Tn5 Transposase Applied in Genomics Research

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
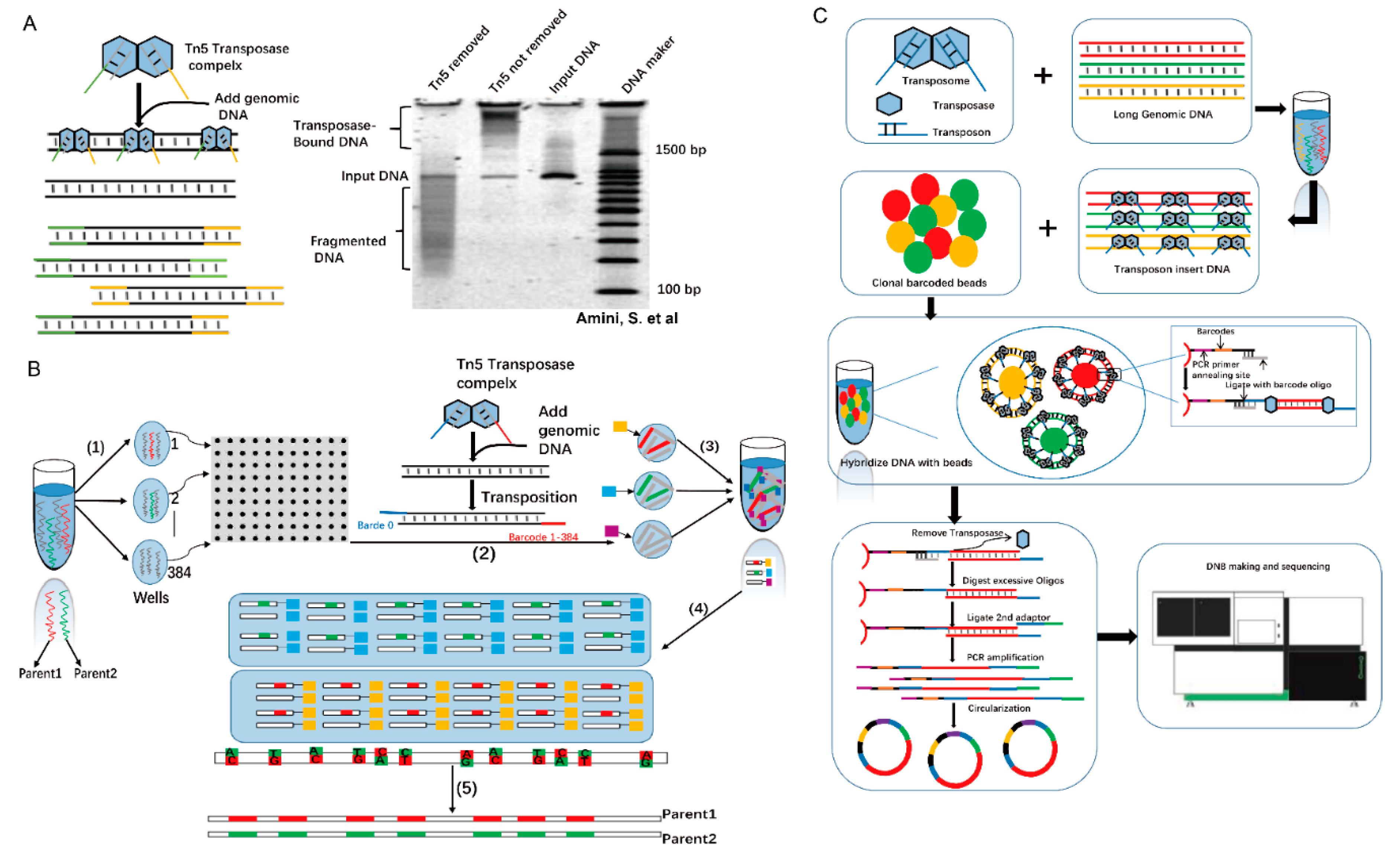{kind=link}
{kind=link}
1. Tn5 Transposition Mechanism
2. Application of Tn5 to 3D Genome Structures
3. Application of Tn5 to Study Genomic Variation
4. Tn5 Application in Open Chromatin
5. The Application of Tn5 in Long Fragments
6. The Application of Tn5 in Epigenetics
7. Challenges and Bottlenecks of Tn5-Related Techniques
8. Conclusions and Perspectives
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Carnell, S.C.; Bowen, A.; Morgan, E.; Maskell, D.J.; Wallis, T.S.; Stevens, M.P. Role in virulence and protective efficacy in pigs of Salmonella enterica serovar Typhimurium secreted components identified by signature-tagged mutagenesis. Microbiology 2007, 153, 1940–1952. [Google Scholar] [CrossRef] [PubMed]
- Blot, M.; Heitman, J.; Arber, W. Tn5-mediated bleomycin resistance in Escherichia coli requires the expression of host genes. Mol. Microbiol. 1993, 8, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Gordenin, D.A.; Malkova, A.L.; Peterzen, A.; Kulikov, V.N.; Pavlov, Y.I.; Perkins, E.; Resnick, M.A. Transposon Tn5 excision in yeast: Influence of DNA polymerases alpha, delta, and epsilon and repair genes. Proc. Natl. Acad. Sci. USA 1992, 89, 3785–3789. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, C.R.; Wardle, S.J.; Haniford, D.B. The global bacterial regulator H-NS promotes transpososome formation and transposition in the Tn5 system. Nucleic Acids Res. 2009, 37, 309–321. [Google Scholar] [CrossRef]
- Zhou, M.; Bhasin, A.; Reznikoff, W.S. Molecular genetic analysis of transposase-end DNA sequence recognition: Cooperativity of three adjacent base-pairs in specific interaction with a mutant Tn5 transposase. J. Mol. Biol. 1998, 276, 913–925. [Google Scholar] [CrossRef]
- Naumann, T.A.; Reznikoff, W.S. Tn5 transposase with an altered specificity for transposon ends. J. Bacteriol. 2002, 184, 233–240. [Google Scholar] [CrossRef]
- Kennedy, A.K.; Guhathakurta, A.; Kleckner, N.; Haniford, D.B. Tn10 transposition via a DNA hairpin intermediate. Cell 1998, 95, 125–134. [Google Scholar] [CrossRef]
- Gradman, R.J.; Ptacin, J.L.; Bhasin, A.; Reznikoff, W.S.; Goryshin, I.Y. A bifunctional DNA binding region in Tn5 transposase. Mol. Microbiol. 2008, 67, 528–540. [Google Scholar] [CrossRef]
- Twining, S.S.; Goryshin, I.Y.; Bhasin, A.; Reznikoff, W.S. Functional characterization of arginine 30, lysine 40, and arginine 62 in Tn5 transposase. J. Biol. Chem. 2001, 276, 23135–23143. [Google Scholar] [CrossRef]
- Davies, D.R.; Goryshin, I.Y.; Reznikoff, W.S.; Rayment, I. Three-dimensional structure of the Tn5 synaptic complex transposition intermediate. Science 2000, 289, 77–85. [Google Scholar] [CrossRef]
- Weinreich, M.D.; Mahnke-Braam, L.; Reznikoff, W.S. A functional analysis of the Tn5 transposase. Identification of domains required for DNA binding and multimerization. J. Mol. Biol. 1994, 241, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A. Alternative mechanisms for tn5 transposition. PLoS Genet. 2009, 5, e1000619. [Google Scholar] [CrossRef]
- Johnson, R.C.; Reznikoff, W.S. DNA sequences at the ends of transposon Tn5 required for transposition. Nature 1983, 304, 280–282. [Google Scholar] [CrossRef]
- Hennig, B.P.; Velten, L.; Racke, I.; Tu, C.S.; Thoms, M.; Rybin, V.; Besir, H.; Remans, K.; Steinmetz, L.M. Large-Scale Low-Cost NGS Library Preparation Using a Robust Tn5 Purification and Tagmentation Protocol. G3 2018, 8, 79–89. [Google Scholar] [CrossRef]
- Cogne, B.; Snyder, R.; Lindenbaum, P.; Dupont, J.B.; Redon, R.; Moullier, P.; Leger, A. NGS library preparation may generate artifactual integration sites of AAV vectors. Nat. Med. 2014, 20, 577–578. [Google Scholar] [CrossRef]
- Herron, P.R.; Hughes, G.; Chandra, G.; Fielding, S.; Dyson, P.J. Transposon Express, a software application to report the identity of insertions obtained by comprehensive transposon mutagenesis of sequenced genomes: Analysis of the preference for in vitro Tn5 transposition into GC-rich DNA. Nucleic Acids Res. 2004, 32, e113. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Xing, D.; Chang, C.H.; Li, H.; Xie, X.S. Three-dimensional genome structures of single diploid human cells. Science 2018, 361, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Arimura, Y.; Kujirai, T.; Harada, A.; Maehara, K.; Nogami, J.; Ohkawa, Y.; Kurumizaka, H. Biochemical analysis of nucleosome targeting by Tn5 transposase. Open Biol. 2019, 9, 190116. [Google Scholar] [CrossRef] [PubMed]
- Adey, A.; Shendure, J. Ultra-low-input, tagmentation-based whole-genome bisulfite sequencing. Genome Res. 2012, 22, 1139–1143. [Google Scholar] [CrossRef]
- Arlt, M.F.; Ozdemir, A.C.; Birkeland, S.R.; Lyons, R.H., Jr.; Glover, T.W.; Wilson, T.E. Comparison of constitutional and replication stress-induced genome structural variation by SNP array and mate-pair sequencing. Genetics 2011, 187, 675–683. [Google Scholar] [CrossRef][Green Version]
- Chen, X.; Shen, Y.; Draper, W.; Buenrostro, J.D.; Litzenburger, U.; Cho, S.W.; Satpathy, A.T.; Carter, A.C.; Ghosh, R.P.; East-Seletsky, A.; et al. ATAC-see reveals the accessible genome by transposase-mediated imaging and sequencing. Nat. Methods 2016, 13, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Xing, D.; Tan, L.; Li, H.; Zhou, G.; Huang, L.; Xie, X.S. Single-cell whole-genome analyses by Linear Amplification via Transposon Insertion (LIANTI). Science 2017, 356, 189–194. [Google Scholar] [CrossRef]
- Corces, M.R.; Granja, J.M.; Shams, S.; Louie, B.H.; Seoane, J.A.; Zhou, W.; Silva, T.C.; Groeneveld, C.; Wong, C.K.; Cho, S.W.; et al. The chromatin accessibility landscape of primary human cancers. Science 2018, 362. [Google Scholar] [CrossRef]
- Cremer, T.; Cremer, C. Chromosome territories, nuclear architecture and gene regulation in mammalian cells. Nat. Rev. Genet. 2001, 2, 292–301. [Google Scholar] [CrossRef]
- Dekker, J.; Rippe, K.; Dekker, M.; Kleckner, N. Capturing chromosome conformation. Science 2002, 295, 1306–1311. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Van Berkum, N.L.; Lieberman-Aiden, E.; Williams, L.; Imakaev, M.; Gnirke, A.; Mirny, L.A.; Dekker, J.; Lander, E.S. Hi-C: A method to study the three-dimensional architecture of genomes. J. Vis. Exp. 2010. [Google Scholar] [CrossRef]
- Huang, J.; Jiang, Y.; Zheng, H.; Ji, X. BAT Hi-C maps global chromatin interactions in an efficient and economical way. Methods 2020, 170, 38–47. [Google Scholar] [CrossRef]
- Wu, J.; Dai, W.; Wu, L.; Wang, J. SALP, a new single-stranded DNA library preparation method especially useful for the high-throughput characterization of chromatin openness states. BMC Genom. 2018, 19, 143. [Google Scholar] [CrossRef]
- Amini, S.; Pushkarev, D.; Christiansen, L.; Kostem, E.; Royce, T.; Turk, C.; Pignatelli, N.; Adey, A.; Kitzman, J.O.; Vijayan, K.; et al. Haplotype-resolved whole-genome sequencing by contiguity-preserving transposition and combinatorial indexing. Nat. Genet. 2014, 46, 1343–1349. [Google Scholar] [CrossRef]
- Chen, S.; Lake, B.B.; Zhang, K. High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat. Biotechnol. 2019, 37, 1452–1457. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, R.D. Chromatin structure: A repeating unit of histones and DNA. Science 1974, 184, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, R.D.; Lorch, Y. Chromatin structure and transcription. Annu. Rev. Cell Biol. 1992, 8, 563–587. [Google Scholar] [CrossRef]
- Mellor, J. The dynamics of chromatin remodeling at promoters. Mol. Cell 2005, 19, 147–157. [Google Scholar] [CrossRef]
- Boyle, A.P.; Davis, S.; Shulha, H.P.; Meltzer, P.; Margulies, E.H.; Weng, Z.; Furey, T.S.; Crawford, G.E. High-resolution mapping and characterization of open chromatin across the genome. Cell 2008, 132, 311–322. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- Song, L.; Crawford, G.E. DNase-seq: A high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb. Protoc. 2010, 2010, pdb prot5384. [Google Scholar] [CrossRef]
- Anderson, S. Shotgun DNA sequencing using cloned DNase I-generated fragments. Nucleic Acids Res. 1981, 9, 3015–3027. [Google Scholar] [CrossRef] [PubMed]
- Waki, H.; Nakamura, M.; Yamauchi, T.; Wakabayashi, K.; Yu, J.; Hirose-Yotsuya, L.; Take, K.; Sun, W.; Iwabu, M.; Okada-Iwabu, M.; et al. Global mapping of cell type-specific open chromatin by FAIRE-seq reveals the regulatory role of the NFI family in adipocyte differentiation. PLoS Genet. 2011, 7, e1002311. [Google Scholar] [CrossRef]
- Bianco, S.; Rodrigue, S.; Murphy, B.D.; Gevry, N. Global Mapping of Open Chromatin Regulatory Elements by Formaldehyde-Assisted Isolation of Regulatory Elements Followed by Sequencing (FAIRE-seq). Methods Mol. Biol. 2015, 1334, 261–272. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Litzenburger, U.M.; Ruff, D.; Gonzales, M.L.; Snyder, M.P.; Chang, H.Y.; Greenleaf, W.J. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 2015, 523, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Sos, B.C.; Fung, H.L.; Gao, D.R.; Osothprarop, T.F.; Kia, A.; He, M.M.; Zhang, K. Characterization of chromatin accessibility with a transposome hypersensitive sites sequencing (THS-seq) assay. Genome Biol. 2016, 17, 20. [Google Scholar] [CrossRef]
- Ponnaluri, V.K.C.; Zhang, G.; Esteve, P.O.; Spracklin, G.; Sian, S.; Xu, S.Y.; Benoukraf, T.; Pradhan, S. NicE-seq: High resolution open chromatin profiling. Genome Biol. 2017, 18, 122. [Google Scholar] [CrossRef] [PubMed]
- Wang, O.; Chin, R.; Cheng, X.; Wu, M.K.Y.; Mao, Q.; Tang, J.; Sun, Y.; Anderson, E.; Lam, H.K.; Chen, D.; et al. Efficient and unique cobarcoding of second-generation sequencing reads from long DNA molecules enabling cost-effective and accurate sequencing, haplotyping, and de novo assembly. Genome Res. 2019, 29, 798–808. [Google Scholar] [CrossRef]
- Peters, B.A.; Kermani, B.G.; Sparks, A.B.; Alferov, O.; Hong, P.; Alexeev, A.; Jiang, Y.; Dahl, F.; Tang, Y.T.; Haas, J.; et al. Accurate whole-genome sequencing and haplotyping from 10 to 20 human cells. Nature 2012, 487, 190–195. [Google Scholar] [CrossRef]
- Chen, H.; Yao, J.; Fu, Y.; Pang, Y.; Wang, J.; Huang, Y. Tagmentation on Microbeads: Restore Long-Range DNA Sequence Information Using Next Generation Sequencing with Library Prepared by Surface-Immobilized Transposomes. ACS Appl. Mater. Interfaces 2018, 10, 11539–11545. [Google Scholar] [CrossRef]
- Van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The Third Revolution in Sequencing Technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef]
- Paulsen, M.; Ferguson-Smith, A.C. DNA methylation in genomic imprinting, development, and disease. J. Pathol. 2001, 195, 97–110. [Google Scholar] [CrossRef]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef]
- Barnett, K.R.; Decato, B.E.; Scott, T.J.; Hansen, T.J.; Chen, B.; Attalla, J.; Smith, A.D.; Hodges, E. ATAC-Me Captures Prolonged DNA Methylation of Dynamic Chromatin Accessibility Loci during Cell Fate Transitions. Mol. Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Schmidl, C.; Rendeiro, A.F.; Sheffield, N.C.; Bock, C. ChIPmentation: Fast, robust, low-input ChIP-seq for histones and transcription factors. Nat. Methods 2015, 12, 963–965. [Google Scholar] [CrossRef] [PubMed]
- Skene, P.J.; Henikoff, J.G.; Henikoff, S. Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc. 2018, 13, 1006–1019. [Google Scholar] [CrossRef]
- Kaya-Okur, H.S.; Wu, S.J.; Codomo, C.A.; Pledger, E.S.; Bryson, T.D.; Henikoff, J.G.; Ahmad, K.; Henikoff, S. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 2019, 10, 1930. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Maehara, K.; Handa, T.; Arimura, Y.; Nogami, J.; Hayashi-Takanaka, Y.; Shirahige, K.; Kurumizaka, H.; Kimura, H.; Ohkawa, Y. A chromatin integration labelling method enables epigenomic profiling with lower input. Nat. Cell Biol. 2019, 21, 287–296. [Google Scholar] [CrossRef]
- Handa, T.; Harada, A.; Maehara, K.; Sato, S.; Nakao, M.; Goto, N.; Kurumizaka, H.; Ohkawa, Y.; Kimura, H. Chromatin integration labeling for mapping DNA-binding proteins and modifications with low input. Nat. Protoc. 2020, 15, 3334–3360. [Google Scholar] [CrossRef]
- Wang, Q.; Xiong, H.; Ai, S.; Yu, X.; Liu, Y.; Zhang, J.; He, A. CoBATCH for High-Throughput Single-Cell Epigenomic Profiling. Mol. Cell 2019, 76, 206–216. [Google Scholar] [CrossRef]
- Ai, S.; Xiong, H.; Li, C.C.; Luo, Y.; Shi, Q.; Liu, Y.; Yu, X.; Li, C.; He, A. Profiling chromatin states using single-cell itChIP-seq. Nat. Cell Biol. 2019, 21, 1164–1172. [Google Scholar] [CrossRef]
- McCommas, S.A.; Syvanen, M. Temporal control of transposition in Tn5. J. Bacteriol. 1988, 170, 889–894. [Google Scholar] [CrossRef]
- Suganuma, R.; Pelczar, P.; Spetz, J.F.; Hohn, B.; Yanagimachi, R.; Moisyadi, S. Tn5 transposase-mediated mouse transgenesis. Biol. Reprod. 2005, 73, 1157–1163. [Google Scholar] [CrossRef]
- Cole, C.; Byrne, A.; Beaudin, A.E.; Forsberg, E.C.; Vollmers, C. Tn5Prime, a Tn5 based 5′ capture method for single cell RNA-seq. Nucleic Acids Res. 2018, 46, e62. [Google Scholar] [CrossRef] [PubMed]
- Gertz, J.; Varley, K.E.; Davis, N.S.; Baas, B.J.; Goryshin, I.Y.; Vaidyanathan, R.; Kuersten, S.; Myers, R.M. Transposase mediated construction of RNA-seq libraries. Genome Res. 2012, 22, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Fu, Y.; Sun, Y.; Li, J.; Liu, L.; Yao, J.; Wang, G.; Wu, Y.; Lao, K.; Lee, R.W.; et al. RNA sequencing by direct tagmentation of RNA/DNA hybrids. Proc. Natl. Acad. Sci. USA 2020, 117, 2886–2893. [Google Scholar] [CrossRef]
- Rubin, A.J.; Parker, K.R.; Satpathy, A.T.; Qi, Y.; Wu, B.; Ong, A.J.; Mumbach, M.R.; Ji, A.L.; Kim, D.S.; Cho, S.W.; et al. Coupled Single-Cell CRISPR Screening and Epigenomic Profiling Reveals Causal Gene Regulatory Networks. Cell 2019, 176, 361–376.e17. [Google Scholar] [CrossRef] [PubMed]
- Dan, L.; Liu, L.; Sun, Y.; Song, J.; Yin, Q.; Zhang, G.; Qi, F.; Hu, Z.; Yang, Z.; Zhou, Z.; et al. The phosphatase PAC1 acts as a T cell suppressor and attenuates host antitumor immunity. Nat. Immunol. 2020, 21, 287–297. [Google Scholar] [CrossRef]
- LaRock, D.L.; Chaudhary, A.; Miller, S.I. Salmonellae interactions with host processes. Nat. Rev. Microbiol. 2015, 13, 191–205. [Google Scholar] [CrossRef]
- Panagi, I.; Jennings, E.; Zeng, J.; Gunster, R.A.; Stones, C.D.; Mak, H.; Jin, E.; Stapels, D.A.C.; Subari, N.Z.; Pham, T.H.M.; et al. Salmonella Effector SteE Converts the Mammalian Serine/Threonine Kinase GSK3 into a Tyrosine Kinase to Direct Macrophage Polarization. Cell Host Microbe 2020, 27, 41–53.e6. [Google Scholar] [CrossRef]
- Song, X.; Guo, J.; Ma, W.X.; Ji, Z.Y.; Zou, L.F.; Chen, G.Y.; Zou, H.S. Identification of seven novel virulence genes from Xanthomonas citri subsp. citri by Tn5-based random mutagenesis. J. Microbiol. 2015, 53, 330–336. [Google Scholar] [CrossRef]
- Davenis, S.V.; Markov, A.P.; Golubev, A.V.; Smirnov, G.B. The effect of mutations in recB and recC genes on the precise excision of Tn5 from pNM1 plasmid genome. Mol. Gen. Mikrobiol. Virusol. 1988, 9, 14–17. [Google Scholar]
- Buchan, B.W.; McLendon, M.K.; Jones, B.D. Identification of differentially regulated francisella tularensis genes by use of a newly developed Tn5-based transposon delivery system. Appl. Environ. Microbiol. 2008, 74, 2637–2645. [Google Scholar] [CrossRef]
- Zhang, W.; Qu, J.; Liu, G.H.; Belmonte, J.C.I. The ageing epigenome and its rejuvenation. Nat. Rev. Mol. Cell Biol. 2020, 21, 137–150. [Google Scholar] [CrossRef]
- Dossin, F.; Pinheiro, I.; Zylicz, J.J.; Roensch, J.; Collombet, S.; Le Saux, A.; Chelmicki, T.; Attia, M.; Kapoor, V.; Zhan, Y.; et al. SPEN integrates transcriptional and epigenetic control of X-inactivation. Nature 2020, 578, 455–460. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, N.; Jin, K.; Bai, Y.; Fu, H.; Liu, L.; Liu, B. Tn5 Transposase Applied in Genomics Research. Int. J. Mol. Sci. 2020, 21, 8329. https://doi.org/10.3390/ijms21218329
Li N, Jin K, Bai Y, Fu H, Liu L, Liu B. Tn5 Transposase Applied in Genomics Research. International Journal of Molecular Sciences. 2020; 21(21):8329. https://doi.org/10.3390/ijms21218329
Chicago/Turabian StyleLi, Niannian, Kairang Jin, Yanmin Bai, Haifeng Fu, Lin Liu, and Bin Liu. 2020. "Tn5 Transposase Applied in Genomics Research" International Journal of Molecular Sciences 21, no. 21: 8329. https://doi.org/10.3390/ijms21218329
APA StyleLi, N., Jin, K., Bai, Y., Fu, H., Liu, L., & Liu, B. (2020). Tn5 Transposase Applied in Genomics Research. International Journal of Molecular Sciences, 21(21), 8329. https://doi.org/10.3390/ijms21218329