–1.43) | 0.124 | 0.023 | 1.13 (1.02–1.27) | 8.39 × 10−7 | 0.002 | 1.21 | 1.21 | 0.122 | 58.2 |
| 36 | 2 | rs58075375 | 17 | 37975592 | T/C | 0.036 | 3.42 × 10−6 | 1.287 (1.157–1.432) | 0.124 | 0.023 | 1.14 (1.02–1.27) | 8.39 × 10−7 | 0.002 | 1.21 | 1.21 | 0.122 | 58.2 |
| 37 | 2 | rs9902621 | 17 | 37973010 | A/G | 0.036 | 3.42 × 10−6 | 1.29 (1.16–1.43) | 0.124 | 0.023 | 1.14 (1.02–1.27) | 8.39 × 10−7 | 0.002 | 1.21 | 1.21 | 0.122 | 58.2 |
| 38 | 2 | rs9898031 | 17 | 37972647 | G/C | 0.036 | 6.45 × 10−6 | 1.28 (1.16–1.42) | 0.124 | 0.023 | 1.14 (1.02–1.27) | 1.38 × 10−6 | 0.001 | 1.21 | 1.20 | 0.147 | 52.4 |
| 39 | 1 | rs112412105 | 17 | 37971635 | G/A | 0.036 | 4.06 × 10−6 | 1.29 (1.16–1.43) | 0.005 | 0.059 | 1.59 (0.979–2.58) | 9.57 × 10−7 | 9.57 × 10−7 | 1.30 | 1.30 | 0.402 | 0 |
| 40 | 1 | rs113115305 | 17 | 37970686 | C/A | 0.036 | 9.37 × 10−6 | 1.27 (1.14–1.42) | 0.005 | 0.059 | 1.59 (0.979–2.58) | 2.28 × 10−6 | 2.28 × 10−6 | 1.29 | 1.29 | 0.380 | 0 |
| 41 | 1 | rs112238900 | 17 | 37968494 | T/C | 0.036 | 4.06 × 10−6 | 1.29 (1.16–1.43) | 0.005 | 0.059 | 1.59 (0.979–2.58) | 9.57 × 10−7 | 9.57 × 10−7 | 1.30 | 1.30 | 0.402 | 0 |
| 42 | 2 | rs67135646 | 17 | 37967871 | G/C | 0.036 | 5.47 × 10−6 | 1.28 (1.15–1.42) | 0.252 | 0.004 | 1.13 (1.04–1.24) | 4.03 × 10−7 | 0.003 | 1.19 | 1.20 | 0.082 | 66.9 |
| 43 | 2 | rs114777282 | 17 | 37967649 | A/C | 0.036 | 5.47 × 10−6 | 1.28 (1.15–1.42) | 0.250 | 0.005 | 1.13 (1.04–1.23) | 4.48 × 10−7 | 0.003 | 1.19 | 1.20 | 0.080 | 67.3 |
| 44 | 2 | rs4337325 | 17 | 37964435 | T/C | 0.036 | 9.22 × 10−6 | 1.27 (1.14–1.41) | 0.250 | 0.005 | 1.13 (1.04–1.23) | 7.12 × 10−7 | 0.002 | 1.18 | 1.19 | 0.095 | 64.2 |
| 45 | 2 | rs9901617 | 17 | 37964175 | C/G | 0.036 | 4.24 × 10−6 | 1.284 (1.15–1.43) | 0.125 | 0.027 | 1.13 (1.01–1.27) | 1.27 × 10−6 | 0.002 | 1.21 | 1.21 | 0.116 | 59.6 |
| 46 | 1 | rs113064843 | 17 | 37960421 | C/T | 0.036 | 5.02 × 10−6 | 1.28 (1.15–1.43) | 0.005 | 0.059 | 1.59 (0.979–2.58) | 1.25 × 10−6 | 1.25 × 10−6 | 1.29 | 1.29 | 0.395 | 0 |
| 47 | 2 | rs7211998 | 17 | 37959788 | G/A | 0.036 | 6.42 × 10−6 | 1.28 (1.15–1.42) | 0.235 | 0.005 | 1.13 (1.04–1.23) | 5.27 × 10−7 | 0.002 | 1.19 | 1.20 | 0.089 | 65.4 |
| 48 | 2 | rs36097841 | 17 | 37958112 | A/G | 0.036 | 6.08 × 10−6 | 1.28 (1.15–1.42) | 0.252 | 0.002 | 1.14 (1.05–1.24) | 2.69 × 10−7 | 0.001 | 1.19 | 1.20 | 0.101 | 62.8 |
| 49 | 2 | rs34988504 | 17 | 37957631 | T/C | 0.036 | 5.47 × 10−6 | 1.28 (1.15–1.42) | 0.252 | 0.004 | 1.14 (1.04–1.24) | 3.14 × 10−7 | 0.002 | 1.19 | 1.20 | 0.089 | 65.4 |
| 50 | 1 | rs16965347 | 17 | 37957566 | C/G | 0.030 | 1.24 × 10−5 | 1.29 (1.15–1.45) | 0.004 | 0.154 | 1.52 (0.852–2.70) | 5.12 × 10−6 | 5.12 × 10−6 | 1.30 | 1.30 | 0.595 | 0 |
| 51 | 2 | rs12937330 | 17 | 37957316 | A/C | 0.036 | 6.02 × 10−6 | 1.28 (1.15–1.42) | 0.268 | 0.009 | 1.12 (1.03–1.22) | 1.33 × 10−6 | 0.008 | 1.18 | 1.19 | 0.056 | 72.7 |
| 52 | 2 | rs34344462 | 17 | 37955193 | G/A | 0.036 | 5.47 × 10−6 | 1.28 (1.15–1.42) | 0.252 | 0.005 | 1.13 (1.04–1.23) | 4.78 × 10−7 | 0.003 | 1.19 | 1.20 | 0.078 | 67.82 |
| 53 | 2 | rs9899345 | 17 | 37954757 | A/G | 0.035 | 2.37 × 10−5 | 1.26 (1.13–1.41) | 0.251 | 0.004 | 1.13 (1.04–1.24) | 1.17 × 10−6 | 0.001 | 1.18 | 1.20 | 0.125 | 57.4 |
| 54 | 1 | rs113369293 | 17 | 37952654 | T/C | 0.036 | 4.51 × 10−6 | 1.28 (1.15–1.43) | 0.007 | 0.287 | 1.27 (0.820–1.95) | 2.63 × 10−6 | 2.63 × 10−6 | 1.28 | 1.28 | 0.948 | 0 |
| 55 | 1 | rs75148376 | 17 | 37952508 | T/C | 0.036 | 4.51 × 10−6 | 1.28 (1.15–1.43) | 0.007 | 0.287 | 1.27 (0.820–1.95) | 2.63 × 10−6 | 2.63 × 10−6 | 1.28 | 1.28 | 0.948 | 0 |
| 56 | 2 | rs73302152 | 17 | 37952350 | C/G | 0.036 | 6.84 × 10−6 | 1.28 (1.15–1.42) | 0.127 | 0.059 | 1.11 (0.996–1.25) | 5.54 × 10−6 | 0.010 | 1.20 | 1.19 | 0.081 | 67.1 |
| 57 | 2 | rs113159227 | 17 | 37952091 | A/G | 0.036 | 3.81 × 10−6 | 1.29 (1.16–1.43) | 0.127 | 0.063 | 1.11 (0.994–1.24) | 4.04 × 10−6 | 0.014 | 1.20 | 1.20 | 0.065 | 70.7 |
| 58 | 2 | rs56928975 | 17 | 37952031 | G/A | 0.048 | 2.38 × 10−7 | 1.28 (1.16–1.40) | 0.250 | 0.014 | 1.11 (1.02–1.22) | 1.18 × 10−7 | 0.011 | 1.19 | 1.19 | 0.034 | 77.7 |
| 59 | 2 | rs12938749 | 17 | 37951847 | T/C | 0.036 | 3.81 × 10−6 | 1.29 (1.16–1.43) | 0.127 | 0.063 | 1.11 (0.994–1.24) | 4.04 × 10−6 | 0.014 | 1.20 | 1.20 | 0.064 | 70.7 |
| 60 | 2 | rs35938199 | 17 | 37950812 | T/C | 0.036 | 4.24 × 10−6 | 1.28 (1.15–1.43) | 0.127 | 0.063 | 1.11 (0.994–1.24) | 4.23 × 10−6 | 0.014 | 1.20 | 1.20 | 0.066 | 70.4 |
| 61 | 2 | rs35105110 | 17 | 37950421 | A/G | 0.036 | 3.24 × 10−6 | 1.29 (1.16–1.43) | 0.127 | 0.063 | 1.11 (0.994–1.24) | 3.63 × 10−6 | 0.014 | 1.20 | 1.20 | 0.062 | 71.3 |
| 62 | 2 | rs35352075 | 17 | 37949790 | C/T | 0.036 | 3.81 × 10−6 | 1.29 (1.16–1.43) | 0.127 | 0.063 | 1.11 (0.994–1.24) | 4.04 × 10−6 | 0.014 | 1.20 | 1.20 | 0.064 | 70.7 |
| 63 | 1 | rs112771646 | 17 | 37945708 | C/A | 0.036 | 4.51 × 10−6 | 1.28 (1.15–1.43) | 0.007 | 0.287 | 1.27 (0.820–1.95) | 2.63 × 10−6 | 2.63 × 10−6 | 1.28 | 1.28 | 0.950 | 0 |
| 64 | 1 | rs112301322 | 17 | 37944518 | G/C | 0.036 | 4.51 × 10−6 | 1.28 (1.15–1.43) | 0.007 | 0.287 | 1.27 (0.820–1.95) | 2.63 × 10−6 | 2.63 × 10−6 | 1.28 | 1.28 | 0.950 | 0 |
| 65 | 2 | rs35088469 | 17 | 37944481 | T/C | 0.036 | 2.21 × 10−6 | 1.29 (1.16–1.44) | 0.119 | 0.096 | 1.10 (0.983–1.24) | 4.27 × 10−6 | 0.024 | 1.20 | 1.20 | 0.048 | 74.4 |
| 66 | 2 | rs34291217 | 17 | 37944410 | A/C | 0.036 | 3.81 × 10−6 | 1.29 (1.16–1.43) | 0.127 | 0.063 | 1.11 (0.994–1.24) | 4.04 × 10−6 | 0.014 | 1.20 | 1.20 | 0.065 | 70.7 |
| 67 | 2 | rs9911688 | 17 | 37943800 | T/C | 0.036 | 4.71 × 10−6 | 1.28 (1.15–1.43) | 0.127 | 0.060 | 1.11 (0.996–1.24) | 4.20 × 10−6 | 0.011 | 1.20 | 1.20 | 0.073 | 69.0 |
| 68 | 2 | rs9911669 | 17 | 37943766 | G/C | 0.036 | 3.81 × 10−6 | 1.29 (1.16–1.43) | 0.127 | 0.063 | 1.11 (0.994–1.24) | 4.04 × 10−6 | 0.014 | 1.20 | 1.20 | 0.065 | 70.7 |
| 69 | 1 | rs111862642 | 17 | 37942983 | G/C | 0.036 | 4.51 × 10−6 | 1.28 (1.15–1.43) | 0.007 | 0.287 | 1.27 (0.820–1.95) | 2.63 × 10−6 | 2.63 × 10−6 | 1.28 | 1.28 | 0.945 | 0 |
| 70 | 2 | rs34599546 | 17 | 37942971 | T/C | 0.036 | 4.93 × 10−6 | 1.28 (1.15–1.42) | 0.255 | 0.010 | 1.12 (1.03–1.22) | 1.10 × 10−6 | 0.008 | 1.18 | 1.19 | 0.055 | 72.8 |
| 71 | 1 | rs112345383 | 17 | 37942017 | T/C | 0.036 | 4.51 × 10−6 | 1.28 (1.15–1.43) | 0.007 | 0.287 | 1.27 (0.82–1.95) | 2.63 × 10−6 | 2.63 × 10−6 | 1.28 | 1.28 | 0.950 | 0 |
| 72 | 2 | rs1510475 | 17 | 37941379 | C/A | 0.036 | 3.81 × 10−6 | 1.29 (1.16–1.43) | 0.127 | 0.063 | 1.11 (0.994–1.24) | 4.04 × 10−6 | 0.014 | 1.20 | 1.20 | 0.065 | 70.7 |
| 73 | 2 | rs113812449 | 17 | 37940167 | C/T | 0.036 | 4.93 × 10−6 | 1.28 (1.15–1.42) | 0.255 | 0.010 | 1.12 (1.03–1.22) | 1.10 × 10−6 | 0.008 | 1.18 | 1.20 | 0.055 | 72.8 |
| 74 | 2 | rs9909365 | 17 | 37939958 | G/A | 0.036 | 4.93 × 10−6 | 1.28 (1.15–1.42) | 0.255 | 0.010 | 1.12 (1.03–1.22) | 1.10 × 10−6 | 0.008 | 1.18 | 1.19 | 0.055 | 72.76 |
| 75 | 2 | rs34016964 | 17 | 37938976 | T/G | 0.036 | 4.93 × 10−6 | 1.28 (1.15–1.42) | 0.255 | 0.010 | 1.12 (1.03–1.22) | 1.10 × 10−6 | 0.008 | 1.18 | 1.19 | 0.055 | 72.8 |
| 76 | 2 | rs67605703 | 17 | 37938496 | C/T | 0.036 | 4.24 × 10−6 | 1.28 (1.15–1.43) | 0.128 | 0.078 | 1.11 (0.989–1.24) | 5.78 × 10−6 | 0.019 | 1.20 | 1.19 | 0.057 | 72.5 |
| 77 | 2 | rs35506518 | 17 | 37938093 | C/T | 0.036 | 2.61 × 10−6 | 1.29 (1.16–1.44) | 0.127 | 0.060 | 1.11 (0.996–1.24) | 2.70 × 10−6 | 0.01404 | 1.20 | 1.20 | 0.060 | 71.8 |
| 78 | 2 | rs13380871 | 17 | 37936248 | C/T | 0.036 | 3.78 × 10−6 | 1.28 (1.16–1.43) | 0.255 | 0.020 | 1.11 (1.02–1.21) | 2.39 × 10−6 | 0.019 | 1.17 | 1.19 | 0.034 | 77.6 |
| 79 | 2 | rs7224641 | 17 | 37934910 | C/T | 0.036 | 2.60 × 10−6 | 1.29 (1.16–1.44) | 0.255 | 0.011 | 1.12 (1.03–1.22) | 9.12 × 10−7 | 0.013 | 1.18 | 1.20 | 0.039 | 76.5 |
| 80 | 2 | rs12709364 | 17 | 37933822 | G/A | 0.036 | 2.22 × 10−6 | 1.29 (1.16–1.44) | 0.128 | 0.079 | 1.11 (0.988–1.24) | 3.83 × 10−6 | 0.023 | 1.20 | 1.20 | 0.046 | 74.9 |
| 81 | 1 | rs113370572 | 17 | 37933467 | C/T | 0.035 | 2.94 × 10−6 | 1.29 (1.16–1.44) | 0.007 | 0.287 | 1.27 (0.820–1.95) | 1.78 × 10−6 | 1.78 × 10-6 | 1.29 | 1.29 | 0.932 | 0 |
| 82 | 2 | rs9901483 | 17 | 37932773 | A/T | 0.036 | 2.60 × 10−6 | 1.29 (1.16–1.44) | 0.255 | 0.011 | 1.12 (1.03–1.22) | 9.12 × 10−7 | 0.013 | 1.18 | 1.20 | 0.039 | 76.5 |
| 83 | 2 | rs9894898 | 17 | 37932220 | C/T | 0.036 | 1.99 × 10−6 | 1.30 (1.16–1.44) | 0.127 | 0.073 | 1.11 (0.991–1.24) | 3.13 × 10−6 | 0.021 | 1.20 | 1.20 | 0.047 | 74.8 |
| 84 | 2 | rs9913596 | 17 | 37932062 | A/G | 0.036 | 1.99 × 10−6 | 1.30 (1.16–1.44) | 0.127 | 0.122 | 1.09 (0.977–1.22) | 6.92 × 10−6 | 0.041 | 1.19 | 1.20 | 0.031 | 78.6 |
| 85 | 2 | rs9652840 | 17 | 37929427 | T/A | 0.037 | 3.32 × 10−5 | 1.25 (1.13–1.39) | 0.201 | 0.045 | 1.1 (1.00–1.21) | 2.24 × 10−5 | 0.015 | 1.16 | 1.17 | 0.074 | 68.7 |
| 86 | 2 | rs71369788 | 17 | 37927144 | A/G | 0.036 | 3.42 × 10−6 | 1.29 (1.16–1.43) | 0.200 | 0.052 | 1.10 (0.999–1.20) | 6.32 × 10−6 | 0.033 | 1.18 | 1.19 | 0.027 | 79.5 |
| 87 | 2 | rs8072612 | 17 | 37927119 | G/A | 0.036 | 3.06 × 10−6 | 1.29 (1.16–1.43) | 0.255 | 0.011 | 1.12 (1.03–1.22) | 9.31 × 10−7 | 0.011 | 1.18 | 1.20 | 0.043 | 75.7 |
| 88 | 2 | rs9894370 | 17 | 37926003 | C/G | 0.037 | 4.77 × 10−7 | 1.31 (1.18–1.45) | 0.318 | 0.018 | 1.10 (1.02–1.20) | 8.05 × 10−7 | 0.037 | 1.17 | 1.20 | 0.011 | 84.6 |
| 89 | 2 | rs34758895 | 17 | 37925467 | T/C | 0.037 | 5.33 × 10-7 | 1.31 (1.18–1.45) | 0.318 | 0.018 | 1.10 (1.02–1.20) | 7.76 × 10-7 | 0.034 | 1.17 | 1.20 | 0.012 | 84.2 |
| 90 | 1 | rs112771360 | 17 | 37923770 | G/A | 0.035 | 2.79 × 10−6 | 1.29 (1.16–1.44) | 0.007 | 0.287 | 1.27 (0.820–1.95) | 1.59 × 10−6 | 1.59 × 10−6 | 1.29 | 1.29 | 0.926 | 0 |
| 91 | 1 | rs112876941 | 17 | 37922803 | T/A | 0.035 | 2.36 × 10−6 | 1.29 (1.16–1.44) | 0.007 | 0.287 | 1.27 (0.820–1.95) | 1.37 × 10−6 | 1.37 × 10−6 | 1.29 | 1.29 | 0.921 | 0 |
| 92 | 2 | rs2941509 | 17 | 37921193 | T/C | 0.037 | 1.30 × 10−5 | 1.27 (1.14–1.41) | 0.244 | 0.052 | 1.09 (0.999–1.19) | 2.07 × 10−5 | 0.034 | 1.16 | 1.17 | 0.034 | 77.9 |
| 93 | 2 | rs67571561 | 17 | 37920846 | C/T | 0.036 | 2.44 × 10−6 | 1.29 (1.16–1.43) | 0.230 | 0.049 | 1.09 (1–1.20) | 6.03 × 10−6 | 0.041 | 1.17 | 1.18 | 0.019 | 81.8 |
| # | Group | rs | Chr | Pos | PC Hi-C Interaction Region | Pos (hg19) | GeneHancer | Pos (hg19) |
|---|---|---|---|---|---|---|---|---|
| 1 | 1 | rs111678394 | 17 | 38021116 | IKZF3-ZPBP2 | 38018444-38027003 | GH17J039859 | 38015831-38025531 |
| 2 | 1 | rs117278702 | 17 | 38020420 | ||||
| 3 | 2 | rs9905881 | 17 | 38018954 | ||||
| 4 | 2 | rs9899336 | 17 | 38017779 | ||||
| 5 | 2 | rs9899006 | 17 | 38017064 | ||||
| 6 | 1 | rs77924338 | 17 | 38016356 | ||||
| 7 | 2 | rs9915797 | 17 | 38014867 | ||||
| 8 | 2 | rs16965367 | 17 | 38014315 | ||||
| 9 | 2 | rs113466546 | 17 | 38012586 | ||||
| 10 | 2 | rs9907291 | 17 | 38010036 | ||||
| 11 | 2 | rs8069531 | 17 | 38009343 | GH17J039852 | 38008382-38009513 | ||
| 12 | 2 | rs8068894 | 17 | 38008999 | ||||
| 13 | 1 | rs113233720 | 17 | 38008190 | ||||
| 14 | 1 | rs112677036 | 17 | 38002152 | ||||
| 15 | 2 | rs67600807 | 17 | 38001558 | ||||
| 16 | 2 | rs9908694 | 17 | 37997771 | ||||
| 17 | 2 | rs9900541 | 17 | 37996070 | ||||
| 18 | 1 | rs111691913 | 17 | 37993238 | ||||
| 19 | 2 | rs28449671 | 17 | 37991630 | ||||
| 20 | 1 | rs111944912 | 17 | 37988476 | GH17J039817 | 37974070-37978821 | ||
| 21 | 2 | rs73304123 | 17 | 37987588 | ||||
| 22 | 2 | rs112141468 | 17 | 37987464 | ||||
| 23 | 1 | rs111734595 | 17 | 37987399 | ||||
| 24 | 1 | rs113479772 | 17 | 37987042 | ||||
| 25 | 1 | rs112797570 | 17 | 37983751 | ||||
| 26 | 1 | rs112437508 | 17 | 37983512 | ||||
| 27 | 2 | rs35130019 | 17 | 37983141 | ||||
| 28 | 1 | rs111469562 | 17 | 37982696 | ||||
| 29 | 2 | rs12942660 | 17 | 37982037 | ||||
| 30 | 2 | rs8076347 | 17 | 37977540 | ||||
| 31 | 2 | rs9908983 | 17 | 37976926 | ||||
| 32 | 2 | rs9911069 | 17 | 37976601 | ||||
| 33 | 2 | rs9901917 | 17 | 37976205 | 5′ (I3) | 37965773-37976506 | ||
| 34 | 1 | rs112743130 | 17 | 37975855 | ||||
| 35 | 2 | rs34053394 | 17 | 37975660 | ||||
| 36 | 2 | rs58075375 | 17 | 37975592 | ||||
| 37 | 2 | rs9902621 | 17 | 37973010 | ||||
| 38 | 2 | rs9898031 | 17 | 37972647 | ||||
| 39 | 1 | rs112412105 | 17 | 37971635 | ||||
| 40 | 1 | rs113115305 | 17 | 37970686 | GH17J039812 | 37968642-37971311 | ||
| 41 | 1 | rs112238900 | 17 | 37968494 | ||||
| 42 | 2 | rs67135646 | 17 | 37967871 | ||||
| 43 | 2 | rs114777282 | 17 | 37967649 | ||||
| 44 | 2 | rs4337325 | 17 | 37964435 | ||||
| 45 | 2 | rs9901617 | 17 | 37964175 | ||||
| 46 | 1 | rs113064843 | 17 | 37960421 | ||||
| 47 | 2 | rs7211998 | 17 | 37959788 | ||||
| 48 | 2 | rs36097841 | 17 | 37958112 | ||||
| 49 | 2 | rs34988504 | 17 | 37957631 | 3′ (E4-7) | 37932293-37957717 | GH17J039798 | 37954998-37957986 |
| 50 | 1 | rs16965347 | 17 | 37957566 | ||||
| 51 | 2 | rs12937330 | 17 | 37957316 | ||||
| 52 | 2 | rs34344462 | 17 | 37955193 | ||||
| 53 | 2 | rs9899345 | 17 | 37954757 | ||||
| 54 | 1 | rs113369293 | 17 | 37952654 | GH17J039790 | 37946728-37952847 | ||
| 55 | 1 | rs75148376 | 17 | 37952508 | ||||
| 56 | 2 | rs73302152 | 17 | 37952350 | ||||
| 57 | 2 | rs113159227 | 17 | 37952091 | ||||
| 58 | 2 | rs56928975 | 17 | 37952031 | ||||
| 59 | 2 | rs12938749 | 17 | 37951847 | ||||
| 60 | 2 | rs35938199 | 17 | 37950812 | ||||
| 61 | 2 | rs35105110 | 17 | 37950421 | ||||
| 62 | 2 | rs35352075 | 17 | 37949790 | ||||
| 63 | 1 | rs112771646 | 17 | 37945708 | ||||
| 64 | 1 | rs112301322 | 17 | 37944518 | ||||
| 65 | 2 | rs35088469 | 17 | 37944481 | ||||
| 66 | 2 | rs34291217 | 17 | 37944410 | ||||
| 67 | 2 | rs9911688 | 17 | 37943800 | ||||
| 68 | 2 | rs9911669 | 17 | 37943766 | ||||
| 69 | 1 | rs111862642 | 17 | 37942983 | ||||
| 70 | 2 | rs34599546 | 17 | 37942971 | ||||
| 71 | 1 | rs112345383 | 17 | 37942017 | ||||
| 72 | 2 | rs1510475 | 17 | 37941379 | ||||
| 73 | 2 | rs113812449 | 17 | 37940167 | ||||
| 74 | 2 | rs9909365 | 17 | 37939958 | ||||
| 75 | 2 | rs34016964 | 17 | 37938976 | GH17J039766 | 37922530-37939749 | ||
| 76 | 2 | rs67605703 | 17 | 37938496 | ||||
| 77 | 2 | rs35506518 | 17 | 37938093 | ||||
| 78 | 2 | rs13380871 | 17 | 37936248 | ||||
| 79 | 2 | rs7224641 | 17 | 37934910 | ||||
| 80 | 2 | rs12709364 | 17 | 37933822 | ||||
| 81 | 1 | rs113370572 | 17 | 37933467 | ||||
| 82 | 2 | rs9901483 | 17 | 37932773 | ||||
| 83 | 2 | rs9894898 | 17 | 37932220 | ||||
| 84 | 2 | rs9913596 | 17 | 37932062 | ||||
| 85 | 2 | rs9652840 | 17 | 37929427 | ||||
| 86 | 2 | rs71369788 | 17 | 37927144 | ||||
| 87 | 2 | rs8072612 | 17 | 37927119 | ||||
| 88 | 2 | rs9894370 | 17 | 37926003 | ||||
| 89 | 2 | rs34758895 | 17 | 37925467 | ||||
| 90 | 1 | rs112771360 | 17 | 37923770 | ||||
| 91 | 1 | rs112876941 | 17 | 37922803 | ||||
| 92 | 2 | rs2941509 | 17 | 37921193 | ||||
| 93 | 2 | rs67571561 | 17 | 37920846 |
| Group I Risk Variants | SNPs in IKZF3-ZPBP2 bi-Directional Promoter | |||||||
|---|---|---|---|---|---|---|---|---|
| Risk SNP | Location | Interact. Fragment | ASTF | Alt-Ref Enrich. | Promoter SNP | Shared Promoter TF | Alt-Ref Enrich. | |
| A | rs193004755 | I1 | no | - | − | - | - | − |
| B | rs115164861 | I1 | no | - | − | - | - | − |
| C | rs142142756 | I1 | no | Foxj1_1 | −2.5 | rs145735506 | Foxj1_1 | 11.8 |
| Foxo_3 | −2.3 | rs184525456 rs138959946 | Foxo_3 | −12 −3 | ||||
| p300_disc3 | 2.0 | rs188089973 rs9907794 rs116467677 rs145275643 rs138461720 rs112745149 rs192412458 | p300_disc5 p300_disc5 p300_disc9 p300_disc10 p300_disc5 p300_disc5 p300_disc1 | 1.9 −5.9 −1.7 11.9 −2.5 3.2 11.9 | ||||
| D | rs145168309 | I2 | no | AP-1_disc8 | 11.2 | rs190729974 rs4795397 rs192412458 rs192412458 rs147224870 rs1453558 rs1453560 rs36111081 rs66565390 | AP-1_disc1 AP-1_disc2 AP-1_disc3/7/9 AP-1_known2/3/4 AP-1_disc7 AP-1_disc2 AP-1_known1 AP-1_disc7 AP-1_disc7 | 12 −6.8 11.9/0.4/12 11.8/4.2/12 −10.9 11.9 −2.5 11.1 −11.1 |
| Irf_known7 | 9.0 | rs9907564 rs188089973 rs75027016 rs138461720 rs112745149 rs184525456 | Irf_known9 Irf_disc5/known9 Irf_known1/2 Irf_disc3 Irf_disc3/known9 Irf_known1/9 | −1.1 11.9/−0.6 11.9/12 5.5 9.6/12 12/11.9 | ||||
| Pax-5_disc4 | 4.7 | - | - | − | ||||
| Pou2f2_disc1 Pou2f2_known10 | 4.7 3.1 | rs202227901 rs191534721 rs9905881 rs193079571 rs140511615 rs4622539 rs184966935 rs145101657 rs145975450 | Pou2f2_known4 Pou2f2_known4 Pou2f2_known2 Pou2f2_known2 Pou2f2_known8 Pou2f2_known8 Pou2f2_known2 Pou2f2_known10 Pou2f2_known2 | −0.2 −0.6 2.6 4.3 −5.4 −5.2 1.9 4.9 1 | ||||
| p300_disc5 | 2.9 | rs188089973 rs9907794 rs116467677 rs145275643 rs138461720 rs112745149 rs192412458 | p300_disc5 p300_disc5 p300_disc9 p300_disc10 p300_disc5 p300_disc5 p300_disc1 | 1.9 −5.9 −1.7 11.9 −2.5 3.2 11.9 | ||||
| E | rs111907649 | I3 | no | AP-1_disc7 | −10.9 | rs190729974 rs4795397 rs192412458 rs192412458 rs147224870 rs1453558 rs1453560 rs36111081 rs66565390 | AP-1_disc1 AP-1_disc2 AP-1_disc3/7/9 AP-1_known2/3/4 AP-1_disc7 AP-1_disc2 AP-1_known1 AP-1_disc7 AP-1_disc7 | 12 −6.8 11.9/0.4/12 11.8/4.2/12 −10.9 11.9 −2.5 11.1 −11.1 |
| BHLHE40_disc2 | −11.2 | rs145275643 rs11557466 | BHLHE40_known1 BHLHE40_known1 | −0.2 1.3 | ||||
| F | rs140386398 | I3 | no | BDP1_disc3 | −12 | rs79042302 | BDP1_disc1 | −5.3 |
| GR_disc5 | −12 | rs199994111 rs183478341 rs192412458 rs190942850 rs192800564 rs11655198 | GR_disc6 GR_disc1 GR_disc2 GR_known3/9 GR_disc6 GR_disc4 | −0.3 6.6 11.8 −0.2/−0.3 −9.2 12 | ||||
| G | rs149317842 | I3 | 3′ E4-7 | Dlx2 | −10.1 | rs191534721 | Dlx2 | −1.9 |
| Dlx3 | −9.4 | rs191534721 | Dlx2 | −1.3 | ||||
| Irx | −5.6 | - | - | − | ||||
| Lhx3_1 | −12 | rs138350717 | Lhx3_1 | −1 | ||||
| Pou3f2_2 | −11 | rs202227901 rs182045388 rs200781948 rs11078924 | Pou3f2_2 Pou3f2_2 Pou3f2_2 Pou3f2_2 | −12 −11 −12 −2.9 | ||||
| SRF_known3 | 4.3 | rs188089973 rs75027016 | SRF_known3 SRF_known3 | −1 1.3 | ||||
| STAT_known3 | 4.9 | rs202227901 rs191534721 rs4622539 rs145275643 rs79042302 rs79042302 rs112745149 rs192412458 rs181849193 rs185870642 rs145975450 rs74805134 | STAT_disc5/known1 STAT_disc5 STAT_disc4 STAT_known13 STAT_disc1 STAT_known10/11/12/15/4/6/7 STAT_disc3 STAT_disc2 STAT_disc6 STAT_known14/15 STAT_known11 STAT_disc7 | 2.2/4.7 −11.8 12 5.2 −4 −11.9/1.2/−1/0.1/−3/−12/−0.9 12 12 11.9 11.9/11.9 −4.3 −11.7 | ||||
| YY1_known6 | 3.7 | rs188089973 rs147224870 rs28661251 | YY1_known6 YY1_disc4 YY1_disc1/known2 | −1.6 −3.1 −3.9/−0.6 | ||||
| H | rs186234194 | I7 | 3′ E4-7 | - | - | - | − | |
| I | rs145335424 | I7 | no | AP-1_disc2 | −12 | rs190729974 rs4795397 rs192412458 rs192412458 rs147224870 rs1453558 rs1453560 rs36111081 rs66565390 | AP-1_disc1 AP-1_disc2 AP-1_disc3/7/9 AP-1_known2/3/4 AP-1_disc7 AP-1_disc2 AP-1_known1 AP-1_disc7 AP-1_disc7 | 12 −6.8 11.9/0.4/12 11.8/4.2/12 −10.9 11.9 −2.5 11.1 −11.1 |
| Gfi1_3 | −12 | - | - | − | ||||
| NF-Y_disc1 | −12 | - | - | − | ||||
| NF-Y_known1 | −5.2 | - | - | − | ||||
| RFX5_disc2 | −11.9 | rs4795397 | RFX5_disc2 | −7.5 | ||||
| TATA_disc6 | −5.4 | rs188089973 rs140511615 rs4622539 rs184966935 rs112745149 rs184525456 rs185009382 rs192678773 | TATA_known4 TATA_disc9 TATA_disc9 TATA_known1 TATA_disc7 TATA_known1 TATA_disc7 TATA_disc7 | 0.7 −5.1 −3.2 −2.1 1.3 −0.6 −2.8 −7 | ||||
| J | rs113730542 | I7 | no | Fox | 8.3 | rs111678394 | Fox | −1 |
| SNP | DNAse HotSpot (ENCODE) | Interaction Region Hi-C | Shared TF between IKZF3-ZPBP2 and 3′ (E4-7) Interaction Regions | Shared DNase HotSpot between IKZF3-ZPBP2 and 3′ (E4-7) Interaction Regions | Source |
|---|---|---|---|---|---|
| rs111678394 | y | IKZF3-ZPBP2 | (Foxi1) Foxo_1, Pax-4_5 | CD20, CD4, CD34+, LCL, Th1, Th2, Treg | Table 2 |
| rs75148376 | y | 3′ (E4-7) | Ncx, Nkx6, Pou4f3, Dbx1, Hoxb4 | LCL, Th1, Th2, Treg | Table 2 |
| rs113370572 | y | 3′ (E4-7) | HDAC2 | LCL, Th1, Th2, Treg | Table 2 |
| rs113730542 * | y | <2kb from 3′ (E4-7) | Fox | CD4, LCL, Th1, Th2, Treg | Table A6 |
| rs112876941 | y | <10kb from 3′ (E4-7) | Foxa, Foxj1, Foxo, HNF1, TCF12 | CD14+, LCL | Table 2 |
References
- Chen, L.; Morris, D.L.; Vyse, T.J. Genetic advances in systemic lupus erythematosus: An update. Curr. Opin. Rheumatol. 2017, 29, 423–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hom, G.; Graham, R.R.; Modrek, B.; Taylor, K.E.; Ortmann, W.; Garnier, S.; Lee, A.T.; Chung, S.A.; Ferreira, R.C.; Pant, P.V.; et al. Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N. Engl. J. Med. 2008, 358, 900–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozyrev, S.V.; Abelson, A.K.; Wojcik, J.; Zaghlool, A.; Linga Reddy, M.V.; Sanchez, E.; Gunnarsson, I.; Svenungsson, E.; Sturfelt, G.; Jonsen, A.; et al. Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus. Nat. Genet. 2008, 40, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.R.; Cotsapas, C.; Davies, L.; Hackett, R.; Lessard, C.J.; Leon, J.M.; Burtt, N.P.; Guiducci, C.; Parkin, M.; Gates, C.; et al. A genome-wide association scan identifies Tumour Necrosis Factor Alpha Inducible Protein 3 (TNFAIP3/A20) as a susceptibility locus for Systemic Lupus Erythematosus. Nat. Genet. 2008, 40, 1059–1061. [Google Scholar] [CrossRef]
- Harley, J.B.; Alarcon-Riquelme, M.E.; Criswell, L.A.; Jacob, C.O.; Kimberly, R.P.; Moser, K.L.; Tsao, B.P.; Vyse, T.J.; Langefeld, C.D.; Nath, S.K.; et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat. Genet. 2008, 40, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.L.; Taylor, K.E.; Fernando, M.M.; Nititham, J.; Alarcon-Riquelme, M.E.; Barcellos, L.F.; Behrens, T.W.; Cotsapas, C.; Gaffney, P.M.; Graham, R.R.; et al. Unraveling multiple MHC gene associations with systemic lupus erythematosus: Model choice indicates a role for HLA alleles and non-HLA genes in Europeans. Am. J. Hum. Genet. 2012, 91, 778–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Shen, N.; Ye, D.Q.; Liu, Q.; Zhang, Y.; Qian, X.X.; Hirankarn, N.; Ying, D.; Pan, H.F.; Mok, C.C.; et al. Genome-wide association study in Asian populations identifies variants in ETS1 and WDFY4 associated with systemic lupus erythematosus. PLoS Genet. 2010, 6, e1000841. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Shimane, K.; Kochi, Y.; Tahira, T.; Suzuki, A.; Higasa, K.; Takahashi, A.; Horita, T.; Atsumi, T.; Ishii, T.; et al. A genome-wide association study identified AFF1 as a susceptibility locus for systemic lupus eyrthematosus in Japanese. PLoS Genet. 2012, 8, e1002455. [Google Scholar] [CrossRef]
- Lee, H.S.; Kim, T.; Bang, S.Y.; Na, Y.J.; Kim, I.; Kim, K.; Kim, J.H.; Chung, Y.J.; Shin, H.D.; Kang, Y.M.; et al. Ethnic specificity of lupus-associated loci identified in a genome-wide association study in Korean women. Ann. Rheum. Dis. 2014, 73, 1240–1245. [Google Scholar] [CrossRef]
- Lessard, C.J.; Sajuthi, S.; Zhao, J.; Kim, K.; Ice, J.A.; Li, H.; Ainsworth, H.; Rasmussen, A.; Kelly, J.A.; Marion, M.; et al. Identification of a Systemic Lupus Erythematosus Risk Locus Spanning ATG16L2, FCHSD2, and P2RY2 in Koreans. Arthritis Rheumatol. 2016, 68, 1197–1209. [Google Scholar]
- Demirci, F.Y.; Wang, X.; Kelly, J.A.; Morris, D.L.; Barmada, M.M.; Feingold, E.; Kao, A.H.; Sivils, K.L.; Bernatsky, S.; Pineau, C.; et al. Identification of a New Susceptibility Locus for Systemic Lupus Erythematosus on Chromosome 12 in Individuals of European Ancestry. Arthritis Rheumatol. 2016, 68, 174–183. [Google Scholar] [CrossRef]
- Bentham, J.; Morris, D.L.; Cunninghame Graham, D.S.; Pinder, C.L.; Tombleson, P.; Behrens, T.W.; Martin, J.; Fairfax, B.P.; Knight, J.C.; Chen, L.; et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. 2015, 47, 1457–1464. [Google Scholar] [CrossRef]
- John, L.B.; Ward, A.C. The Ikaros gene family: Transcriptional regulators of hematopoiesis and immunity. Mol. Immunol. 2011, 48, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Avitahl, N.; Cariappa, A.; Friedrich, C.; Ikeda, T.; Renold, A.; Andrikopoulos, K.; Liang, L.; Pillai, S.; Morgan, B.A.; et al. Aiolos regulates B cell activation and maturation to effector state. Immunity 1998, 9, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Ng, S.Y.; Zuniga-Pflucker, J.C.; Georgopoulos, K. Early hematopoietic lineage restrictions directed by Ikaros. Nat. Immunol 2006, 7, 382–391. [Google Scholar] [CrossRef]
- Schmitt, C.; Tonnelle, C.; Dalloul, A.; Chabannon, C.; Debre, P.; Rebollo, A. Aiolos and Ikaros: Regulators of lymphocyte development, homeostasis and lymphoproliferation. Apoptosis 2002, 7, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Franke, A.; McGovern, D.P.; Barrett, J.C.; Wang, K.; Radford-Smith, G.L.; Ahmad, T.; Lees, C.W.; Balschun, T.; Lee, J.; Roberts, R.; et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 2010, 42, 1118–1125. [Google Scholar] [CrossRef] [Green Version]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [Green Version]
- International Multiple Sclerosis Genetics Consortium; Beecham, A.H.; Patsopoulos, N.A.; Xifara, D.K.; Davis, M.F.; Kemppinen, A.; Cotsapas, C.; Shah, T.S.; Spencer, C.; Booth, D.; et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013, 45, 1353–1360. [Google Scholar] [CrossRef]
- Swafford, A.D.; Howson, J.M.; Davison, L.J.; Wallace, C.; Smyth, D.J.; Schuilenburg, H.; Maisuria-Armer, M.; Mistry, T.; Lenardo, M.J.; Todd, J.A. An allele of IKZF1 (Ikaros) conferring susceptibility to childhood acute lymphoblastic leukemia protects against type 1 diabetes. Diabetes 2011, 60, 1041–1044. [Google Scholar] [CrossRef] [Green Version]
- Stahl, E.A.; Raychaudhuri, S.; Remmers, E.F.; Xie, G.; Eyre, S.; Thomson, B.P.; Li, Y.; Kurreeman, F.A.; Zhernakova, A.; Hinks, A.; et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet. 2010, 42, 508–514. [Google Scholar] [CrossRef]
- Liu, J.Z.; Almarri, M.A.; Gaffney, D.J.; Mells, G.F.; Jostins, L.; Cordell, H.J.; Ducker, S.J.; Day, D.B.; Heneghan, M.A.; Neuberger, J.M.; et al. Dense fine-mapping study identifies new susceptibility loci for primary biliary cirrhosis. Nat. Genet. 2012, 44, 1137–1141. [Google Scholar] [CrossRef] [Green Version]
- Anderson, C.A.; Boucher, G.; Lees, C.W.; Franke, A.; D’Amato, M.; Taylor, K.D.; Lee, J.C.; Goyette, P.; Imielinski, M.; Latiano, A.; et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 2011, 43, 246–252. [Google Scholar] [CrossRef] [Green Version]
- Moffatt, M.F.; Kabesch, M.; Liang, L.; Dixon, A.L.; Strachan, D.; Heath, S.; Depner, M.; von Berg, A.; Bufe, A.; Rietschel, E.; et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature 2007, 448, 470–473. [Google Scholar] [CrossRef]
- Barrett, J.C.; Clayton, D.G.; Concannon, P.; Akolkar, B.; Cooper, J.D.; Erlich, H.A.; Julier, C.; Morahan, G.; Nerup, J.; Nierras, C.; et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat.Genet. 2009, 41, 703–707. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, Y.; Kosek, J.; Capone, L.; Hur, E.M.; Schafer, P.H.; Ringheim, G.E. Aiolos Overexpression in Systemic Lupus Erythematosus B Cell Subtypes and BAFF-Induced Memory B Cell Differentiation Are Reduced by CC-220 Modulation of Cereblon Activity. J. Immunol. 2017, 199, 2388–2407. [Google Scholar] [CrossRef]
- Roadmap Epigenomics, C.; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Javierre, B.M.; Burren, O.S.; Wilder, S.P.; Kreuzhuber, R.; Hill, S.M.; Sewitz, S.; Cairns, J.; Wingett, S.W.; Varnai, C.; Thiecke, M.J.; et al. Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters. Cell 2016, 167, 1369–1384.e19. [Google Scholar] [CrossRef] [PubMed]
- Ward, L.D.; Kellis, M. HaploReg v4: Systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 2016, 44, D877–D881. [Google Scholar] [CrossRef]
- Han, J.W.; Zheng, H.F.; Cui, Y.; Sun, L.D.; Ye, D.Q.; Hu, Z.; Xu, J.H.; Cai, Z.M.; Huang, W.; Zhao, G.P.; et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat. Genet. 2009, 41, 1234–1237. [Google Scholar] [CrossRef]
- Morris, D.L.; Sheng, Y.; Zhang, Y.; Wang, Y.F.; Zhu, Z.; Tombleson, P.; Chen, L.; Cunninghame Graham, D.S.; Bentham, J.; Roberts, A.L.; et al. Genome-wide association meta-analysis in Chinese and European individuals identifies ten new loci associated with systemic lupus erythematosus. Nat. Genet. 2016, 48, 940–946. [Google Scholar] [CrossRef]
- Langefeld, C.D.; Ainsworth, H.C.; Cunninghame Graham, D.S.; Kelly, J.A.; Comeau, M.E.; Marion, M.C.; Howard, T.D.; Ramos, P.S.; Croker, J.A.; Morris, D.L.; et al. Transancestral mapping and genetic load in systemic lupus erythematosus. Nat. Commun. 2017, 8, 16021. [Google Scholar] [CrossRef]
- Wu, C.; Orozco, C.; Boyer, J.; Leglise, M.; Goodale, J.; Batalov, S.; Hodge, C.L.; Haase, J.; Janes, J.; Huss, J.W., 3rd; et al. BioGPS: An extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009, 10, R130. [Google Scholar] [CrossRef] [PubMed]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Iny Stein, T.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database (Oxford) 2017, 2017, bax028. [Google Scholar] [CrossRef] [Green Version]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Consortium, G.T. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, R.; Hottenga, J.J.; Nivard, M.G.; Abdellaoui, A.; Laport, B.; de Geus, E.J.; Wright, F.A.; Penninx, B.; Boomsma, D.I. Conditional eQTL analysis reveals allelic heterogeneity of gene expression. Hum. Mol. Genet. 2017, 26, 1444–1451. [Google Scholar] [CrossRef]
- Wang, Y.; Song, F.; Zhang, B.; Zhang, L.; Xu, J.; Kuang, D.; Li, D.; Choudhary, M.N.K.; Li, Y.; Hu, M.; et al. The 3D Genome Browser: A web-based browser for visualizing 3D genome organization and long-range chromatin interactions. Genome Biol. 2018, 19, 151. [Google Scholar] [CrossRef] [Green Version]
- Nolis, I.K.; McKay, D.J.; Mantouvalou, E.; Lomvardas, S.; Merika, M.; Thanos, D. Transcription factors mediate long-range enhancer-promoter interactions. Proc. Natl. Acad. Sci. USA 2009, 106, 20222–20227. [Google Scholar] [CrossRef] [Green Version]
- Sanyal, A.; Lajoie, B.R.; Jain, G.; Dekker, J. The long-range interaction landscape of gene promoters. Nature 2012, 489, 109–113. [Google Scholar] [CrossRef]
- Singh, P.; Han, E.H.; Endrizzi, J.A.; O’Brien, R.M.; Chi, Y.I. Crystal structures reveal a new and novel FoxO1 binding site within the human glucose-6-phosphatase catalytic subunit 1 gene promoter. J. Struct. Biol. 2017, 198, 54–64. [Google Scholar] [CrossRef] [Green Version]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-Andre, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [Green Version]
- Absher, D.M.; Li, X.; Waite, L.L.; Gibson, A.; Roberts, K.; Edberg, J.; Chatham, W.W.; Kimberly, R.P. Genome-wide DNA methylation analysis of systemic lupus erythematosus reveals persistent hypomethylation of interferon genes and compositional changes to CD4+ T-cell populations. PLoS Genet. 2013, 9, e1003678. [Google Scholar] [CrossRef] [Green Version]
- Ulff-Moller, C.J.; Asmar, F.; Liu, Y.; Svendsen, A.J.; Busato, F.; Gronbaek, K.; Tost, J.; Jacobsen, S. Twin DNA Methylation Profiling Reveals Flare-Dependent Interferon Signature and B Cell Promoter Hypermethylation in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2018, 70, 878–890. [Google Scholar] [CrossRef] [Green Version]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinfomatics 2005, 21, 263–265. [Google Scholar] [CrossRef] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Blankenberg, D.; Von Kuster, G.; Coraor, N.; Ananda, G.; Lazarus, R.; Mangan, M.; Nekrutenko, A.; Taylor, J. Galaxy: A web-based genome analysis tool for experimentalists. Curr. Protoc. Mol. Biol. 2010, 89, 10–19. [Google Scholar] [CrossRef]
- Burren, O.S.; Rubio Garcia, A.; Javierre, B.M.; Rainbow, D.B.; Cairns, J.; Cooper, N.J.; Lambourne, J.J.; Schofield, E.; Castro Dopico, X.; Ferreira, R.C.; et al. Chromosome contacts in activated T cells identify autoimmune disease candidate genes. Genome Biol. 2017, 18, 165. [Google Scholar] [CrossRef]
- Hoffman, M.M.; Ernst, J.; Wilder, S.P.; Kundaje, A.; Harris, R.S.; Libbrecht, M.; Giardine, B.; Ellenbogen, P.M.; Bilmes, J.A.; Birney, E.; et al. Integrative annotation of chromatin elements from ENCODE data. Nuc. Acids Res. 2013, 41, 827–841. [Google Scholar] [CrossRef] [Green Version]
- Zhbannikov, I.Y.; Arbeev, K.; Ukraintseva, S.; Yashin, A.I. haploR: An R package for querying web-based annotation tools. F1000Res 2017, 6, 97. [Google Scholar]
- Hahne, F.; Ivanek, R. Visualizing Genomic Data Using Gviz and Bioconductor. Methods Mol. Biol. 2016, 1418, 335–351. [Google Scholar]


| Enhancer Region (Enh) | Promoter/TSS Region (PC Hi-C) | GeneHancer Promoter Region (GH07J050293) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Risk SNP | TF Showing Allele-Specific Binding (ASTF) | Alt-Ref Enrichment | TSS SNP with Same TF Binding Site as Risk Allele | TF Binding to TSS SNP | Alt-Ref Enrichment | TSS SNP with Same TF Binding Site as Risk Allele | TF Binding to TSS SNP | Alt-Ref Enrichment | |
| rs11185603 * | A | RXRA_disc4 | −11.1 | rs146295095 | RXRA_known1 | 3 | |||
| rs141865623 | RXRA_disc2 | −0.8 | |||||||
| rs11765436 # | RXRA_disc2 | 5.7 | rs11765436 # | RXRA_disc2 | 5.7 | ||||
| rs187496825 | RXRA_known2 | 12 | |||||||
| rs180969166 ^ | RXRA_known6 | 0 | |||||||
| rs183264036 ^ | RXRA_disc1 | 0.2 | rs7802443 # | RXRA_disc2 | 11.4 | ||||
| B | PU.1_disc3 | −11.9 | rs191336126 rs80161560 | PU.1_disc2 | 0.8 | rs9886239 * | PU.1_disc2 | −12 | |
| PU.1_disc3 | 1.6 | ||||||||
| C | TATA_disc7 | −6.3 | rs142010565 | TATA_known1 | −1.9 | rs7777365 | TATA_known3 | −2.4 | |
| rs142762599 | TATA_known1 | 0.1 | |||||||
| rs79391891 | TATA_disc2 | −12 | |||||||
| rs186224998 | TATA_disc9 | −4 | |||||||
| rs62447182 | TATA_disc9 | −5.1 | |||||||
| rs876036 | D | ERalpha-a_disc4 | 10.5 | rs180969166 ^ rs183264036 rs151114892 rs145086785 | ERalpha-a_disc2/4 | −2/−0.3 | |||
| ERalpha-a_disc4 | 3.3 | ||||||||
| ERalpha-a_disc4 | −3.6 | ||||||||
| ERalpha-a_disc4 | 0.5 | ||||||||
| D | VDR_2/3 | −7.8, −3.9 | rs180969166 ^ | VDR_4 | 12 | ||||
| rs151114892 | VDR_4 | −11.5 | |||||||
| E | RXRA_known4 | −10.6 | rs11765436 # | RXRA_disc2 | 5.7 | ||||
| rs7802443 # | RXRA_disc2 | 11.4 | |||||||
| rs876038 * | A | XBP-1_1 | −12 | rs184933329 | XBP-1_2 | −11.9 | |||
| rs74607523 | XBP-1_2 | −2.3 | |||||||
| B | BDP1_disc1 | −0.6 | rs11761922 * | BDP1_disc1 | 12 | ||||
| rs7781977 # | BDP1_disc1 | 12 | |||||||
| C | Brachyury_1 | −3.2 | rs10269380 * | Brachyury_1 | 4.8 | ||||
| rs876039 | Foxa_known2,3 | 1.1, 0.6 | rs7777365 # | Foxa_known1 | −2.7 | ||||
| Group I Risk Variants | SNPs in IKZF3-ZPBP2 Bi-Directional Promoter | ||||||
|---|---|---|---|---|---|---|---|
| Risk SNP | Interaction Fragment | ASTF * | Alt-Ref Enrichment | Promoter SNP | Shared Promoter TF | Alt-Ref Enrichment | |
| 1 | rs111678394 | IKZF3-ZPBP2 | Foxi1 | −3.9 | - | - | - |
| Foxo_1 | −2.1 | rs138959946 a | Foxo1 | −2.4 | |||
| Pax-4_5 | −2.3 | rs189743120 a | Pax_4_5 | 1 | |||
| 2 | rs117278702 | IKZF3-ZPBP2 | - | - | - | - | - |
| 3 | rs77924338 | no | VDR_4 | −9.1 | rs74805134 b | VDR_2 | −11.5 |
| 4 | rs113233720 | no | DMRT4 | −11.5 | rs147630723 a | DMRT4 | 11.9 |
| 5 | rs112677036 | no | Mef2_known5 | 11.5 | rs73985223 b | Mef2_known6 | 11.9 |
| rs73985223 b | Mef2_disc1 | 6.4 | |||||
| rs4622539 b | Mef2_known5 | −3.2 | |||||
| rs192412458 a | Mef2_disc3 | 11.9 | |||||
| rs188089973 | Mef2_known5 | −3.8 | |||||
| rs185330833 a | Mef2_known6 | 11.7 | |||||
| rs184966935 a | Mef2_known1 | −10 | |||||
| rs184525456 a | Mef2_known5 | −3.1 | |||||
| rs140511615 a | Mef2_known5 | −11.8 | |||||
| 6 | rs111691913 | no | Zntb3 | 8.0 | - | - | - |
| 7 | rs111944912 | no | Hoxa13 | 2 | rs12150079 | Hoxa13 | 0.7 |
| 8 | rs111734595 | no | - | - | - | - | - |
| 9 | rs113479772 | no | - | - | - | - | - |
| 10 | rs112797570 | no | - | - | - | - | - |
| 11 | rs111734595 | no | SETDB1 Zfx | 8.2 −5.7 | rs201229892 rs117064469 | SETDB1 Zfx | −0.6 −1.4 |
| 12 | rs111469562 | no | Obox6 | 4.6 | rs11078925 | Obox3 | −6.7 |
| Dmbx1 | 4.1 | rs11078925 | Dmbx1 | −9 | |||
| 13 | rs112743130 | 5′ (I3) | - | - | - | - | - |
| 14 | rs112412105 | 5′ (I3) | GR_disc4 | −12 | rs183478341 u/k | GR_disc1 | 6.6 |
| 15 | rs113115305 | 3′ (E4-7) | - | - | - | - | - |
| 16 | rs112238900 | 3′ (E4-7) | - | - | - | - | - |
| 17 | rs113064843 | 3′ (E4-7) | - | - | - | - | - |
| 18 | rs16965347 | 3′ (E4-7) | Pou6f1_2 | - | - | - | |
| 19 | rs113369293 | 3′ (E4-7) | Irf_disc3 | 2.3 | rs138461720 u/k | Irf_disc3 | 5.5 |
| Irf_disc3 | 2.3 | rs112745149 u/k | Irf_disc3 | 9.6 | |||
| 20 | rs75148376 | 3′ (E4-7) | Ncx_2 | 4 | rs9905881 b | Ncx_2 | 3.2 |
| Nkx6-1_2 | 6.7 | rs149800216 a | Nkx6-1_3 | −9.7 | |||
| Nkx6-1_2 | 6.7 | rs149800216 a | Nkx6-1_2 | −10.2 | |||
| Nkx6-1_2 | 6.7 | rs149800216 a | Nkx6-1_1 | −12 | |||
| Ncx_2 | 4 | rs149800216 a | Ncx_2 | −6.4 | |||
| Pou4f3 | 5.6 | rs138350717 a | Pou4f3 | 5.9 | |||
| Nkx6-1_2 | 6.7 | rs138350717 a | Nkx6-1_2 | 3.5 | |||
| Nkx6-1_2 | 6.7 | rs138350717 a | Nkx6-1_1 | 7.9 | |||
| Dbx1 | 2.2 | rs202227901 b | Dbx1 | −0.1 | |||
| Dbx1 | 2.2 | rs138350717 a | Dbx1 | 0.6 | |||
| Dbx1 | 2.2 | rs145735506 a | Dbx1 | 1.4 | |||
| Dbx1 | 2.2 | rs185330833 a | Dbx1 | −1.2 | |||
| Hoxb4 | 2.1 | rs202227901 b | Hoxb4 | −0.5 | |||
| 21 | rs112771646 | 3′ (E4-7) | GR_disc5 | −3.8 | rs192800564 a | GR_disc6 | −9.2 |
| GR_disc5 | −3.8 | rs192412458 a | GR_disc2 | 11.8 | |||
| GR_disc5 | −3.8 | rs11655198 | GR_disc4 | 12 | |||
| 22 | rs112301322 | 3′ (E4-7) | NF-E2_disc1 | 11.9 | rs201229892 a | NF-E2_disc1 | 12 |
| Rad21_disc10 | −11.5 | rs187549822 a | Rad21_disc2 | −4.2 | |||
| 23 | rs111862642 | 3′ (E4-7) | Sin3Ak-20_disc1 | −2.9 | rs116467677 a | Sin3Ak-20_disc6 | −0.6 |
| 24 | rs112345383 | 3′ (E4-7) | HNF1_2 | 6.1 | rs202236981 a | HNF1_2 | −1.8 |
| 25 | rs113370572 T | 3′ (E4-7) | HDAC2_disc5 | 9.6 | rs202227901 b | HDAC2_disc6 | 10.6 |
| HDAC2_disc5 | 9.6 | rs200781948 a | HDAC2_disc6 | −3.9 | |||
| 26 | rs112771360 | no | - | - | - | - | - |
| 27 | rs112876941 | no | HNF1_7 | 3.5 | - | - | - |
| HNF1_6 | 3.1 | rs9905881 b | HNF1_6 | −2.7 | |||
| HNF1_6 | 3.1 | rs9907564 b | HNF1_6 | −1.1 | |||
| HNF1_6 | 3.1 | rs138350717 a | HNF1_6 | 0.7 | |||
| HNF1_1 | 4.3 | rs9905881 b | HNF1_1 | −4.3 | |||
| Foxo_2 | 11.9 | rs184525456 a | Foxo_2 | −12 | |||
| Foxa_disc2 | −10.6 | rs145895912 a | Foxa_disc3 | 11.7 | |||
| Foxj1_1 | 4.6 | rs145735506 a | Foxj1_1 | 11.8 | |||
| Foxo_2 | 11.9 | rs138959946 a | Foxo_2 | −12 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vyse, T.J.; Cunninghame Graham, D.S. Trans-Ancestral Fine-Mapping and Epigenetic Annotation as Tools to Delineate Functionally Relevant Risk Alleles at IKZF1 and IKZF3 in Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2020, 21, 8383. https://doi.org/10.3390/ijms21218383
Vyse TJ, Cunninghame Graham DS. Trans-Ancestral Fine-Mapping and Epigenetic Annotation as Tools to Delineate Functionally Relevant Risk Alleles at IKZF1 and IKZF3 in Systemic Lupus Erythematosus. International Journal of Molecular Sciences. 2020; 21(21):8383. https://doi.org/10.3390/ijms21218383
Chicago/Turabian StyleVyse, Timothy J., and Deborah S. Cunninghame Graham. 2020. "Trans-Ancestral Fine-Mapping and Epigenetic Annotation as Tools to Delineate Functionally Relevant Risk Alleles at IKZF1 and IKZF3 in Systemic Lupus Erythematosus" International Journal of Molecular Sciences 21, no. 21: 8383. https://doi.org/10.3390/ijms21218383
APA StyleVyse, T. J., & Cunninghame Graham, D. S. (2020). Trans-Ancestral Fine-Mapping and Epigenetic Annotation as Tools to Delineate Functionally Relevant Risk Alleles at IKZF1 and IKZF3 in Systemic Lupus Erythematosus. International Journal of Molecular Sciences, 21(21), 8383. https://doi.org/10.3390/ijms21218383