Molecular Mechanisms Underlying Muscle Wasting in Huntington’s Disease


Abstract
1. Introduction
2. Loss of Body Weight and Body Mass Index
3. Physiology of HD Skeletal Muscle
4. Morphology of HD Skeletal Muscle and Protein Aggregates
5. Does Mutant Huntingtin Affect Differentiation of Skeletal Muscle Cells?
6. Activation of Cellular Stress Response
6.1. Protein Synthesis and Degradation
6.2. Inflammation
6.3. Apoptosis
7. Mitochondrial Dysfunction in HD Skeletal Muscle
7.1. Deficits of the Energy Metabolism
7.2. Aberrations of the Mitochondrial Morphology
7.3. Impairment of the Mitochondrial Electron Transport Chain
7.4. Role of PGC1-α in the Mitochondrial Anomalies and Abnormal Distribution of Fiber Types
8. Muscle Fiber Type Transition
9. Defects of Motoneurons
10. Therapies for HD Treatment
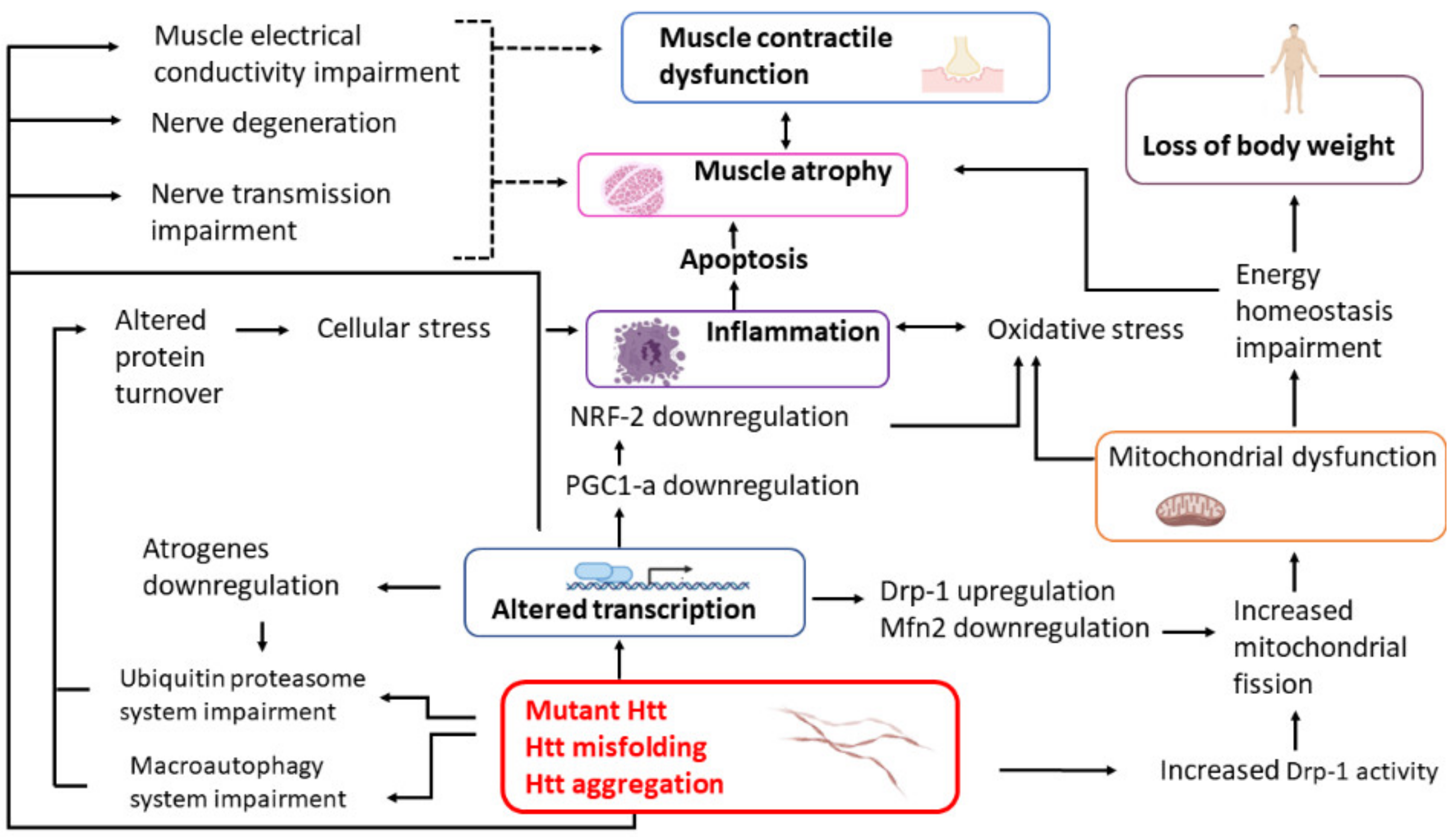
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Li, W.; Serpell, L.C.; Carter, W.J.; Rubinsztein, D.C.; Huntington, J.A. Expression and characterization of full-length human huntingtin, an elongated HEAT repeat protein. J. Biol. Chem. 2006, 281, 15916–15922. [Google Scholar] [CrossRef] [PubMed]
- Tartari, M.; Gissi, C.; Lo Sardo, V.; Zuccato, C.; Picardi, E.; Pesole, G.; Cattaneo, E. Phylogenetic comparison of huntingtin homologues reveals the appearance of a primitive PolyQ in sea urchin. Mol. Biol. Evol. 2008, 25, 330–338. [Google Scholar] [CrossRef]
- Duyao, M.P.; Auerbach, A.B.; Ryan, A.; Persichetti, F.; Barnes, G.T.; McNeil, S.M.; Ge, P.; Vonsattel, J.P.; Gusella, J.F.; Joyner, A.L.; et al. Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science 1995, 269, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Nasir, J.; Floresco, S.B.; O’Kusky, J.R.; Diewert, V.M.; Richman, J.M.; Zeisler, J.; Borowski, A.; Marth, J.D.; Phillips, A.G.; Hayden, M.R. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell 1995, 81, 811–823. [Google Scholar] [CrossRef]
- Zeitlin, S.; Liu, J.P.; Chapman, D.L.; Papaioannou, V.E.; Efstratiadis, A. Increased apoptosis and early embryonic lethality in mice nullizygous for the huntington’s disease gene homologue. Nat. Genet. 1995, 11, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Shirasaki, D.I.; Greiner, E.R.; Al-Ramahi, I.; Gray, M.; Boontheung, P.; Geschwind, D.H.; Botas, J.; Coppola, G.; Horvath, S.; Loo, J.A.; et al. Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron 2012, 75, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Cantle, J.P.; Chatzopoulou, D.; Wang, N.; Gao, F.; Al-Ramahi, I.; Lu, X.H.; Ramos, E.M.; El-Zein, K.; Zhao, Y.; et al. Integrated genomics and proteomics define huntingtin cag length-dependent networks in mice. Nat. Neurosci. 2016, 19, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Saudou, F.; Humbert, S. The biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.E.; Barnes, G.; Srinidhi, J.; Duyao, M.P.; Ambrose, C.M.; Myers, R.H.; Gray, J.; Conneally, P.M.; Young, A.; Penney, J.; et al. Gametic but not somatic instability of CAG repeat length in Huntington’s disease. J. Med. Genet. 1993, 30, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Gusella, J.F.; MacDonald, M.E. Molecular genetics: Unmasking polyglutamine triggers in neurodegenerative disease. Nat. Rev. Neurosci. 2000, 1, 109–115. [Google Scholar] [CrossRef]
- Orr, H.T.; Zoghbi, H.Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007, 30, 575–621. [Google Scholar] [CrossRef]
- Kremer, B.; Goldberg, P.; Andrew, S.E.; Theilmann, J.; Telenius, H.; Zeisler, J.; Squitieri, F.; Lin, B.; Bassett, A.; Almqvist, E.; et al. A worldwide study of the Huntington’s disease mutation: The sensitivity and specificity of measuring CAG repeats. N. Engl. J. Med. 1994, 330, 1401–1406. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Aylward, E.H.; Wild, E.J.; Langbehn, D.R.; Long, J.D.; Warner, J.H.; Scahill, R.I.; Leavitt, B.R.; Stout, J.C.; Paulsen, J.S.; et al. Huntington disease: Natural history, biomarkers and prospects for therapeutics. Nat. Rev. Neurol. 2014, 10, 204–216. [Google Scholar] [CrossRef]
- Lieberman, A.P.; Shakkottai, V.G.; Albin, R.L. Polyglutamine repeats in neurodegenerative diseases. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 1–27. [Google Scholar] [CrossRef]
- Wanker, E.E.; Ast, A.; Schindler, F.; Trepte, P.; Schnoegl, S. The pathobiology of perturbed mutant huntingtin protein–protein interactions in Huntington’s disease. J. Neurochem. 2019, 151, 507–519. [Google Scholar] [CrossRef]
- Arrasate, M.; Mitra, S.; Schweitzer, E.S.; Segal, M.R.; Finkbeiner, S. Inclusion body formation reduces levels of mutant Huntingtin and the risk of neuronal death. Nature 2004, 431, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Mende-Mueller, L.M.; Toneff, T.; Hwang, S.R.; Chesselet, M.F.; Hook, V.Y.H. Tissue-specific proteolysis of Huntingtin (Htt) in human brain: Evidence of enhanced levels of N-and C-terminal Htt fragments in Huntington’s disease striatum. J. Neurosci. 2001, 21, 1830–1837. [Google Scholar] [CrossRef]
- Wellington, C.L.; Ellerby, L.M.; Gutekunst, C.A.; Rogers, D.; Warby, S.; Graham, R.K.; Loubser, O.; Van Raamsdonk, J.; Singaraja, R.; Yang, Y.Z.; et al. Caspase cleavage of mutant Huntingtin precedes neurodegeneration in Huntington’s disease. J. Neurosci. 2002, 22, 7862–7872. [Google Scholar] [CrossRef]
- Wang, C.E.; Tydlacka, S.; Orr, A.L.; Yang, S.H.; Graham, R.K.; Hayden, M.R.; Li, S.; Chan, A.W.S.; Li, X.J. Accumulation of N-terminal mutant huntingtin in mouse and monkey models implicated as a pathogenic mechanism in huntington’s disease. Hum. Mol. Genet. 2008, 17, 2738–2751. [Google Scholar] [CrossRef]
- Miller, J.P.; Holcomb, J.; Al-Ramahi, I.; de Haro, M.; Gafni, J.; Zhang, N.; Kim, E.; Sanhueza, M.; Torcassi, C.; Kwak, S.; et al. Matrix metalloproteinases are modifiers of huntingtin proteolysis and toxicity in huntington’s disease. Neuron 2010, 67, 199–212. [Google Scholar] [CrossRef]
- Steffan, J.S.; Kazantsev, A.; Spasic-Boskovic, O.; Greenwald, M.; Zhu, Y.Z.; Gohler, H.; Wanker, E.E.; Bates, G.P.; Housman, D.E.; Thompson, L.M. The huntington’s disease protein interacts with P53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA 2000, 97, 6763–6768. [Google Scholar] [CrossRef] [PubMed]
- Warby, S.C.; Doty, C.N.; Graham, R.K.; Carroll, J.B.; Yang, Y.Z.; Singaraja, R.R.; Overall, C.M.; Hayden, M.R. Activated caspase-6 and caspase-6-cleaved fragments of huntingtin specifically colocalize in the nucleus. Hum. Mol. Genet. 2008, 17, 2390–2404. [Google Scholar] [CrossRef]
- Vonsattel, J.P.G.; DiFiglia, M. Huntington disease. J. Neuropathol. Exp. Neurol. 1998, 57, 369–384. [Google Scholar] [CrossRef]
- Cruickshank, T.; Reyes, A.; Peñailillo, L.; Thompson, J.; Ziman, M. Factors that contribute to balance and mobility impairments in individuals with Huntington’s disease. Basal Ganglia 2014, 4, 67–70. [Google Scholar] [CrossRef]
- Strong, T.V.; Tagle, D.A.; Valdes, J.M.; Elmer, L.W.; Boehm, K.; Swaroop, M.; Kaatz, K.W.; Collins, F.S.; Albin, R.L. Widespread expression of the human and rat Huntington’s disease gene in brain and nonneural tissues. Nat. Genet. 1993, 5, 259–265. [Google Scholar] [CrossRef]
- Sharp, A.H.; Loev, S.J.; Schilling, G.; Li, S.H.; Li, X.J.; Bao, J.; Wagster, M.V.; Kotzuk, J.A.; Steiner, J.P.; Lo, A.; et al. Widespread expression of Huntington’s disease gene (IT15) protein product. Neuron 1995, 14, 1065–1074. [Google Scholar] [CrossRef]
- Trottier, Y.; Devys, D.; Imbert, G.; Saudou, F.; An, I.; Lutz, Y.; Weber, C.; Agid, Y.; Hirsch, E.C.; Mandel, J.L. Cellular localization of the Huntington’s disease protein and discrimination of the normal and mutated form. Nat. Genet. 1995, 10, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Sathasivam, K.; Hobbs, C.; Turmaine, M.; Mangiarini, L.; Mahal, A.; Bertaux, F.; Wanker, E.E.; Doherty, P.; Davies, S.W.; Bates, G.P. Formation of polyglutamine inclusions in non-CNS tissue. Hum. Mol. Genet. 1999, 8, 813–822. [Google Scholar] [CrossRef]
- Luthi-Carter, R.; Hanson, S.A.; Strand, A.D.; Bergstrom, D.A.; Chun, W.; Peters, N.L.; Woods, A.M.; Chan, E.Y.; Kooperberg, C.; Krainc, D.; et al. Dysregulation of gene expression in the R6/2 model of polyglutamine disease: Parallel changes in muscle and brain. Hum. Mol. Genet. 2002, 11, 1911–1926. [Google Scholar] [CrossRef]
- Sassone, J.; Colciago, C.; Cislaghi, G.; Silani, V.; Ciammola, A. Huntington’s disease: The current state of research with peripheral tissues. Exp. Neurol. 2009, 219, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Ciammola, A.; Sassone, J.; Alberti, L.; Meola, G.; Mancinelli, E.; Russo, M.A.; Squitieri, F.; Silani, V. Increased apoptosis, huntingtin inclusions and altered differentiation in muscle cell cultures from Huntington’s disease subjects. Cell Death Differ. 2006, 13, 2068–2078. [Google Scholar] [CrossRef]
- Almeida, S.; Sarmento-Ribeiro, A.B.; Januário, C.; Rego, A.C.; Oliveira, C.R. Evidence of apoptosis and mitochondrial abnormalities in peripheral blood cells of Huntington’s disease patients. Biochem. Biophys. Res. Commun. 2008, 374, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Kosinski, C.M.; Schlangen, C.; Gellerich, F.N.; Gizatullina, Z.; Deschauer, M.; Schiefer, J.; Young, A.B.; Landwehrmeyer, G.B.; Toyka, K.V.; Sellhaus, B.; et al. Myopathy as a first symptom of Huntington’s disease in a marathon runner. Mov. Disord. 2007, 22, 1637–1640. [Google Scholar] [CrossRef]
- Busse, M.E.; Hughes, G.; Wiles, C.M.; Rosser, A.E. Use of hand-held dynamometry in the evaluation of lower limb muscle strength in people with Huntington’s disease. J. Neurol. 2008, 255, 1534–1540. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.M.; Wolfson, T.; Peavy, G.M.; Jacobson, M.W.; Corey-Bloom, J. Rate and correlates of weight change in Huntington’s disease. J. Neurol. Neurosurg. Psychiatry 2004, 75, 209–212. [Google Scholar] [CrossRef]
- Turner, C.; Cooper, J.M.; Schapira, A.H.V. Clinical correlates of mitochondrial function in Huntington’s disease muscle. Mov. Disord. 2007, 22, 1715–1721. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.B.; Bates, G.P.; Steffan, J.; Saft, C.; Tabrizi, S.J. Treating the whole body in Huntington’s disease. Lancet Neurol. 2015, 14, 1135–1142. [Google Scholar] [CrossRef]
- Van der Burg, J.M.; Björkqvist, M.; Brundin, P. Beyond the brain: Widespread pathology in Huntington’s disease. Lancet Neurol. 2009, 8, 765–774. [Google Scholar] [CrossRef]
- Hickey, M.A.; Chesselet, M.F. The use of transgenic and knock-in mice to study Huntington’s disease. Cytogenet. Genome Res. 2003, 100, 276–286. [Google Scholar] [CrossRef]
- Rubinsztein, D.C. Lessons from animal models of Huntington’s disease. Trends Genet. 2002, 18, 202–209. [Google Scholar] [CrossRef]
- Menalled, L.B.; Chesselet, M.F. Mouse models of Huntington’s disease. Trends Pharmacol. Sci. 2002, 23, 32–39. [Google Scholar] [CrossRef]
- Menalled, L.B. Knock-in mouse models of Huntington’s disease. NeuroRx 2005, 2, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; et al. Exon I of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef]
- Schilling, G.; Savonenko, A.V.; Klevytska, A.; Morton, J.L.; Tucker, S.M.; Poirier, M.; Gale, A.; Chan, N.; Gonzales, V.; Slunt, H.H.; et al. Nuclear-targeting of mutant huntingtin fragments produces huntington’s disease-like phenotypes in transgenic mice. Hum. Mol. Genet. 2004, 13, 1599–1610. [Google Scholar] [CrossRef]
- Schilling, G.; Becher, M.W.; Sharp, A.H.; Jinnah, H.A.; Duan, K.; Kotzuk, J.A.; Slunt, H.H.; Ratovitski, T.; Cooper, J.K.; Jenkins, N.A.; et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of Huntingtin. Hum. Mol. Genet. 1999, 8, 397–407. [Google Scholar] [CrossRef]
- Li, H.; Li, S.H.; Cheng, A.L.; Mangiarini, L.; Bates, G.P.; Li, X.J. Ultrastructural localization and progressive formation of neuropil aggregates in Huntington’s disease transgenic mice. Hum. Mol. Genet. 1999, 8, 1227–1236. [Google Scholar] [CrossRef]
- Davies, S.W.; Turmaine, M.; Cozens, B.A.; DiFiglia, M.; Sharp, A.H.; Ross, C.A.; Scherzinger, E.; Wanker, E.E.; Mangiarini, L.; Bates, G.P. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 1997, 90, 537–548. [Google Scholar] [CrossRef]
- Sathasivam, K.; Neueder, A.; Gipson, T.A.; Landles, C.; Benjamin, A.C.; Bondulich, M.K.; Smith, D.L.; Faull, R.L.M.; Roos, R.A.C.; Howland, D.; et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. USA 2013, 110, 2366–2370. [Google Scholar] [CrossRef]
- Woodman, B.; Butler, R.; Landles, C.; Lupton, M.K.; Tse, J.; Hockly, E.; Moffitt, H.; Sathasivam, K.; Bates, G.P. The HdhQ150/Q150 Knock-in mouse model of HD and the R6/2 Exon 1 model develop comparable and widespread molecular phenotypes. Brain Res. Bull. 2007, 72, 83–97. [Google Scholar] [CrossRef]
- Yang, X.W.; Model, P.; Heintz, N. Homologous recombination based modification in esherichia coli and germline transmission in transgenic mice of a bacterial artificial chromsome. Nat. Biotechnol. 1997, 15, 859–865. [Google Scholar] [CrossRef]
- Gray, M.; Shirasaki, D.I.; Cepeda, C.; André, V.M.; Wilburn, B.; Lu, X.H.; Tao, J.; Yamazaki, I.; Li, S.H.; Sun, Y.E.; et al. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J. Neurosci. 2008, 28, 6182–6195. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, J.G.; Smith, D.J.; McCutcheon, K.; Koide, H.B.; Nishiyama, K.; Dinulos, M.B.; Stevens, M.E.; Bissada, N.; Nasir, J.; Kanazawa, I.; et al. Human huntingtin derived from YAC transgenes compensates for loss of murine huntingtin by rescue of the embryonic lethal phenotype. Hum. Mol. Genet. 1996, 5, 1875–1885. [Google Scholar] [CrossRef]
- Hodgson, J.G.; Agopyan, N.; Gutekunst, C.A.; Leavitt, B.R.; Lepiane, F.; Singaraja, R.; Smith, D.J.; Bissada, N.; McCutcheon, K.; Nasir, J.; et al. A YAC mouse model for huntington’s disease with full-length mutant Huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron 1999, 23, 181–192. [Google Scholar] [CrossRef]
- Slow, E.J.; van Raamsdonk, J.; Rogers, D.; Coleman, S.H.; Graham, R.K.; Deng, Y.; Oh, R.; Bissada, N.; Hossain, S.M.; Yang, Y.Z.; et al. Selective striatal neuronal loss in a YAC128 mouse model of huntington disease. Hum. Mol. Genet. 2003, 12, 1555–1567. [Google Scholar] [CrossRef]
- Van Raamsdonk, J.M.; Murphy, Z.; Slow, E.J.; Leavitt, B.R.; Hayden, M.R. Selective degeneration and nuclear localization of mutant huntingtin in the YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 2005, 14, 3823–3835. [Google Scholar] [CrossRef]
- Baxa, M.; Hruska-Plochan, M.; Juhas, S.; Vodicka, P.; Pavlok, A.; Juhasova, J.; Miyanohara, A.; Nejime, T.; Klima, J.; Macakova, M.; et al. A transgenic minipig model of Huntington’s disease. J. Huntington’s Dis. 2013, 2, 47–68. [Google Scholar] [CrossRef]
- Djoussé, L.; Knowlton, B.; Cupples, L.A.; Marder, K.; Shoulson, I.; Myers, R.H. Weight loss in early stage of Huntington’s disease. Neurology 2002, 59, 1325–1330. [Google Scholar] [CrossRef]
- Robbins, A.O.; Ho, A.K.; Barker, R.A. Weight Changes in Huntington’s disease. Eur. J. Neurol. 2006, 13, e7. [Google Scholar] [CrossRef]
- Aziz, N.A.; Van Der Burg, J.M.M.; Landwehrmeyer, G.B.; Brundin, P.; Stijnen, T.; Roos, R.A.C. Weight loss in Huntington disease increases with higher CAG repeat number. Neurology 2008, 71, 1506–1513. [Google Scholar] [CrossRef] [PubMed]
- Mochel, F.; Charles, P.; Seguin, F.; Barritault, J.; Coussieu, C.; Perin, L.; Le Bouc, Y.; Gervais, C.; Carcelain, G.; Vassault, A.; et al. Early energy deficit in huntingdon disease: Identification of a plasma biomarker traceable during disease progression. PLoS ONE 2007, 2, e647. [Google Scholar] [CrossRef]
- Marder, K.; Zhao, H.; Eberly, S.; Tanner, C.M.; Oakes, D.; Shoulson, I. Dietary intake in adults at risk for Huntington disease: Analysis of PHAROS research participants. Neurology 2009, 73, 385–392. [Google Scholar] [CrossRef]
- Lee, J.K.; Mathews, K.; Schlaggar, B.; Perlmutter, J.; Paulsen, J.S.; Epping, E.; Burmeister, L.; Nopoulos, P. Measures of growth in children at risk for Huntington disease. Neurology 2012, 79, 668–674. [Google Scholar] [CrossRef]
- Ribchester, R.R.; Thomson, D.; Wood, N.I.; Hinks, T.; Gillingwater, T.H.; Wishart, T.M.; Court, F.A.; Morton, A.J. Progressive abnormalities in skeletal muscle and neuromuscular junctions of transgenic mice expressing the Huntington’s disease mutation. Eur. J. Neurosci. 2004, 20, 3092–3114. [Google Scholar] [CrossRef]
- Van der Burg, J.M.M.; Bacos, K.; Wood, N.I.; Lindqvist, A.; Wierup, N.; Woodman, B.; Wamsteeker, J.I.; Smith, R.; Deierborg, T.; Kuhar, M.J.; et al. Increased metabolism in the R6/2 mouse model of Huntington’s disease. Neurobiol. Dis. 2008, 29, 41–51. [Google Scholar] [CrossRef] [PubMed]
- She, P.; Zhang, Z.; Marchionini, D.; Diaz, W.C.; Jetton, T.J.; Kimball, S.R.; Vary, T.C.; Lang, C.H.; Lynch, C.J. Molecular characterization of skeletal muscle atrophy in the R6/2 mouse model of Huntington’s disease. Am. J. Physiol. Endocrinol. Metab. 2011, 301, 49–61. [Google Scholar] [CrossRef]
- Aziz, N.A.; Roos, R.A. Characteristics, pathophysiology and clinical management of weight loss in Huntington’s disease. Neurodegener. Dis. Manag. 2013, 3, 253–266. [Google Scholar] [CrossRef]
- Skene, D.J.; Middleton, B.; Fraser, C.K.; Pennings, J.L.A.; Kuchel, T.R.; Rudiger, S.R.; Bawden, C.S.; Morton, A.J. Metabolic profiling of presymptomatic Huntington’s disease sheep reveals novel biomarkers. Sci. Rep. 2017, 7, 43030–43045. [Google Scholar] [CrossRef]
- Bondulich, M.K.; Jolinon, N.; Osborne, G.F.; Smith, E.J.; Rattray, I.; Neueder, A.; Sathasivam, K.; Ahmed, M.; Ali, N.; Benjamin, A.C.; et al. Myostatin inhibition prevents skeletal muscle pathophysiology in Huntington’s disease mice. Sci. Rep. 2017, 7, 14275–14288. [Google Scholar] [CrossRef]
- Miranda, D.R.; Wong, M.; Romer, S.H.; McKee, C.; Garza-Vasquez, G.; Medina, A.C.; Bahn, V.; Steele, A.D.; Talmadge, R.J.; Voss, A.A. Progressive Cl-channel defects reveal disrupted skeletal muscle maturation in R6/2 Huntington’s mice. J. Gen. Physiol. 2016, 149, 55–74. [Google Scholar] [CrossRef]
- Moffitt, H.; McPhail, G.D.; Woodman, B.; Hobbs, C.; Bates, G.P. Formation of polyglutamine inclusions in a wide range of non-cns tissues in the HdhQ150 Knock-in mouse model of Huntington’s disease. PLoS ONE 2009, 4, e8025. [Google Scholar] [CrossRef]
- Van der Burg, J.M.M.; Gardiner, S.L.; Ludolph, A.C.; Landwehrmeyer, G.B.; Roos, R.A.C.; Aziz, N.A. Body weight is a robust predictor of clinical progression in Huntington disease. Ann. Neurol. 2017, 82, 479–483. [Google Scholar] [CrossRef]
- Tereshchenko, A.; McHugh, M.; Lee, J.K.; Gonzalez-Alegre, P.; Crane, K.; Dawson, J.; Nopoulos, P. Abnormal weight and body mass index in children with juvenile Huntington’s disease. J. Huntington’s Dis. 2015, 4, 231–238. [Google Scholar] [CrossRef]
- Mielcarek, M.; Toczek, M.; Smeets, C.J.L.M.; Franklin, S.A.; Bondulich, M.K.; Jolinon, N.; Muller, T.; Ahmed, M.; Dick, J.R.T.; Piotrowska, I.; et al. HDAC4-myogenin axis as an important marker of HD-related skeletal muscle atrophy. PLoS Genet. 2015, 11, e1005021. [Google Scholar] [CrossRef]
- Strand, A.D.; Aragaki, A.K.; Shaw, D.; Bird, T.; Holton, J.; Turner, C.; Tapscott, S.J.; Tabrizi, S.J.; Schapira, A.H.; Kooperberg, C.; et al. Gene expression in Huntington’s disease skeletal muscle: A potential biomarker. Hum. Mol. Genet. 2005, 14, 1863–1876. [Google Scholar] [CrossRef]
- Zong, H.; Ren, J.M.; Young, L.H.; Pypaert, M.; Mu, J.; Birnbaum, M.J.; Shulman, G.I. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc. Natl. Acad. Sci. USA 2002, 99, 15983–15987. [Google Scholar] [CrossRef]
- Chaturvedi, R.K.; Adhihetty, P.; Shukla, S.; Hennessy, T.; Calingasan, N.; Yang, L.; Starkov, A.; Kiaei, M.; Cannella, M.; Sassone, J.; et al. Impaired PGC-1α function in muscle in Huntington’s disease. Hum. Mol. Genet. 2009, 18, 3048–3065. [Google Scholar] [CrossRef]
- Menalled, L.; El-Khodor, B.F.; Patry, M.; Suárez-Fariñas, M.; Orenstein, S.J.; Zahasky, B.; Leahy, C.; Wheeler, V.; Yang, X.W.; MacDonald, M.; et al. Systematic behavioral evaluation of Huntington’s disease transgenic and knock-in mouse models. Neurobiol. Dis. 2009, 35, 319–336. [Google Scholar] [CrossRef]
- Mantovani, S.; Gordon, R.; Li, R.; Christie, D.C.; Kumar, V.; Woodruff, T.M. Motor deficits associated with Huntington’s disease occur in the absence of striatal degeneration in BACHD transgenic mice. Hum. Mol. Genet. 2016, 25, 1780–1791. [Google Scholar] [CrossRef]
- Valadão, P.A.C.; de Aragão, B.C.; Andrade, J.N.; Magalhães-Gomes, M.P.S.; Foureaux, G.; Joviano-Santos, J.V.; Nogueira, J.C.; Machado, T.C.G.; de Jesus, I.C.G.; Nogueira, J.M.; et al. Abnormalities in the motor unit of a fast-twitch lower limb skeletal muscle in Huntington’s disease. ASN Neuro 2019, 11. [Google Scholar] [CrossRef]
- Braubach, P.; Orynbayev, M.; Andronache, Z.; Hering, T.; Landwehrmeyer, G.B.; Lindenberg, K.S.; Melzer, W. Altered Ca2+ signaling in skeletal muscle fibers of the R6/2 mouse, a model of Huntington’s disease. J. Gen. Physiol. 2014, 144, 393–413. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.M.; Dutka, T.L.; Horvath, D.; Bell, J.R.; Delbridge, L.M.; Lamb, G.D. Ca2+-dependent proteolysis of junctophilin-1 and junctophilin-2 in skeletal and cardiac muscle. J. Physiol. 2013, 591, 719–729. [Google Scholar] [CrossRef]
- Saft, C.; Zange, J.; Andrich, J.; Müller, K.; Lindenberg, K.; Landwehrmeyer, B.; Vorgerd, M.; Kraus, P.H.; Przuntek, H.; Schöls, L. Mitochondrial impairment in patients and asymptomatic mutation carriers of Huntington’s disease. Mov. Disord. 2005, 20, 674–679. [Google Scholar] [CrossRef]
- Baldo, B.; Paganetti, P.; Grueninger, S.; Marcellin, D.; Kaltenbach, L.S.; Lo, D.C.; Semmelroth, M.; Zivanovic, A.; Abramowski, D.; Smith, D.; et al. TR-FRET-based duplex immunoassay reveals an inverse correlation of soluble and aggregated mutant Huntingtin in huntington’s disease. Chem. Biol. 2012, 19, 264–275. [Google Scholar] [CrossRef]
- Kojer, K.; Hering, T.; Bazenet, C.; Weiss, A.; Herrmann, F.; Taanman, J.W.; Orth, M. Huntingtin aggregates and mitochondrial pathology in skeletal muscle but not heart of late-stage R6/2 mice. J. Huntington’s. Dis. 2019, 8, 145–159. [Google Scholar] [CrossRef]
- Disatnik, M.H.; Joshi, A.U.; Saw, N.L.; Shamloo, M.; Leavitt, B.R.; Qi, X.; Mochly-Rosen, D. Potential biomarkers to follow the progression and treatment response of Huntington’s disease. J. Exp. Med. 2016, 213, 2655–2669. [Google Scholar] [CrossRef]
- Sjögren, M.; Duarte, A.I.; McCourt, A.C.; Shcherbina, L.; Wierup, N.; Björkqvist, M. Ghrelin rescues skeletal muscle catabolic profile in the R6/2 mouse model of Huntington’s disease. Sci. Rep. 2017, 7, 13896–13908. [Google Scholar] [CrossRef]
- Nath, S.R.; Lieberman, M.L.; Yu, Z.; Marchioretti, C.; Jones, S.T.; Danby, E.C.E.; Van Pelt, K.M.; Sorarù, G.; Robins, D.M.; Bates, G.P.; et al. MEF2 impairment underlies skeletal muscle atrophy in polyglutamine disease. Acta Neuropathol. 2020, 140, 63–80. [Google Scholar] [CrossRef]
- Orth, M.; Cooper, J.M.; Bates, G.P.; Schapira, A.H.V. Inclusion formation in Huntington’s disease R6/2 mouse muscle cultures. J. Neurochem. 2003, 87, 1–6. [Google Scholar] [CrossRef]
- Ooi, J.; Langley, S.R.; Xu, X.; Utami, K.H.; Sim, B.; Huang, Y.; Harmston, N.P.; Tay, Y.L.; Ziaei, A.; Zeng, R.; et al. Unbiased profiling of isogenic Huntington disease HPSC-derived CNS and peripheral cells reveals strong cell-type specificity of CAG length effects. Cell Rep. 2019, 26, 2494–2508. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. MTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Bell, R.A.V.; Al-Khalaf, M.; Megeney, L.A. The beneficial role of proteolysis in skeletal muscle growth and stress adaptation. Skelet. Muscle 2016, 6, 16–28. [Google Scholar] [CrossRef]
- Vainshtein, A.; Sandri, M. Signaling pathways that control muscle mass. Int. J. Mol. Sci. 2020, 21, 4759. [Google Scholar] [CrossRef] [PubMed]
- Scalabrin, M.; Adams, V.; Labeit, S.; Bowen, T.S. Emerging strategies targeting catabolic muscle stress relief. Int. J. Mol. Sci. 2020, 21, 4681. [Google Scholar] [CrossRef]
- Kumar, D.; Ambasta, R.K.; Kumar, P. Ubiquitin biology in neurodegenerative disorders: From impairment to therapeutic strategies. Ageing Res Rev. 2020, 61, 101078–101100. [Google Scholar] [CrossRef]
- Zhou, J.; Chong, S.Y.; Lim, A.; Singh, B.K.; Sinha, R.A.; Salmon, A.B.; Yen, P.M. Changes in macroautophagy, chaperone-mediated autophagy, and mitochondrial metabolism in murine skeletal and cardiac muscle during aging. Aging 2017, 9, 583–599. [Google Scholar] [CrossRef]
- Carter, H.N.; Kim, Y.; Erlich, A.T.; Zarrin-khat, D.; Hood, D.A. Autophagy and mitophagy flux in young and aged skeletal muscle following chronic contractile activity. J. Physiol. 2018, 596, 3567–3584. [Google Scholar] [CrossRef]
- Jiao, J.; Demontis, F. Skeletal muscle autophagy and its role in sarcopenia and organismal aging. Curr. Opin. Pharmacol. 2017, 34, 1–6. [Google Scholar] [CrossRef]
- Paré, M.F.; Baechler, B.L.; Fajardo, V.A.; Earl, E.; Wong, E.; Campbell, T.L.; Tupling, A.R.; Quadrilatero, J. Effect of acute and chronic autophagy deficiency on skeletal muscle apoptotic signaling, morphology, and function. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 708–718. [Google Scholar] [CrossRef]
- Zhao, J.; Brault, J.J.; Schild, A.; Cao, P.; Sandri, M.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007, 6, 472–483. [Google Scholar] [CrossRef]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef]
- Mammucari, C.; Milan, G.; Romanello, V.; Masiero, E.; Rudolf, R.; Del Piccolo, P.; Burden, S.J.; Di Lisi, R.; Sandri, C.; Zhao, J.; et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007, 6, 458–471. [Google Scholar] [CrossRef]
- Juenemann, K.; Schipper-Krom, S.; Wiemhoefer, A.; Kloss, A.; Sanz, A.S.; Reits, E.A.J. Expanded polyglutamine-containing n-terminal Huntingtin fragments are entirely degraded by mammalian proteasomes. J. Biol. Chem. 2013, 288, 27068–27084. [Google Scholar] [CrossRef]
- Holmberg, C.I.; Staniszewski, K.E.; Mensah, K.N.; Matouschek, A.; Morimoto, R.I. Inefficient degradation of truncated polyglutamine proteins by the proteasome. EMBO J. 2004, 23, 4307–4318. [Google Scholar] [CrossRef] [PubMed]
- Ochaba, J.; Lukacsovich, T.; Csikos, G.; Zheng, S.; Margulis, J.; Salazar, L.; Mao, K.; Lau, A.L.; Yeung, S.Y.; Humbert, S.; et al. Potential function for the huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 16889–16894. [Google Scholar] [CrossRef]
- Rui, Y.N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262–275. [Google Scholar] [CrossRef]
- Rui, Y.N.; Xu, Z.; Patel, B.; Cuervo, A.M.; Zhang, S. HTT/Huntingtin in selective autophagy and Huntington disease: A foe or a friend within? Autophagy 2015, 11, 858–860. [Google Scholar] [CrossRef]
- El-Daher, M.; Hangen, E.; Bruyère, J.; Poizat, G.; Al-Ramahi, I.; Pardo, R.; Bourg, N.; Souquere, S.; Mayet, C.; Pierron, G.; et al. Huntingtin proteolysis releases non-polyq fragments that cause toxicity through dynamin 1 dysregulation. EMBO J. 2015, 34, 2255–2271. [Google Scholar] [CrossRef]
- Martin, D.D.O.; Ladha, S.; Ehrnhoefer, D.E.; Hayden, M.R. Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 2015, 38, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Bentzinger, C.F.; Lin, S.; Romanino, K.; Castets, P.; Guridi, M.; Summermatter, S.; Handschin, C.; Tintignac, L.A.; Hall, M.N.; Rüegg, M.A. Differential response of skeletal muscles to MTORC1 signaling during atrophy and hypertrophy. Skelet. Muscle 2013, 3, 6. [Google Scholar] [CrossRef]
- You, J.S.; Anderson, G.B.; Dooley, M.S.; Hornberger, T.A. The Role of MTOR Signaling in the regulation of protein synthesis and muscle mass during immobilization in mice. DMM Dis. Model. Mech. 2015, 8, 1059–1069. [Google Scholar] [CrossRef]
- Tang, H.; Inoki, K.; Lee, M.; Wright, E.; Khuong, A.; Khuong, A.; Sugiarto, S.; Garner, M.; Paik, J.; DePinho, R.A.; et al. MTORC1 promotes denervation-induced muscle atrophy through a mechanism involving the activation of FoxO and E3 ubiquitin ligases. Sci. Sign. 2014, 7, ra18. [Google Scholar] [CrossRef] [PubMed]
- Skotte, N.H.; Pouladi, M.A.; Ehrnhoefer, D.E.; Huynh, K.; Qiu, X.; Nielsen, S.M.B.; Nielsen, T.T.; Nørremølle, A.; Hayden, M.R. Compromised IGF signaling causes caspase-6 activation in Huntington disease. Exp. Neurol. 2020, 332, 113396–113404. [Google Scholar] [CrossRef]
- Pouladi, M.A.; Xie, Y.; Skotte, N.H.; Ehrnhoefer, D.E.; Graham, R.K.; Kim, J.E.; Bissada, N.; Yang, X.W.; Paganetti, P.; Friedlander, R.M.; et al. Full-length huntingtin levels modulate body weight by influencing insulin-like growth factor 1 expression. Hum. Mol. Genet. 2010, 19, 1528–1538. [Google Scholar] [CrossRef] [PubMed]
- Forcina, L.; Miano, C.; Scicchitano, B.M.; Rizzuto, E.; Berardinelli, M.G.; De Benedetti, F.; Pelosi, L.; Musarò, A. Increased circulating levels of interleukin-6 affect the redox balance in skeletal muscle. Oxid. Med. Cell. Longev. 2019, 2019, 3018584–3018596. [Google Scholar] [CrossRef]
- Judge, A.R.; Koncarevic, A.; Hunter, R.B.; Liou, H.C.; Jackman, R.W.; Kandarian, S.C. Role for IκBα, but not c-Rel, in skeletal muscle atrophy. Am. J. Physiol. Cell. Physiol. 2007, 292, C372–C382. [Google Scholar] [CrossRef]
- Mourkioti, F.; Kratsios, P.; Luedde, T.; Song, Y.H.; Delafontaine, P.; Adami, R.; Parente, V.; Bottinelli, R.; Pasparakis, M.; Rosenthal, N. Erratum: Targeted ablation of IKK2 improves skeletal muscle strength, maintains mass, and promotes regeneration. J. Clin. Investig. 2006, 116, 2945–2954. [Google Scholar] [CrossRef]
- Magnusson-Lind, A.; Davidsson, M.; Silajdžić, E.; Hansen, C.; McCourt, A.C.; Tabrizi, S.J.; Björkqvist, M. Skeletal muscle atrophy in R6/2 mice-altered circulating skeletal muscle markers and gene expression profile changes. J. Huntington’s. Dis. 2014, 3, 13–24. [Google Scholar] [CrossRef] [PubMed]
- He, W.A.; Berardi, E.; Cardillo, V.M.; Acharyya, S.; Aulino, P.; Thomas-Ahner, J.; Wang, J.; Bloomston, M.; Muscarella, P.; Nau, P.; et al. NF-ΚB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J. Clin. Investig. 2013, 123, 4821–4835. [Google Scholar] [CrossRef]
- Björkqvist, M.; Wild, E.J.; Thiele, J.; Silvestroni, A.; Andre, R.; Lahiri, N.; Raibon, E.; Lee, R.V.; Benn, C.L.; Soulet, D.; et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J. Exp. Med. 2008, 205, 1869–1877. [Google Scholar] [CrossRef]
- Visser, M.; Deeg, D.J.H.; Lips, P. Low vitamin D and high parathyroid hormone levels as determinants of loss of muscle strength and muscle mass (sarcopenia): The longitudinal aging study Amsterdam. J. Clin. Endocrinol. Metab. 2003, 88, 5766–5772. [Google Scholar] [CrossRef]
- Schaap, L.A.; Pluijm, S.M.F.; Deeg, D.J.H.; Harris, T.B.; Kritchevsky, S.B.; Newman, A.B.; Colbert, L.H.; Pahor, M.; Rubin, S.M.; Tylavsky, F.A.; et al. Higher inflammatory marker levels in older persons: Associations with 5-year change in muscle mass and muscle strength. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2009, 64, 1183–1189. [Google Scholar] [CrossRef]
- Stewart, R.; Flechner, L.; Montminy, M.; Berdeaux, R. Creb Is activated by muscle injury and promotes muscle regeneration. PLoS ONE 2011, 6, e24714. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Skotte, N.H.; Ladha, S.; Nguyen, Y.T.N.; Qiu, X.; Deng, Y.; Huynh, K.T.; Engemann, S.; Nielsen, S.M.; Becanovic, K.; et al. P53 increases caspase-6 expression and activation in muscle tissue expressing mutant huntingtin. Hum. Mol. Genet. 2014, 23, 717–729. [Google Scholar] [CrossRef]
- Koroshetz, W.J.; Jenkins, B.G.; Rosen, B.R.; Flint Beal, M. Energy metabolism defects in Huntington’s disease and effects of coenzyme Q10. Ann. Neurol. 1997, 41, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Lodi, R.; Schapira, A.H.V.; Manners, D.; Styles, P.; Wood, N.W.; Taylor, D.J.; Warner, T.T. Abnormal in vivo skeletal muscle energy metabolism in Huntington’s disease and dentatorubropallidoluysian atrophy. Ann. Neurol. 2000, 48, 72–76. [Google Scholar] [CrossRef]
- Ciammola, A.; Sassone, J.; Sciacco, M.; Mencacci, N.E.; Ripolone, M.; Bizzi, C.; Colciago, C.; Moggio, M.; Parati, G.; Silani, V.; et al. Low anaerobic threshold and increased skeletal muscle lactate production in subjects with Huntington’s disease. Mov. Disord. 2011, 26, 130–137. [Google Scholar] [CrossRef]
- Anzell, A.R.; Maizy, R.; Przyklenk, K.; Sanderson, T.H. Mitochondrial quality control and disease: Insights into ischemia-reperfusion injury. Mol. Neurobiol. 2018, 55, 2547–2564. [Google Scholar] [CrossRef]
- Twig, G.; Elorza, A.; Molina, A.J.A.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef]
- van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 2013, 5, a011072. [Google Scholar] [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 2006, 22, 79–99. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar]
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion is required for mtdna stability in skeletal muscle and tolerance of MtDNA mutations. Cell 2010, 141, 280–289. [Google Scholar] [CrossRef]
- Tezze, C.; Romanello, V.; Desbats, M.A.; Fadini, G.P.; Albiero, M.; Favaro, G.; Ciciliot, S.; Soriano, M.E.; Morbidoni, V.; Cerqua, C.; et al. Age-associated loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metab. 2017, 25, 1374–1389. [Google Scholar] [CrossRef]
- Sebastián, D.; Sorianello, E.; Segalés, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Muñoz, J.P.; Sánchez-Feutrie, M.; et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef]
- Romanello, V.; Guadagnin, E.; Gomes, L.; Roder, I.; Sandri, C.; Petersen, Y.; Milan, G.; Masiero, E.; Del Piccolo, P.; Foretz, M.; et al. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 2010, 29, 1774–1785. [Google Scholar] [CrossRef]
- Touvier, T.; De Palma, C.; Rigamonti, E.; Scagliola, A.; Incerti, E.; Mazelin, L.; Thomas, J.L.; D’Antonio, M.; Politi, L.; Schaeffer, L.; et al. Muscle-specific Drp1 overexpression impairs skeletal muscle growth via translational attenuation. Cell Death Dis. 2015, 6, e1663. [Google Scholar] [CrossRef]
- Favaro, G.; Romanello, V.; Varanita, T.; Andrea Desbats, M.; Morbidoni, V.; Tezze, C.; Albiero, M.; Canato, M.; Gherardi, G.; De Stefani, D.; et al. DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat. Commun. 2019, 10, 2576–2592. [Google Scholar] [CrossRef]
- Moore, T.M.; Zhou, Z.; Cohn, W.; Norheim, F.; Lin, A.J.; Kalajian, N.; Strumwasser, A.R.; Cory, K.; Whitney, K.; Ho, T.; et al. The impact of exercise on mitochondrial dynamics and the role of Drp1 in exercise performance and training adaptations in skeletal muscle. Mol. Metab. 2019, 21, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Dulac, M.; Leduc-Gaudet, J.P.; Reynaud, O.; Ayoub, M.B.; Guérin, A.; Finkelchtein, M.; Hussain, S.N.A.; Gouspillou, G. Drp1 knockdown induces severe muscle atrophy and remodelling, mitochondrial dysfunction, autophagy impairment and denervation. J. Physiol. 2020, 598, 3691–3710. [Google Scholar] [CrossRef]
- Song, W.; Chen, J.; Petrilli, A.; Liot, G.; Klinglmayr, E.; Poquiz, P.; Tjong, J.; Pouladi, M.A.; Hayden, M.R.; Masliah, E.; et al. Mutant huntingtin binds the mitochondrial fission. Nat. Med. 2011, 17, 377–382. [Google Scholar] [CrossRef]
- Shirendeb, U.; Reddy, A.P.; Manczak, M.; Calkins, M.J.; Mao, P.; Tagle, D.A.; Reddy, P.H. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: Implications for selective neuronal damage. Hum. Mol. Genet. 2011, 20, 1438–1455. [Google Scholar] [CrossRef]
- Shirendeb, U.P.; Calkins, M.J.; Manczak, M.; Anekonda, V.; Dufour, B.; McBride, J.L.; Mao, P.; Reddy, P.H. Mutant huntingtin’s interaction with mitochondrial protein drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum. Mol. Genet. 2012, 21, 406–420. [Google Scholar] [CrossRef]
- Arenas, J.; Campos, Y.; Ribacoba, R.; Martín, M.A.; Rubio, J.C.; Ablanedo, P.; Cabello, A. Complex I defect in muscle from patients with Huntington’s disease. Ann. Neurol. 1998, 43, 397–400. [Google Scholar] [CrossRef]
- Squitieri, F.; Falleni, A.; Cannella, M.; Orobello, S.; Fulceri, F.; Lenzi, P.; Fornai, F. Abnormal morphology of peripheral cell tissues from patients with Huntington disease. J. Neural Transm. 2010, 117, 77–83. [Google Scholar] [CrossRef]
- Johri, A.; Calingasan, N.Y.; Hennessey, T.M.; Sharma, A.; Yang, L.; Wille, E.; Chandra, A.; Beal, M.F. Pharmacologic activation of mitochondrial biogenesis exerts widespread beneficial effects in a transgenic mouse model of Huntington’s disease. Hum. Mol. Genet. 2012, 21, 1124–1137. [Google Scholar] [CrossRef]
- Gizatullina, Z.Z.; Lindenberg, K.S.; Harjes, P.; Chen, Y.; Kosinski, C.M.; Landwehrmeyer, B.G.; Ludolph, A.C.; Striggow, F.; Zierz, S.; Gellerich, F.N. Low stability of huntington muscle mitochondria against Ca 2+ in R6/2 mice. Ann. Neurol. 2006, 59, 407–411. [Google Scholar] [CrossRef]
- Rodinova, M.; Krizova, J.; Stufkova, H.; Bohuslavova, B.; Askeland, G.; Dosoudilova, Z.; Juhas, S.; Juhasova, J.; Ellederova, Z.; Zeman, J.; et al. Deterioration of mitochondrial bioenergetics and ultrastructure impairment in skeletal muscle of a transgenic minipig model in the early stages of Huntington’s disease. DMM Dis. Model. Mech. 2019, 12, dmm038737. [Google Scholar] [CrossRef]
- Buck, E.; Zügel, M.; Schumann, U.; Merz, T.; Gumpp, A.M.; Witting, A.; Steinacker, J.M.; Landwehrmeyer, G.B.; Weydt, P.; Calzia, E.; et al. High-resolution respirometry of fine-needle muscle biopsies in pre-manifest Huntington’s disease expansion mutation carriers shows normal mitochondrial respiratory function. PLoS ONE 2017, 12, 1–21. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Workman, J.; Hart, P.E.; Mangiarini, L.; Mahal, A.; Bates, G.; Cooper, J.M.; Schapira, A.H.V. Mitochondrial dysfunction and free radical damage in the huntington R6/2 transgenic mouse. Ann. Neurol. 2000, 47, 80–86. [Google Scholar] [CrossRef]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional Co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nature 2002, 418, 797–801. [Google Scholar] [CrossRef]
- Lin, J.; Wu, P.H.; Tarr, P.T.; Lindenberg, K.S.; St-Pierre, J.; Zhang, C.Y.; Mootha, V.K.; Jäger, S.; Vianna, C.R.; Reznick, R.M.; et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1α null mice. Cell 2004, 119, 121–135. [Google Scholar] [CrossRef]
- Leone, T.C.; Lehman, J.J.; Finck, B.N.; Schaeffer, P.J.; Wende, A.R.; Boudina, S.; Courtois, M.; Wozniak, D.F.; Sambandam, N.; Bernal-Mizrachi, C.; et al. PGC-1α deficiency causes multi-system energy metabolic derangements: Muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 2005, 3, 0672–0687. [Google Scholar] [CrossRef] [PubMed]
- Talmadge, R.J. Myosin heavy chain isoform expression following reduced neuromuscular activity: Potential regulatory mechanisms. Muscle Nerve 2000, 23, 661–679. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef]
- Agbulut, O.; Noirez, P.; Beaumont, F.; Butler-Browne, G. Myosin heavy chain isoforms in postnatal muscle development of mice. Biol. Cell 2003, 95, 399–406. [Google Scholar] [CrossRef]
- Pette, D.; Vrbová, G. Adaptation of mammalian skeletal muscle fibers to chronic electrical stimulation. Rev. Physiol. Biochem. Pharmacol. 1992, 120, 115–202. [Google Scholar]
- Hering, T.; Braubach, P.; Landwehrmeyer, G.B.; Lindenberg, K.S.; Melzer, W. Fast-to-slow transition of skeletal muscle contractile function and corresponding changes in myosin heavy and light chain formation in the R6/2 mouse model of Huntington’s disease. PLoS ONE 2016, 11, e0166106. [Google Scholar] [CrossRef]
- Rozas, J.L.; Gómez-Sánchez, L.; Tomás-Zapico, C.; Lucas, J.J.; Fernández-Chacón, R. Increased neurotransmitter release at the neuromuscular junction in a mouse model of polyglutamine disease. J. Neurosci. 2011, 31, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Li, S.H.; Li, X.J. Huntingtin-protein interactions and the pathogenesis of huntington’s disease. Trends Genet. 2004, 20, 146–154. [Google Scholar] [CrossRef]
- Zuccato, C.; Valenza, M.; Cattaneo, E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol. Rev. 2010, 90, 905–981. [Google Scholar] [CrossRef]
- Romero, E.; Cha, G.H.; Verstreken, P.; Ly, C.V.; Hughes, R.E.; Bellen, H.J.; Botas, J. Suppression of neurodegeneration and increased neurotransmission caused by expanded full-length huntingtin accumulating in the cytoplasm. Neuron 2008, 57, 27–40. [Google Scholar] [CrossRef]
- Khedraki, A.; Reed, E.J.; Romer, S.H.; Wang, Q.; Romine, W.; Rich, M.M.; Talmadge, R.J.; Voss, A.A. Depressed synaptic transmission and reduced vesicle release sites in Huntington’s disease neuromuscular junctions. J. Neurosci. 2017, 37, 8077–8091. [Google Scholar] [CrossRef] [PubMed]
- De Aragão, B.C.; Rodrigues, H.A.; Valadão, P.A.C.; Camargo, W.; Naves, L.A.; Ribeiro, F.M.; Guatimosim, C. Changes in structure and function of diaphragm neuromuscular junctions from BACHD mouse model for Huntington’s disease. Neurochem. Int. 2016, 93, 64–72. [Google Scholar] [CrossRef]
- Waters, C.W.; Varuzhanyan, G.; Talmadge, R.J.; Voss, A.A. Huntington disease skeletal muscle is hyperexcitable owing to chloride and potassium channel dysfunction. Proc. Natl. Acad. Sci. USA 2013, 110, 9160–9165. [Google Scholar] [CrossRef]
- Miranda, D.R.; Reed, E.; Jama, A.; Bottomley, M.; Ren, H.; Rich, M.M.; Voss, A.A. Mechanisms of altered skeletal muscle action potentials in the R6/2 mouse model of Huntington’s disease. Am. J. Physiol. Cell Physiol. 2020, 319, C218–C232. [Google Scholar] [CrossRef]
- Wade, A.; Jacobs, P.; Morton, A.J. Atrophy and degeneration in sciatic nerve of presymptomatic mice carrying the Huntington’s disease mutation. Brain Res. 2008, 1188, 61–68. [Google Scholar] [CrossRef]
- Valadão, P.A.C.; de Aragão, B.C.; Andrade, J.N.; Magalhães-Gomes, M.P.S.; Foureaux, G.; Joviano-Santos, J.V.; Nogueira, J.C.; Ribeiro, F.M.; Tapia, J.C.; Guatimosim, C. Muscle atrophy is associated with cervical spinal motoneuron loss in BACHD mouse model for Huntington’s disease. Eur. J. Neurosci. 2017, 45, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Mielcarek, M.; Rattray, I.; Osborne, G.; Jolinon, N.; Dick, J.; Bondulich, M.; Franklin, S.; Ahmed, M.; Benjamin, A.; Goodwin, D.; et al. M09 myostatin inhibition as a novel approach to targeting muscle pathology in HD. J. Neurol. Neurosurg. Psychiatry 2014, 85, A97-A97. [Google Scholar] [CrossRef]
- Dickey, A.S.; La Spada, A.R. Therapy development in huntington disease: From current strategies to emerging opportunities. Am. J. Med. Genet. Part A 2018, 176, 842–861. [Google Scholar] [CrossRef] [PubMed]
- Dash, D.; Mestre, T.A. Therapeutic update on Huntington’s disease: Symptomatic treatments and emerging disease-modifying therapies. Neurotherapeutics 2020. [Google Scholar] [CrossRef]
- Lee, S.J.; McPherron, A.C. Regulation of myostatin activity and muscle growth. Proc. Natl. Acad. Sci. USA 2001, 98, 9306–9311. [Google Scholar] [CrossRef]
- McPherron, A.C.; Lawler, A.M.; Lee, S.J. Regulation of skeletal muscle mass in mice by a new TGF-β superfamily member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef]
- Fão, L.; Rego, A.C. Mitochondrial and redox-based therapeutic strategies in Huntington’s disease. Antioxid. Redox Sign. 2020, 1–24. [Google Scholar] [CrossRef]
- Matthews, R.T.; Yang, L.; Browne, S.; Baik, M.; Beal, M.F. Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc. Natl. Acad. Sci. USA 1998, 95, 8892–8897. [Google Scholar] [CrossRef]
- Schilling, G.; Coonfield, M.L.; Ross, C.A.; Borchelt, D.R. Coenzyme Q10 and remacemide hydrochloride ameliorate motor deficits in a Huntington’s disease transgenic mouse model. Neurosci. Lett. 2001, 315, 149–153. [Google Scholar] [CrossRef]
- Smith, K.M.; Matson, S.; Matson, W.R.; Cormier, K.; Del Signore, S.J.; Hagerty, S.W.; Stack, E.C.; Ryu, H.; Ferrante, R.J. Dose ranging and efficacy study of high-dose coenzyme Q10 formulations in Huntington’s disease mice. Biochim. Biophys. Acta Mol. Basis Dis. 2006, 1762, 616–626. [Google Scholar] [CrossRef]
- McGarry, A.; McDermott, M.; Kieburtz, K.; DeBlieck, E.A.; Beal, F.; Marder, K.; Ross, C.; Shoulson, I.; Gilbert, P.; Mallonee, W.M.; et al. A randomized, double-blind, placebo-controlled trial of coenzyme Q10 in Huntington disease. Neurology 2017, 88, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Menalled, L.B.; Patry, M.; Ragland, N.; Lowden, P.A.S.; Goodman, J.; Minnich, J.; Zahasky, B.; Park, L.; Leeds, J.; Howland, D.; et al. Comprehensive behavioral testing in the R6/2 mouse model of Huntington’s disease shows no benefit from CoQ10 or minocycline. PLoS ONE 2010, 5. [Google Scholar] [CrossRef]
- Verbessem, P.; Lemiere, J.; Eijnde, B.O.; Swinnen, S.; Vanhees, L.; Van Leemputte, M.; Hespel, P.; Dom, R. Creatine supplementation in Huntington’s disease: A placebo-controlled pilot trial. Neurology 2003, 61, 925–930. [Google Scholar] [CrossRef]
- Puri, B.K.; Leavitt, B.R.; Hayden, M.R.; Ross, C.A.; Rosenblatt, A.; Greenamyre, J.T.; Hersch, S.; Vaddadi, K.S.; Sword, A.; Horrobin, D.F.; et al. Ethyl-EPA in Huntington disease: A double-blind, randomized, placebo-controlled trial. Neurology 2005, 65, 286–292. [Google Scholar] [CrossRef]
- Dobson, L.; Träger, U.; Farmer, R.; Hayardeny, L.; Loupe, P.; Hayden, M.R.; Tabrizi, S.J. Laquinimod Dampens hyperactive cytokine production in Huntington’s disease patient myeloid cells. J. Neurochem. 2016, 137, 782–794. [Google Scholar] [CrossRef]
- Ellrichmann, G.; Blusch, A.; Fatoba, O.; Brunner, J.; Hayardeny, L.; Hayden, M.; Sehr, D.; Winklhofer, K.F.; Saft, C.; Gold, R. Laquinimod treatment in the R6/2 mouse model. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Southwell, A.L.; Franciosi, S.; Villanueva, E.B.; Xie, Y.; Winter, L.A.; Veeraraghavan, J.; Jonason, A.; Felczak, B.; Zhang, W.; Kovalik, V.; et al. Anti-semaphorin 4D immunotherapy ameliorates neuropathology and some cognitive impairment in the YAC128 mouse model of Huntington disease. Neurobiol. Dis. 2015, 76, 46–56. [Google Scholar] [CrossRef]
- Fatoba, O.; Ohtake, Y.; Itokazu, T.; Yamashita, T. Immunotherapies in Huntington’s disease and α-synucleinopathies. Front. Immunol. 2020, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, X.; Li, Y.; Tang, T.S.; Bezprozvanny, I. Tetrabenazine is neuroprotective in Huntington’s disease mice. Mol. Neurodegener. 2010, 5, 18. [Google Scholar] [CrossRef]
- Coppen, E.M.; Roos, R.A.C. Current pharmacological approaches to reduce chorea in Huntington’s disease. Drugs 2017, 77, 29–46. [Google Scholar] [CrossRef] [PubMed]
- De Tommaso, M.; Serpino, C.; Sciruicchio, V. Management of Huntington’s disease: Role of tetrabenazine. Ther. Clin. Risk Manag. 2011, 7, 123–129. [Google Scholar]
- Claassen, D.O.; Carroll, B.; De Boer, L.M.; Wu, E.; Ayyagari, R.; Gandhi, S.; Stamler, D. Indirect tolerability comparison of deutetrabenazine and tetrabenazine for Huntington disease. J. Clin. Mov. Disord. 2017, 4, 3. [Google Scholar] [CrossRef]
- Shacham, T.; Sharma, N.; Lederkremer, G.Z. Protein misfolding and ER stress in Huntington’s disease. Front. Mol. Biosci. 2019, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Ramsingh, A.I.; Manley, K.; Rong, Y.; Reilly, A.; Messer, A. Transcriptional dysregulation of inflammatory/immune pathways after active vaccination against Huntington′s disease. Hum. Mol. Genet. 2015, 24, 6186–6197. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.W.; Shirley, T.L.; Wolfgang, W.J.; Kang, X.; Messer, A. DNA vaccination against mutant huntingtin ameliorates the HDR6/2 diabetic phenotype. Mol. Ther. 2003, 7, 572–579. [Google Scholar] [CrossRef]
- Shannon, K.M. Recent Advances in the treatment of Huntington’s disease: Targeting DNA and RNA. CNS Drugs 2020, 34, 219–228. [Google Scholar] [CrossRef]
- Bennett, C.F.; Baker, B.F.; Pham, N.; Swayze, E.; Geary, R.S. Pharmacology of antisense drugs. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 81–105. [Google Scholar] [CrossRef]
- Smith, A.V.; Tabrizi, S.J. Therapeutic antisense targeting of huntingtin. DNA Cell Biol. 2020, 39, 154–158. [Google Scholar] [CrossRef]


{kind=link}
| Name | Type | DNA Construct | CAG Repeats Length | AGE of Onset | Mutant HTT Expression* | Reference |
|---|---|---|---|---|---|---|
| R6/2 mouse | Transgenic | Promoter and exon 1 of human Htt gene | 144 | 9–11 weeks (juvenile HD) | 75% | [43] |
| R6/1 mouse | Transgenic | Promoter and exon 1 of human Htt gene | 116 | 4–5 months | 30% | [43] |
| NLS-N171-82Q mouse | Transgenic | Mouse prion promoter and human sequence for the first 171 N-terminal amino acids of Htt | 82 | 9–11 weeks (juvenile HD) | 10–20% | [44,45,46,47] |
| HdhQn mouse | Knock-in | Knock-in of variable number of CAG repeats into mouse Htt locus | 92–200 | Depending on the length of the CAG repeats | 100% | [40,41,42,48,49] |
| BACHD mouse | Transgenic | Full-length human Htt gene under human Htt promoter | 97 | Slow disease progression and normal life span | 100% | [50,51] |
| YACQn mouse | Transgenic | Full-length human Htt gene with variable number of CAG repeats under human Htt promoter | 48–128 | Depending on the length of the CAG repeats | 30–100% | [52,53,54,55] |
| Mini pigs | Transgenic | Human promoter and human sequence for the first 548 N-terminal amino acids of Htt | 124 | Slow progression of the disease | 100% | [56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bozzi, M.; Sciandra, F. Molecular Mechanisms Underlying Muscle Wasting in Huntington’s Disease. Int. J. Mol. Sci. 2020, 21, 8314. https://doi.org/10.3390/ijms21218314
Bozzi M, Sciandra F. Molecular Mechanisms Underlying Muscle Wasting in Huntington’s Disease. International Journal of Molecular Sciences. 2020; 21(21):8314. https://doi.org/10.3390/ijms21218314
Chicago/Turabian StyleBozzi, Manuela, and Francesca Sciandra. 2020. "Molecular Mechanisms Underlying Muscle Wasting in Huntington’s Disease" International Journal of Molecular Sciences 21, no. 21: 8314. https://doi.org/10.3390/ijms21218314
APA StyleBozzi, M., & Sciandra, F. (2020). Molecular Mechanisms Underlying Muscle Wasting in Huntington’s Disease. International Journal of Molecular Sciences, 21(21), 8314. https://doi.org/10.3390/ijms21218314