Vinylene-Bridged Cyclic Dipyrrin and BODIPY Trimers


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
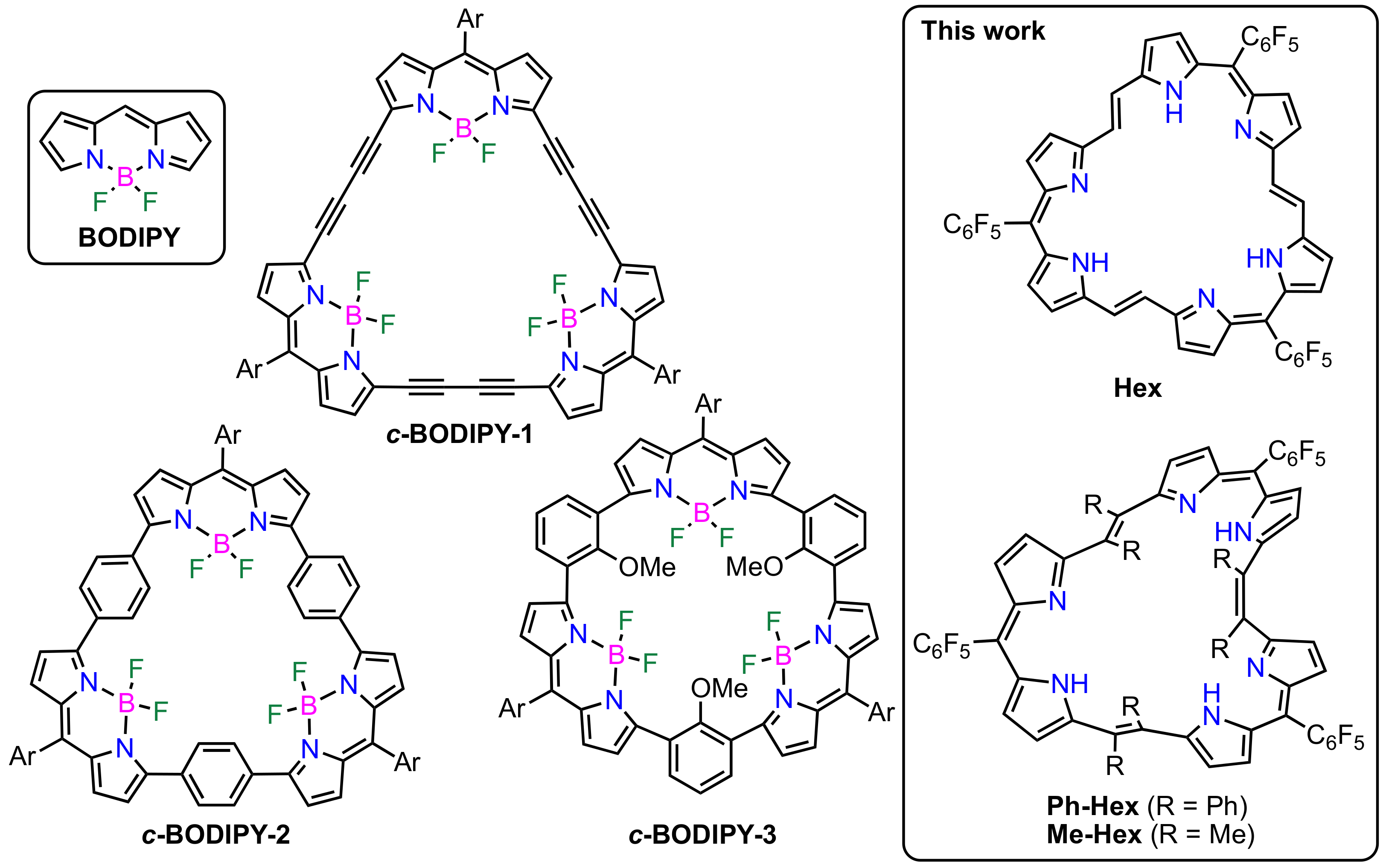
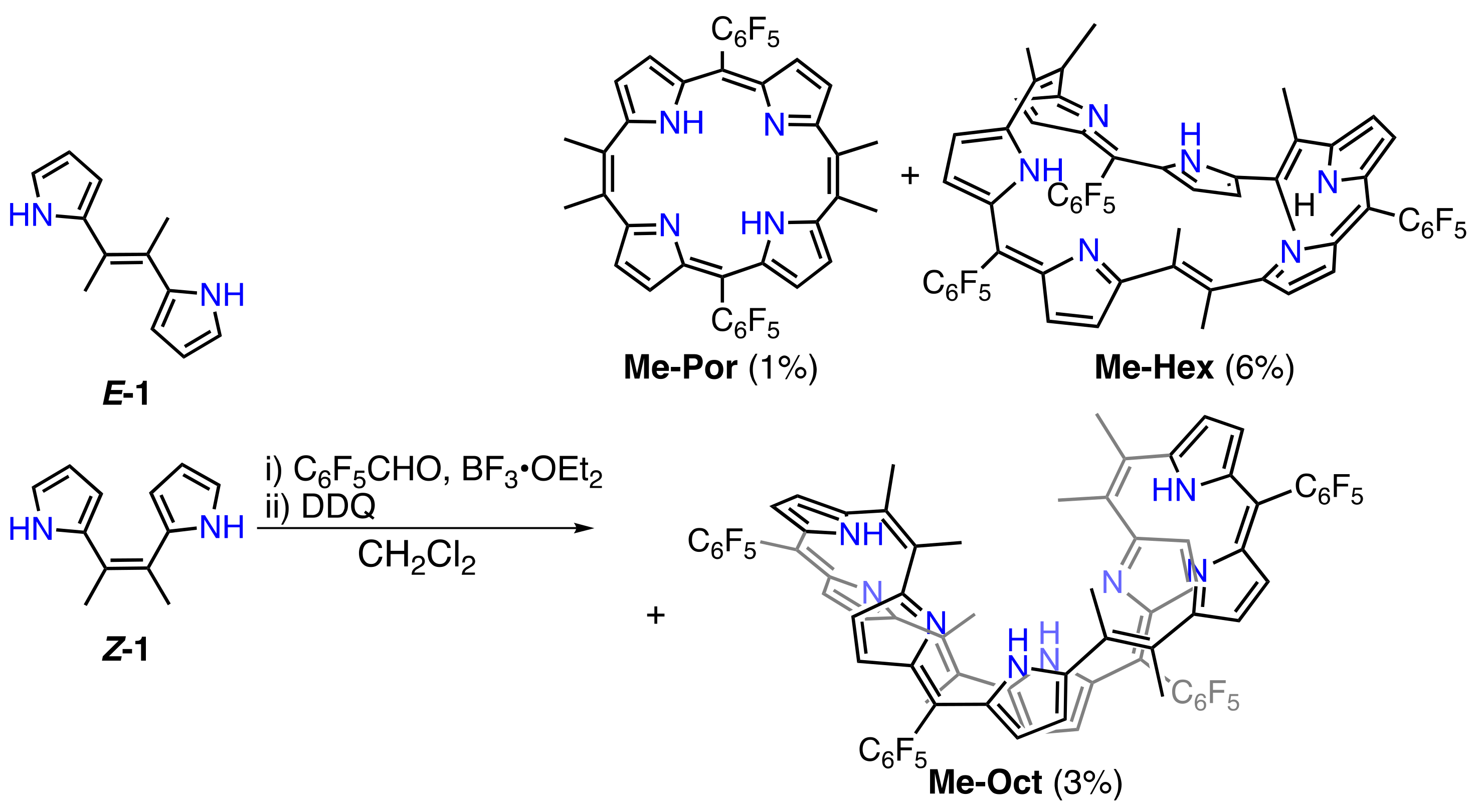
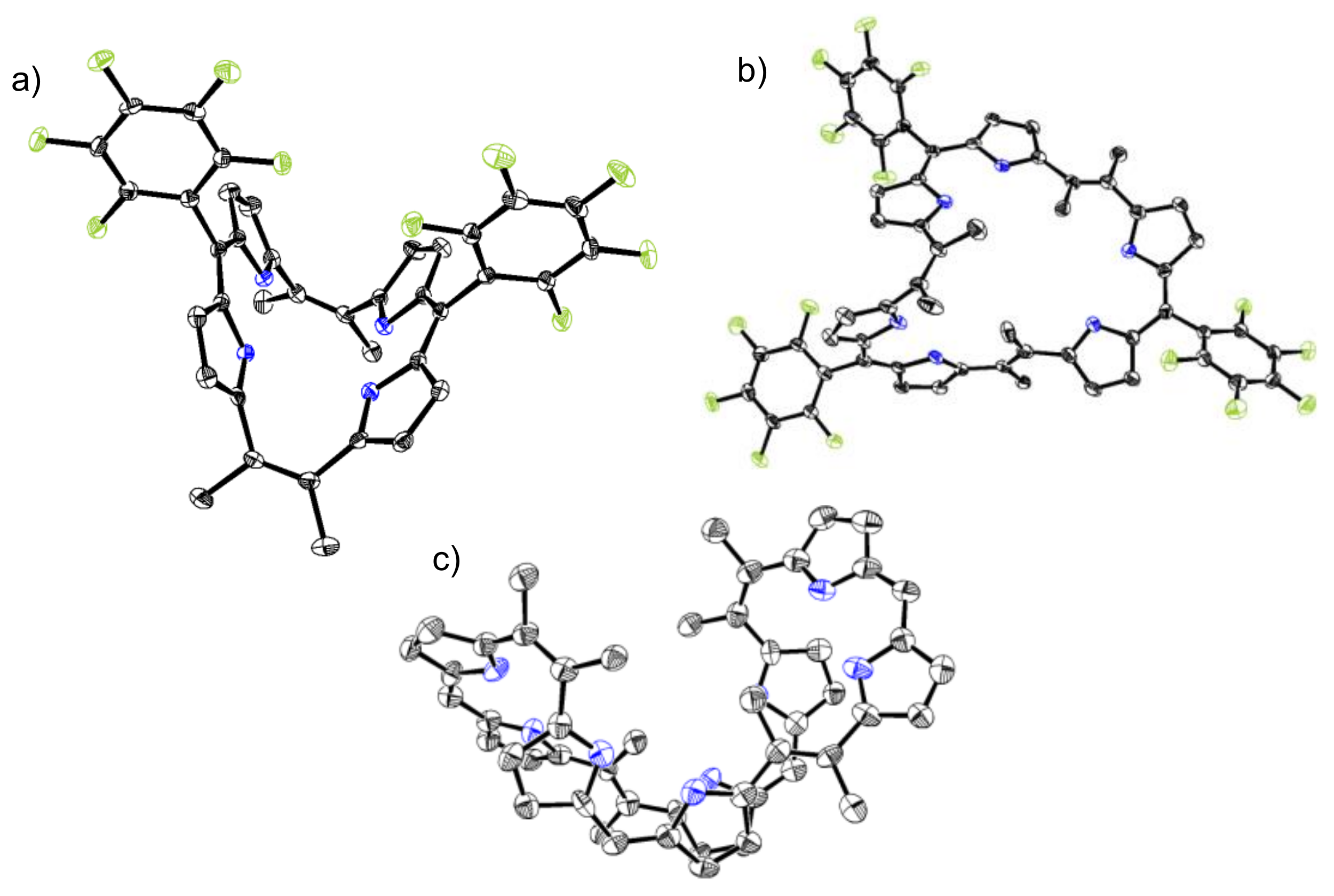
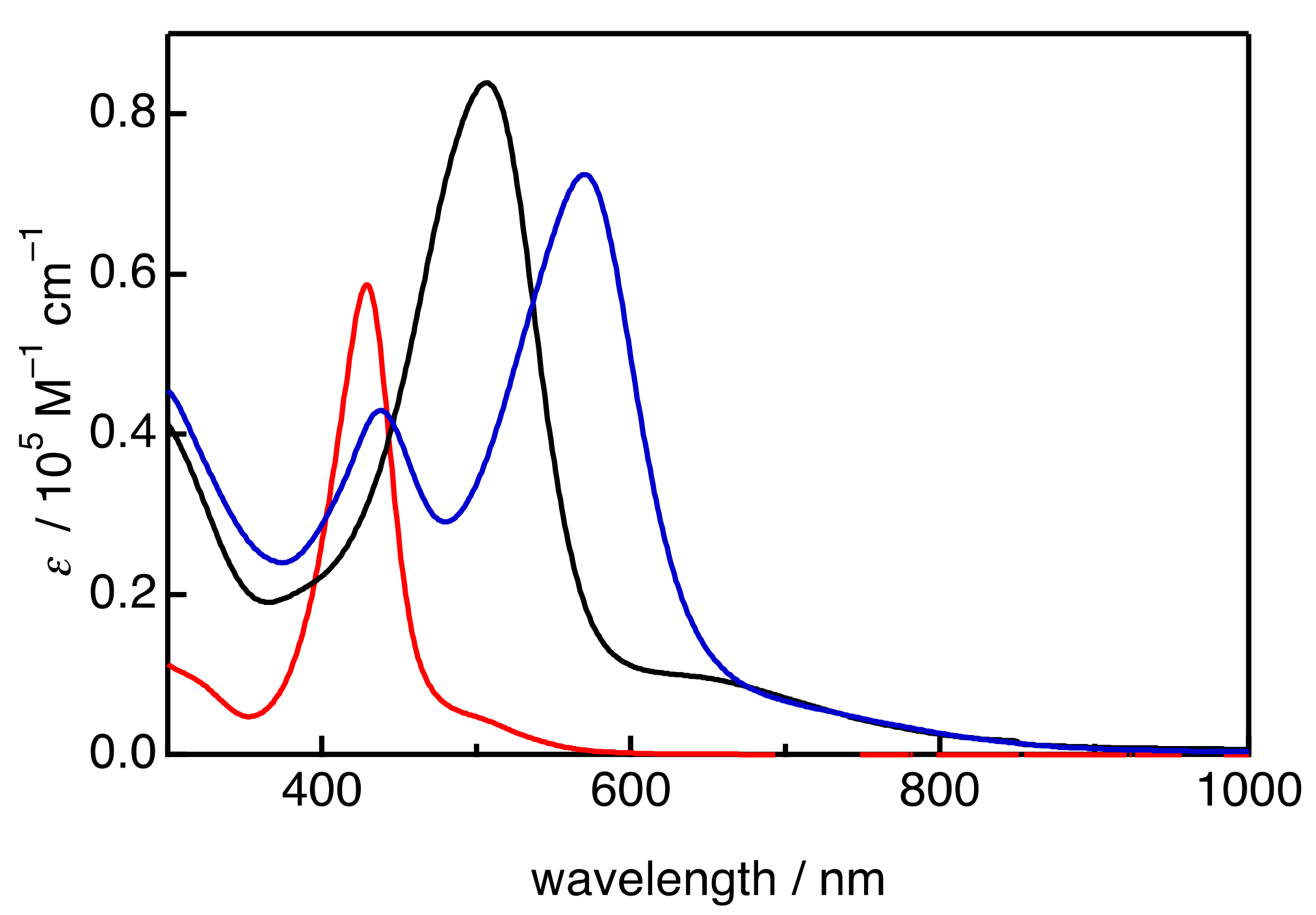
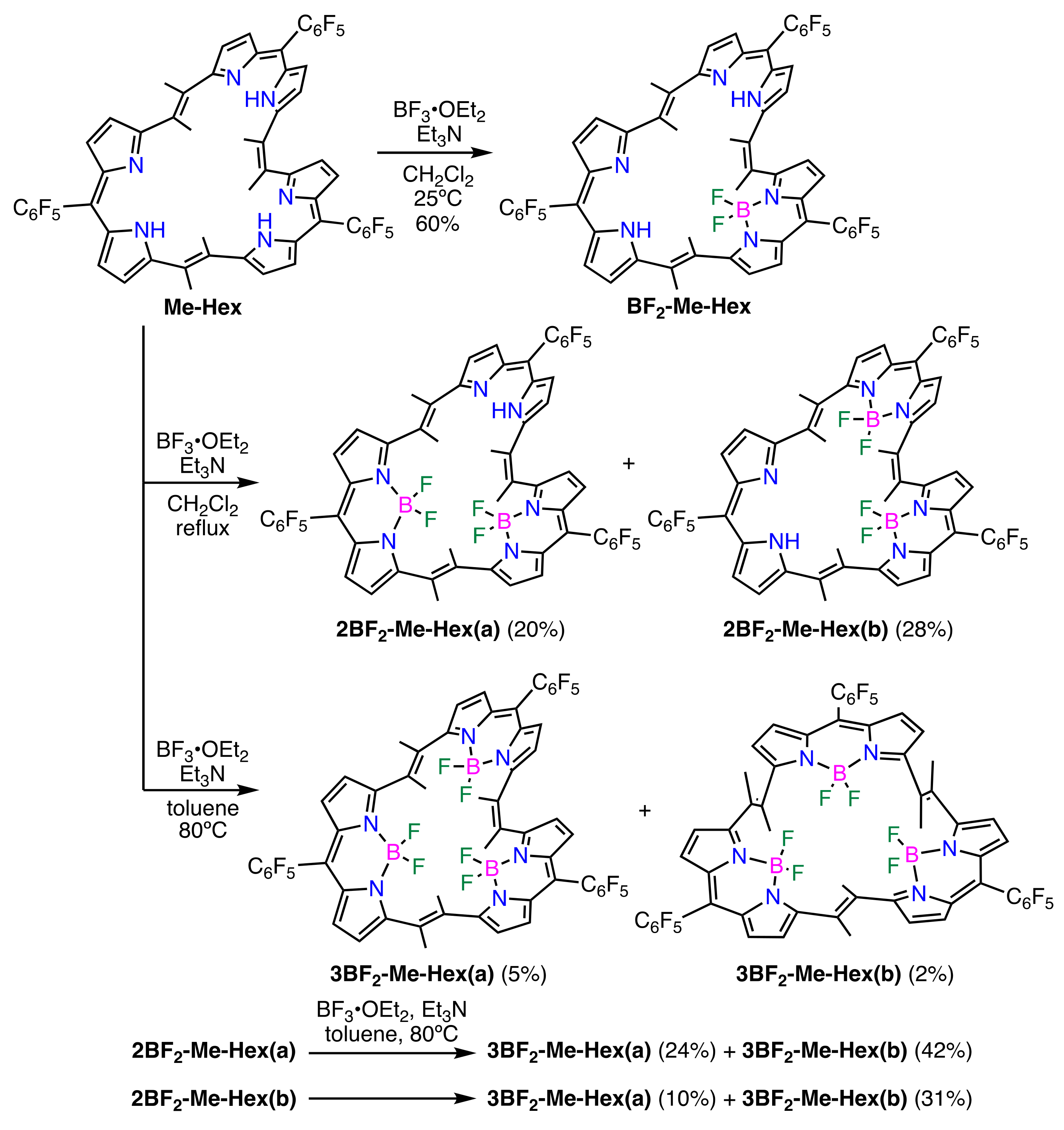
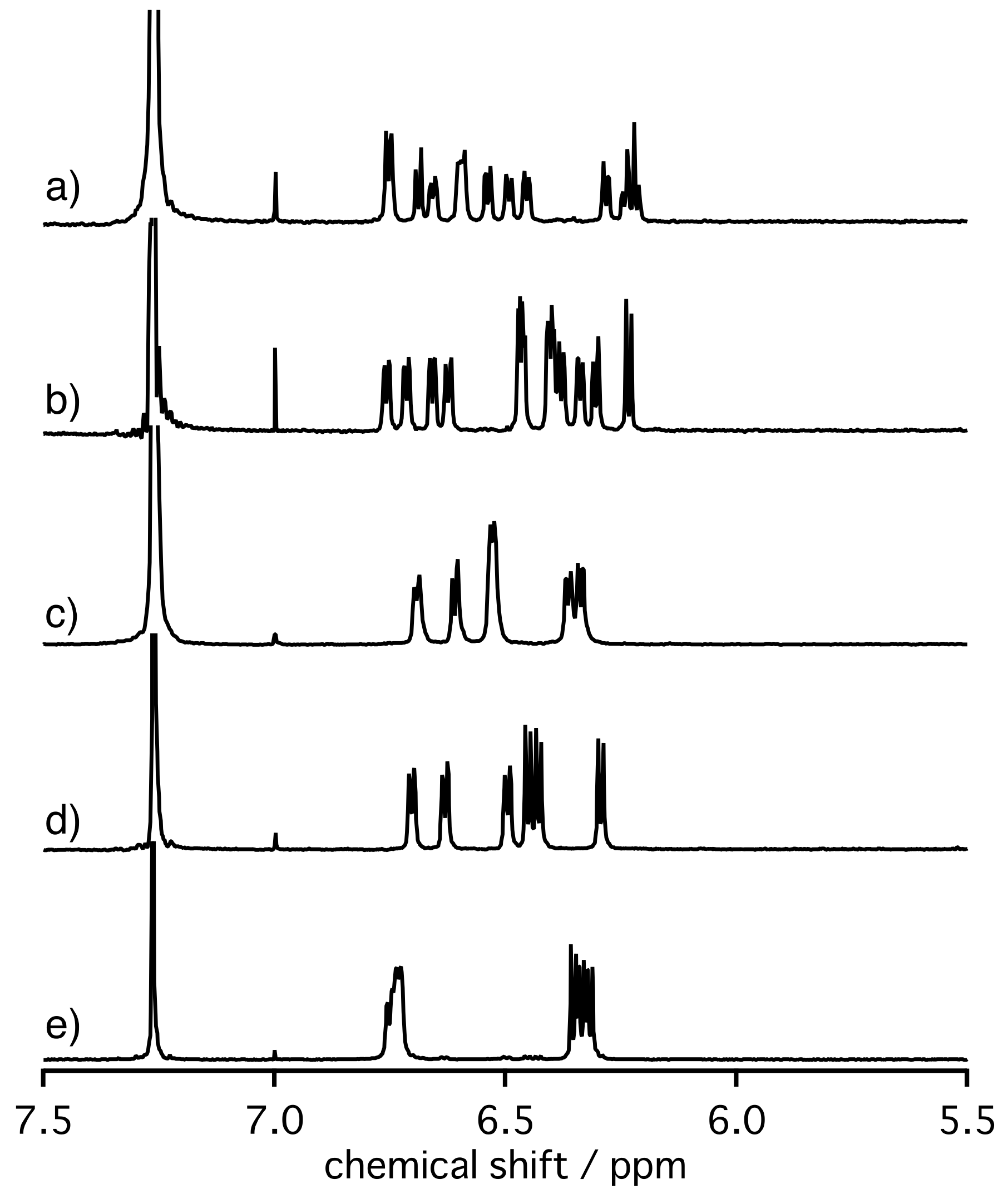
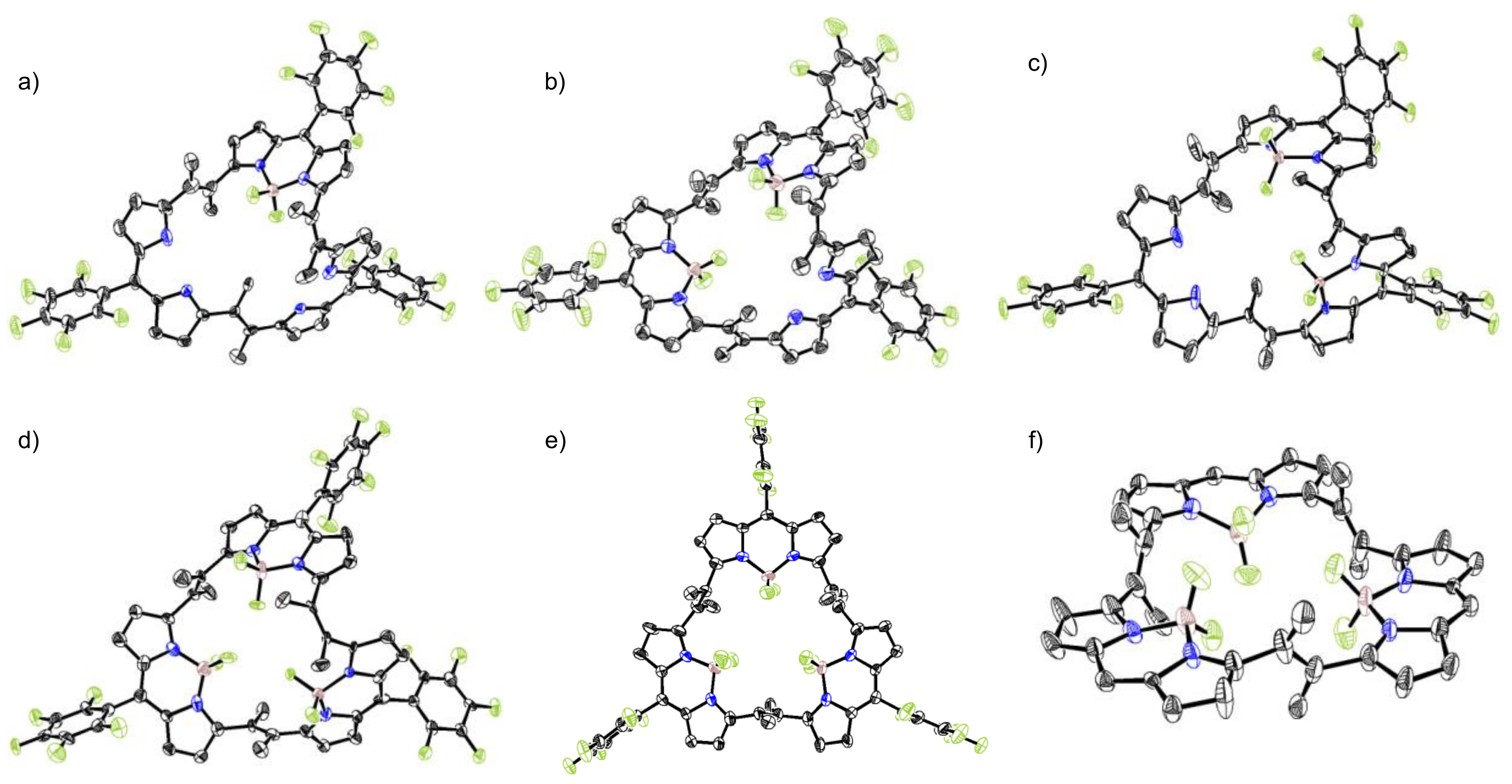
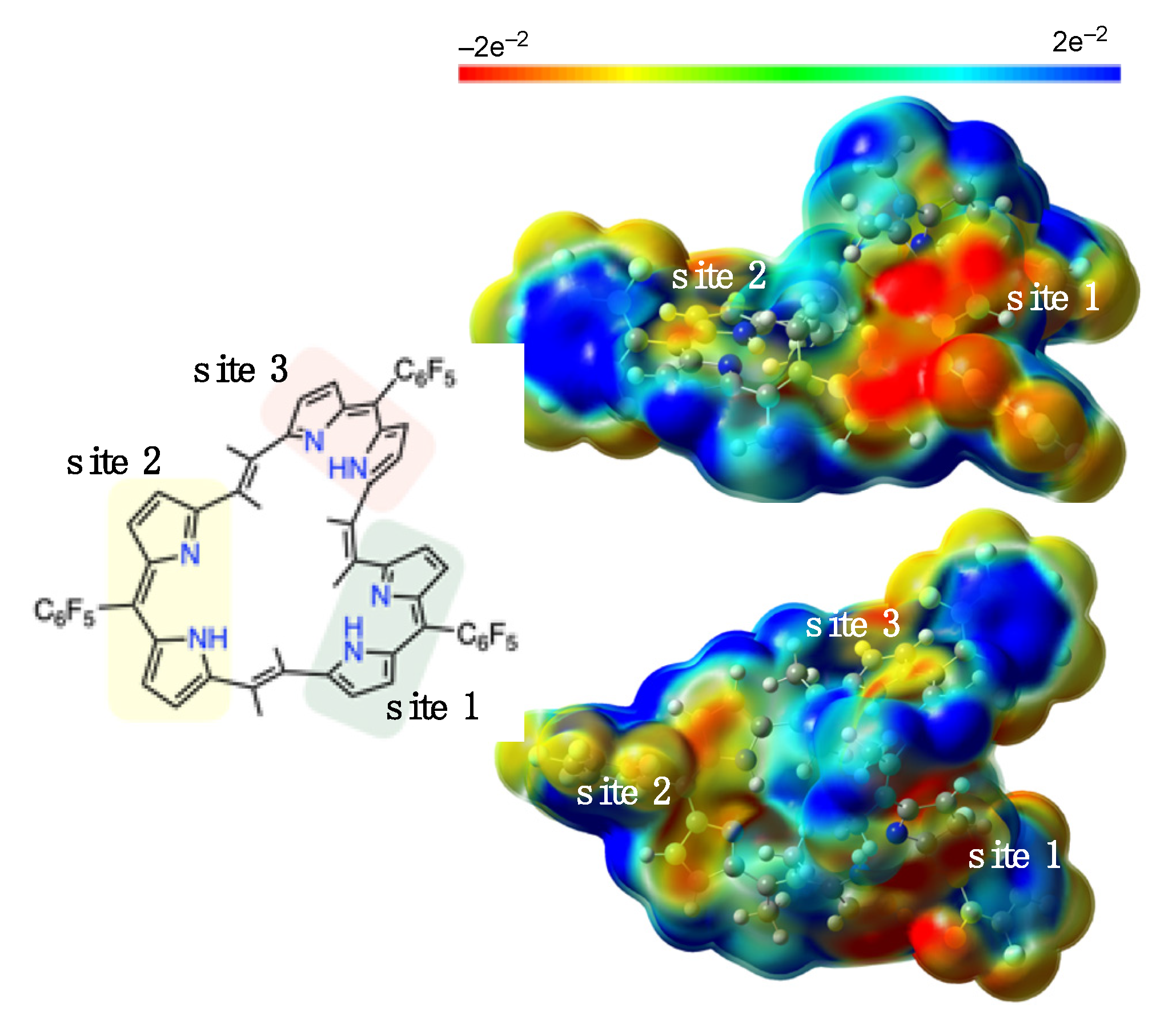
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. X-ray Analysis
3.3. Theoretical Calculations
3.4. Synthesis of Dimethyl-Dipyrrolylethane (E/Z-1)
3.5. Synthesis of Me-Por, Me-Hex, and Me-Oct
3.6. Synthesis of Boron Complexes of Me-Hex
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Loudet, A.; Burgess, K. BODIPY Dyes and Their Derivatives: Syntheses and Spectroscopic Properties. Chem. Rev. 2007, 107, 4891–4932. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Mack, J.; Yang, Y.; Shen, Z. Structural modification strategies for the rational design of red/NIR region BODIPYs. Chem. Soc. Rev. 2014, 43, 4778–4823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, Y.; Yamaguchi, S.; Cha, W.; Kim, D.; Shinokubo, H. Synthesis of directly connected BODIPY oligomers through Suzuki–Miyaura coupling. Org. Lett. 2011, 13, 2992–2995. [Google Scholar] [CrossRef] [PubMed]
- Cakmak, Y.; Akkaya, E. Phenylethynyl-BODIPY oligomers: Bright dyes and fluorescent building blocks. Org. Lett. 2009, 11, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, T.; Cravino, A.; Bura, T.; Ulrich, G.; Ziessel, R.; Roncali, J. BODIPY derivatives as donor materials for bulk heterojunction solar cells. Chem. Commun. 2009, 13, 1673–1675. [Google Scholar] [CrossRef]
- Terai, T.; Nagano, T. Small-molecule fluorophores and fluorescent probes for bioimaging. J. Physiol. 2013, 465, 347–359. [Google Scholar] [CrossRef]
- Kamkaew, A.; Lim, S.; Lee, H.; Kiew, L.; Chung, L.; Burgess, K. BODIPY dyes in photodynamic therapy. Chem. Soc. Rev. 2013, 42, 77–88. [Google Scholar] [CrossRef]
- Kohler, T.; Hodgson, M.; Seidel, D.; Veauthier, J.; Meter, S.; Lynch, V.; Boyd, P.; Brothers, P.; Sessler, J. Octaethylporphyrin and expanded porphyrin complexes containing coordinated BF2 groups. Chem. Commun. 2004, 9, 1060–1061. [Google Scholar] [CrossRef]
- Sakida, T.; Yamaguchi, S.; Shinokubo, H. Metal-Mediated Synthesis of Antiaromatic Porphyrinoids from a BODIPY Precursor. Angew. Chem. Int. Ed. 2011, 50, 2280–2283. [Google Scholar] [CrossRef]
- Ishida, M.; Omagari, T.; Hirosawa, R.; Jono, K.; Sung, Y.; Yasutake, Y.; Uno, H.; Toganoh, M.; Nakanotani, H.; Fukatsu, S.; et al. Boron Difluoride Complexes of Expanded N-Confused Calix[n]phyrins That Demonstrate Unique Luminescent and Lasing Properties. Angew. Chem. Int. Ed. 2016, 55, 12045–12049. [Google Scholar] [CrossRef]
- Sakamoto, N.; Ikeda, C.; Nabeshima, T. Cation recognition and pseudorotaxane formation of tris-dipyrrin BF2 macrocycles. Chem. Commun. 2010, 46, 6732–6734. [Google Scholar] [CrossRef]
- Nakamura, T.; Yamaguchi, G.; Nabeshima, T. Unidirectional Threading into a Bowl-Shaped Macrocyclic Trimer of Boron–Dipyrrin Complexes through Multipoint Recognition. Angew. Chem. Int. Ed. 2016, 128, 9758–9761. [Google Scholar] [CrossRef]
- Sakamoto, N.; Ikeda, C.; Yamamura, M.; Nabeshima, T. α-Bridged BODIPY oligomers with switchable near-IR photoproperties by external-stimuli-induced foldamer formation and disruption. Chem. Commun. 2012, 48, 4818–4820. [Google Scholar] [CrossRef] [PubMed]
- Uchida, J.; Nakamura, T.; Yamamura, M.; Yamaguchi, G.; Nabeshima, T. m-Phenylene-Linked Dipyrrins and Their Boron–Difluoride Complexes as Variously Shaped Macrocyclic Oligomers. Org. Lett. 2016, 18, 5380–5383. [Google Scholar] [CrossRef]
- Hojo, T.; Nakamura, T.; Matsuoka, R.; Nabeshima, T. Uniquely folded shapes, photophysical properties, and recognition abilities of macrocyclic BODIPY oligomers. Heteroat. Chem. 2018, 29, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Kuzuhara, D.; Furukawa, W.; Kitashiro, A.; Aratani, N.; Yamada, H. Synthesis and Metalation of Doubly o-Phenylene-Bridged Cyclic Bis(dipyrrin)s with Highly Bent Skeleton of Dibenzoporphyrin(2.1. 2.1). Chem. Eur. J. 2016, 22, 10671–10678. [Google Scholar] [CrossRef] [Green Version]
- Xue, S.; Kuzuhara, D.; Aratani, N.; Yamada, H. [30]Hexaphyrin(2.1. 2.1. 2.1) as Aromatic Planar Ligand and Its Trinuclear Rhodium(I) Complex. Inorg. Chem. 2018, 57, 9902–9906. [Google Scholar] [CrossRef]
- Xue, S.; Kuzuhara, D.; Aratani, N.; Yamada, H. Control of Aromaticity and cis-/trans-Isomeric Structure of Non-Planar Hexaphyrin(2.1.2.1.2.1) and Metal Complexes. Angew. Chem. Int. Ed. 2019, 58, 12524–12528. [Google Scholar] [CrossRef]
- Xue, S.; Kuzuhara, D.; Aratani, N.; Yamada, H. Synthesis of a Porphyrin(2.1.2.1) Nanobelt and Its Ability To Bind Fullerene. Org. Lett. 2019, 21, 2069–2072. [Google Scholar] [CrossRef]
- Garg, K.; Ganapathi, E.; Rajakannu, P.; Ravikanth, M. Stereochemical modulation of emission behaviour in E/Z isomers of diphenyldipyrroethene from aggregation induced emission to crystallization induced emission. Phys. Chem. Chem. Phys. 2015, 17, 19465–19473. [Google Scholar] [CrossRef]
- Anju, K.; Ramakrishnan, S.; Srinivasan, A. meso-Aryl Triphyrin(2.1.1). Org. Lett. 2011, 13, 2498–2501. [Google Scholar] [CrossRef]
- Gaussian 09, Revision B.01; Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. (Eds.) Gaussian Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Bhyrappa, P.; Sankar, M.; Varghese, B.; Bhavana, P. meso-Tetrathienylporphyrins: Steady-state emission and structural properties. J. Chem. Sci. 2006, 118, 393–397. [Google Scholar] [CrossRef]
- Kubát, P.; Mosinger, J. Photophysical properties of metal complexes of meso-tetrakis (4-sulphonatophenyl) porphyrin. J. Photoch. Photobio. A 1996, 96, 93–98. [Google Scholar] [CrossRef]
- Chen, X.; Zhou, Y.; Peng, X.; Yoon, J. Fluorescent and colorimetric probes for detection of thiols. Chem. Soc. Rev. 2010, 39, 2120–2135. [Google Scholar] [CrossRef]
- Guo, H.; Jing, Y.; Yuan, X.; Ji, S.; Zhao, J.; Li, X.; Kan, Y. Highly selective fluorescent OFF–ON thiol probes based on dyads of BODIPY and potent intramolecular electron sink 2, 4-dinitrobenzenesulfonyl subunits. Org. Biomol. Chem. 2011, 9, 3844–3853. [Google Scholar] [CrossRef] [PubMed]
- Hisamune, Y.; Kim, T.; Nishimura, K.; Ishida, M.; Toganoh, M.; Mori, S.; Kim, D.; Furuta, H. Switch-ON Near IR Fluorescent Dye Upon Protonation: Helically Twisted Bis(Boron Difluoride) Complex of π-Extended Corrorin. Chem. Eur. J. 2018, 24, 4628–4634. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, S.; Kuzuhara, D.; Aratani, N.; Yamada, H. Vinylene-Bridged Cyclic Dipyrrin and BODIPY Trimers. Int. J. Mol. Sci. 2020, 21, 8041. https://doi.org/10.3390/ijms21218041
Xue S, Kuzuhara D, Aratani N, Yamada H. Vinylene-Bridged Cyclic Dipyrrin and BODIPY Trimers. International Journal of Molecular Sciences. 2020; 21(21):8041. https://doi.org/10.3390/ijms21218041
Chicago/Turabian StyleXue, Songlin, Daiki Kuzuhara, Naoki Aratani, and Hiroko Yamada. 2020. "Vinylene-Bridged Cyclic Dipyrrin and BODIPY Trimers" International Journal of Molecular Sciences 21, no. 21: 8041. https://doi.org/10.3390/ijms21218041