Review of Single-Cell RNA Sequencing in the Heart



Abstract
1. Introduction
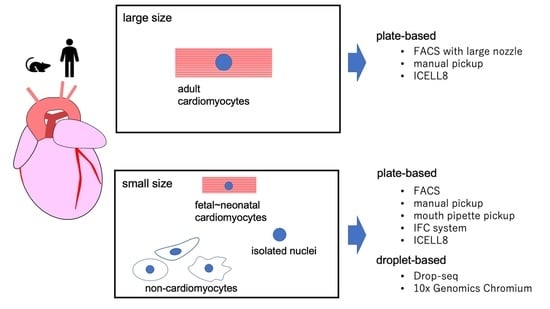
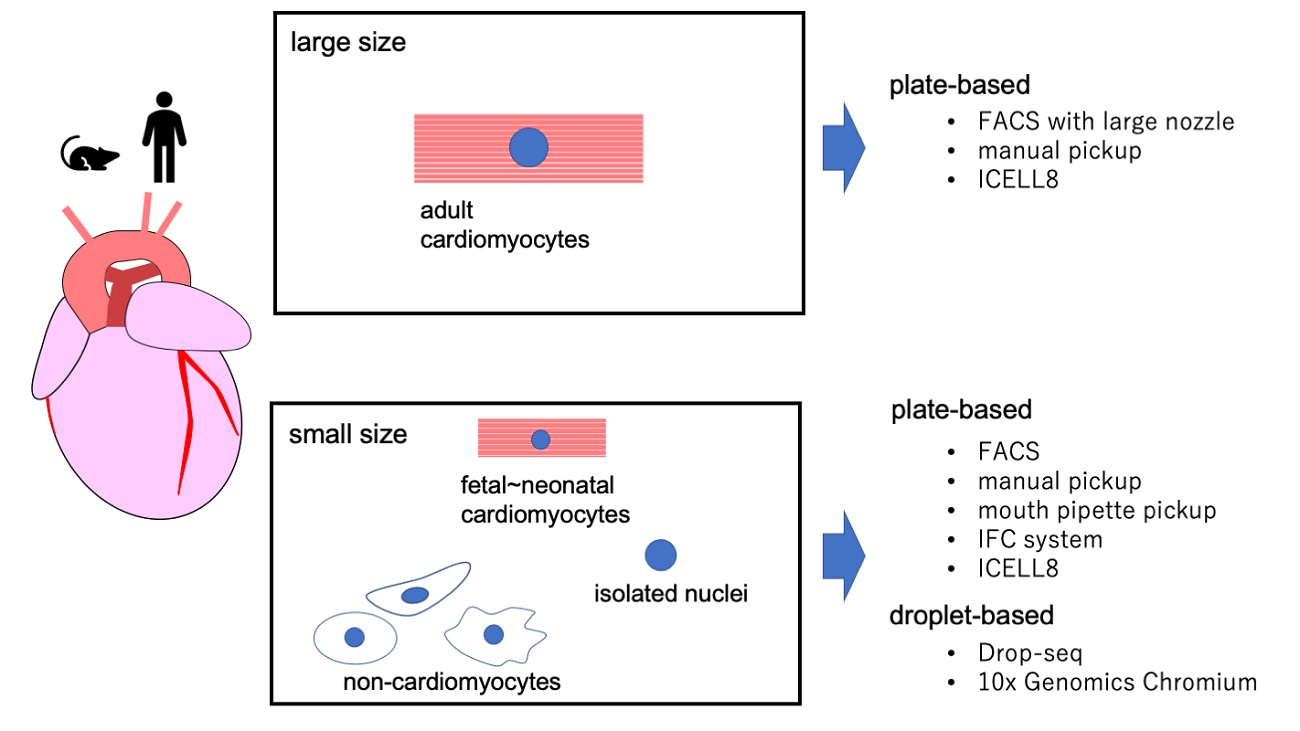
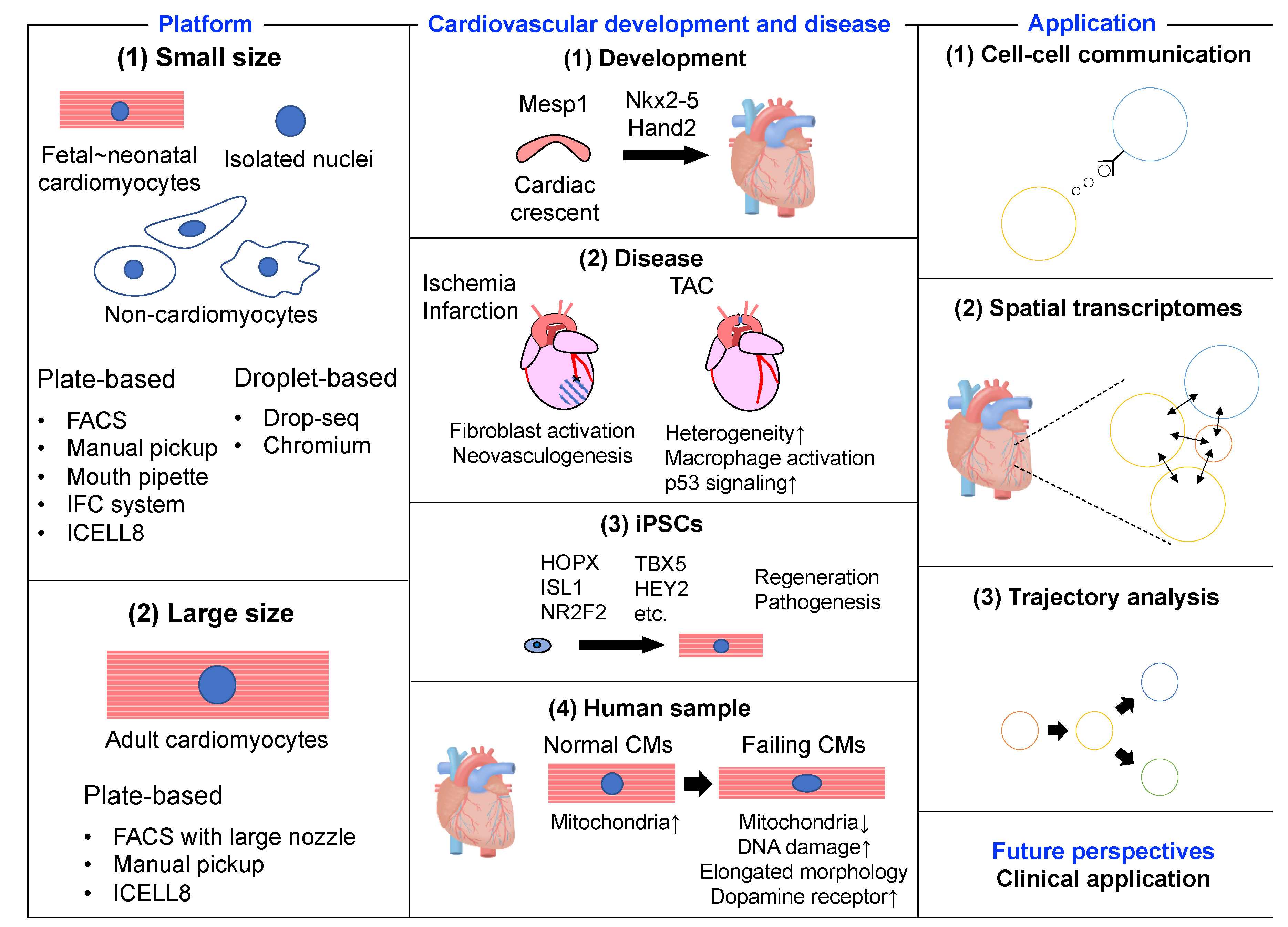
2. scRNA-seq Platforms
2.1. Platforms for Small Cells
2.2. Platforms for Large Cells
3. scRNA-seq and Cardiovascular Development and Disease
3.1. Murine Embryonic and Neonatal Heart
3.2. Murine Adult Heart
3.3. Human-Induced Pluripotent Stem Cells
3.4. Human Heart
4. scRNA-seq and Applications
4.1. Cell–Cell Interactions
4.2. Spatial Transcriptomes
4.3. Trajectory Analysis
5. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| scRNA-seq | single-cell RNA sequencing |
| CMs | cardiomyocytes |
| IFC | integrated fluidic circuit |
| FACS | fluorescence-activated cell sorting |
| snRNA-seq | single-nucleus RNA-seq |
| MI | myocardial infarction |
| LAD | the left anterior descending artery |
| I/R | ischemia/reperfusion |
| TAC | transverse aortic constriction |
| hiPSCs | human induced pluripotent stem cells |
| seqFISH | sequential fluorescence in situ hybridization |
| CODEX | Co-Detection by Indexing |
References
- Lafzi, A.; Moutinho, C.; Picelli, S.; Heyn, H. Tutorial: Guidelines for the experimental design of single-cell RNA sequencing studies. Nat. Protoc. 2018, 13, 2742–2757. [Google Scholar] [CrossRef] [PubMed]
- Kornfeld, O.S.; Hwang, S.; Disatnik, M.H.; Chen, C.H.; Qvit, N.; Mochly-Rosen, D. Mitochondrial reactive oxygen species at the heart of the matter: New therapeutic approaches for cardiovascular diseases. Circ. Res. 2015, 116, 1783–1799. [Google Scholar] [CrossRef]
- Komuro, I. Molecular mechanism of mechanical stress-induced cardiac hypertrophy. Jpn. Heart J. 2000, 41, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.F.; van den Engh, G. Flow cytometry and cell sorting. Adv. Biochem. Eng. Biotechnol. 2007, 106, 19–39. [Google Scholar]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef]
- Gladka, M.M.; Molenaar, B.; de Ruiter, H.; van der Elst, S.; Tsui, H.; Versteeg, D.; Lacraz, G.P.A.; Huibers, M.M.H.; van Oudenaarden, A.; van Rooij, E. Single-Cell Sequencing of the Healthy and Diseased Heart Reveals Cytoskeleton-Associated Protein 4 as a New Modulator of Fibroblasts Activation. Circulation 2018, 138, 166–180. [Google Scholar] [CrossRef]
- DeLaughter, D.M.; Bick, A.G.; Wakimoto, H.; McKean, D.; Gorham, J.M.; Kathiriya, I.S.; Hinson, J.T.; Homsy, J.; Gray, J.; Pu, W.; et al. Single-Cell Resolution of Temporal Gene Expression during Heart Development. Dev. Cell 2016, 39, 480–490. [Google Scholar] [CrossRef]
- Li, G.; Xu, A.; Sim, S.; Priest, J.R.; Tian, X.; Khan, T.; Quertermous, T.; Zhou, B.; Tsao, P.S.; Quake, S.R.; et al. Transcriptomic Profiling Maps Anatomically Patterned Subpopulations among Single Embryonic Cardiac Cells. Dev. Cell 2016, 39, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Luo, Y.; Yue, Y.; Zhang, J.; Ai, S.; Li, X.; Wang, X.; Zhang, Y.L.; Wei, Y.; Li, H.H.; et al. Single-Cell Transcriptomics Reveals Chemotaxis-Mediated Intraorgan Crosstalk During Cardiogenesis. Circ. Res. 2019, 125, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Lescroart, F.; Wang, X.; Lin, X.; Swedlund, B.; Gargouri, S.; Sànchez-Dànes, A.; Moignard, V.; Dubois, C.; Paulissen, C.; Kinston, S.; et al. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science 2018, 359, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Hu, Y.; Fan, X.; Wu, X.; Mao, Y.; Hu, B.; Guo, H.; Wen, L.; Tang, F. Single-cell RNA-seq analysis unveils a prevalent epithelial/mesenchymal hybrid state during mouse organogenesis. Genome Biol. 2018, 19, 31. [Google Scholar] [CrossRef]
- De Soysa, T.Y.; Ranade, S.S.; Okawa, S.; Ravichandran, S.; Huang, Y.; Salunga, H.T.; Schricker, A.; del Sol, A.; Gifford, C.A.; Srivastava, D. Single-cell analysis of cardiogenesis reveals basis for organ-level developmental defects. Nature 2019, 572, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Zhang, J.; Liu, Y.; Fan, X.; Ai, S.; Luo, Y.; Li, X.; Jin, H.; Luo, S.; Zheng, H.; et al. The lncRNA Hand2os1/Uph locus orchestrates heart development through regulation of precise expression of Hand2. Development 2019, 146, dev176198. [Google Scholar] [CrossRef]
- Hu, P.; Liu, J.; Zhao, J.; Wilkins, B.J.; Lupino, K.; Wu, H.; Pei, L. Single-nucleus transcriptomic survey of cell diversity and functional maturation in postnatal mammalian hearts. Genes Dev. 2018, 32, 1344–1357. [Google Scholar] [CrossRef]
- Goodyer, W.R.; Beyersdorf, B.M.; Paik, D.T.; Tian, L.; Li, G.; Buikema, J.W.; Chirikian, O.; Choi, S.; Venkatraman, S.; Adams, E.L.; et al. Transcriptomic Profiling of the Developing Cardiac Conduction System at Single-Cell Resolution. Circ. Res. 2019, 125, 379–397. [Google Scholar] [CrossRef]
- Hulin, A.; Hortells, L.; Gomez-Stallons, M.V.; O’Donnell, A.; Chetal, K.; Adam, M.; Lancellotti, P.; Oury, C.; Potter, S.S.; Salomonis, N.; et al. Maturation of heart valve cell populations during postnatal remodeling. Development 2019, 146, dev173047. [Google Scholar] [CrossRef]
- Liu, X.; Chen, W.; Li, W.; Li, Y.; Priest, J.R.; Zhou, B.; Wang, J.; Zhou, Z. Single-Cell RNA-Seq of the Developing Cardiac Outflow Tract Reveals Convergent Development of the Vascular Smooth Muscle Cells. Cell Rep. 2019, 28, 1346–1361. [Google Scholar] [CrossRef]
- Yekelchyk, M.; Guenther, S.; Preussner, J.; Braun, T. Mono- and multi-nucleated ventricular cardiomyocytes constitute a transcriptionally homogenous cell population. Basic Res. Cardiol. 2019, 114, 36. [Google Scholar] [CrossRef]
- Kretzschmar, K.; Post, Y.; Bannier-Hélaouët, M.; Mattiotti, A.; Drost, J.; Basak, O.; Li, V.S.W.; van den Born, M.; Gunst, Q.D.; Versteeg, D.K.; et al. Profiling proliferative cells and their progeny in damaged murine hearts. Proc. Natl. Acad. Sci. USA 2018, 115, E12245–E12254. [Google Scholar] [CrossRef]
- Ren, Z.; Yu, P.; Li, D.; Li, Z.; Liao, Y.; Wang, Y.; Zhou, B.; Wang, L. Single-Cell Reconstruction of Progression Trajectory Reveals Intervention Principles in Pathological Cardiac Hypertrophy. Circulation 2020, 141, 1704–1719. [Google Scholar] [CrossRef]
- Wang, Y.; Yao, F.; Wang, L.; Li, Z.; Ren, Z.; Li, D.; Zhang, M.; Han, L.; Wang, S.Q.; Zhou, B.; et al. Single-cell analysis of murine fibroblasts identifies neonatal to adult switching that regulates cardiomyocyte maturation. Nat. Commun. 2020, 11, 2585. [Google Scholar] [CrossRef]
- Nomura, S.; Satoh, M.; Fujita, T.; Higo, T.; Sumida, T.; Ko, T.; Yamaguchi, T.; Tobita, T.; Naito, A.T.; Ito, M.; et al. Cardiomyocyte gene programs encoding morphological and functional signatures in cardiac hypertrophy and failure. Nat. Commun. 2018, 9, 4435. [Google Scholar] [CrossRef]
- Vidal, R.; Wagner, J.U.G.; Braeuning, C.; Fischer, C.; Patrick, R.; Tombor, L.; Muhly-Reinholz, M.; John, D.; Kliem, M.; Conrad, T.; et al. Transcriptional heterogeneity of fibroblasts is a hallmark of the aging heart. JCI Insight 2019, 4, e131092. [Google Scholar] [CrossRef]
- Zhang, Y.; Gago-Lopez, N.; Li, N.; Zhang, Z.; Alver, N.; Liu, Y.; Martinson, A.M.; Mehri, A.; MacLellan, W.R. Single-cell imaging and transcriptomic analyses of endogenous cardiomyocyte dedifferentiation and cycling. Cell Discov. 2019, 5, 30. [Google Scholar] [CrossRef]
- See, K.; Tan, W.L.W.; Lim, E.H.; Tiang, Z.; Lee, L.T.; Li, P.Y.Q.; Luu, T.D.A.; Ackers-Johnson, M.; Foo, R.S. Single cardiomyocyte nuclear transcriptomes reveal a lincRNA-regulated de-differentiation and cell cycle stress-response in vivo. Nat. Commun. 2017, 8, 225. [Google Scholar] [CrossRef]
- Linscheid, N.; Logantha, S.J.R.J.; Poulsen, P.C.; Zhang, S.; Schrölkamp, M.; Egerod, K.L.; Thompson, J.J.; Kitmitto, A.; Galli, G.; Humphries, M.J.; et al. Quantitative proteomics and single-nucleus transcriptomics of the sinus node elucidates the foundation of cardiac pacemaking. Nat. Commun. 2019, 10, 2889. [Google Scholar] [CrossRef]
- Farbehi, N.; Patrick, R.; Dorison, A.; Xaymardan, M.; Janbandhu, V.; Wystub-Lis, K.; Ho, J.W.; Nordon, R.E.; Harvey, R.P. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife 2019, 8, e43882. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Solomonidis, E.G.; Meloni, M.; Taylor, R.S.; Duffin, R.; Dobie, R.; Magalhaes, M.S.; Henderson, B.E.P.; Louwe, P.A.; D’Amico, G.; et al. Single-cell transcriptome analyses reveal novel targets modulating cardiac neovascularization by resident endothelial cells following myocardial infarction. Eur. Heart J. 2019, 40, 2507–2520. [Google Scholar] [CrossRef] [PubMed]
- Friedman, C.E.; Nguyen, Q.; Lukowski, S.W.; Helfer, A.; Chiu, H.S.; Miklas, J.; Levy, S.; Suo, S.; Han, J.J.; Osteil, P.; et al. Single-Cell Transcriptomic Analysis of Cardiac Differentiation from Human PSCs Reveals HOPX-Dependent Cardiomyocyte Maturation. Cell Stem Cell 2018, 23, 586–598. [Google Scholar] [CrossRef]
- Churko, J.M.; Garg, P.; Treutlein, B.; Venkatasubramanian, M.; Wu, H.; Lee, J.; Wessells, Q.N.; Chen, S.Y.; Chen, W.Y.; Chetal, K.; et al. Defining human cardiac transcription factor hierarchies using integrated single-cell heterogeneity analysis. Nat. Commun. 2018, 9, 4906. [Google Scholar] [CrossRef]
- Sahara, M.; Santoro, F.; Sohlmér, J.; Zhou, C.; Witman, N.; Leung, C.Y.; Mononen, M.; Bylund, K.; Gruber, P.; Chien, K.R. Population and Single-Cell Analysis of Human Cardiogenesis Reveals Unique LGR5 Ventricular Progenitors in Embryonic Outflow Tract. Dev. Cell 2019, 48, 475–490. [Google Scholar] [CrossRef]
- Selewa, A.; Dohn, R.; Eckart, H.; Lozano, S.; Xie, B.; Gauchat, E.; Elorbany, R.; Rhodes, K.; Burnett, J.; Gilad, Y.; et al. Systematic Comparison of High-throughput Single-Cell and Single-Nucleus Transcriptomes during Cardiomyocyte Differentiation. Sci. Rep. 2020, 10, 1535. [Google Scholar] [CrossRef]
- Gambardella, L.; McManus, S.A.; Moignard, V.; Sebukhan, D.; Delaune, A.; Andrews, S.; Bernard, W.G.; Morrison, M.A.; Riley, P.R.; Göttgens, B.; et al. BNC1 regulates cell heterogeneity in human pluripotent stem cell-derived epicardium. Development 2019, 146, dev174441. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, Z.; Welch, J.D.; Gao, X.; Wang, L.; Garbutt, T.; Keepers, B.; Ma, H.; Prins, J.F.; Shen, W.; et al. Single-Cell Transcriptomic Analyses of Cell Fate Transitions during Human Cardiac Reprogramming. Cell Stem Cell. 2019, 25, 149–164. [Google Scholar] [CrossRef]
- Cui, Y.; Zheng, Y.; Liu, X.; Yan, L.; Fan, X.; Yong, J.; Hu, Y.; Dong, J.; Li, Q.; Wu, X.; et al. Single-Cell Transcriptome Analysis Maps the Developmental Track of the Human Heart. Cell Rep. 2019, 26, 1934–1950. [Google Scholar] [CrossRef]
- Asp, M.; Giacomello, S.; Larsson, L.; Wu, C.; Fürth, D.; Qian, X.; Wärdell, E.; Custodio, J.; Reimegård, J.; Salmén, F.; et al. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell 2019, 179, 1647–1660. [Google Scholar] [CrossRef]
- Suryawanshi, H.; Clancy, R.; Morozov, P.; Halushka, M.K.; Buyon, J.P.; Tuschl, T. Cell atlas of the fetal human heart and implications for autoimmune-mediated congenital heart block. Cardiovasc. Res. 2020, 116, 1446–1457. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yu, P.; Zhou, B.; Song, J.; Li, Z.; Zhang, M.; Guo, G.; Wang, Y.; Chen, X.; Han, L.; et al. Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat. Cell Biol. 2020, 22, 108–119. [Google Scholar] [CrossRef]
- Katoh, M.; Nomura, S.; Yamada, S.; Aburatani, H.; Issei, K. Single-Cardiomyocyte RNA sequencing to dissect the molecular pathophysiology of the heart. Methods Mol. Biol. 2020, in press. [Google Scholar]
- Aoi, T. 10th anniversary of iPS cells: The challenges that lie ahead. J. Biochem. 2016, 160, 121–129. [Google Scholar] [CrossRef]
- Cohen, M.; Giladi, A.; Gorki, A.D.; Solodkin, D.G.; Zada, M.; Hladik, A.; Miklosi, A.; Salame, T.M.; Halpern, K.B.; David, E.; et al. Lung Single-Cell Signaling Interaction Map Reveals Basophil Role in Macrophage Imprinting. Cell 2018, 175, 1031–1044. [Google Scholar] [CrossRef]
- Vento-Tormo, R.; Efremova, M.; Botting, R.A.; Turco, M.Y.; Vento-Tormo, M.; Meyer, K.B.; Park, J.E.; Stephenson, E.; Polański, K.; Goncalves, A.; et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 2018, 563, 347–353. [Google Scholar] [CrossRef]
- Ma, S.; Sun, S.; Geng, L.; Song, M.; Wang, W.; Ye, Y.; Ji, Q.; Zou, Z.; Wang, S.; He, X.; et al. Caloric Restriction Reprograms the Single-Cell Transcriptional Landscape of Rattus Norvegicus Aging. Cell 2020, 180, 984–1001. [Google Scholar] [CrossRef]
- Marinković, G.; Koenis, D.S.; de Camp, L.; Jablonowski, R.; Graber, N.; de Waard, V.; de Vries, C.J.; Goncalves, I.; Nilsson, J.; Jovinge, S.; et al. S100A9 Links Inflammation and Repair in Myocardial Infarction. Circ. Res. 2020, 127, 664–676. [Google Scholar] [CrossRef]
- Mueller, K.; Thiel, F.; Beutner, F.; Teren, A.; Frisch, S.; Ballarini, T.; Möller, H.E.; Ihle, K.; Thiery, J.; Schuler, G.A.; et al. Brain Damage With Heart Failure: Cardiac Biomarker Alterations and Gray Matter Decline. Circ. Res. 2020, 126, 750–764. [Google Scholar] [CrossRef]
- Lubeck, E.; Cai, L. Single-cell systems biology by super-resolution imaging and combinatorial labeling. Nat. Methods 2012, 9, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.L.; Lawson, M.; Zhu, Q.; Dries, R.; Koulena, N.; Takei, Y.; Yun, J.; Cronin, C.; Karp, C.; Yuan, G.C.L. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature 2019, 568, 235–239. [Google Scholar] [CrossRef]
- Goltsev, Y.; Samusik, N.; Kennedy-Darling, J.; Bhate, S.; Hale, M.; Vazquez, G.S.; Nolan, G.P. Deep Profiling of Mouse Splenic Architecture with CODEX Multiplexed Imaging. Cell 2018, 174, 968–981. [Google Scholar] [CrossRef]
- Jackson, H.W.; Fischer, J.R.; Zanotelli, V.R.T.; Ali, H.R.; Mechera, R.; Soysal, S.D.; Moch, H.; Muenst, S.; Varga, Z.; Weber, W.P.; et al. The single-cell pathology landscape of breast cancer. Nature 2020, 578, 615–620. [Google Scholar] [CrossRef]
- Satoh, M.; Nomura, S.; Harada, M.; Yamaguchi, T.; Ko, T.; Sumida, T.; Toko, H.; Naito, A.T.; Takeda, N.; Tobita, T.; et al. High-throughput single-molecule RNA imaging analysis reveals heterogeneous responses of cardiomyocytes to hemodynamic overload. J. Mol. Cell Cardiol. 2019, 128, 77–89. [Google Scholar] [CrossRef]
- Luecken, M.D.; Theis, F.J. Current best practices in single-cell RNA-seq analysis: A tutorial. Mol. Syst. Biol. 2019, 15, e8746. [Google Scholar] [CrossRef]
- Qiu, X.; Mao, Q.; Tang, Y.; Wang, L.; Chawla, R.; Pliner, H.A.; Trapnell, C. Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 2017, 14, 979–982. [Google Scholar] [CrossRef] [PubMed]
- Bendall, S.C.; Davis, K.L.; el Amir, A.D.; Tadmor, M.D.; Simonds, E.F.; Chen, T.J.; Shenfeld, D.K.; Nolan, G.P.; Pe’er, D. Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell 2014, 157, 714–725. [Google Scholar] [CrossRef]
- Street, K.; Risso, D.; Fletcher, R.B.; Das, D.; Ngai, J.; Yosef, N.; Purdom, E.; Dudoit, S. Slingshot: Cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genom. 2018, 19, 477. [Google Scholar] [CrossRef]
- Wolf, F.A.; Hamey, F.K.; Plass, M.; Solana, J.; Dahlin, J.S.; Göttgens, B.; Rajewsky, N.; Simon, L.; Theis, F.J. PAGA: Graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 2019, 20, 59. [Google Scholar] [CrossRef]
- La Manno, G.; Soldatov, R.; Zeisel, A.; Braun, E.; Hochgerner, H.; Petukhov, V.; Lidschreiber, K.; Kastriti, M.E.; Lönnerberg, P.; Furlan, A.; et al. RNA velocity of single cells. Nature 2018, 560, 494–498. [Google Scholar] [CrossRef]
- Pei, W.; Feyerabend, T.B.; Rössler, J.; Wang, X.; Postrach, D.; Busch, K.; Rode, I.; Klapproth, K.; Dietlein, N.; Quedenau, C.; et al. Polylox barcoding reveals haematopoietic stem cell fates realized in vivo. Nature 2017, 548, 456–460. [Google Scholar] [CrossRef]
- Biddy, B.A.; Kong, W.; Kamimoto, K.; Guo, C.; Waye, S.E.; Sun, T.; Morris, S.A. Single-cell mapping of lineage and identity in direct reprogramming. Nature 2018, 564, 219–224. [Google Scholar] [CrossRef]
- Weinreb, C.; Rodriguez-Fraticelli, A.; Camargo, F.D.; Klein, A.M. Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science 2020, 367, eaaw3381. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.M.; Smith, Z.D.; Grosswendt, S.; Kretzmer, H.; Norman, T.M.; Adamson, B.; Jost, M.; Quinn, J.J.; Yang, D.; Jones, M.G.; et al. Molecular recording of mammalian embryogenesis. Nature 2019, 570, 77–82. [Google Scholar] [CrossRef]
- Kalhor, R.; Kalhor, K.; Mejia, L.; Leeper, K.; Graveline, A.; Mali, P.; Church, G.M. Developmental barcoding of whole mouse via homing CRISPR. Science 2018, 361, eaat9804. [Google Scholar] [CrossRef] [PubMed]
- Alemany, A.; Florescu, M.; Baron, C.S.; Peterson-Maduro, J.; van Oudenaarden, A. Whole-organism clone tracing using single-cell sequencing. Nature 2018, 556, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Lázár, E.; Sadek, H.A.; Bergmann, O. Cardiomyocyte renewal in the human heart: Insights from the fall-out. Eur. Heart J. 2017, 38, 2333–2342. [Google Scholar] [CrossRef]
- Zhou, B.; Pu, W.T. Epicardial epithelial-to-mesenchymal transition in injured heart. J. Cell. Mol. Med. 2011, 15, 2781–2783. [Google Scholar] [CrossRef]
- Velmeshev, D.; Schirmer, L.; Jung, D.; Haeussler, M.; Perez, Y.; Mayer, S.; Bhaduri, A.; Goyal, N.; Rowitch, D.H.; Kriegstein, A.R. Single-cell genomics identifies cell type-specific molecular changes in autism. Science 2019, 364, 685–689. [Google Scholar] [CrossRef]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; de Rubeis, S.; An, J.Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584. [Google Scholar] [CrossRef]
- Kim, D.; Kobayashi, T.; Voisin, B.; Jo, J.H.; Sakamoto, K.; Jin, S.P.; Kelly, M.; Pasieka, H.B.; Naff, J.L.; Meyerle, J.H.; et al. Targeted therapy guided by single-cell transcriptomic analysis in drug-induced hypersensitivity syndrome: A case report. Nat. Med. 2020, 26, 236–243. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Sumida, T.S.; Nomura, S.; Satoh, M.; Higo, T.; Ito, M.; Ko, T.; Fujita, K.; Sweet, M.E.; Sanbe, A.; et al. Cardiac dopamine D1 receptor triggers ventricular arrhythmia in chronic heart failure. Nat. Commun. 2020, 11, 4364. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Species | Vivo / Vitro | Age | Cells or Nuclei | Model | Device | Number of Cells for Analysis | Findings | Ref. |
|---|---|---|---|---|---|---|---|---|
| mouse | vivo | fetus | cells from whole heart | healthy development | IFC system | 2233 cells | chamber-specific genes in the embryonic mouse heart | [8] |
| fetus | cells from whole heart | healthy development and Hand2 knock out | Chromium | 73,926 cells | Hand2 is a specifier of outflow tract cells | [12] | ||
| fetus | cells from whole heart | healthy development and Hand2os1 knock out | Chromium | 3600 cells | lncRNA Hand2os1/Uph regulates Hand2 | [13] | ||
| fetus | cells from whole heart and other 7 organs | healthy development | mouth pipette | 1819 cells | mutual interactions between epithelial and mesenchymal cells | [11] | ||
| fetus | Mesp1 positive or null cardiac progenitors | healthy development and Mesp1 knock out | FACS | 598cells | Mesp1 is required for the exit from the pluripotent state | [10] | ||
| fetus | Nkx2.5 or Isl1 expressing cardiac progenitors | healthy development | FACS | 1231 cells | Cxcr2 regulates chemotaxis during development | [9] | ||
| fetus | cells from cardiac conduction system | healthy development | Chromium | 22,462 cells | transcriptional profiles of cardiac conduction system | [15] | ||
| fetus | cells from cardiac outflow tract | healthy development | Chromium | 55,611cells | cellular transitions in cardiac outflow tract | [17] | ||
| fetus ~ neonate | cells from whole heart | healthy development | IFC system | >1200 cells | temporal and chamber-specific markers during development | [7] | ||
| neonate | nuclei from whole heart | healthy development and pediatric mitochondrial cardiomyopathy | Chromium | 15,083 nuclei | heterogeneity of various cell types | [14] | ||
| neonate | cells from left ventricles | healthy development | ICELL8 | 4231 cells | transcriptomes of mono- or multi-nucleated cardiomyocytes are highly similar | [18] | ||
| neonate ~ juvenile | cells from aortic valve and mitral valve | healthy development | Drop-seq | 2840 cells | Interstitial cell subpopulations undergo changes in gene expression during development | [16] | ||
| neonate, adult | cells from ventricles | healthy, I/R and MI | FACS | 1939 cells | Cycling CMs are few adult mouse | [19] | ||
| mouse | vivo | adult | cells from whole heart | healthy condition and ischemia reperfusion | FACS | 935 cells | Ckap4 is a modulator of fibroblasts activation | [6] |
| adult | cells from whole heart | healthy and TAC | ICELL8 | 11,492 cells | Macrophage activation is a key factor of hypertrophy | [20] | ||
| adult | cells from left ventricles | healthy development | ICELL8 | 2497 cells | Fibroblast regulates CM maturation | [21] | ||
| adult | CMs from ventricles | healthy and TAC | ICELL 8 | <1015 cells | heterogeneity among CMs after TAC | [18] | ||
| adult | CMs from whole heart | healthy and TAC | manual pickup | 396 cells | p53 induces molecular and morphological remodeling | [22] | ||
| adult | nuclei from whole heart | healthy aging | Chromium | 27,808 nuclei | heterogeneity of fibroblasts with aging | [23] | ||
| adult | nuclei from ventricles | healthy and MI | Chromium | 31,542 nuclei | dedifferentiation in cycling CMs after MI | [24] | ||
| adult | nuclei of CMs from left ventricles | healthy and TAC | IFC system | 243 nuclei | lincRNA regulates dedifferentiation and cell cycle genes | [25] | ||
| adult | cells from sinus node | healthy pacemaking | Chromium | 5357 nuclei | Membrane clock underpins pacemaking | [26] | ||
| adult | non-CMs | healthy and MI | Chromium | 13,331 cells | transcriptome changes of non-CMs after MI | [27] | ||
| adult | fibroblasts | healthy and MI | IFC system | 104 cells | transcriptome changes of fibroblast after MI | [27] | ||
| adult | endothelial cells | healthy and MI | Chromium | 28,598 cells | Plvap regulates endothelial proliferation | [28] | ||
| neonate, adult | neonatal CMs, and neonatal and adult fibroblasts | healthy development | ICELL8 | 1580 cells | Fibroblast regulates CM maturation | [21] | ||
| human | vitro | hiPSC-CMs | differentiation | Chromium | 43,168 cells | Hopx is a key regulator of CM maturation | [29] | |
| hiPSC-CMs | differentiation | Chromium | 10,376 cells | ISL1, NR2F2, TBX5, HEY2, or HOPX are makers of hiPSC-CMs | [30] | |||
| hiPSC-CMs | differentiation | IFC system | 43 cells | ISL1, NR2F2, TBX5, HEY2, or HOPX are makers of hiPSC-CMs | [30] | |||
| CMs derived from embryonic stem cells | differentiation | FACS | 366 cells | LGR5 is a marker of cardiac progenitors in embryonic outflow tract | [31] | |||
| hiPSC-CMs | differentiation | Drop-seq | 23,554 cells | the comparison with DroNc-seq | [32] | |||
| nuclei of hiPSC-CMs | differentiation | DroNc-seq | 24,318 nuclei | Inclusion of reads from intronic regions increases the sensitivity | [32] | |||
| epicardium hiPSC-CMs | differentiation | FACS | 232 cells | BNC1 regulates cell heterogeneity | [33] | |||
| CMs reprogrammed from human fibroblasts | differentiation | IFC system | 704 cells | cell fate transitions during reprogramming | [34] | |||
| human | vivo | fetus | cells from free wall | healthy development | mouth pipette | 3842 cells | Atrial and ventricular CMs acquires distinct features early in heart development | [35] |
| fetus | cells from whole heart | healthy development | Chromium | 4026 cells | cell atlas of the developing human heart | [36] | ||
| fetus | cells from whole heart | healthy development | FACS | 458 cells | LGR5 is a marker of cardiac progenitors in embryonic outflow tract | [31] | ||
| fetus | cells from whole heart | healthy and autoimmune-associated congenital heart block | Chromium | 17,747 cells | heterogeneous interferon responses in congenital heart block heart | [37] | ||
| adult | cells from whole heart | healthy, HF and functional recovery from HF after treatment with LVAD | ICELL8 | 21,422 cells | CM contractility and metabolism are prominent aspects that are correlated with changes in heart function. | [38] | ||
| adult | CMs from left ventricles | healthy and DCM | manual pickup | 411 cells | heterogeneity in DCM CMs | [22] | ||
| adult | nuclei from whole heart | healthy | DroNuc-seq | 1491 nuclei | the usefulness of DroNc-seq in adult human CMs | [32] | ||
| adult | nuclei from CMs | healthy and DCM | IFC system | 116 nuclei | lincRNA regulates dedifferentiation and cell cycle genes | [25] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamada, S.; Nomura, S. Review of Single-Cell RNA Sequencing in the Heart. Int. J. Mol. Sci. 2020, 21, 8345. https://doi.org/10.3390/ijms21218345
Yamada S, Nomura S. Review of Single-Cell RNA Sequencing in the Heart. International Journal of Molecular Sciences. 2020; 21(21):8345. https://doi.org/10.3390/ijms21218345
Chicago/Turabian StyleYamada, Shintaro, and Seitaro Nomura. 2020. "Review of Single-Cell RNA Sequencing in the Heart" International Journal of Molecular Sciences 21, no. 21: 8345. https://doi.org/10.3390/ijms21218345
APA StyleYamada, S., & Nomura, S. (2020). Review of Single-Cell RNA Sequencing in the Heart. International Journal of Molecular Sciences, 21(21), 8345. https://doi.org/10.3390/ijms21218345