Toxicant-Induced Metabolic Alterations in Lipid and Amino Acid Pathways Are Predictive of Acute Liver Toxicity in Rats

 ,
,
Abstract
:1. Introduction
2. Results
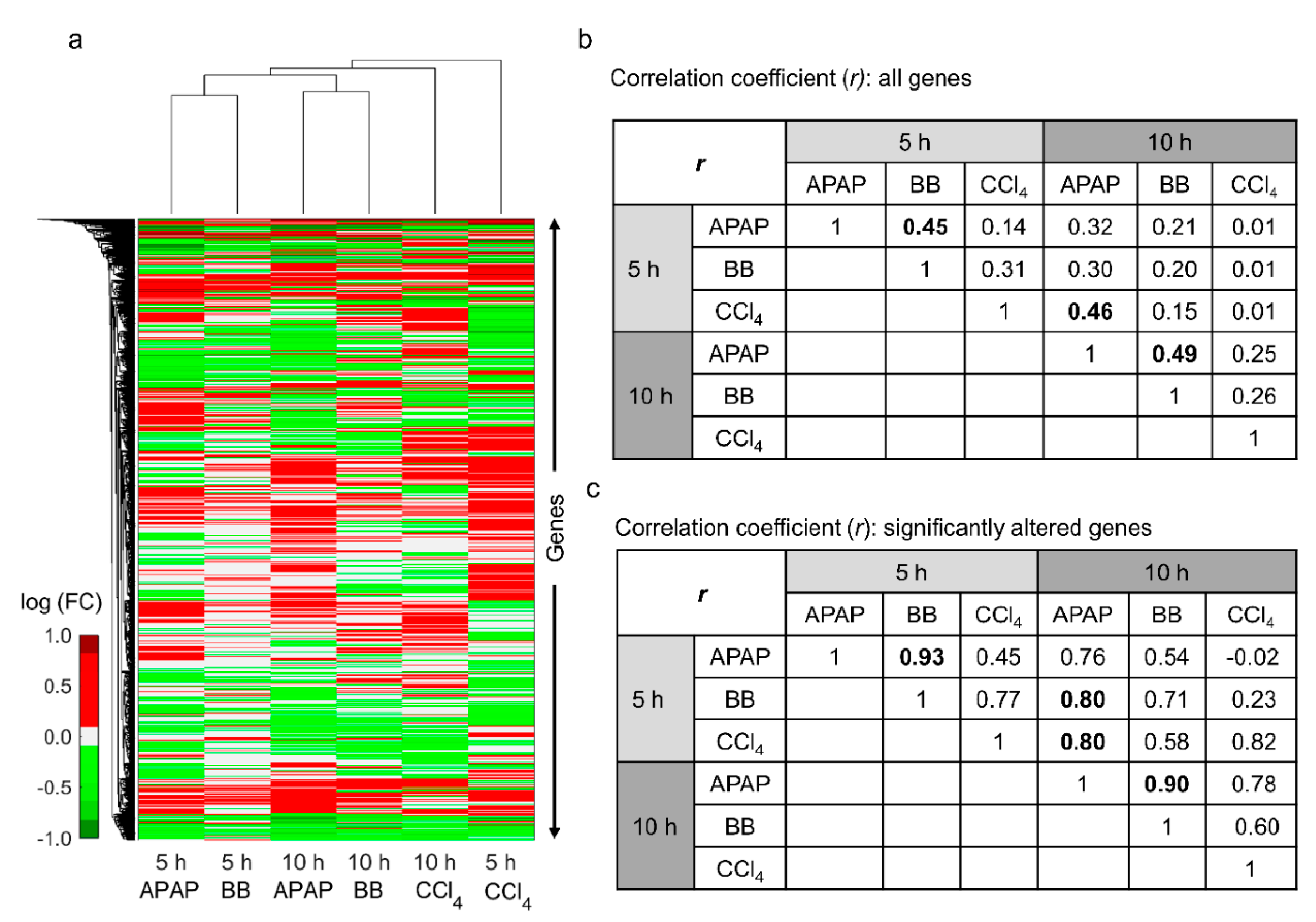
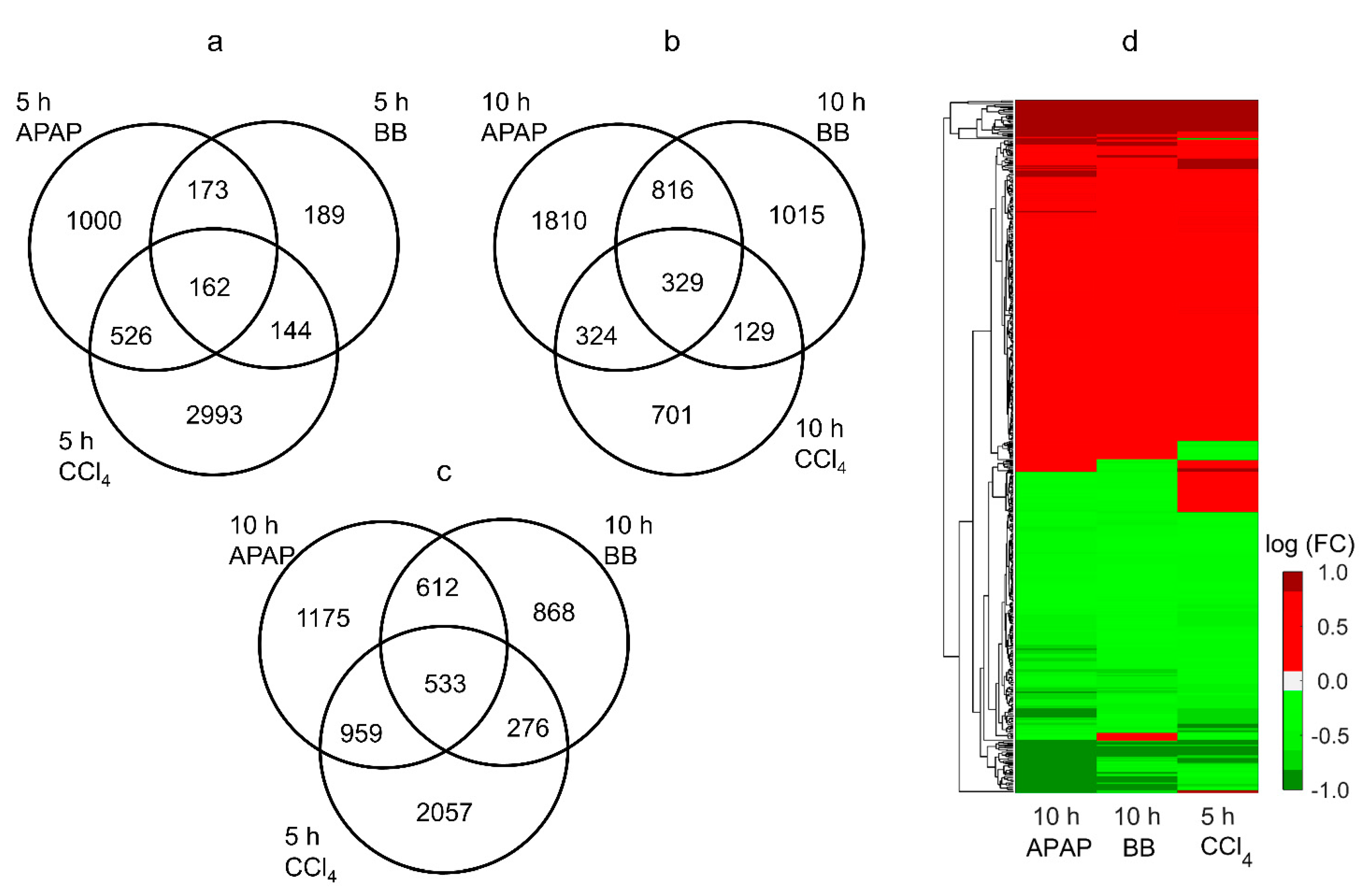
2.1. Common Genes That Changed Significantly Are Highly Correlated between Toxicants
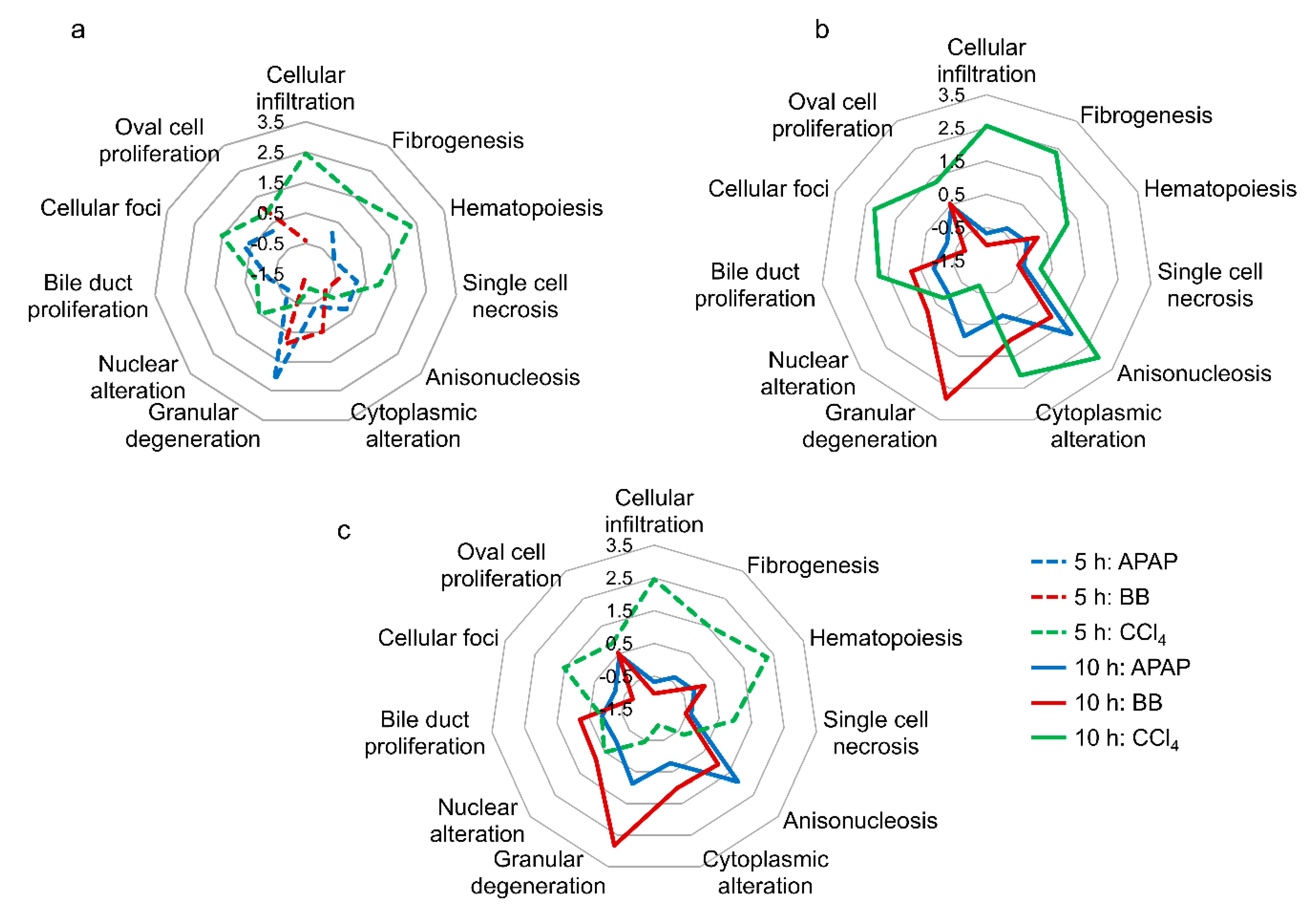
2.2. Toxicant-Induced Changes in Gene Expression Predict Liver Injury Phenotypes
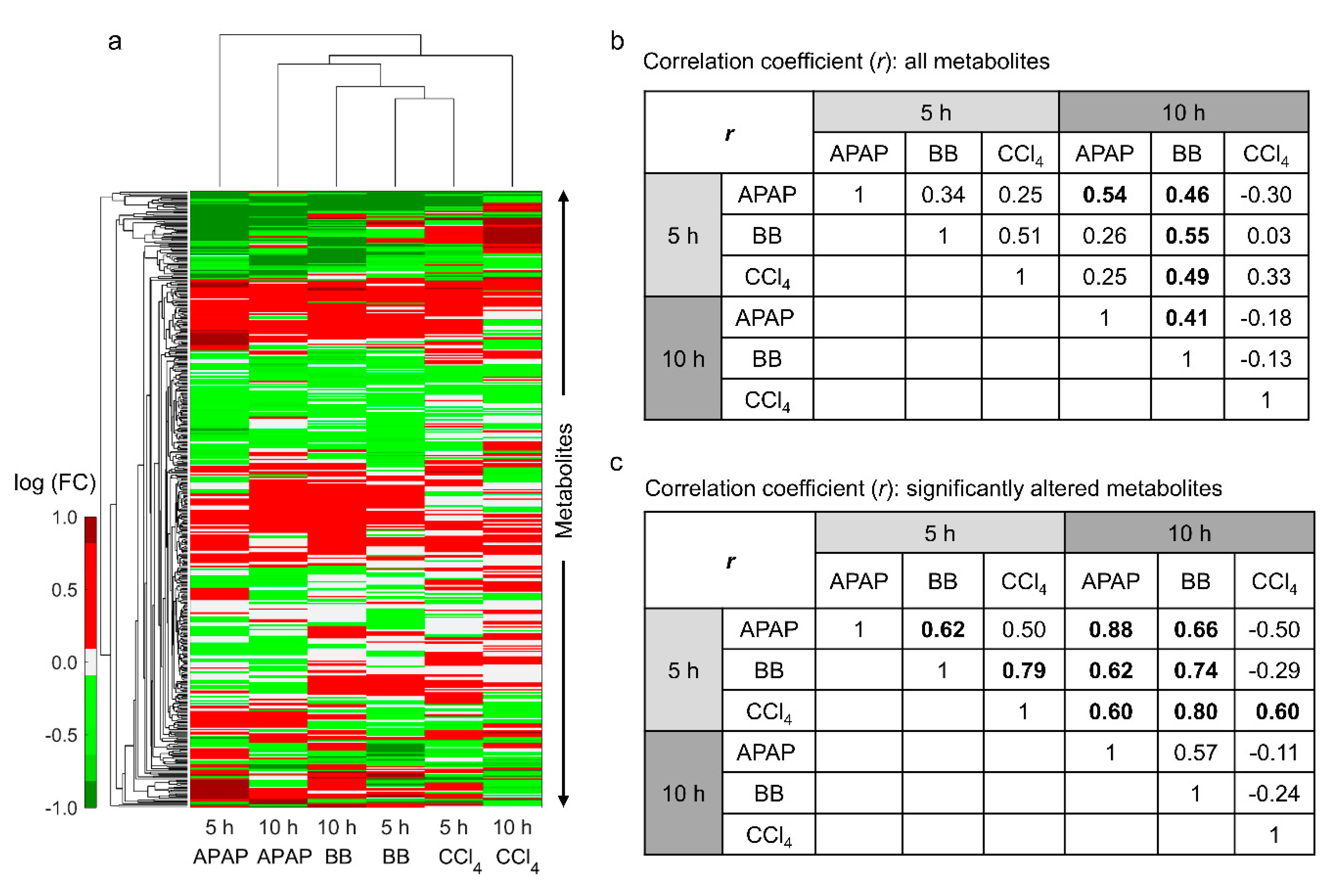
2.3. Correlation between Plasma Metabolite Profiles
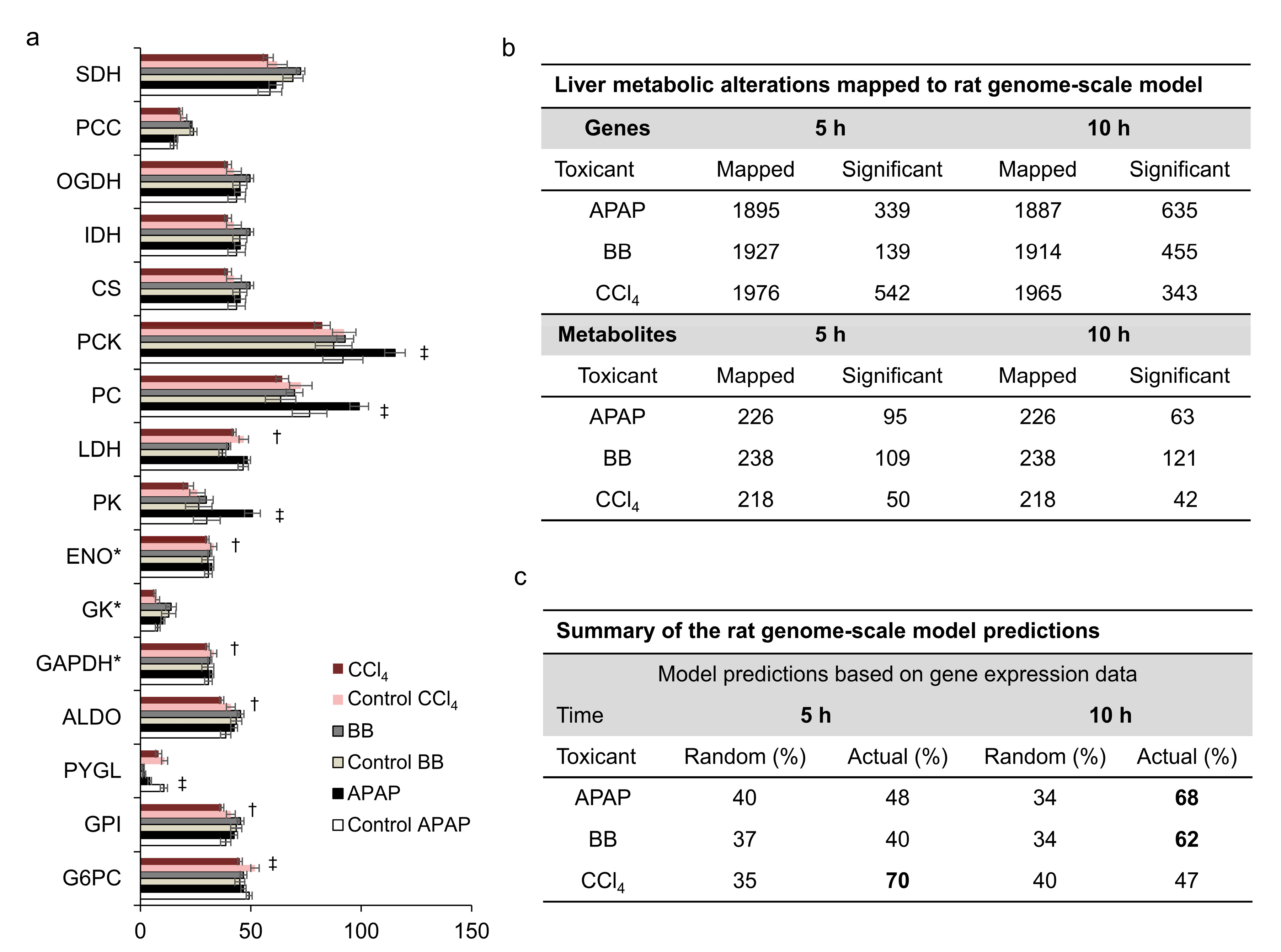
2.4. Toxicants Differed in Altering Hepatic Fluxes in Central Carbon Metabolism
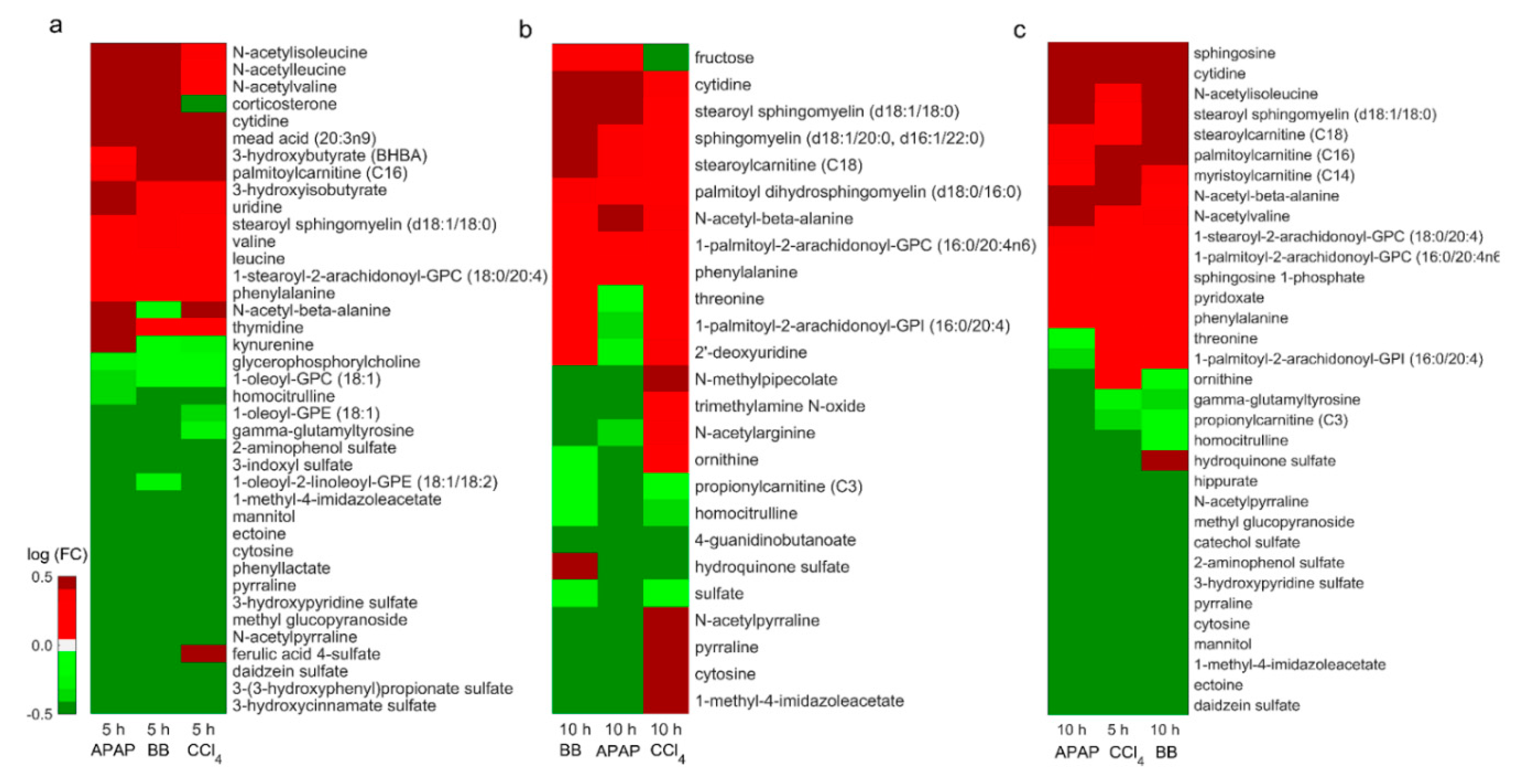
2.5. Identification of Metabolic Changes in Liver Metabolism Associated with Liver Toxicity
2.6. Common Changes in Amino Acid and Lipid Metabolism as Potential Signatures of Liver Injury
3. Discussion
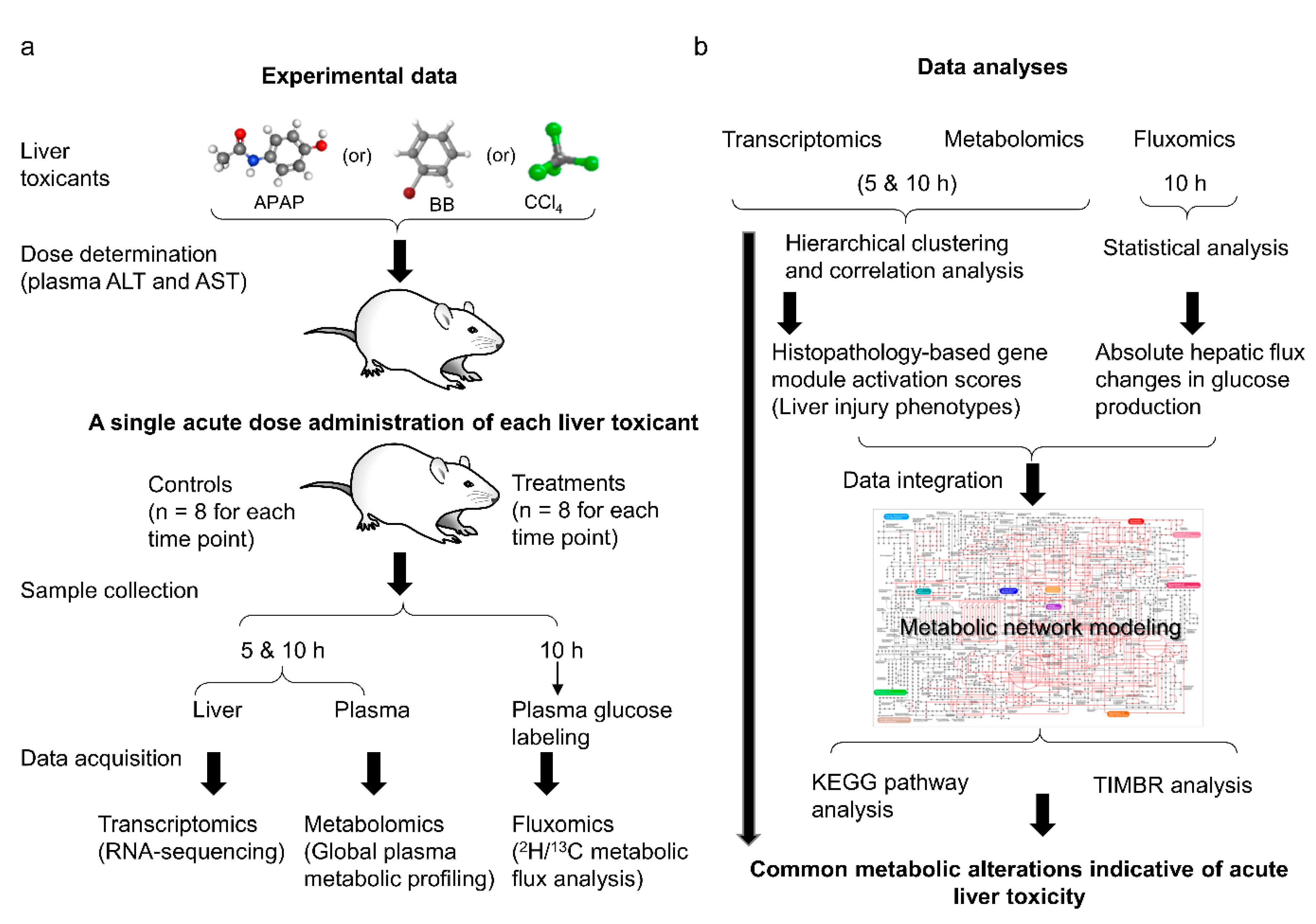
4. Materials and Methods
4.1. Animals and Toxicant Dose Determination
4.2. Studies for Measuring Changes in Liver Gene-Expression and Plasma Metabolic Profiles
4.3. Analysis of RNA-Sequencing and Metabolomic Data
4.4. Tracer Labeling Studies for Measuring Metabolic Flux and Metabolic Flux Analysis
4.5. Rat Metabolic Network and Algorithm for Data Integration and Metabolite Predictions
4.6. KEGG Pathway Analysis
4.7. Liver Injury Module Activation Score
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Benedict, M.; Zhang, X. Non-alcoholic fatty liver disease: An expanded review. World J. Hepatol. 2017, 9, 715–732. [Google Scholar] [CrossRef] [PubMed]
- Byass, P. The global burden of liver disease: A challenge for methods and for public health. BMC Med. 2014, 12, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell Death and Cell Death Responses in Liver Disease: Mechanisms and Clinical Relevance. Gastroenterology 2014, 147, 765–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minemura, M.; Tajiri, K.; Shimizu, Y. Systemic abnormalities in liver disease. World J. Gastroenterol. 2009, 15, 2960–2974. [Google Scholar] [CrossRef] [PubMed]
- Mokhtari, Z.; Gibson, D.L.; Hekmatdoost, A. Nonalcoholic Fatty Liver Disease, the Gut Microbiome, and Diet. Adv. Nutr. 2017, 8, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Marsh, S.; Hu, J.; Feng, W.; Wu, C. The Pathogenesis of Nonalcoholic Fatty Liver Disease: Interplay between Diet, Gut Microbiota, and Genetic Background. Gastroenterol. Res. Pract. 2016, 2016, 2862173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dash, A.; Figler, R.A.; Sanyal, A.J.; Wamhoff, B.R. Drug-induced steatohepatitis. Expert Opin. Drug Metab. Toxicol. 2017, 13, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Rabinowich, L.; Shibolet, O. Drug Induced Steatohepatitis: An Uncommon Culprit of a Common Disease. BioMed Res. Int. 2015, 2015, 168905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlang, B.; Beier, J.I.; Clair, H.B.; Bellis-Jones, H.J.; Falkner, K.C.; McClain, C.J.; Cave, M.C. Toxicant-associated Steatohepatitis. Toxicol. Pathol. 2013, 41, 343–360. [Google Scholar] [CrossRef] [Green Version]
- Al-Eryani, L.; Wahlang, B.; Falkner, K.C.; Guardiola, J.J.; Clair, H.B.; Prough, R.A.; Cave, M. Identification of Environmental Chemicals Associated with the Development of Toxicant-associated Fatty Liver Disease in Rodents. Toxicol. Pathol. 2015, 43, 482–497. [Google Scholar] [CrossRef]
- Heindel, J.J.; Blumberg, B.; Cave, M.; Machtinger, R.; Mantovani, A.; Mendez, M.A.; Nadal, A.; Palanza, P.; Panzica, G.; Sargis, R.; et al. Metabolism disrupting chemicals and metabolic disorders. Reprod. Toxicol. 2017, 68, 3–33. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, J.P.; Lipscomb, J.C.; Wesselkamper, S.C. Putative Mechanisms of Environmental Chemical–Induced Steatosis. Int. J. Toxicol. 2012, 31, 551–563. [Google Scholar] [CrossRef] [Green Version]
- Kneeman, J.M.; Misdraji, J.; Corey, K.E. Secondary causes of nonalcoholic fatty liver disease. Ther. Adv. Gastroenterol. 2011, 5, 199–207. [Google Scholar] [CrossRef] [Green Version]
- McDyre, B.C.; AbdulHameed, M.D.M.; Permenter, M.G.; Dennis, W.E.; Baer, C.E.; Koontz, J.M.; Boyle, M.H.; Wallqvist, A.; Lewis, J.A.; Ippolito, D.L. Comparative Proteomic Analysis of Liver Steatosis and Fibrosis after Oral Hepatotoxicant Administration in Sprague-Dawley Rats. Toxicol. Pathol. 2018, 46, 202–223. [Google Scholar] [CrossRef]
- Jaeschke, H.; Xie, Y.; McGill, M.R. Acetaminophen-induced Liver Injury: From Animal Models to Humans. J. Clin. Transl. Hepatol. 2014, 2, 153–161. [Google Scholar] [CrossRef]
- Weber, L.W.D.; Boll, M.; Stampfl, A. Hepatotoxicity and Mechanism of Action of Haloalkanes: Carbon Tetrachloride as a Toxicological Model. Crit. Rev. Toxicol. 2003, 33, 105–136. [Google Scholar] [CrossRef]
- Chen, I.-S.; Chen, Y.-C.; Chou, C.-H.; Chuang, R.-F.; Sheen, L.-Y.; Chiu, C.-H. Hepatoprotection of silymarin against thioacetamide-induced chronic liver fibrosis. J. Sci. Food Agric. 2012, 92, 1441–1447. [Google Scholar] [CrossRef]
- Blais, E.M.; Rawls, K.D.; Dougherty, B.V.; Li, Z.I.; Kolling, G.L.; Ye, P.; Wallqvist, A.; Papin, J.A. Reconciled rat and human metabolic networks for comparative toxicogenomics and biomarker predictions. Nat. Commun. 2017, 8, 14250. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Agren, R.; Kampf, C.; Asplund, A.; Uhlén, M.; Nielsen, J.J. Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 3083. [Google Scholar] [CrossRef] [Green Version]
- Jerby, L.; Shlomi, T.; Ruppin, E. Computational reconstruction of tissue-specific metabolic models: Application to human liver metabolism. Mol. Syst. Biol. 2010, 6, 401. [Google Scholar] [CrossRef]
- Rawls, K.; Blais, E.M.; Dougherty, B.V.; Vinnakota, K.C.; Pannala, V.R.; Wallqvist, A.; Kolling, G.L.; Papin, J.A. Genome-Scale Characterization of Toxicity-Induced Metabolic Alterations in Primary Hepatocytes. Toxicol. Sci. 2019, 172, 279–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pannala, V.R.; Vinnakota, K.C.; Rawls, K.D.; Estes, S.K.; O’Brien, T.P.; Printz, R.L.; Papin, J.A.; Reifman, J.; Shiota, M.; Young, J.D.; et al. Mechanistic identification of biofluid metabolite changes as markers of acetaminophen-induced liver toxicity in rats. Toxicol. Appl. Pharmacol. 2019, 372, 19–32. [Google Scholar] [CrossRef]
- Beger, R.D.; Bhattacharyya, S.; Yang, X.; Gill, P.S.; Schnackenberg, L.K.; Sun, J.; James, L.P. Translational biomarkers of acetaminophen-induced acute liver injury. Arch. Toxicol. 2015, 89, 1497–1522. [Google Scholar] [CrossRef] [Green Version]
- Minami, K.; Saito, T.; Narahara, M.; Tomita, H.; Kato, H.; Sugiyama, H.; Katoh, M.; Nakajima, M.; Yokoi, T. Relationship between Hepatic Gene Expression Profiles and Hepatotoxicity in Five Typical Hepatotoxicant-Administered Rats. Toxicol. Sci. 2005, 87, 296–305. [Google Scholar] [CrossRef] [Green Version]
- Pannala, V.R.; Estes, S.K.; Rahim, M.; Trenary, I.; O’Brien, T.P.; Shiota, C.; Printz, R.L.; Reifman, J.; Oyama, T.; Shiota, M.; et al. Mechanism-based identification of plasma metabolites associated with liver toxicity. Toxicology 2020, 441, 152493. [Google Scholar] [CrossRef]
- Pannala, V.R.; Wall, M.L.; Estes, S.K.; Trenary, I.; O’Brien, T.P.; Printz, R.L.; Vinnakota, K.C.; Reifman, J.; Shiota, M.; Young, J.D.; et al. Metabolic network-based predictions of toxicant-induced metabolite changes in the laboratory rat. Sci. Rep. 2018, 8, 11678. [Google Scholar] [CrossRef]
- Schyman, P.; Printz, R.L.; Estes, S.K.; O’Brien, T.P.; Shiota, M.; Wallqvist, A. Assessing Chemical-Induced Liver Injury In Vivo From In Vitro Gene Expression Data in the Rat: The Case of Thioacetamide Toxicity. Front. Genet. 2019, 10, 1233. [Google Scholar] [CrossRef]
- Te, J.A.; AbdulHameed, M.D.M.; Wallqvist, A. Systems toxicology of chemically induced liver and kidney injuries: Histopathology-associated gene co-expression modules. J. Appl. Toxicol. 2016, 36, 1137–1149. [Google Scholar] [CrossRef] [Green Version]
- Pannala, V.R.; Vinnakota, K.C.; Estes, S.K.; Trenary, I.; O’brien, T.P.; Printz, R.L.; Papin, J.A.; Reifman, J.; Oyama, T.; Shiota, M.; et al. Genome-Scale Model-Based Identification of Metabolite Indicators for Early Detection of Kidney Toxicity. Toxicol. Sci. 2020, 173, 293–312. [Google Scholar] [CrossRef]
- Jang, J.; Kang, K.; Kim, Y.; Kang, Y.; Lee, I. Reevaluation of Experimental Model of Hepatic Fibrosis Induced by Hepatotoxic Drugs: An Easy, Applicable, and Reproducible Model. Transplant. Proc. 2008, 40, 2700–2703. [Google Scholar] [CrossRef]
- Dong, S.; Chen, Q.-L.; Song, Y.-N.; Sun, Y.; Wei, B.; Li, X.-Y.; Hu, Y.-Y.; Liu, P.; Su, S.-B. Mechanisms of CCl4-induced liver fibrosis with combined transcriptomic and proteomic analysis. J. Toxicol. Sci. 2016, 41, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Hinson, J.A.; Roberts, D.W.; James, L.P. Mechanisms of Acetaminophen-Induced Liver Necrosis. Handb. Exp. Pharmacol. 2010, 196, 369–405. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, J.R.; Jollow, D.J.; Potter, W.Z.; Gillette, J.R.; Brodie, B.B. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J. Pharmacol. Exp. Ther. 1973, 187, 211–217. [Google Scholar]
- Jollow, D.J.; Mitchell, J.R.; Zampaglione, N.; Gillette, J.R. Bromobenzene-Induced Liver Necrosis. Protective Role of Glutathione and Evidence for 3,4-Bromobenzene Oxide as the Hepatotoxic Metabolite. Pharmacology 1974, 11, 151–169. [Google Scholar] [CrossRef]
- Gao, Y.; Cao, Z.; Yang, X.; Abdelmegeed, M.A.; Sun, J.; Chen, S.; Beger, R.D.; Davis, K.; Salminen, W.F.; Song, B.-J.; et al. Proteomic analysis of acetaminophen-induced hepatotoxicity and identification of heme oxygenase 1 as a potential plasma biomarker of liver injury. Proteomics Clin. Appl. 2017, 11, 1–2. [Google Scholar] [CrossRef]
- Schwender, J.; Konig, C.; Klapperstuck, M.; Heinzel, N.; Munz, E.; Hebbelmann, I.; Hay, J.O.; Denolf, P.; De Bodt, S.; Redestig, H.; et al. Transcript abundance on its own cannot be used to infer fluxes in central metabolism. Front. Plant Sci. 2014, 5, 668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, A.; Jaeschke, H. Mechanisms of acetaminophen hepatotoxicity and their translation to the human pathophysiology. J. Clin. Transl. Res. 2017, 3 (Suppl. 1), 157–169. [Google Scholar] [CrossRef]
- Heijne, W.H.M.; Stierum, R.H.; Slijper, M.; Van Bladeren, P.J.; Van Ommen, B. Toxicogenomics of bromobenzene hepatotoxicity: A combined transcriptomics and proteomics approach. Biochem. Pharmacol. 2003, 65, 857–875. [Google Scholar] [CrossRef]
- Jeong, H.G. Inhibition of cytochrome P450 2E1 expression by oleanolic acid: Hepatoprotective effects against carbon tetrachloride-induced hepatic injury. Toxicol. Lett. 1999, 105, 215–222. [Google Scholar] [CrossRef]
- Krahenbuhl, S.; Reichen, J. Carnitine metabolism in patients with chronic liver disease. Hepatology 1997, 25, 148–153. [Google Scholar] [CrossRef]
- Amodio, P.; Angeli, P.; Merkel, C.; Menon, F.; Gatta, A. Plasma Carnitine Levels in Liver Cirrhosis: Relationship with Nutritional Status and Liver Damage. Clin. Chem. Lab. Med. 1990, 28, 619–626. [Google Scholar] [CrossRef] [Green Version]
- Yoo, H.J.; Jung, K.J.; Kim, M.; Kim, M.; Kang, M.; Jee, S.H.; Choi, Y.; Lee, J.H. Liver Cirrhosis Patients Who Had Normal Liver Function Before Liver Cirrhosis Development Have the Altered Metabolic Profiles Before the Disease Occurrence Compared to Healthy Controls. Front. Physiol. 2019, 10, 1421. [Google Scholar] [CrossRef]
- Ishay, Y.; Nachman, D.; Khoury, T.; Ilan, Y. The role of the sphingolipid pathway in liver fibrosis: An emerging new potential target for novel therapies. Am. J. Physiol. Physiol. 2020, 318, C1055–C1064. [Google Scholar] [CrossRef]
- Kleuser, B. Divergent Role of Sphingosine 1-Phosphate in Liver Health and Disease. Int. J. Mol. Sci. 2018, 19, 722. [Google Scholar] [CrossRef] [Green Version]
- Adeva-Andany, M.M.; Carneiro-Freire, N.; Seco-Filgueira, M.; Fernandez-Fernandez, C.; Mourino-Bayolo, D. Mitochondrial beta-oxidation of saturated fatty acids in humans. Mitochondrion 2019, 46, 73–90. [Google Scholar] [CrossRef]
- Satapathy, S.K.; Sanyal, A. Epidemiology and Natural History of Nonalcoholic Fatty Liver Disease. Semin. Liver Dis. 2015, 35, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.; Ouro, A.; Ala-Ibanibo, L.; Presa, N.; Delgado, T.C.; Martínez-Chantar, M.L. Sphingolipids in Non-Alcoholic Fatty Liver Disease and Hepatocellular Carcinoma: Ceramide Turnover. Int. J. Mol. Sci. 2019, 21, 40. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Zheng, S.; You, H.; Liu, X.; Lin, M.; Yang, L.; Li, L. Sphingosine 1-phosphate (S1P)/S1P receptors are involved in human liver fibrosis by action on hepatic myofibroblasts motility. J. Hepatol. 2011, 54, 1205–1213. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Pimentel, H.; Bray, N.L.; Puente, S.; Melsted, P.; Pachter, L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods 2017, 14, 687–690. [Google Scholar] [CrossRef]
- Yates, A.D.; Achuthan, P.; Akanni, W.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; et al. Ensembl 2020. Nucleic Acids Res. 2020, 48, D682–D688. [Google Scholar] [CrossRef]
- Hasenour, C.M.; Wall, M.L.; Ridley, D.E.; Hughey, C.C.; James, F.D.; Wasserman, D.H.; Young, J.D. Mass spectrometry-based microassay of 2H and 13C plasma glucose labeling to quantify liver metabolic fluxes in vivo. Am. J. Physiol. Metab. 2015, 309, E191–E203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, J.D. INCA: A computational platform for isotopically non-stationary metabolic flux analysis. Bioinformatics 2014, 30, 1333–1335. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Woo, H.J.; Yu, X.; Oyama, T.; Wallqvist, A.; Reifman, J. A strategy for evaluating pathway analysis methods. BMC Bioinform. 2017, 18, 453. [Google Scholar] [CrossRef] [Green Version]
- Schyman, P.; Printz, R.L.; Estes, S.K.; Boyd, K.L.; Shiota, M.; Wallqvist, A. Identification of the Toxicity Pathways Associated With Thioacetamide-Induced Injuries in Rat Liver and Kidney. Front. Pharmacol. 2018, 9, 1272. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Main Pathway | Subordinate Pathway | 10 h: APAP | 10 h: BB | 5 h: CCl4 | |||
|---|---|---|---|---|---|---|---|
| Genes | AFC z-score | Genes | AFC z-score | Genes | AFC z-score | ||
| Lipid | Glycerophospholipid metabolism | 28 | 1.42 | 26 | 1.75 | 21 | 0.10 |
| Glycerolipid metabolism | 18 | 0.31 | 15 | 2.84 | 14 | −0.97 | |
| Steroid biosynthesis | 12 | −4.42 | 13 | −4.09 | 6 | −1.58 | |
| Steroid hormone biosynthesis | 26 | −3.49 | 11 | −1.01 | 11 | −0.90 | |
| Fatty acid metabolism | 14 | −1.89 | 17 | −1.83 | 12 | −1.96 | |
| Biosynthesis of unsaturated fatty acids | 10 | −1.23 | 13 | −0.21 | 7 | −1.63 | |
| Fatty acid degradation | 14 | −0.03 | 12 | −0.07 | 8 | −2.06 | |
| Arachidonic acid metabolism | 15 | −0.42 | 12 | −0.71 | |||
| Fatty acid elongation | 8 | 1.08 | 9 | 1.30 | |||
| Sphingolipid metabolism | 12 | 0.96 | 13 | −1.81 | |||
| Ether lipid metabolism | 7 | −0.96 | 12 | 0.03 | |||
| Fatty acid biosynthesis | 5 | −1.57 | 5 | −0.93 | |||
| Primary bile acid biosynthesis | 5 | 0.57 | 5 | −1.49 | |||
| AA | Glycine, serine, and threonine metabolism | 17 | 1.63 | 7 | 1.15 | 14 | 1.51 |
| Cysteine and methionine metabolism | 17 | 2.16 | 13 | 1.47 | 7 | 1.26 | |
| Arginine and proline metabolism | 12 | 0.45 | 12 | 0.40 | 12 | 1.30 | |
| Glutathione metabolism | 17 | 0.42 | 8 | 0.76 | 13 | 2.94 | |
| Alanine, aspartate, and glutamate metabolism | 11 | 2.28 | 7 | 2.15 | 11 | 0.55 | |
| Tryptophan metabolism | 15 | −3.63 | 10 | −1.46 | 11 | −1.99 | |
| Arginine biosynthesis | 6 | 2.40 | 5 | 0.06 | |||
| Selenocompound metabolism | 8 | −1.03 | 5 | 1.29 | |||
| Valine, leucine, and isoleucine degradation | 10 | −2.33 | 16 | −2.16 | |||
| beta-Alanine metabolism | 6 | 0.71 | 11 | 0.75 | |||
| Carb | Amino sugar and nucleotide sugar metabolism | 15 | −1.40 | 15 | −2.64 | 12 | 2.46 |
| Phosphatidylinositol signaling system | 14 | −0.10 | 13 | 1.54 | 19 | −1.73 | |
| Fructose and mannose metabolism | 10 | 0.31 | 12 | 0.19 | 8 | 2.85 | |
| Starch and sucrose metabolism | 9 | −1.97 | 8 | −2.03 | 9 | −0.44 | |
| Glyoxylate and dicarboxylate metabolism | 11 | −1.56 | 11 | −2.21 | 10 | −1.44 | |
| Glycolysis/Gluconeogenesis | 18 | 0.18 | 18 | −2.22 | 13 | 1.17 | |
| Pyruvate metabolism | 17 | −0.67 | 15 | −2.36 | 7 | 0.13 | |
| Ascorbate and aldarate metabolism | 8 | −1.12 | 6 | −0.40 | |||
| Propanoate metabolism | 7 | −1.52 | 9 | −1.39 | |||
| Pentose phosphate pathway | 10 | −0.41 | 7 | 1.46 | |||
| Nucleotide | Purine metabolism | 44 | 0.39 | 24 | 0.52 | 43 | 0.09 |
| Pyrimidine metabolism | 29 | −0.57 | 15 | −0.15 | 24 | 0.08 | |
| 10 h: APAP | 10 h: BB | 5 h: CCl4 | |||||
|---|---|---|---|---|---|---|---|
| Pathway | Metabolite | log(FC) | TIMBR Score | log(FC) | TIMBR Score | log(FC) | TIMBR Score |
| Lipid | L-palmitoylcarnitine | 0.52 | 0.18 | 0.68 | 0.58 | 0.70 | 0.70 |
| stearoyl sphingomyelin (d18:1/18:0) | 0.83 | 0.75 | 0.78 | 0.69 | 0.39 | 0.07 | |
| sphingosine | 0.62 | 1.81 | 1.36 | 0.43 | 1.20 | 0.15 | |
| sphingosine-1-phosphate | 0.37 | 1.73 | 0.41 | 0.43 | 0.32 | 0.02 | |
| stearoylcarnitine | 0.46 | 0.27 | 0.83 | 0.61 | 0.45 | 0.75 | |
| O-propanoylcarnitine | −0.69 | −0.32 | −0.27 | 0.61 | −0.54 | 0.73 | |
| arachidonate | 0.51 | −0.29 | 0.34 | 0.54 | - | ||
| cholesterol | 0.28 | 0.20 | 0.48 | −2.68 | |||
| choline | 0.29 | 2.46 | 0.16 | 0.81 | |||
| L-carnitine | −0.38 | −0.35 | −0.23 | 0.63 | |||
| O-butanoylcarnitine | −0.60 | −0.24 | −0.40 | 0.61 | |||
| chenodeoxycholic acid | −1.64 | −1.04 | −1.43 | −1.68 | |||
| (R)-3-hydroxybutanoate | 1.50 | 0.47 | 0.77 | 0.95 | |||
| 16-hydroxyhexadecanoic acid | 0.45 | 0.46 | 0.56 | 0.81 | |||
| 3-hydroxyisobutyrate | 1.03 | 0.62 | 0.53 | 0.72 | |||
| 3-methyl-2-oxobutyrate | 0.30 | 0.61 | 0.29 | 0.69 | |||
| sphinganine-1-phosphate | 1.32 | 0.46 | 0.79 | 0.04 | |||
| L-oleoylcarnitine | 0.55 | 0.60 | 0.34 | 0.72 | |||
| mead acid | 0.43 | −0.36 | 0.78 | −0.60 | |||
| tauroursodeoxycholate | 0.55 | −1.30 | 0.97 | −1.96 | |||
| Amino Acid | methylimidazoleacetic acid | −1.02 | −0.49 | −3.64 | 0.45 | −2.12 | 0.94 |
| creatine | 0.43 | 1.35 | 0.38 | 0.70 | |||
| GSSG | −1.15 | −0.60 | −0.58 | 0.53 | |||
| aspartate | −0.38 | −0.49 | −0.17 | 0.48 | |||
| citrulline | −0.20 | −0.67 | −0.29 | 0.63 | |||
| cysteine | −0.56 | −0.53 | −0.47 | 0.62 | |||
| guanidinoacetate | −0.79 | −0.39 | −0.45 | 0.36 | |||
| ornithine | −0.64 | −0.79 | −0.23 | 0.62 | |||
| arginine | −0.27 | 0.94 | −0.17 | 0.67 | |||
| 5-oxoproline | 0.24 | 0.53 | 0.23 | 1.34 | |||
| glutamine | 0.14 | 0.51 | 0.24 | 1.15 | |||
| serotonin | 3.31 | 0.58 | 2.21 | 0.62 | |||
| urocanate | 0.40 | 0.43 | 1.23 | 0.85 | |||
| kynurenine | −0.30 | 0.59 | −0.45 | 0.80 | |||
| Carb | fructose | 0.53 | 0.36 | 0.56 | 0.55 | ||
| D-glucitol | −2.47 | 0.61 | −2.32 | 0.69 | |||
| Nucleo-tide | cytidine | 0.99 | 1.18 | 1.14 | 0.38 | 0.64 | −0.05 |
| spermidine | 1.10 | 0.82 | 0.96 | 0.14 | |||
| uracil | 0.51 | 0.42 | 0.41 | 0.55 | |||
| thymidine | 0.33 | 0.36 | 0.31 | 0.14 | |||
| urate | 0.38 | 0.80 | 0.34 | 0.01 | |||
| uridine | 0.10 | 0.26 | 0.46 | 0.12 | |||
| thymine | 0.33 | 0.46 | 0.61 | −0.30 | |||
| Cofactor | 4-pyridoxate | 0.39 | −0.67 | 0.34 | 0.61 | 0.39 | 0.69 |
| D-gluconic acid | 1.16 | 0.33 | 0.57 | 0.51 | |||
| oxalate | 0.23 | 0.18 | 0.43 | 0.40 | |||
| threonate | 0.42 | 0.00 | 0.58 | 0.35 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pannala, V.R.; Estes, S.K.; Rahim, M.; Trenary, I.; O’Brien, T.P.; Shiota, C.; Printz, R.L.; Reifman, J.; Shiota, M.; Young, J.D.; et al. Toxicant-Induced Metabolic Alterations in Lipid and Amino Acid Pathways Are Predictive of Acute Liver Toxicity in Rats. Int. J. Mol. Sci. 2020, 21, 8250. https://doi.org/10.3390/ijms21218250
Pannala VR, Estes SK, Rahim M, Trenary I, O’Brien TP, Shiota C, Printz RL, Reifman J, Shiota M, Young JD, et al. Toxicant-Induced Metabolic Alterations in Lipid and Amino Acid Pathways Are Predictive of Acute Liver Toxicity in Rats. International Journal of Molecular Sciences. 2020; 21(21):8250. https://doi.org/10.3390/ijms21218250
Chicago/Turabian StylePannala, Venkat R., Shanea K. Estes, Mohsin Rahim, Irina Trenary, Tracy P. O’Brien, Chiyo Shiota, Richard L. Printz, Jaques Reifman, Masakazu Shiota, Jamey D. Young, and et al. 2020. "Toxicant-Induced Metabolic Alterations in Lipid and Amino Acid Pathways Are Predictive of Acute Liver Toxicity in Rats" International Journal of Molecular Sciences 21, no. 21: 8250. https://doi.org/10.3390/ijms21218250