The Tumor Microenvironment of Primitive and Metastatic Breast Cancer: Implications for Novel Therapeutic Strategies

 ,
,  , ,
, , 
Abstract
:1. Introduction
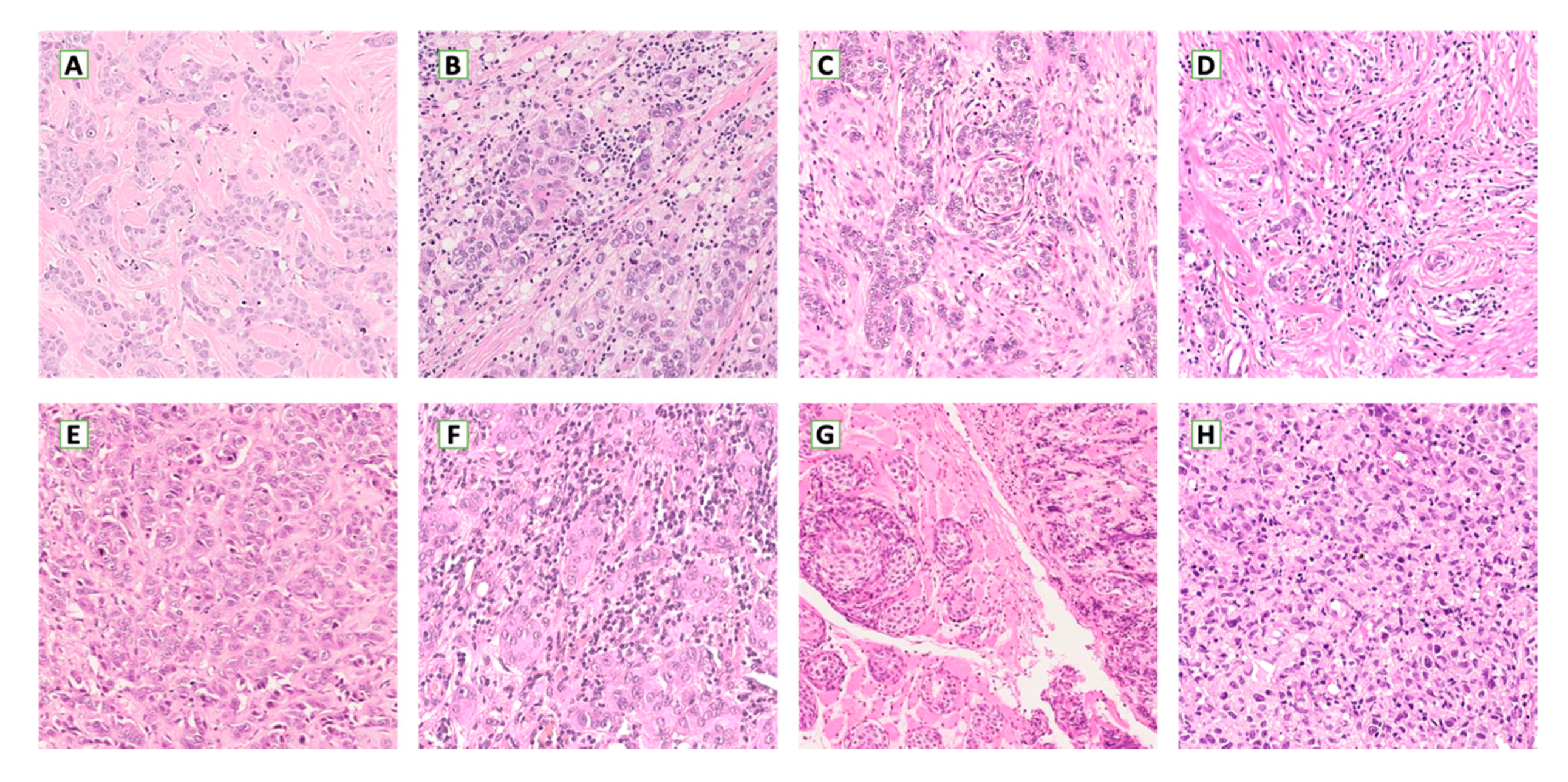
2. Primary Breast Cancer Microenvironment
2.1. Breast Cancer-Associated Fibroblasts
2.1.1. TGF-β1 (Transforming Growth Factor β1)
2.1.2. HGF (Hepatocyte Growth Factor)
2.1.3. IL32 (Interleukin 32)
2.1.4. IL6 (Interleukin 6)
2.2. Breast Cancer-Associated Leukocytes
2.3. Breast Cancer-Associated Endothelium
2.4. Breast Cancer-Associated Mesenchymal Stem Cells
2.5. Breast Cancer-Associated Extracellular Matrix
3. Engineered Breast Cancer Models

{kind=link}
{kind=link}
| TME Component | Main Functions | Ref. |
|---|---|---|
| Fibroblasts | Promotion of EMT. Enhancement of proliferation rate. Induction of ECM remodeling. Lowering of apoptotic rate. Promotion of angiogenesis. | [10,11,12,13,14,15,16,17,18] |
| Macrophages | Promotion of angiogenesis. Immunosuppression. Promotion of EMT. Enhancement of cancer motility. | [21,22,23,24] |
| Lymphocytes | Deregulation of immune checkpoints in favor of immunosuppression. | [24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53] |
| Endothelial cells | Angiogenesis Enhanced proliferation rate and metastatic potential through paracrine signaling. | [57,58,59] |
| Mesenchymal stem cells | Enhancement of proliferation rate via exosomes. Immunosuppression. Induction of dormancy through cannibalization. | [60,61,62,63,64,65,66,67,68,69] |
| Extracellular matrix | Enhancement of cancer motility. | [71,72,73,74,75,76,77] |
4. Interaction Mechanisms in the Breast Cancer Tumor Microenvironment
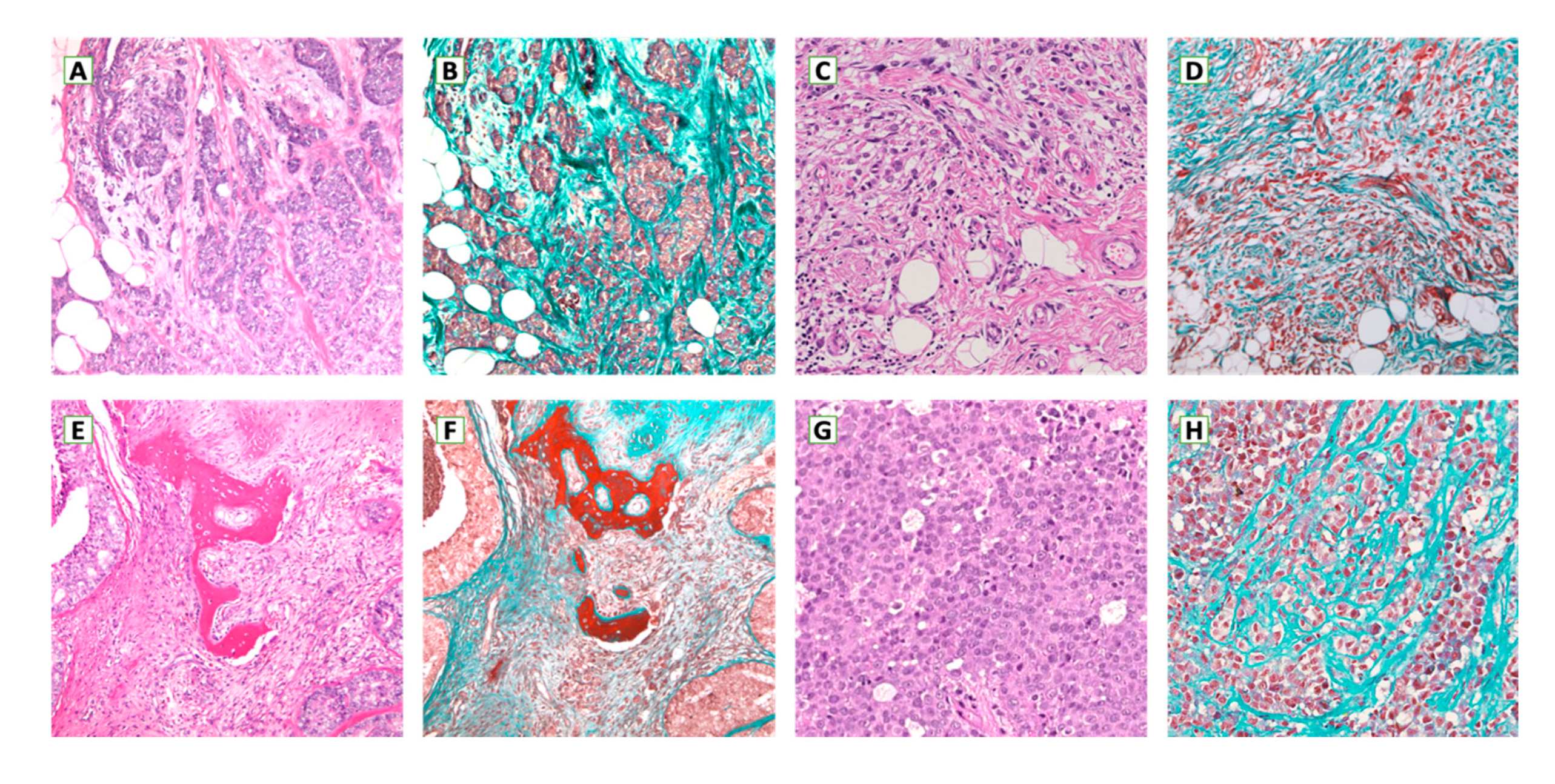
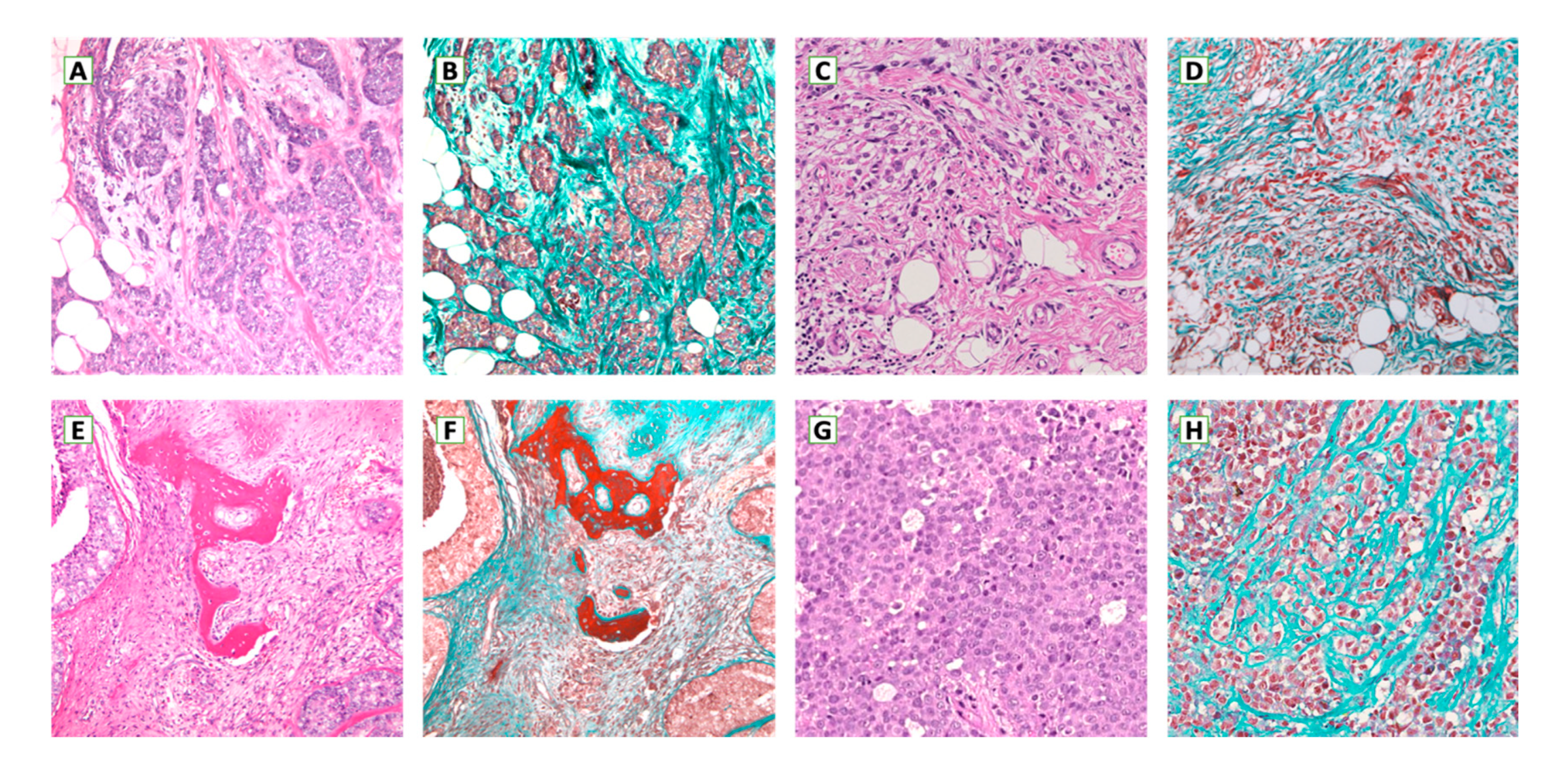
5. Metastatic Breast Cancer Microenvironment
6. Relationship between the Microenvironment and the Molecular Subtypes of Breast Cancer
7. Therapeutic Implications
8. Conclusions
Funding
Conflicts of Interest
References
- Bray, F.; Me, J.F.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soysal, S.D.; Tzankov, A.; Muenst, S.E. Role of the Tumor Microenvironment in Breast Cancer. Pathobiology 2015, 82, 142–152. [Google Scholar] [CrossRef]
- Wang, J.-J.; Lei, K.-F.; Han, F. Tumor microenvironment: Recent advances in various cancer treatments. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 3855–3864. [Google Scholar] [PubMed]
- Paget, S. The distribution of secondary growths in cancer of the breast. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; Macdonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nat. Cell Biol. 2005, 438, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conklin, M.W.; Keely, P.J. Why the stroma matters in breast cancer: Insights into breast cancer patient outcomes through the examination of stromal biomarkers. Cell Adhes. Migr. 2012, 6, 249–260. [Google Scholar] [CrossRef] [Green Version]
- Polyak, K.; Kalluri, R. The Role of the Microenvironment in Mammary Gland Development and Cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a003244. [Google Scholar] [CrossRef] [Green Version]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model. Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef] [Green Version]
- Roche, J. The Epithelial-to-Mesenchymal Transition in Cancer. Cancers 2018, 10, 52. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Xiao, C.-H.; Tan, L.-D.; Wang, Q.-S.; Li, X.-Q.; Feng, Y.-M. Cancer-associated fibroblasts induce epithelial–mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. Br. J. Cancer 2013, 110, 724–732. [Google Scholar] [CrossRef] [Green Version]
- Jedeszko, C.; Victor, B.C.; Podgorski, I.; Sloane, B.F. Fibroblast Hepatocyte Growth Factor Promotes Invasion of Human Mammary Ductal Carcinoma In situ. Cancer Res. 2009, 69, 9148–9155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brauer, H.A.; Makowski, L.; Hoadley, K.A.; Casbas-Hernandez, P.; Lang, L.J.; Roman-Perez, E.; D’Arcy, M.; Freemerman, A.J.; Perou, C.M.; Troestere, M.A. Impact of tumor microenvironment and epithelial phenotypes on metabolism in breast cancer. Clin. Cancer Res. 2013, 19, 571–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, S.; Hou, Y.; Fu, L.; Xi, L.; Yang, D.; Zhao, M.; Qin, Y.; Sun, K.; Teng, Y.; Liu, M. Cancer-associated fibroblast (CAF)-derived IL32 promotes breast cancer cell invasion and metastasis via integrin β3–p38 MAPK signalling. Cancer Lett. 2019, 442, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chen, F.; Tang, L. IL-32 promotes breast cancer cell growth and invasiveness. Oncol. Lett. 2014, 9, 305–307. [Google Scholar] [CrossRef] [Green Version]
- Masjedi, A.; Hashemi, V.; Hojjat-Farsangi, M.; Ghalamfarsa, G.; Azizi, G.; Yousefi, M.; Jadidi-Niaragh, F. The significant role of interleukin-6 and its signaling pathway in the immunopathogenesis and treatment of breast cancer. Biomed. Pharmacother. 2018, 108, 1415–1424. [Google Scholar] [CrossRef]
- Banerjee, K.; Resat, H. Constitutive activation of STAT3 in breast cancer cells: A review. Int. J. Cancer 2016, 138, 2570–2578. [Google Scholar] [CrossRef]
- Sun, X.; Qu, Q.; Lao, Y.; Zhang, M.; Yin, X.; Zhu, H.; Wang, Y.; Yang, J.; Yi, J.; Hao, M. Tumor suppressor HIC1 is synergistically compromised by cancer-associated fibroblasts and tumor cells through the IL-6/pSTAT3 axis in breast cancer. BMC Cancer 2019, 19, 1180. [Google Scholar] [CrossRef]
- Ravelli, A.; Roviello, G.; Cretella, D.; Cavazzoni, A.; Biondi, A.; Cappelletti, M.R.; Zanotti, L.; Ferrero, G.; Ungari, M.; Zanconati, F.; et al. Tumor-infiltrating lymphocytes and breast cancer: Beyond the prognostic and predictive utility. Tumor Biol. 2017, 39, 1010428317695023. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Qu, J.; Sun, Y.; Wang, J.; Liu, X.; Wang, F.; Zhang, H.; Wang, W.; Ma, X.; Gao, X.; et al. Prognostic significance of tumor-associated macrophages in breast cancer: A meta-analysis of the literature. Oncotarget 2017, 8, 30576–30586. [Google Scholar] [CrossRef] [Green Version]
- Wyckoff, J.; Wang, W.; Lin, E.Y.; Wang, Y.; Pixley, F.; Stanley, E.R.; Graf, T.; Pollard, J.W.; Segall, J.; Condeelis, J. A Paracrine Loop between Tumor Cells and Macrophages Is Required for Tumor Cell Migration in Mammary Tumors. Cancer Res. 2004, 64, 7022–7029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.; Liu, Q.; Chen, J.; Chen, J.; Chen, F.; He, C.; Huang, D.; Wu, W.; Lin, L.; Huang, W.; et al. A Positive Feedback Loop between Mesenchymal-like Cancer Cells and Macrophages Is Essential to Breast Cancer Metastasis. Cancer Cell 2014, 25, 605–620. [Google Scholar] [CrossRef] [Green Version]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 Blocks CD8+ T Cell-Dependent Responses to Chemotherapy by Suppressing IL-12 Expression in Intratumoral Dendritic Cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef] [Green Version]
- Marigo, I.; Trovato, R.; Hofer, F.; Ingangi, V.; DeSantis, G.; Leone, K.; De Sanctis, F.; Ugel, S.; Cane, S.; Simonelli, A.; et al. The Disabled homolog 2 controls pro-metastatic activity of tumor-associated macrophages. Cancer Discov. 2020. [Google Scholar] [CrossRef] [PubMed]
- Dieci, M.V.; Radosevic-Robin, N.; Fineberg, S.; Eynden, G.V.D.; Ternes, N.; Penault-Llorca, F.; Pruneri, G.; D’Alfonso, T.M.; DeMaria, S.; Castaneda, C.; et al. Update on tumor-infiltrating lymphocytes (TILs) in breast cancer, including recommendations to assess TILs in residual disease after neoadjuvant therapy and in carcinoma in situ: A report of the International Immuno-Oncology Biomarker Working Group on Breast Cancer. Semin. Cancer Biol. 2018, 52, 16–25. [Google Scholar] [CrossRef]
- Folgueira, M.A.A.K.; Maistro, S.; Katayama, M.L.H.; Roela, R.A.; Mundim, F.G.L.; Nanogaki, S.; De Bock, G.H.; Brentani, M.M. Markers of breast cancer stromal fibroblasts in the primary tumour site associated with lymph node metastasis: A systematic review including our case series. Biosci. Rep. 2013, 33, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Eiro, N.; Gonzalez, L.O.; Fraile, M.; Cid, S.; Schneider, J.; Vizoso, F.J. Breast Cancer Tumor Stroma: Cellular Components, Phenotypic Heterogeneity, Intercellular Communication, Prognostic Implications and Therapeutic Opportunities. Cancers 2019, 11, 664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kos, Z.; The International Immuno-Oncology Biomarker Working Group; Roblin, E.; Kim, R.S.; Michiels, S.; Gallas, B.D.; Chen, W.; Van De Vijver, K.K.; Goel, S.; Adams, S.; et al. Pitfalls in assessing stromal tumor infiltrating lymphocytes (sTILs) in breast cancer. NPJ Breast Cancer 2020, 6, 1–16. [Google Scholar] [CrossRef]
- Bohling, S.D.; Allison, K.H. Immunosuppressive regulatory T cells are associated with aggressive breast cancer phenotypes: A potential therapeutic target. Mod. Pathol. 2008, 21, 1527–1532. [Google Scholar] [CrossRef]
- Salgado, R.; Denkert, C.; DeMaria, S.; Sirtaine, N.; Klauschen, F.; Pruneri, G.; Wienert, S.; Eynden, G.V.D.; Baehner, F.L.; Penault-Llorca, F.; et al. The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: Recommendations by an International TILs Working Group 2014. Ann. Oncol. 2015, 26, 259–271. [Google Scholar] [CrossRef]
- Loi, S.; Drubay, D.; Adams, S.; Pruneri, G.; Francisl, P.A.; Lacroix-Triki, M.; Joensuu, H.; Dieci, M.V.; Badve, S.; DeMaria, S.; et al. Tumor-Infiltrating Lymphocytes and Prognosis: A Pooled Individual Patient Analysis of Early-Stage Triple-Negative Breast Cancers. J. Clin. Oncol. 2019, 37, 559–569. [Google Scholar] [CrossRef]
- Michiels, S.; Jonas, S.F.; Bataillon, G.; Criscitiello, C.; Salgado, R.; Loi, S.; Viale, G.; Lee, H.J.; Dieci, M.V.; Kim, S.-B.; et al. Prognostic value of tumor-infiltrating lymphocytes in patients with early-stage triple-negative breast cancers (TNBC) who did not receive adjuvant chemotherapy. Ann. Oncol. 2019, 30, 1941–1949. [Google Scholar] [CrossRef]
- Dieci, M.; Conte, P.; Bisagni, G.; Brandes, A.; Frassoldati, A.; Cavanna, L.; Musolino, A.; Giotta, F.; Rimanti, A.; Garrone, O.; et al. Association of tumor-infiltrating lymphocytes with distant disease-free survival in the ShortHER randomized adjuvant trial for patients with early HER2+ breast cancer. Ann. Oncol. 2019, 30, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Dieci, M.V.; Criscitiello, C.; Goubar, A.; Viale, G.; Conte, P.; Guarneri, V.; Ficarra, G.; Mathieu, M.C.; Delaloge, S.; Curigliano, G.; et al. Prognostic value of tumor-infiltrating lymphocytes on residual disease after primary chemotherapy for triple-negative breast cancer: A retrospective multicenter study. Ann. Oncol. 2014, 25, 611–618. [Google Scholar] [CrossRef]
- Denkert, C.; Von Minckwitz, G.; Darb-Esfahani, S.; Lederer, B.; I Heppner, B.; E Weber, K.; Budczies, J.; Huober, J.; Klauschen, F.; Furlanetto, J.; et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: A pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. 2018, 19, 40–50. [Google Scholar] [CrossRef]
- Kim, R.S.; Song, N.; Gavin, P.G.; Salgado, R.; Bandos, H.; Kos, Z.; Floris, G.; Eynden, G.G.G.M.V.D.; Badve, S.; DeMaria, S.; et al. Stromal Tumor-infiltrating Lymphocytes in NRG Oncology/NSABP B-31 Adjuvant Trial for Early-Stage HER2-Positive Breast Cancer. J. Natl. Cancer Inst. 2019, 111, 867–871. [Google Scholar] [CrossRef]
- Cardoso, F.; Kyriakides., S.; Ohno, S.; Penault-Llorca, F.; Poortmans, P.; Rubio, I.T.; Zackrisson, S.; Senkus, E.; ESMO Guidelines Committee. Early breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 1194–1220. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Di, G. Role of tumor microenvironment in triple-negative breast cancer and its prognostic significance. Chin. J. Cancer Res. 2017, 29, 237–252. [Google Scholar] [CrossRef]
- Lee, S.; Cho, E.Y.; Park, Y.H.; Ahn, J.S.; Im, Y.-H. Prognostic impact of FOXP3 expression in triple-negative breast cancer. Acta Oncol. 2012, 52, 73–81. [Google Scholar] [CrossRef]
- Mahmoud, S.M.; Paish, E.C.; Powe, D.G.; Macmillan, R.D.; Grainge, M.J.; Lee, A.H.S.; Ellis, I.O.; Green, A.R. Tumor-Infiltrating CD8+ Lymphocytes Predict Clinical Outcome in Breast Cancer. J. Clin. Oncol. 2011, 29, 1949–1955. [Google Scholar] [CrossRef]
- Dieci, M.V.; Tsvetkova, V.; Griguolo, G.; Miglietta, F.; Tasca, G.; Giorgi, C.A.; Cumerlato, E.; Massa, D.; Mele, M.L.; Orvieto, E.; et al. Integration of tumour infiltrating lymphocytes, programmed cell-death ligand-1, CD8 and FOXP3 in prognostic models for triple-negative breast cancer: Analysis of 244 stage I–III patients treated with standard therapy. Eur. J. Cancer 2020, 136, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Doedens, A.L.; Stockmann, C.; Rubinstein, M.P.; Liao, D.; Zhang, N.; DeNardo, D.G.; Coussens, L.M.; Karin, M.; Goldrath, A.W.; Johnson, R.S. Macrophage Expression of Hypoxia-Inducible Factor-1 Suppresses T-Cell Function and Promotes Tumor Progression. Cancer Res. 2010, 70, 7465–7475. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Qu, Q.; Chen, X.; Huang, O.; Wu, J.; Shen, K. The Prognostic Value of Tumor-Infiltrating Lymphocytes in Breast Cancer: A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0152500. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, M.; Sasano, H.; Tamaki, K.; Hirakawa, H.; Takahashi, Y.; Nakagawa, S.; Watanabe, G.; Tada, H.; Suzuki, A.; Ohuchi, N.; et al. Prognostic significance of tumor-infiltrating CD8+ and FOXP3+ lymphocytes in residual tumors and alterations in these parameters after neoadjuvant chemotherapy in triple-negative breast cancer: A retrospective multicenter study. Breast Cancer Res. 2015, 17, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bottai, G.; Raschioni, C.; Losurdo, A.; Di Tommaso, L.; Tinterri, C.; Torrisi, R.; Reis-Filho, J.S.; Roncalli, M.; Sotiriou, C.; Santoro, A.; et al. An immune stratification reveals a subset of PD-1/LAG-3 double-positive triple-negative breast cancers. Breast Cancer Res. 2016, 18, 121. [Google Scholar] [CrossRef] [Green Version]
- Yeong, J.; Thike, A.A.; Lim, J.C.T.; Lee, B.; Li, H.; Wong, S.-C.; Hue, S.S.S.; Tan, P.H.; Iqbal, J. Higher densities of Foxp3+ regulatory T cells are associated with better prognosis in triple-negative breast cancer. Breast Cancer Res. Treat. 2017, 163, 21–35. [Google Scholar] [CrossRef] [PubMed]
- West, N.R.; E Kost, S.; Martin, S.D.; Milne, K.; DeLeeuw, R.J.; Nelson, B.H.; Watson, P.H. Tumour-infiltrating FOXP3+ lymphocytes are associated with cytotoxic immune responses and good clinical outcome in oestrogen receptor-negative breast cancer. Br. J. Cancer 2012, 108, 155–162. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Foulkes, W.D.; Leung, S.; Gao, D.; Lau, S.; Kos, Z.; O Nielsen, T. Prognostic significance of FOXP3+ tumor-infiltrating lymphocytes in breast cancer depends on estrogen receptor and human epidermal growth factor receptor-2 expression status and concurrent cytotoxic T-cell infiltration. Breast Cancer Res. 2014, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Weyer-Elberich, V.; Hengstler, J.G.; Heimes, A.-S.; Almstedt, K.; Gerhold-Ay, A.; Lebrecht, A.; Battista, M.J.; Hasenburg, A.; Sahin, U.; et al. Prognostic impact of CD4-positive T cell subsets in early breast cancer: A study based on the FinHer trial patient population. Breast Cancer Res. 2018, 20, 1–10. [Google Scholar] [CrossRef]
- Schalper, K.A.; Velcheti, V.; Carvajal, D.; Wimberly, H.; Brown, J.; Pusztai, L.; Rimm, D. In Situ Tumor PD-L1 mRNA Expression Is Associated with Increased TILs and Better Outcome in Breast Carcinomas. Clin. Cancer Res. 2014, 20, 2773–2782. [Google Scholar] [CrossRef] [Green Version]
- Kythreotou, A.; Siddique, A.; Mauri, F.A.; Bower, M.; Pinato, D.J. PD-L1. J. Clin. Pathol. 2018, 7, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Leite, M.S.M.; Van De Vijver, K.K.; Michaut, M.; Van Der Linden, R.; Hooijer, G.K.; Horlings, H.M.; Severson, T.M.; Mulligan, A.M.; Weerasooriya, N.; Sanders, J.; et al. Assessment of PD-L1 expression across breast cancer molecular subtypes, in relation to mutation rate, BRCA1-like status, tumor-infiltrating immune cells and survival. OncoImmunology 2018, 7, e150982015. [Google Scholar] [CrossRef] [Green Version]
- Miglietta, F.; Griguolo, G.; Guarneri, V.; Dieci, M.V. Programmed Cell Death Ligand 1 in Breast Cancer: Technical Aspects, Prognostic Implications, and Predictive Value. Oncology 2019, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Seock-Ah IMpassion130 Trial Investigators; Wright, G.S.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. New Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Muenst, S.; Schaerli, A.R.; Gao, F.; Däster, S.; Trella, E.; Droeser, R.A.; Muraro, M.G.; Zajac, P.; Zanetti, R.; Gillanders, W.E.; et al. Expression of programmed death ligand 1 (PD-L1) is associated with poor prognosis in human breast cancer. Breast Cancer Res. Treat. 2014, 146, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Sun, H.; Zhao, S.; Wang, Y.; Pu, H.; Zhang, Q. Expression of PD-L1 and prognosis in breast cancer: A meta-analysis. Oncotarget 2017, 8, 31347–31354. [Google Scholar] [CrossRef] [Green Version]
- Sa-Nguanraksa, D.; O-Charoenrat, P. The Role of Vascular Endothelial Growth Factor A Polymorphisms in Breast Cancer. Int. J. Mol. Sci. 2012, 13, 14845–14864. [Google Scholar] [CrossRef] [Green Version]
- Ghiabi, P.; Jiang, J.; Pasquier, J.; Maleki, M.; Abu-Kaoud, N.; Rafii, S.; Rafii, A. Endothelial Cells Provide a Notch-Dependent Pro-Tumoral Niche for Enhancing Breast Cancer Survival, Stemness and Pro-Metastatic Properties. PLoS ONE 2014, 9, e112424. [Google Scholar] [CrossRef]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; De Almeida, D.L.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.R.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef]
- Hill, B.S.; Sarnella, A.; D’Avino, G.; Zannetti, A. Recruitment of stromal cells into tumour microenvironment promote the metastatic spread of breast cancer. Semin. Cancer Biol. 2020, 60, 202–213. [Google Scholar] [CrossRef]
- Melzer, C.; Von Der Ohe, J.; Hass, R. Enhanced metastatic capacity of breast cancer cells after interaction and hybrid formation with mesenchymal stroma/stem cells (MSC). Cell Commun. Signal. 2018, 16, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Dwyer, R.; Potter-Beirne, S.; Harrington, K.; Lowery, A.; Hennessy, E.; Murphy, J.; Barry, F.; O’Brien, T.; Kerin, M. Monocyte Chemotactic Protein-1 Secreted by Primary Breast Tumors Stimulates Migration of Mesenchymal Stem Cells. Clin. Cancer Res. 2007, 13, 5020–5027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Chen, Y.; Wang, Q.; Jayasinghe, U.; Luo, X.; Wei, Q.; Wang, J.; Xiong, H.; Chen, C.; Xu, B.; et al. Exosome: Emerging biomarker in breast cancer. Oncotarget 2017, 8, 41717–41733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Su, X.; Xu, M.; Xiao, X.; Li, X.; Li, H.; Keating, A.; Zhao, R.C. Exosomes secreted by mesenchymal stromal/stem cell-derived adipocytes promote breast cancer cell growth via activation of Hippo signaling pathway. Stem Cell Res. Ther. 2019, 10, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.; Mandal, G.; Chowdhury, S.R.; Purohit, S.; Payne, K.K.; Anadon, C.; Gupta, A.; Swanson, P.; Yu, X.; Conejo-Garcia, J.R.; et al. Exosomes Produced by Mesenchymal Stem Cells Drive Differentiation of Myeloid Cells into Immunosuppressive M2-Polarized Macrophages in Breast Cancer. J. Immunol. 2019, 203, 3447–3460. [Google Scholar] [CrossRef] [PubMed]
- Rőszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015, 2015, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Bartosh, T.J.; Ullah, M.; Zeitouni, S.; Beaver, J.; Prockop, D.J. Cancer cells enter dormancy after cannibalizing mesenchymal stem/stromal cells (MSCs). Proc. Natl. Acad. Sci. USA 2016, 113, E6447–E6456. [Google Scholar] [CrossRef] [Green Version]
- Ghajar, C.M. Metastasis prevention by targeting the dormant niche. Nat. Rev. Cancer 2015, 15, 238–247. [Google Scholar] [CrossRef]
- He, M.-F.; Wang, S.; Wang, Y.; Wang, X.-N. Modeling cell-in-cell structure into its biological significance. Cell Death Dis. 2013, 4, e630. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-C.; Gonzalez, M.E.; Burman, B.; Zhao, X.; Anwar, T.; Tran, M.; Medhora, N.; Hiziroglu, A.B.; Lee, W.; Cheng, Y.-H.; et al. Mesenchymal Stem/Stromal Cell Engulfment Reveals Metastatic Advantage in Breast Cancer. Cell Rep. 2019, 27, 3916–3926.e5. [Google Scholar] [CrossRef] [Green Version]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [Green Version]
- Gole, L.; Yeong, J.; Lim, J.C.T.; Ong, K.H.; Han, H.; Thike, A.A.; Poh, Y.C.; Yee, S.; Iqbal, J.; Hong, W.; et al. Quantitative stain-free imaging and digital profiling of collagen structure reveal diverse survival of triple negative breast cancer patients. Breast Cancer Res. 2020, 22, 42. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, T. Extracellular matrix components in breast cancer progression and metastasis. Breast 2013, 22, S66–S72. [Google Scholar] [CrossRef]
- Duffy, M.J.; Maguire, T.M.; Hill, A.; McDermott, E.; O’Higgins, N. Metalloproteinases: Role in breast carcinogenesis, invasion and metastasis. Breast Cancer Res. 2000, 2, 252–257. [Google Scholar] [CrossRef] [Green Version]
- Radisky, E.S.; Radisky, D.C. Matrix metalloproteinases as breast cancer drivers and therapeutic targets. Front. Biosci. 2015, 20, 1144–1163. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Eliceiri, K.W.; Campbell, J.M.; Inman, D.R.; White, J.G.; Keely, P.J. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006, 4, 38. [Google Scholar] [CrossRef] [Green Version]
- Conklin, M.W.; Eickhoff, J.C.; Riching, K.M.; Pehlke, C.A.; Eliceiri, K.W.; Provenzano, P.P.; Friedl, A.; Keely, P.J. Aligned Collagen Is a Prognostic Signature for Survival in Human Breast Carcinoma. Am. J. Pathol. 2011, 178, 1221–1232. [Google Scholar] [CrossRef]
- Guiro, K.; Arinzeh, T.L. Bioengineering Models for Breast Cancer Research. Breast Cancer: Basic Clin. Res. 2015, 9, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Nath, S.; Devi, G.R. Three-dimensional culture systems in cancer research: Focus on tumor spheroid model. Pharmacol. Ther. 2016, 163, 94–108. [Google Scholar] [CrossRef] [Green Version]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Weiswald, L.-B.; Bellet, D.; Dangles-Marie, V. Spherical Cancer Models in Tumor Biology. Neoplasia 2015, 17, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Vunjak-Novakovic, G. Modeling tumor microenvironments using custom-designed biomaterial scaffolds. Curr. Opin. Chem. Eng. 2016, 11, 94–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavallaro, S. CXCR4/CXCL12 in non-small-cell lung cancer metastasis to the brain. Int. J. Mol. Sci. 2013, 14, 1713–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Q.; Xu, C.; Zhao, H.; Chen, H. CXCR4 in breast cancer: Oncogenic role and therapeutic targeting. Drug Des. Dev. Ther. 2015, 9, 4953–4964. [Google Scholar] [CrossRef] [Green Version]
- Dewan, M.; Ahmed, S.; Iwasaki, Y.; Ohba, K.; Toi, M.; Yamamoto, N. Stromal cell-derived factor-1 and CXCR4 receptor interaction in tumor growth and metastasis of breast cancer. Biomed. Pharmacother. 2006, 60, 273–276. [Google Scholar] [CrossRef]
- Angst, B.D.; Marcozzi, C.; I Magee, A. The cadherin superfamily: Diversity in form and function. J. Cell Sci. 2001, 114, 629–641. [Google Scholar]
- Andrews, J.L.; Kim, A.C.; Hens, J. The role and function of cadherins in the mammary gland. Breast Cancer Res. 2012, 14, 203. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.H.; Miller, P.; Garcia-Contreras, M.; Ao, Z.; Machlin, L.; Issa, E.; El-Ashry, D. Hierarchical paracrine interaction of breast cancer associated fibroblasts with cancer cells via hMAPK-microRNAs to drive ER-negative breast cancer phenotype. Cancer Biol. Ther. 2015, 16, 1671–1681. [Google Scholar] [CrossRef] [Green Version]
- Jaalouk, D.E.; Lammerding, J. Mechanotransduction gone awry. Nat. Rev. Mol. Cell Biol. 2009, 10, 63–73. [Google Scholar] [CrossRef] [Green Version]
- Chin, L.; Xia, Y.; E Discher, D.; A Janmey, P. Mechanotransduction in cancer. Curr. Opin. Chem. Eng. 2016, 11, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Ingber, D.E. Cell tension, matrix mechanics, and cancer development. Cancer Cell 2005, 8, 175–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, K.; Wu, Y.I.; Liu, Y.; Geiger, J.; Tam, E.; Overall, C.M.; Stack, M.S.; Friedl, P. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat. Cell Biol. 2007, 9, 893–904. [Google Scholar] [CrossRef]
- Suresh, S. Biomechanics and biophysics of cancer cells. Acta Biomater. 2007, 3, 413–438. [Google Scholar] [CrossRef] [PubMed]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Hebner, C.; Weaver, V.M.; Debnath, J. Modeling Morphogenesis and Oncogenesis in Three-Dimensional Breast Epithelial Cultures. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 313–339. [Google Scholar] [CrossRef] [PubMed]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef]
- Calses, P.C.; Crawford, J.J.; Lill, J.R.; Dey, A. Hippo Pathway in Cancer: Aberrant Regulation and Therapeutic Opportunities. Trends Cancer 2019, 5, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Meng, Z.; Chen, R.; Guan, K.-L. The Hippo Pathway: Biology and Pathophysiology. Annu. Rev. Biochem. 2019, 88, 577–604. [Google Scholar] [CrossRef] [Green Version]
- Harvey, K.F.; Tapon, N. The Salvador–Warts–Hippo pathway—An emerging tumour-suppressor network. Nat. Rev. Cancer 2007, 7, 182–191. [Google Scholar] [CrossRef]
- Zhao, B.; Li, L.; Tumaneng, K.; Wang, C.Y.; Guan, K.L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 2010, 24, 72–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Totaro, A.; Panciera, T.; Piccolo, S. YAP/TAZ upstream signals and downstream responses. Nat. Cell Biol. 2018, 20, 888–899. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP and TAZ: A signalling hub of the tumour microenvironment. Nat. Rev. Cancer 2019, 19, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Yang, X. Targeting the Hippo Pathway for Breast Cancer Therapy. Cancers 2018, 10, 422. [Google Scholar] [CrossRef] [Green Version]
- Lamar, J.M.; Stern, P.; Liu, H.; Schindler, J.W.; Jiang, Z.-G.; Hynes, R.O. The Hippo pathway target, YAP, promotes metastasis through its TEAD-interaction domain. Proc. Natl. Acad. Sci. USA 2012, 109, E2441–E2450. [Google Scholar] [CrossRef] [Green Version]
- Overholtzer, M.; Zhang, J.; Smolen, G.A.; Muir, B.; Li, W.; Sgroi, D.C.; Deng, C.-X.; Brugge, J.S.; Haber, D.A. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc. Natl. Acad. Sci. USA 2006, 103, 12405–12410. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Chang, J.K.; Dominguez, A.A.; Lee, H.-P.; Nam, S.; Chang, J.; Varma, S.; Qi, L.S.; West, R.B.; Chaudhuri, O. YAP-independent mechanotransduction drives breast cancer progression. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Zhang, N.; Gray, R.S.; Li, H.; Ewald, A.J.; Zahnow, C.A.; Pan, D. A temporal requirement for Hippo signaling in mammary gland differentiation, growth, and tumorigenesis. Genes Dev. 2014, 28, 432–437. [Google Scholar] [CrossRef] [Green Version]
- Bartucci, M.; Dattilo, R.; Moriconi, C.; Pagliuca, A.; Mottolese, M.; Federici, G.; Di Benedetto, A.; Todaro, M.; Stassi, G.; Sperati, F.; et al. TAZ is required for metastatic activity and chemoresistance of breast cancer stem cells. Oncogene 2014, 34, 681–690. [Google Scholar] [CrossRef]
- Díaz-Martín, J.; López-García, M.Á.; Romero-Pérez, L.; Atienza-Amores, M.R.; Pecero, M.L.; Castilla, M.Á.; Biscuola, M.; Santón, A.; Palacios, J.; Atienza, M.R. Nuclear TAZ expression associates with the triple-negative phenotype in breast cancer. Endocr. Relat. Cancer 2015, 22, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.-W.; Shen, H.; Frangou, C.; Yang, N.; Guo, J.; Xu, B.; Bshara, W.; Shepherd, L.; Zhu, Q.; Wang, J.; et al. Characterization of TAZ domains important for the induction of breast cancer stem cell properties and tumorigenesis. Cell Cycle 2015, 14, 146–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skibinski, A.; Breindel, J.L.; Prat, A.; Galván, P.; Smith, E.; Rolfs, A.; Gupta, P.B.; LaBaer, J.; Kuperwasser, C. The Hippo Transducer TAZ Interacts with the SWI/SNF Complex to Regulate Breast Epithelial Lineage Commitment. Cell Rep. 2014, 6, 1059–1072. [Google Scholar] [CrossRef]
- Vici, P.; Mottolese, M.; Pizzuti, L.; Barba, M.; Sperati, F.; Terrenato, I.; Di Benedetto, A.; Natoli, C.; Gamucci, T.; Angelucci, D.; et al. The Hippo transducer TAZ as a biomarker of pathological complete response in HER2-positive breast cancer patients treated with trastuzumab-based neoadjuvant therapy. Oncotarget 2014, 5, 9619–9625. [Google Scholar] [CrossRef] [Green Version]
- Yoon, A.-R.; Stasinopoulos, I.; Kim, J.H.; Yong, H.M.; Kilic, O.; Wirtz, D.; Bhujwalla, Z.M.; An, S.S. COX-2 dependent regulation of mechanotransduction in human breast cancer cells. Cancer Biol. Ther. 2015, 16, 430–437. [Google Scholar] [CrossRef] [Green Version]
- Ao, Z.; Shah, S.H.; Machlin, L.M.; Parajuli, R.; Miller, P.C.; Rawal, S.; Williams, A.J.; Cote, R.J.; Lippman, M.E.; Datar, R.H.; et al. Identification of Cancer-Associated Fibroblasts in Circulating Blood from Patients with Metastatic Breast Cancer. Cancer Res. 2015, 75, 4681–4687. [Google Scholar] [CrossRef] [Green Version]
- Eynden, G.G.V.D.; Majeed, A.W.; Illemann, M.; Vermeulen, P.; Bird, N.C.; Høyer-Hansen, G.; Eefsen, R.L.; Reynolds, A.R.; Brodt, P. The Multifaceted Role of the Microenvironment in Liver Metastasis: Biology and Clinical Implications. Cancer Res. 2013, 73, 2031–2043. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.; Feng, Y.-L.; Lin, S.; Chen, J.; Lin, H.; Liang, X.; Zheng, H.; Cai, X. Mechanisms involved in breast cancer liver metastasis. J. Transl. Med. 2015, 13, 64. [Google Scholar] [CrossRef] [Green Version]
- Saunus, J.M.; Reed, A.E.M.; Lim, Z.L.; Lakhani, S.R. Breast Cancer Brain Metastases: Clonal Evolution in Clinical Context. Int. J. Mol. Sci. 2017, 18, 152. [Google Scholar] [CrossRef] [PubMed]
- Neman, J.; Choy, C.; Kowolik, C.M.; Anderson, A.; Duenas, V.J.; Waliany, S.; Chen, B.T.; Chen, M.Y.; Jandial, R. Co-evolution of breast-to-brain metastasis and neural progenitor cells. Clin. Exp. Metastasis 2013, 30, 753–768. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y. Dissecting Tumor-Stromal Interactions in Breast Cancer Bone Metastasis. Endocrinol. Metab. 2016, 31, 206–212. [Google Scholar] [CrossRef] [Green Version]
- Kolb, A.D.; Bussard, K.M. The Bone Extracellular Matrix as an Ideal Milieu for Cancer Cell Metastases. Cancers 2019, 11, 1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Network, T.C.G.A. Comprehensive molecular portraits of human breast tumours. Nat. Cell Biol. 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-Negative Breast Cancer: Clinical Features and Patterns of Recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; A Rees, C.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nat. Cell Biol. 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Kreike, B.; Van Kouwenhove, M.; Horlings, H.; Weigelt, B.; Peterse, H.; Bartelink, H.; Van De Vijver, M.J. Gene expression profiling and histopathological characterization of triple-negative/basal-like breast carcinomas. Breast Cancer Res. 2007, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Alluri, P.; Newman, L.A. Basal-Like and Triple-Negative Breast Cancers. Surg. Oncol. Clin. North Am. 2014, 23, 567–577. [Google Scholar] [CrossRef] [Green Version]
- Weigelt, B.; Baehner, F.L.; Reis-Filho, J.S. The contribution of gene expression profiling to breast cancer classification, prognostication and prediction: A retrospective of the last decade. J. Pathol. 2010, 220, 263–280. [Google Scholar] [CrossRef]
- Dieci, M.V.; Mathieu, M.C.; Guarneri, V.; Conte, P.; Delaloge, S.; Andre, F.; Goubar, A. Prognostic and predictive value of tumor-infiltrating lymphocytes in two phase III randomized adjuvant breast cancer trials. Ann. Oncol. 2015, 26, 1698–1704. [Google Scholar] [CrossRef]
- Stanton, S.E.; Disis, M.L. Clinical significance of tumor-infiltrating lymphocytes in breast cancer. J. Immunother. Cancer 2016, 4, 59. [Google Scholar] [CrossRef] [Green Version]
- Rakha, E.A.; Ellis, I.O. Triple-negative/basal-like breast cancer: Review. Pathology 2009, 41, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Stanton, S.E.; Adams, S.; Disis, M.L. Variation in the Incidence and Magnitude of Tumor-Infiltrating Lymphocytes in Breast Cancer Subtypes. JAMA Oncol. 2016, 2, 1354–1360. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lang, R.; Zhao, J.; Zhang, X.; Pringle, G.A.; Fan, Y.; Yin, D.; Gu, F.; Yao, Z.; Fu, F. CD8+ cytotoxic T cell and FOXP3+ regulatory T cell infiltration in relation to breast cancer survival and molecular subtypes. Breast Cancer Res. Treat. 2011, 130, 645–655. [Google Scholar] [CrossRef]
- Miyan, M.; Schmidt-Mende, J.; Kiessling, R.; Poschke, I.; De Boniface, J. Differential tumor infiltration by T-cells characterizes intrinsic molecular subtypes in breast cancer. J. Transl. Med. 2016, 14, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Glajcar, A.; Szpor, J.; Pacek, A.; Tyrak, K.E.; Chan, F.; Streb, J.; Hodorowicz-Zaniewska, D.; Okoń, K. The relationship between breast cancer molecular subtypes and mast cell populations in tumor microenvironment. Virchows Arch. 2017, 470, 505–515. [Google Scholar] [CrossRef] [Green Version]
- Medrek, C.; Pontén, F.; Jirström, K.; Leandersson, K. The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. BMC Cancer 2012, 12, 306. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.; Brion, R.; Lintunen, M.; Kronqvist, P.; Sandholm, J.; Mönkkönen, J.; Kellokumpu-Lehtinen, P.-L.; Lauttia, S.; Tynninen, O.; Joensuu, H.; et al. Human breast cancer cells educate macrophages toward the M2 activation status. Breast Cancer Res. 2015, 17, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollmén, M.; Karaman, S.; Schwager, S.; Lisibach, A.; Christiansen, A.J.; Maksimow, M.; Varga, Z.; Jalkanen, S.; Detmar, M. G-CSF regulates macrophage phenotype and associates with poor overall survival in human triple-negative breast cancer. OncoImmunology 2015, 5, e1115177. [Google Scholar] [CrossRef] [Green Version]
- Levano, K.S.; Jung, E.H.; Kenny, P.A. Breast cancer subtypes express distinct receptor repertoires for tumor-associated macrophage derived cytokines. Biochem. Biophys. Res. Commun. 2011, 411, 107–110. [Google Scholar] [CrossRef]
- Niemiec, J.A.; Adamczyk, A.; Ambicka, A.; Mucha-Małecka, A.; Wysocki, W.M.; Ryś, J. Triple-negative, Basal Marker-expressing, and High-grade Breast Carcinomas are Characterized by High Lymphatic Vessel Density and the Expression of Podoplanin in Stromal Fibroblasts. Appl. Immunohistochem. Mol. Morphol. 2014, 22, 10–16. [Google Scholar] [CrossRef]
- Linderholm, B.K.; Hellborg, H.; Johansson, U.; Elmberger, G.; Skoog, L.; Lehtiö, J.; Lewensohn, R. Significantly higher levels of vascular endothelial growth factor (VEGF) and shorter survival times for patients with primary operable triple-negative breast cancer. Ann. Oncol. 2009, 20, 1639–1646. [Google Scholar] [CrossRef]
- Sa-Nguanraksa, D.; Chuangsuwanich, T.; Pongpruttipan, T.; O-Charoenrat, P. High vascular endothelial growth factor gene expression predicts poor outcome in patients with non-luminal A breast cancer. Mol. Clin. Oncol. 2015, 3, 1103–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, B.P.; Gray, R.J.; Radovich, M.; Shen, F.; Vance, G.; Li, L.; Jiang, G.; Miller, K.D.; Gralow, J.R.; Dickler, M.N.; et al. Prognostic and predictive value of tumor vascular endothelial growth factor gene amplification in metastatic breast cancer treated with paclitaxel with and without bevacizumab; results from ECOG 2100 trial. Clin. Cancer Res. 2013, 19, 1281–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sihto, H.; Lundin, J.; Lundin, M.; Lehtimäki, T.; Ristimäki, A.; Holli, K.; Sailas, L.; Kataja, V.; Turpeenniemi-Hujanen, T.; Isola, J.; et al. Breast cancer biological subtypes and protein expression predict for the preferential distant metastasis sites: A nationwide cohort study. Breast Cancer Res. 2011, 13, R87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smid, M.; Wang, Y.; Zhang, Y.; Sieuwerts, A.M.; Yu, J.; Klijn, J.G.M.; Foekens, J.A.; Martens, J.W.M. Subtypes of Breast Cancer Show Preferential Site of Relapse. Cancer Res. 2008, 68, 3108–3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.U.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic Behavior of Breast Cancer Subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef] [PubMed]
- Soni, D.A.; Ren, Z.; Hameed, O.; Chanda, D.; Morgan, C.J.; Siegal, G.P.; Wei, S. Breast Cancer Subtypes Predispose the Site of Distant Metastases. Am. J. Clin. Pathol. 2015, 143, 471–478. [Google Scholar] [CrossRef]
- Emens, L.A.; Nanda, R. Breast Cancer Immunotherapy. Cancer Immunother. Princ. Pract. 2018, 24. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Carroll, K.D.; Policarpio, D.; Osborn, C.; Gregory, M.; Bassi, R.; Jimenez, X.; Prewett, M.; Liebisch, G.; Persaud, K.; et al. Anti-transforming growth factor beta receptor II antibody has therapeutic efficacy against primary tumor growth and metastasis through multieffects on cancer, stroma, and immune cells. Clin. Cancer Res. 2010, 16, 1191–1205. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Yang, Y.; Zhou, Q.; Weiss, J.M.; Howard, O.Z.; McPherson, J.M.; Wakefield, L.M.; Oppenheim, J.J. Effective Chemoimmunotherapy with Anti-TGFβ Antibody and Cyclophosphamide in a Mouse Model of Breast Cancer. PLoS ONE 2014, 9, e85398. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Fletcher, R.; Yu, J.; Zhang, L. Immunogenic effects of chemotherapy-induced tumor cell death. Genes Dis. 2018, 5, 194–203. [Google Scholar] [CrossRef] [PubMed]
- DeMaria, S.; Ng, B.; Devitt, M.L.; Babb, J.S.; Kawashima, N.; Liebes, L.; Formenti, S.C. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int. J. Radiat. Oncol. 2004, 58, 862–870. [Google Scholar] [CrossRef]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; Van De Vijver, K.K.; De Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.; et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Svensson, S.; Abrahamsson, A.; Rodriguez, G.V.; Olsson, A.-K.; Jensen, L.; Cao, Y.; Dabrosin, C. CCL2 and CCL5 Are Novel Therapeutic Targets for Estrogen-Dependent Breast Cancer. Clin. Cancer Res. 2015, 21, 3794–3805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Generali, D.; Bates, G.; Berruti, A.; Brizzi, M.P.; Campo, L.; Bonardi, S.; Bersiga, A.; Allevi, G.; Milani, M.; Aguggini, S.; et al. Immunomodulation of FOXP3+ Regulatory T Cells by the Aromatase Inhibitor Letrozole in Breast Cancer Patients. Clin. Cancer Res. 2009, 15, 1046–1051. [Google Scholar] [CrossRef] [Green Version]
- Chan, M.S.M.; Wang, L.; A Felizola, S.J.; Ueno, T.; Toi, M.; Loo, W.; Chow, L.W.C.; Suzuki, T.; Sasano, H. Changes of tumor infiltrating lymphocyte subtypes before and after neoadjuvant endocrine therapy in estrogen receptor-positive breast cancer patients–an immunohistochemical study of cd8+ and foxp3+ using double immunostaining with correlation to the pathobiological response of the patients. Int. J. Biol. Markers 2012, 27, 295–304. [Google Scholar] [CrossRef]
- Nalbandian, G.; Paharkova-Vatchkova, V.; Mao, A.; Nale, S.; Kovats, S. The Selective Estrogen Receptor Modulators, Tamoxifen and Raloxifene, Impair Dendritic Cell Differentiation and Activation. J. Immunol. 2005, 175, 2666–2675. [Google Scholar] [CrossRef] [Green Version]
- Komi, J.; Lassila, O. Nonsteroidal anti-estrogens inhibit the functional differentiation of human monocyte-derived dendritic cells. Blood 2000, 95, 2875–2882. [Google Scholar] [CrossRef]
- Behjati, S.; Frank, M.H. The Effects of Tamoxifen on Immunity. Curr. Med. Chem. 2009, 16, 3076–3080. [Google Scholar] [CrossRef] [Green Version]
- EBCTCG. Adjuvant bisphosphonate treatment in early breast cancer: Meta-analyses of individual patient data from randomised trials. Lancet 2015, 386, 1353–1361. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yang, K.H.; Wanyan, P.; Tian, J.H. Comparison of the efficacy and safety of denosumab versus bisphosphonates in breast cancer and bone metastases treatment: A meta-analysis of randomized controlled trials. Oncol. Lett. 2014, 7, 1997–2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zarrilli, G.; Businello, G.; Dieci, M.V.; Paccagnella, S.; Carraro, V.; Cappellesso, R.; Miglietta, F.; Griguolo, G.; Guarneri, V.; Lo Mele, M.; et al. The Tumor Microenvironment of Primitive and Metastatic Breast Cancer: Implications for Novel Therapeutic Strategies. Int. J. Mol. Sci. 2020, 21, 8102. https://doi.org/10.3390/ijms21218102
Zarrilli G, Businello G, Dieci MV, Paccagnella S, Carraro V, Cappellesso R, Miglietta F, Griguolo G, Guarneri V, Lo Mele M, et al. The Tumor Microenvironment of Primitive and Metastatic Breast Cancer: Implications for Novel Therapeutic Strategies. International Journal of Molecular Sciences. 2020; 21(21):8102. https://doi.org/10.3390/ijms21218102
Chicago/Turabian StyleZarrilli, Giovanni, Gianluca Businello, Maria Vittoria Dieci, Silvia Paccagnella, Valentina Carraro, Rocco Cappellesso, Federica Miglietta, Gaia Griguolo, Valentina Guarneri, Marcello Lo Mele, and et al. 2020. "The Tumor Microenvironment of Primitive and Metastatic Breast Cancer: Implications for Novel Therapeutic Strategies" International Journal of Molecular Sciences 21, no. 21: 8102. https://doi.org/10.3390/ijms21218102