Abstract
In spite of being a preventable disease, cervical cancer (CC) remains at high incidence, and it has a significant mortality rate. Although hijacking of the host cellular pathway is fundamental for developing a better understanding of the human papillomavirus (HPV) pathogenesis, a major obstacle is identifying the central molecular targets involved in HPV-driven CC. The aim of this study is to investigate transcriptomic patterns of HPV-infected and normal tissues to identify novel prognostic markers. Analyses of functional enrichment and interaction networks reveal that altered genes are mainly involved in cell cycle, DNA damage, and regulated cell-to-cell signaling. Analysis of The Cancer Genome Atlas (TCGA) data has suggested that patients with unfavorable prognostics are more likely to have DNA repair defects attributed, in most cases, to the presence of HPV. However, further studies are needed to fully unravel the molecular mechanisms of such genes involved in CC.
1. Introduction
Although cervical cancer (CC) is a preventable disease, it remains the fourth most common type of malignant cancer in women [1,2]. It accounts for higher incidence among all types of female genital tract malignant tumors, particularly detected among relatively young age groups at diagnosis [1,2]. The latest data reveal incidence of 807,860 new cases and over 200,000 deaths per year due to CC [3]. Although CC is attributed to squamous cell carcinomas (SCC) and adenocarcinomas (ADC), SCC accounts for 90% of all CC cases [3], whereas the least common subtype is that of adenosquamous carcinoma (ADSC) [4].
A large number of factors have been associated with CC, including premature onset of sexual activity, genetic background, infection, long-term administration of oral contraceptives, immunosuppressive medication, and poor health circumstances [5]. Currently, treatment for CC primarily involves surgery and radiotherapy, but sometimes chemotherapy is also used for patients with either metastasis or recurrence [6]. However, patient prognosis remains unfavorable, particularly in patients with advanced CC. Hence, it is critical that the fundamental mechanism of CC is investigated and better understood in order to develop novel therapeutic targets for CC [6], particularly those related to epithelial–mesenchymal transition (EMT). EMT is a key biological process that is involved in the activation of invasion and the metastasis of cancer cells, including those of CC [7,8,9].
Oncogenic human papillomavirus (HPV) represents an important but not a sufficient risk factor involved in the process of promoting cervical carcinogenesis [4], particularly of high-risk (HR) HPV, wherein HPV-16 and 18 are the most common types. Therefore, HPV detection is important for both the screening and prevention of cervical cancer [4]. It is reported that viral oncogenic proteins encoded by HPV are known to target and degrade the DNA repair mechanism, which, in turn, contributes to cervical cancer development [10]. Thus, hijacking of the host cellular metabolic pathway is fundamental for virus pathogenesis [11,12]. Therefore, it is critical to understand how such a hijacking mechanism recognizes central molecular targets, as well as how inputs of such targeted cellular machinery contribute to HPV-driven CC [13].
The recent boom in knowledge that shows the importance of coding and non-coding genes (particular miRNA) in the regulation of multiple major biological processes, which affect tumor genesis and tumor progression, have brought these up-until-now-neglected molecular players to the forefront [14,15,16]. The integration of high-throughput technologies and bioinformatics analysis can provide researchers with valuable data that can be exploited as biomarkers and therapeutic targets, using public data sets [17,18,19,20]. The Cancer Genome Atlas (TCGA) is a comprehensive data set that provides a unified data analysis pipeline, which can be used for additional exploration of the altered oncogenic signaling and its related implication in CC patients prognostic [21]. The scope of this study was to evaluate altered mRNA and miRNA patterns in CC, based on TCGA data following gene enrichment, to make miRNA target gene prediction and to obtain a comprehensive analysis of miRNA–mRNA regulatory networks and their correlation with overall survival of key hub transcripts.
2. Results
2.1. Differentially Expressed mRNAs and miRNA in CC Based on TCGA Data
A total of 304 CC cancer samples and three analogous organ-matched normal tissues were obtained following data preprocessing. In these tissues, a total of 3988 differentially expressed RNAs were identified, wherein 2178 genes were upregulated and 1810 were downregulated (Figure 1A heatmap representation). Moreover, 207 differentially expressed miRNAs were also identified, wherein 183 miRNAs were overexpressed and 24 were underexpressed. Among altered miRNA transcripts, the top 10 overexpressed transcripts were related to EMT. Gene enrichment using Panther for those genes with altered expression levels in CC is presented in Figure 1B,C.
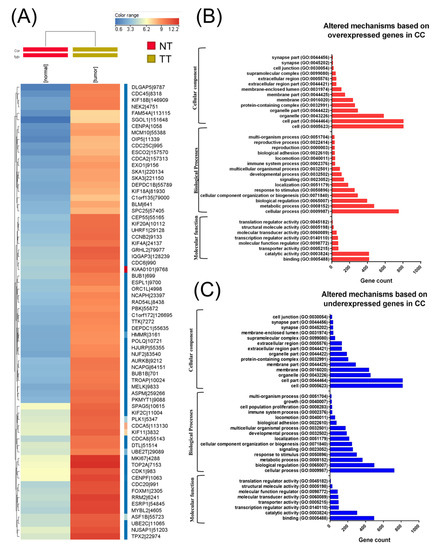
Figure 1.
Biological interpretation of the cervical cancer (CC) altered gene expression signature. (A) A heatmap representation of The Cancer Genome Atlas (TGCA) gene expression data in CC, (B) gene ontology (GO) analysis of overexpressed genes, and (C) GO analysis of underexpressed genes using the Panther Gene ontology tool (available online: http://pantherdb.org); NT: normal tissue TT: tumoral tissue.
2.2. Functional Enrichment Analysis
An ingenuity pathway analysis (IPA) of the canonical signaling pathway, for disease and function, was subsequently performed to identify enrichment of genes altered in CC. This analysis revealed involvement of several cell cycle related pathways, including G2/M DNA damage check-point regulation, G1/S check-point regulation, and BRCA1 DNA damage response. Additional altered signaling related to either TP53 signaling or to those related to both cellular adhesion and tight junction proteins was observed. Gene enrichment analysis of differentially expressed genes revealed the most important altered canonical pathways (Figure 2A), top molecular function (Figure 2B), and the most specific differentially expressed pathways related to CC (Figure 2C), based on mRNA patterns.
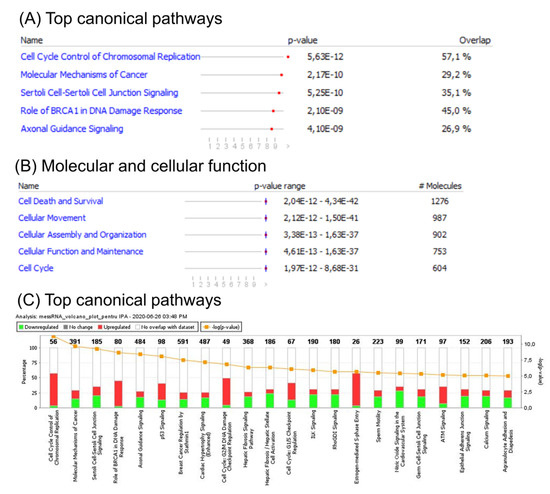
Figure 2.
Functional analysis of mRNA altered genes in CC. (A) Top canonical pathways; (B) top molecular function; (C) ingenuity pathway analysis (IPA) of differentially expressed pathways specific to CC. A log Benjamini–Hochberg value higher than 5, p-value ≥ 0.0001, corresponds to a significantly altered pathway. The solid yellow line in the bar graph corresponds to ratios of number of molecules from the dataset that map to the pathway over the total number of molecules that map to the canonical pathway from the IPA knowledge base. The Z score denotes the activation score for either a pathway, disease, or function. This score corresponds to levels of activation (red) or deactivation (green) of a pathway or function due to changes in expression of genes involved in pathways or functions.
2.3. Gene Network Analysis
In addition to the delineation of main pathways and cellular functions, gene networks have been constructed to link key genes and enriched categories of diseases and functions based on associations between differentially expressed genes. These gene networks along with their top associated diseases and functions are presented in Table 1.

Table 1.
List of the top 10 altered networks in CC based on IPA analysis.
Following identification of hub genes that might be involved in CC progression, a Kaplan–Meier plotter (http://www.oncolnc.org) was performed to identify those genes with significant effects. As a result, six hub genes for survival (SERINC1, TOM1L1, DUOX1, CENPH, AURKB, and CKAP2) related to cell cycle, cellular assembly and organization, DNA replication, recombination, and repair, as well as another three hub genes for survival (SLBP, NOVA1, and SPTBN1) related to cellular assembly and organization, were identified, and these genes were graphically presented using OncoLnc (Figure 3). Similarly, two additional hubs (networks 7 and 8) were observed for overall survival for genes ZNF582 and DEF6, as well as for genes ESD and MYO10, respectively (Figure 4).
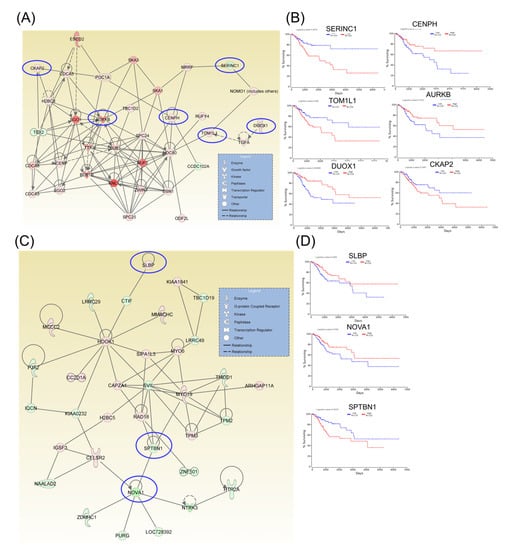
Figure 3.
A signaling network analysis using the IPA software for differentially expressed genes in CC. (A) Network 1: Cell Cycle; Cellular Assembly and Organization; and DNA Replication, Recombination, and Repair. (B) Genes predicting overall survival in network 1. (C) Network 2: Cellular Assembly and Organization. (D) Genes predicting overall survival in Network 2; red: overexpressed genes, green: downregulated genes, blue circles are for genes predicting overall survial rate in CC
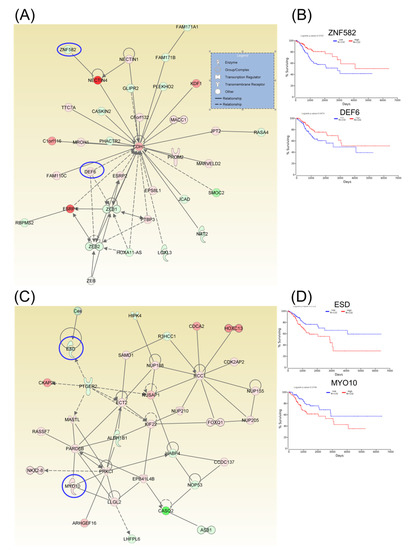
Figure 4.
A signaling network analysis using IPA software for differentially expressed genes in CC. (A) Network 7: Cell-To-Cell Signaling and Interaction, Cellular Assembly and Organization, as well as Cellular Development. (B) Genes predicting overall survival in network 7. (C) Network 8: Cell-To-Cell Signaling and Interaction, Cellular Assembly and Organization, as well as Cellular Development. (D) Genes predicting overall survival in Network 8; red: overexpressed genes, green: downregulated genes, blue circles are for genes predicting overall survial rate in CC
Furthermore, Kaplan–Meier plotter online tools were used to identify prognostic information related to gene networks of differentially expressed genes involved in TP53 signaling (Figure 5). This revealed that four genes (DNA-PK, E2F1, BCL2, and PCNAR) correlated well with overall survival, cell cycle control, and chromosomal replication, while two other genes (ORC1 and CDC45) correlated with overall survival. As for altered genes related to molecular mechanisms for cancer (Figure 6), a total of six genes from network 1 and three genes from network 2 were correlated with overall survival.
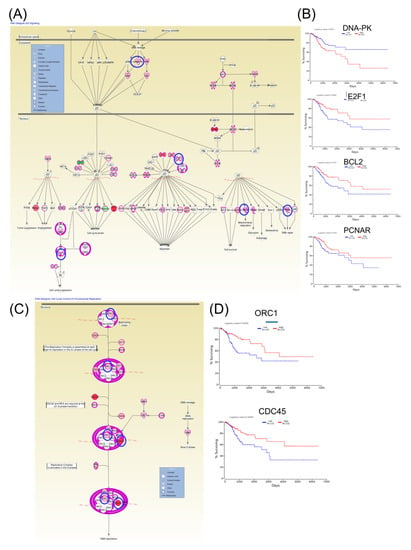
Figure 5.
An IPA gene interconnected network based on altered genes in CC. (A) IPA generated networks focusing on altered genes in CC related to TP53 signaling. (B) Genes predicting overall survival related to TP53 signaling. (C) IPA generated networks focusing on altered genes in CC related to cell cycle control of chromosomal replication (D) Genes predicting overall survival related to cell cycle control of chromosomal replication; overexpressed genes are displayed in red, while downregulated are in green; blue circles are for genes predictin overall survial rate in CC
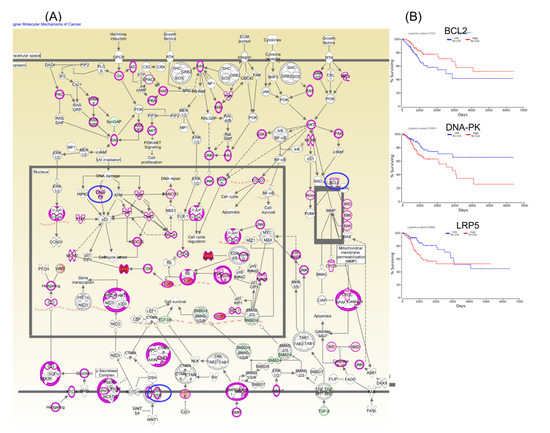
Figure 6.
A molecular mechanism for cancer pathogenesis based on altered genes in CC (Panel A), wherein the key genes BCL2, DNA-PK, and LRP5 predict overall survival rate (Panel B); red: overexpressed genes, green: downregulated genes, blue circles are for genes predicting overall survial rate in CC.
Taken together, these data have demonstrated that these candidate genes are undoubtedly associated with the prognosis of CC patients, whereas, Kaplan–Meier plotter online tools have identified a single gene, vascular endothelial growth factor (VEGF), involved in ILK signaling capable of predicting overall survival rate (Figure 7).
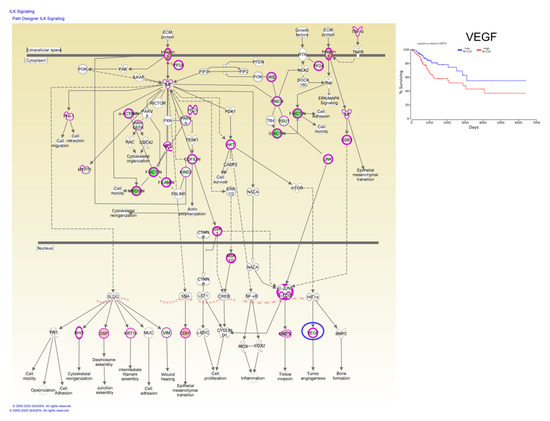
Figure 7.
ILK signaling based on altered genes in CC, wherein the VEGF gene predicts the overall survival rate; red: overexpressed genes, green: downregulated genes, blue circles are for genes predicting overall survial rate in CC.
2.4. mRNA-miRNA Interactions in CC.
The gene list with prognostic values in CC was used for assessing interactions with miRNA using miRtargetlink online software. This represented only the strong connection nodes. It was found that BCL2 and E2F1 genes were connected with key overexpressed altered transcripts (miR-205-5p, miR-17-5p, miR-21-5p, miR-34a-5p, and miR-20a-5p) (Figure 8). Furthermore, it was observed that the multifunctional miR-155 targeted both Nova1 and Myo10. However, none of these miRNAs were capable of predicting the overall survival rate in CC, although it was observed above that miRNAs were capable of predicting overall survival rate in CC (Figure 9). When performing analysis for interconnectedness of miRNAs predicting overall survival rate, it was found that these transcripts targeted the key genes BCL2, VEGFA, and KRAS (Figure 10).
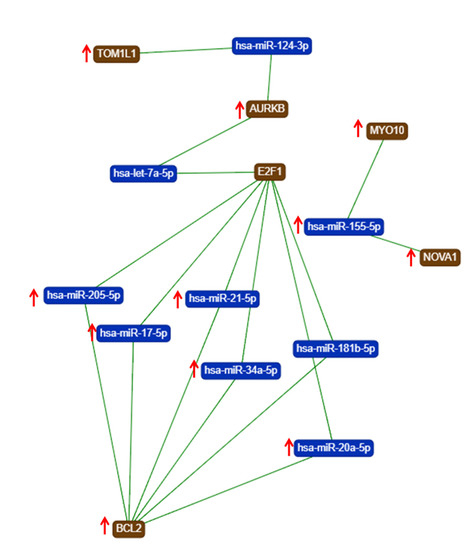
Figure 8.
An mRNA–miRNA iinteraction map for each of the hub genes with prognostic values for CC, generated using miRtargelink (Available online: https://ccb-web.cs.uni-saarland.de/mirtargetlink/). Only those strong interactions are herein presented (↑ overexpressed transcript).
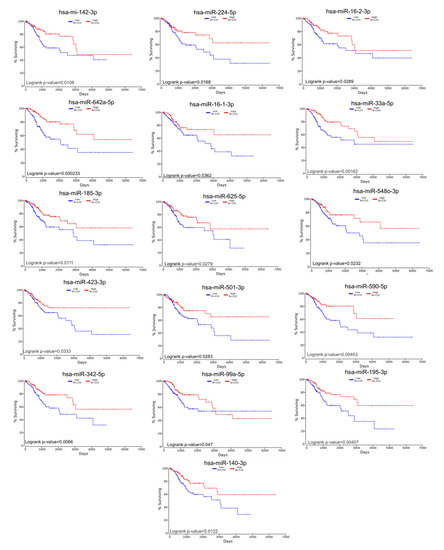
Figure 9.
Altered miRNAs in CC predicting overall survival rate.
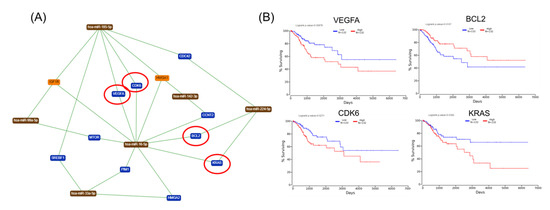
Figure 10.
mRNA-mRNA network interaction in CC. (A) An mRNA–miRNA interaction map based on miRNAs predicting overall survival in CC, generated using miRtargelink (Available online: https://ccb-web.cs.uni-saarland.de/mirtargetlink/). Only those strong interactions are herein presented; (B) network genes predicting overall survival rate in CC; red circles are for genes predicting overall survial rate in CC.
3. Discussion
Although earlier studies have reported on progress in elucidating the potential molecular mechanism of CC development [22,23], the fundamental knowledge of this mechanism remains problematic. As HPVs are DNA viruses with epithelial tropisms, HPV infection results in activation of the DNA damage repair mechanism [13]. It has been reported that activation of factors related to DNA damage correlates with CIN progression [24]. As of now, there is no targeted therapy; therefore, DNA damage-related genes may serve as novel biomarkers for predicting those CC patients who are more likely to have a poorer prognosis [10,25].
Previous studies have described an abnormal regulation of the cell cycle mechanism that plays a vital role in both tumorigenesis and progression, including that detected in CC [26]. In this study, a more complex alteration mechanism at both mRNA and miRNA is observed. This interconnected mechanism is capable of identifying some key hub genes with prognostic values in CC. This interconnection among the prognostic mRNA-miRNA is presented in Figure 10.
In recent years, DNA damage and repair have increased in interest, yielding opportunities for exploring the mechanistic basis underlying potential therapeutic vulnerabilities [12,27,28,29,30]. Therapeutic targeting of these repair constituents may be uniquely advantageous in CC, particularly to be most effective in combination with standard therapy. For example, it has been reported that overexpression of AURKB is correlated with poor clinical prognosis in CC patients, suggesting its interference with a vital function, and thus serving as a potential therapeutic target [31].
A distinct aspect of the initiation of tumorigenesis is the case of HPV negative CC patients. Indeed, in recent years a subset of HPV-negative CC has been highlighted, thus contradicting the widely accepted hypothesis that HPV infection is necessary for the onset of CC [32]. These cancers are more commonly non-squamous, and represent rare subtypes [32]. As the pathogenetic mechanism for tumorigenesis may differ in these forms, a broader and more comprehensive analysis should include a significantly higher numbers of patients since the current numbers of reported cases are rather limited. Therefore, it would be valuable to analyze potential triggers of carcinogenesis in non-HPV CC.
Although TOM1L1 regulates the oncoprotein ERBB2-driven cell invasion with metastatic phenotypes in breast cancer [33], there is no available information on the role of this gene in CC. Another gene predicting overall survival is DOXO1 (Dual oxidase 1), whose overexpression has been reported to promote favorable effects in CC patients via immune response activation [34]. In this study, we have observed that increased expression levels for this dual gene are related to a better prognostic.
Furthermore, the association of HPV infection with overexpression of both TP53 and BCL2 proteins in pre-malign lesion of the uterine cervix has been previously reported [35]. In contrast in this study, we have noted inhibition of BCL2 expression, whereas BCL2 overexpression is observed to be associated with better prognostic [36,37]. Furthermore, in this study, we have observed that high expression levels of BCL2 predict a better survival rate.
It is well known that cell cycle genes play important roles in cell proliferation, as well as in cell growth [24,25]. In this study, it is revealed that alteration of cell cycle signaling pathways has allowed for identification of important candidate genes related to cell cycle loss of control, thus promoting CC pathogenesis, as it has been previously reported for both CDC45 and ORC1 [38]. CDC45 plays a key role in late G1, as it is a key element of the DNA replication machinery; thus, it is deemed as a key element of the pathogenic network in CC [38]. Furthermore, DNA-PK, a dynamic enzyme involved in DNA double-stranded breaks repair pathway [39], has been proposed as a therapeutic target [40,41], while E2F1 plays critical roles in both cell cycle regulation and chromosome segregation. It has been reported that E2F1 may promote DNA replication and cancer cell proliferation. Furthermore, both viral E6 and E7 decrease NOVA1 expression, but only E7 increases expression of RNASEH2A in an E2F1-dependent manner [42]. Therefore, all these genes are worthy of further testing for their roles in HPV infection, including tumor initiation and CC progression [43]. In this study, we have observed that CDC45, DBA-PK, E2F1, and NOVA1 are expressed in CC patients, and therefore, they can be used as prognostic markers for CC.
In this study, CKAP2 has been observed to have an oncogenic function in CC development, based on TCGA data, and this is likely accomplished via its interference with FAK-ERK2 signaling [44]. Another gene detected in this study to be involved in CC pathogenesis is DUOX1. This gene plays a significant role in host mucosal immunity by producing hydrogen peroxide, thus serving as the first line of defense to HPV invasion, particularly in cervical cancer [34]. Yet another critical gene identified in this study is VEGF. The expression pattern of this gene in CC patients suggests that VEGF is mainly related to the response to neoadjuvant chemotherapy, and to an unfavorable prognosis [11,45]. Therefore, the proangiogenic factor VEFG is not only an important modulator of immune responses but it also interferes in key oncogenic signaling in CC, as angiogenesis is a key mechanism activated in CC [25].
Biologically significant major genes related to CC are summarized in Table 2 (all these genes can be included in further studies for validation as biomarkers). These should not be considered as individual genes, but as a part of the complex miRNA–mRNA interconnections, as can be observed in Figure 10. VEGFA, a proangiogenic factor, and BCL2, a key apoptosis regulator, should be considered as therapeutic targets.
Table 2.
Major functions of important prognostic genes related to CC.
4. Materials and Methods
4.1. TCGA Data Collection
RNAseq data and corresponding clinical data of 304 cervical squamous cell carcinoma (CESC) samples and three analogous organ-matched normal tissues samples were downloaded from The Cancer Genome Atlas (TCGA) database (http://firebrowse.org/). All clinical information for patients included in this study is presented in Table 3.
Table 3.
Clinical data for cervical squamous cell carcinoma (CESC) patients (TCGA).
4.2. Differentially Expressed Analysis and Survival Analysis
Identification of differentially expressed genes and miRNAs was performed using GeneSpring version 14.5 (Agilent Technologies, Santa Clara USA). Thresholds for both gene expression and miRNA analysis were as follows: fold-change (FC) ±2 and false discovery rate (FDR) ≤ 0.05.
4.3. OncoLnc
OncoLnc is a Kaplan–Meier plotter tool, accessible online, commonly used to link TCGA survival data to mRNA, miRNA, and lncRNA expression levels (http://www.oncolnc.org). The log rank value and hazard ratio (HR) with 95% confidence intervals and 50% percentile were computed, and presented along the plot.
4.4. Gene Enrichment Analysis
Screened altered genes in CC were submitted to the PANTHER online tool (http://www.pantherdb.org) for displaying Gene Ontology (GO) classifications of these genes based on molecular function, biological processes, and cellular function.
4.5. Gene Network analysis
A gene network analysis was performed using the Ingenuity Pathway Analysis (IPA) software (Ingenuity Systems, Redwood City, CA, USA). All altered genes in CC networks were algorithmically generated based on their connectivity, and scores were assigned. Each score was presented as a numerical value, based on the relevance of altered genes overlapping with the database and taking into account relevance of this network to the original list of target genes. A canonical pathways analysis identified the most significant pathways, from the IPA library of canonical pathways, which were most relevant based on the input dataset.
5. Conclusions
In this study, analysis of TCGA has revealed that patients with unfavorable prognostics are more likely to have DNA repair defects, and that in most cases, this is most likely due to presence of HPV. Furthermore, key genes with prognostic values involved in CC are identified. These genes could be potentially used in pursuing molecular diagnosis or treatment of CC. However, additional studies should be conducted to fully decipher the molecular mechanisms of these key genes involved in CC.
Author Contributions
Conceptualisation, M.D.-S., D.M., I.B.-N.; methodology, formal analysis and visualisation R.C., R.O., C.B., writing orginal draft- M.D.-S., D.M., C.B., A.I.; writing—review and editing, A.I., S.S.K. and I.B.-N. All authors have read and agreed to the published version of the manuscript.
Funding
Dudea-Simon received an internal grant for PhD student no. 1300/21/13/01/2017.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Luo, F.; Wen, Y.; Zhou, H.; Li, Z. Roles of long non-coding RNAs in cervical cancer. Life Sci. 2020, 256, 117981. [Google Scholar] [CrossRef]
- Han, C.; Zhao, F.; Wan, C.; He, Y.; Chen, Y. Associations between the expression of SCCA, MTA1, P16, Ki-67 and the infection of high-risk HPV in cervical lesions. Oncol. Lett. 2020, 20, 884–892. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- He, J.; Huang, B.; Zhang, K.; Liu, M.; Xu, T. Long non-coding RNA in cervical cancer: From biology to therapeutic opportunity. Biomed. Pharm. 2020, 127, 110209. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kim, Y.S.; Shin, J.W.; Osong, B.; Lee, S.H. Prediction scoring system based on clinicohematologic parameters for cervical cancer patients undergoing chemoradiation. Int. J. Gynecol. Cancer 2020. [CrossRef]
- Zhang, H.; Chen, R.; Shao, J. MicroRNA-96-5p Facilitates the Viability, Migration, and Invasion and Suppresses the Apoptosis of Cervical Cancer Cells byNegatively Modulating SFRP4. Technol. Cancer Res. Treat. 2020, 19. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-Y.; Shen, M.-R. Epithelial-mesenchymal transition in cervical carcinoma. Am. J. Transl. Res. 2012, 4, 1–13. [Google Scholar]
- Gulei, D.; Mehterov, N.; Ling, H.; Stanta, G.; Braicu, C.; Berindan-Neagoe, I. The “good-cop bad-cop” TGF-beta role in breast cancer modulated by non-coding RNAs. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1661–1675. [Google Scholar] [CrossRef]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef]
- Sample, K.M. DNA repair gene expression is associated with differential prognosis between HPV16 and HPV18 positive cervical cancer patients following radiation therapy. Sci. Rep. 2020, 10, 2774. [Google Scholar] [CrossRef]
- Zhu, P.; Ou, Y.; Dong, Y.; Xu, P.; Yuan, L. Expression of VEGF and HIF-1α in locally advanced cervical cancer: Potential biomarkers for predicting preoperative radiochemotherapy sensitivity and prognosis. Onco Targets 2016, 9, 3031–3037. [Google Scholar]
- Obasi, T.C.; Braicu, C.; Iacob, B.C.; Bodoki, E.; Jurj, A.; Raduly, L.; Oniga, I.; Berindan-Neagoe, I.; Oprean, R. Securidaca-saponins are natural inhibitors of AKT, MCL-1, and BCL2L1 in cervical cancer cells. Cancer Manag. Res. 2018, 10, 5709–5724. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Munger, K. Expression of the Long Noncoding RNA DINO in Human Papillomavirus-Positive Cervical Cancer Cells Reactivates the Dormant TP53 Tumor Suppressor through ATM/CHK2 Signaling. mBio 2020, 11. [Google Scholar] [CrossRef]
- Braicu, C.; Zimta, A.A.; Harangus, A.; Iurca, I.; Irimie, A.; Coza, O.; Berindan-Neagoe, I. The Function of Non-Coding RNAs in Lung Cancer Tumorigenesis. Cancers 2019, 11, 605. [Google Scholar] [CrossRef] [PubMed]
- Sonea, L.; Buse, M.; Gulei, D.; Onaciu, A.; Simon, I.; Braicu, C.; Berindan-Neagoe, I. Decoding the Emerging Patterns Exhibited in Non-coding RNAs Characteristic of Lung Cancer with Regard to their Clinical Significance. Curr. Genom. 2018, 19, 258–278. [Google Scholar] [CrossRef]
- Zimta, A.A.; Tigu, A.B.; Braicu, C.; Stefan, C.; Ionescu, C.; Berindan-Neagoe, I. An Emerging Class of Long Non-coding RNA With Oncogenic Role Arises From the snoRNA Host Genes. Front. Oncol. 2020, 10, 389. [Google Scholar] [CrossRef]
- Tomuleasa, C.; Braicu, C.; Irimie, A.; Craciun, L.; Berindan-Neagoe, I. Nanopharmacology in translational hematology and oncology. Int. J. Nanomed. 2014, 9, 3465–3479. [Google Scholar]
- Saftencu, M.; Braicu, C.; Cojocneanu, R.; Buse, M.; Irimie, A.; Piciu, D.; Berindan-Neagoe, I. Gene Expression Patterns Unveil New Insights in Papillary Thyroid Cancer. Medicina 2019, 55, 500. [Google Scholar] [CrossRef]
- Irimie, A.I.; Braicu, C.; Sonea, L.; Zimta, A.A.; Cojocneanu-Petric, R.; Tonchev, K.; Mehterov, N.; Diudea, D.; Buduru, S.; Berindan-Neagoe, I. A Looking-Glass of Non-coding RNAs in oral cancer. Int. J. Mol. Sci. 2017, 18, 2620. [Google Scholar] [CrossRef]
- Irimie, A.I.; Braicu, C.; Cojocneanu-Petric, R.; Berindan-Neagoe, I.; Campian, R.S. Novel technologies for oral squamous carcinoma biomarkers in diagnostics and prognostics. Acta Odontol. Scand. 2015, 73, 161–168. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, S.D.; Balakrishnan, V.; Oon, C.E.; Kaur, G. Key Molecular Events in Cervical Cancer Development. Medicina 2019, 55, 384. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Ye, M.; Zhou, J.; Wang, Z.P.; Zhu, X. Recent Advances on the Molecular Mechanism of Cervical Carcinogenesis Based on Systems Biology Technologies. Comput. Struct. Biotechnol. J. 2019, 17, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Spriggs, C.C.; Blanco, L.Z.; Maniar, K.P.; Laimins, L.A. Expression of HPV-induced DNA Damage Repair Factors Correlates with CIN Progression. Int. J. Gynecol. Pathol. 2019, 38, 1–10. [Google Scholar] [CrossRef]
- Tudoran, O.; Soritau, O.; Balacescu, O.; Balacescu, L.; Braicu, C.; Rus, M.; Gherman, C.; Virag, P.; Irimie, F.; Berindan-Neagoe, I. Early transcriptional pattern of angiogenesis induced by EGCG treatment in cervical tumour cells. J. Cell Mol. Med. 2012, 16, 520–530. [Google Scholar] [CrossRef]
- Zhao, Q.; Li, H.; Zhu, L.; Hu, S.; Xi, X.; Liu, Y.; Liu, J.; Zhong, T. Bioinformatics analysis shows that TOP2A functions as a key candidate gene in the progression of cervical cancer. Biomed. Rep. 2020, 13, 21. [Google Scholar] [CrossRef]
- Aldea, M.D.; Petrushev, B.; Soritau, O.; Tomuleasa, C.I.; Berindan-Neagoe, I.; Filip, A.G.; Chereches, G.; Cenariu, M.; Craciun, L.; Tatomir, C.; et al. Metformin plus sorafenib highly impacts temozolomide resistant glioblastoma stem-like cells. J. BUON 2014, 19, 502–511. [Google Scholar]
- Aldea, M.; Craciun, L.; Tomuleasa, C.; Berindan-Neagoe, I.; Kacso, G.; Florian, I.S.; Crivii, C. Repositioning metformin in cancer: Genetics, drug targets, and new ways of delivery. Tumor Biol. 2014, 35, 5101–5110. [Google Scholar] [CrossRef]
- Sugumaran, A.; Mathialagan, V. Colloidal Nanocarriers a Versatile Targeted Delivery System for Cervical Cancer. Curr. Pharm. Des. 2020. [Google Scholar] [CrossRef]
- Turgeon, M.-O.; Perry, N.J.S.; Poulogiannis, G. DNA Damage, Repair, and Cancer Metabolism. Front. Oncol. 2018, 8, 15. [Google Scholar] [CrossRef]
- Wei, J.; Wang, Y.; Shi, K.; Wang, Y. Identification of Core Prognosis-Related Candidate Genes in Cervical Cancer via Integrated Bioinformatical Analysis. Biomed. Res. Int. 2020, 2020, 8959210. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Carunchio, L.; Soveral, I.; Steenbergen, R.D.; Torné, A.; Martinez, S.; Fusté, P.; Pahisa, J.; Marimon, L.; Ordi, J.; del Pino, M. HPV-negative carcinoma of the uterine cervix: A distinct type of cervical cancer with poor prognosis. BJOG 2015, 122, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, C.; Collin, G.; Descamps, S.; Touaitahuata, H.; Simon, V.; Reymond, N.; Fernandez, L.; Milhiet, P.-E.; Georget, V.; Urbach, S.; et al. TOM1L1 drives membrane delivery of MT1-MMP to promote ERBB2-induced breast cancer cell invasion. Nat. Commun. 2016, 7, 10765. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.Y.; Kim, S.; Son, M.-J.; Kim, G.; Singh, P.; Kim, H.N.; Choi, H.-G.; Yoo, H.J.; Ko, Y.B.; Lee, B.S.; et al. Dual oxidase 1 and NADPH oxidase 2 exert favorable effects in cervical cancer patients by activating immune response. BMC Cancer 2019, 19, 1078. [Google Scholar] [CrossRef]
- Shukla, S.; Dass, J.; Pujani, M. p53 and bcl2 expression in malignant and premalignant lesions of uterine cervix and their correlation with human papilloma virus 16 and 18. South Asian J. Cancer 2014, 3, 48–53. [Google Scholar] [PubMed]
- Crawford, R.A.; Caldwell, C.; Iles, R.K.; Lowe, D.; Shepherd, J.H.; Chard, T. Prognostic significance of the bcl-2 apoptotic family of proteins in primary and recurrent cervical cancer. Br. J. Cancer 1998, 78, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Leisching, G.; Loos, B.; Botha, M.; Engelbrecht, A.-M. Bcl-2 confers survival in cisplatin treated cervical cancer cells: Circumventing cisplatin dose-dependent toxicity and resistance. J. Transl. Med. 2015, 13, 328. [Google Scholar] [CrossRef]
- Zhang, Y.-X.; Zhao, Y.-L. Pathogenic Network Analysis Predicts Candidate Genes for Cervical Cancer. Comput. Math. Methods Med. 2016. [Google Scholar] [CrossRef]
- Davis, A.J.; Chen, B.P.C.; Chen, D.J. DNA-PK: A dynamic enzyme in a versatile DSB repair pathway. DNA Repair 2014, 17, 21–29. [Google Scholar] [CrossRef]
- Mohiuddin, I.S.; Kang, M.H. DNA-PK as an Emerging Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 635. [Google Scholar] [CrossRef]
- Deshpande, R.A.; Myler, L.R.; Soniat, M.M.; Makharashvili, N.; Lee, L.; Lees-Miller, S.P.; Finkelstein, I.J.; Paull, T.T. DNA-dependent protein kinase promotes DNA end processing by MRN and CtIP. Sci. Adv. 2020, 6, eaay0922. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Liu, H.; Yang, Y.; Wang, X.; Liu, P.; Li, Y.; Meyers, C.; Banerjee, N.S.; Wang, H.-K.; Cam, M.; et al. Genome-Wide Profiling of Cervical RNA-Binding Proteins Identifies Human Papillomavirus Regulation of RNASEH2A Expression by Viral E7 and E2F1. mBio 2019, 10, e02687. [Google Scholar] [CrossRef] [PubMed]
- Narayan, G.; Murty, V.V. Integrative genomic approaches in cervical cancer: Implications for molecular pathogenesis. Future Oncol. 2010, 6, 1643–1652. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.-S.; Song, Y.; Hua, K.-Q.; Gao, S.-J. Involvement of FAK-ERK2 signaling pathway in CKAP2-induced proliferation and motility in cervical carcinoma cell lines. Sci. Rep. 2017, 7, 2117. [Google Scholar] [CrossRef]
- Choi, C.H.; Song, S.Y.; Choi, J.-J.; Ae Park, Y.; Kang, H.; Kim, T.-J.; Lee, J.-W.; Kim, B.-G.; Lee, J.-H.; Bae, D.-S. Prognostic significance of VEGF expression in patients with bulky cervical carcinoma undergoing neoadjuvant chemotherapy. BMC Cancer 2008, 8, 295. [Google Scholar] [CrossRef]
- Wu, W.; Wu, F.; Wang, Z.; Di, J.; Yang, J.; Gao, P.; Jiang, B.; Su, X. CENPH Inhibits Rapamycin Sensitivity by Regulating GOLPH3-dependent mTOR Signaling Pathway in Colorectal Cancer. J. Cancer 2017, 8, 2163–2172. [Google Scholar] [CrossRef]
- Zhang, M.; Zhao, L. CKAP2 Promotes Ovarian Cancer Proliferation and Tumorigenesis through the FAK-ERK Pathway. DNA Cell Biol. 2017, 36, 983–990. [Google Scholar] [CrossRef]
- Chen, X.; Xiong, D.; Ye, L.; Wang, K.; Huang, L.; Mei, S.; Wu, J.; Chen, S.; Lai, X.; Zheng, L.; et al. Up-regulated lncRNA XIST contributes to progression of cervical cancer via regulating miR-140-5p and ORC1. Cancer Cell Int. 2019, 19, 45. [Google Scholar] [CrossRef]
- Gaffney, D.K.; Haslam, D.; Tsodikov, A.; Hammond, E.; Seaman, J.; Holden, J.; Lee, R.J.; Zempolich, K.; Dodson, M. Epidermal growth factor receptor (EGFR) and vascular endothelial growth factor (VEGF) negatively affect overall survival in carcinoma of the cervix treated with radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2003, 56, 922–928. [Google Scholar] [CrossRef]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).