Protein Homeostasis Networks and the Use of Yeast to Guide Interventions in Alzheimer’s Disease



Abstract
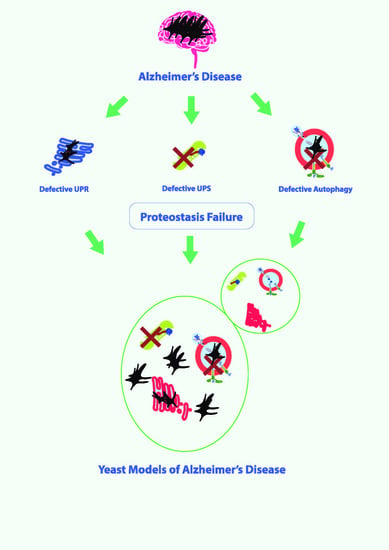
1. Introduction
2. Protein Homeostasis Network in Yeast
2.1. Unfolded Protein Response Conserved in Yeast
2.1.1. Yeast Unfolded Protein Response
2.1.2. Human Unfolded Protein Response
2.2. Ubiquitin Proteasome System
2.3. Autophagy
2.3.1. Core Autophagy Machinery in Yeast
2.3.2. Conservation of Autophagy Regulation in Yeast Models
3. Proteostasis Failure in AD
4. Future Perspectives of Using Yeast as a Model Organism for Alzheimer’s Disease
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s Disease |
| Aβ | Amyloid beta |
| EOAD | Early Onset Alzheimer’s Disease |
| LOAD | Late Onset Alzheimer’s Disease |
| APP | Amyloid precursor protein |
| PSEN | Presenilin |
| GWAS | Genome Wide Association Studies |
| ER | Endoplasmic reticulum |
| UPR | Unfolded protein response |
| eIF2α | Eukaryotic translational initiation factor 2 alpha |
| mRNA | Messenger ribonucleic acid |
| Ire1 | Inositol requiring element 1 |
| Bip | Binding immunoglobulin protein |
| ATF | Activating transcription factor |
| CREB | cAMP response element binding |
| HAC1 | Homologous to Atf/CREB1 |
| Ypt1 | Yeast protein two 1 |
| Rlg | tRNA ligase |
| bZIP | Basic leucine zipper |
| UPRE | Unfolded protein response element |
| ERAD | Endoplasmic reticulum associated degradation |
| PERK | PKR like ER kinase |
| XBP1 | X-box binding protein 1 |
| SP1 | Specificity protein 1 |
| SP2 | Specificity protein 2 |
| CHOP | C/EBP homologous protein |
| GADD34 | Growth arrest and DNA Damage 34 |
| UPS | Ubiquitin proteasome system |
| ATP | Adenosine triphosphate |
| PACE | Proteasomal associated control element |
| Rpn | Regulatory particle non-ATPase |
| Yap1 | Yeast AP-1 |
| Pdr1 | Pleotropic drug resistance 1 |
| SAR | Specific autophagy receptor |
| Atg | Autophagy related |
| PAS | Phagophore assembly site |
| TORC1 | Target of rapamycin complex 1 |
| PI3K | Phosphatidyl inositol-3-kinase |
| PI3P | Phosphatidyl inositiol-3-phosphate |
| SNARE | Soluble N-ethylmaleimide-sensitive fusion attachment protein receptor |
| V-ATPase | Vacuolar adenosine triphosphatase |
| HSF | Heat shock factor |
| HSP | Heat shock protein |
| Snf1 | Sucrose non-fermenting 1 |
| AMPK | Adenosine monophosphate kinase |
| AMP | Adenosine monophosphate |
| ULK | Unc-51 like autophagy activating kinase |
| NAD | Nicotinamide adenosine dinucleotide |
| SIRT1 | Sirtuin 1 |
| TFEB | Transcription factor EB |
| LAMP1 | Lysosome associated membrane protein 1 |
| ZKSCAN3 | Zinc finger with KRAB and SCAN domain 3 |
| Ras | Rat Sarcoma |
| Akt | Protein kinase B |
| cAMP | Cyclic AMP |
| TSC | Tuberous sclerosis complex |
| CAMKKβ | Ca2+/calmodulin dependent protein kinase β |
| BECN1 | Beclin 1 |
| Fkh | Fork head |
| FOX | Fork head box |
| Hcm1 | High-copy suppressor of calmodulin 1 |
| Sesn3 | Sestrin 3 |
| MAP1LC3B | Microtubule associated protein 1 light chain 3 beta |
| GABARAPL1 | γ-amino butyric acid type A receptor associated protein like 1 |
| Rab7 | Member RAS Oncogene family |
| ROS | Reactive oxygen species |
| RNS | Reactive nitrogen species |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| Skn7 | Supressor of Kre Null 7 |
| ARE | Antioxidant response element |
| MAPK | Mitogen activated protein kinase |
| MAPKK | MAPK kinase |
| MAPKKK | MAPK kinase kinase |
| JNK | c-Jun N-terminal kinase |
| PKA | Protein kinase A |
| RAVE | Regulator of ATPase of vacuoles and endosomes |
| Rheb | Ras homolog enriched in brain |
| AXIN | Axis inhibitor |
| LKB1 | Liver kinase B1 |
| DUB | Deubiquitinating enzyme |
| GSK3β | Glycogen synthase kinase 3 beta |
| βCTF | Beta C-terminal fragment |
| TLN | Telencephalin |
References
- Alzheimer’s Association. 2020 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020, 16, 391–460. [Google Scholar] [CrossRef]
- Dhakal, S.; Kushairi, N.; Phan, C.W.; Adhikari, B.; Sabaratnam, V.; Macreadie, I. Dietary polyphenols: A multifactorial strategy to target Alzheimer’s disease. Int. J. Mol. Sci. 2019, 20, 5090. [Google Scholar] [CrossRef]
- Panza, F.; Lozupone, M.; Logroscino, G.; Imbimbo, B.P. A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2019, 15, 73–88. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Cuyvers, E.; Sleegers, K. Genetic variations underlying Alzheimer’s disease: Evidence from genome-wide association studies and beyond. Lancet. Neurol. 2016, 15, 857–868. [Google Scholar] [CrossRef]
- Brouwers, N.; Sleegers, K.; Van Broeckhoven, C. Molecular genetics of Alzheimer’s disease: An update. Ann. Med. 2008, 40, 562–583. [Google Scholar] [CrossRef] [PubMed]
- Bertram, L.; Tanzi, R.E. Alzheimer disease risk genes: 29 and counting. Nat. Rev. Neurol. 2019, 15, 191–192. [Google Scholar] [CrossRef] [PubMed]
- Bettens, K.; Sleegers, K.; Van Broeckhoven, C. Genetic insights in Alzheimer’s disease. Lancet Neurol. 2013, 12, 92–104. [Google Scholar] [CrossRef]
- Xie, Y.; Li, J.; Kang, R.; Tang, D. Interplay between lipid metabolism and autophagy. Front Cell Dev Biol 2020, 8, 431. [Google Scholar] [CrossRef] [PubMed]
- Birgisdottir, Å.B.; Johansen, T. Autophagy and endocytosis – interconnections and interdependencies. J. Cell Sci. 2020, 133, jcs228114. [Google Scholar] [CrossRef] [PubMed]
- Nikoletopoulou, V.; Tavernarakis, N. Regulation and roles of autophagy at synapses. Trends Cell Biol. 2018, 28, 646–661. [Google Scholar] [PubMed]
- Reddy, P.H.; Oliver, D.M. Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer’s disease. Cells 2019, 8, 488. [Google Scholar]
- Loureiro, J.; Ploegh, H.L. Antigen presentation and the ubiquitin—Proteasome system in host–pathogen interactions. In Advances in Immunology; Academic Press: Cambridge, MA, USA, 2006; Volume 92, pp. 225–305. [Google Scholar]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [PubMed]
- Khurana, V.; Lindquist, S. Modelling neurodegeneration in Saccharomyces cerevisiae: Why cook with baker’s yeast? Nat. Rev. Neurosci. 2010, 11, 436–449. [Google Scholar] [PubMed]
- Seynnaeve, D.; Vecchio, M.D.; Fruhmann, G.; Verelst, J.; Cools, M.; Beckers, J.; Mulvihill, D.P.; Winderickx, J.; Franssens, V. Recent insights on Alzheimer’s disease originating from yeast models. Int. J. Mol. Sci. 2018, 19, 1947. [Google Scholar]
- Caine, J.; Sankovich, S.; Antony, H.; Waddington, L.; Macreadie, P.; Varghese, J.; Macreadie, I. Alzheimer’s Abeta fused to green fluorescent protein induces growth stress and a heat shock response. FEMS Yeast Res. 2007, 7, 1230–1236. [Google Scholar] [PubMed]
- Porzoor, A.; Alford, B.; Hügel, H.M.; Grando, D.; Caine, J.; Macreadie, I. Anti-amyloidogenic properties of some phenolic compounds. Biomolecules 2015, 5, 505–527. [Google Scholar]
- Chen, X.; Petranovic, D. Amyloid-β peptide-induced cytotoxicity and mitochondrial dysfunction in yeast. FEMS Yeast Res. 2015, 15, fov061. [Google Scholar]
- Chen, X.; Bisschops, M.M.M.; Agarwal, N.R.; Ji, B.; Shanmugavel, K.P.; Petranovic, D. Interplay of energetics and ER stress exacerbates Alzheimer’s amyloid-β (Aβ) toxicity in yeast. Front. Mol. Neurosci. 2017, 10, 232. [Google Scholar]
- Chen, X.; Ji, B.; Hao, X.; Li, X.; Eisele, F.; Nyström, T.; Petranovic, D. FMN reduces Amyloid-β toxicity in yeast by regulating redox status and cellular metabolism. Nat. Commun. 2020, 11, 867. [Google Scholar]
- Bharadwaj, P.R.; Martins, R.N. Autophagy modulates Aβ accumulation and formation of aggregates in yeast. Mol. Cell. Neurosci. 2020, 104, 103466. [Google Scholar] [CrossRef] [PubMed]
- Macreadie, I.G.; Arvanitis, C.; Bharadwaj, P. Finding chemopreventatives to reduce amyloid beta in yeast. Neural Regen Res. 2016, 11, 244–245. [Google Scholar] [CrossRef]
- Macreadie, I.; Dhakal, S. Insights from yeast on oxidative stress in Alzheimer’s disease, focusing on Ahp1p/Prx5. OBM Geriartrics 2019, 3, 10. [Google Scholar] [CrossRef]
- Mcdonald, J.B.; Dhakal, S.; Macreadie, I.G. Yeast contributions to Alzheimer’s Disease. J. Human. Clin. Gen. 2020, 2, 1–19. [Google Scholar] [CrossRef]
- Webster, B.M.; Gildea, H.K.; Dillin, A. Protein homeostasis from the outside in. Nat. Cell Biol. 2020, 22, 911–912. [Google Scholar] [CrossRef]
- Liberek, K.; Lewandowska, A.; Zietkiewicz, S. Chaperones in control of protein disaggregation. EMBO J. 2008, 27, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Verghese, J.; Abrams, J.; Wang, Y.; Morano, K.A. Biology of the heat shock response and protein chaperones: Budding yeast (Saccharomyces cerevisiae) as a model system. Microbiol. Mol. Biol. Rev. 2012, 76, 115–158. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef]
- Morawe, T.; Hiebel, C.; Kern, A.; Behl, C. Protein homeostasis, aging and Alzheimer’s disease. Mol. Neurobiol. 2012, 46, 41–54. [Google Scholar] [CrossRef]
- Cheng, J.; North, B.J.; Zhang, T.; Dai, X.; Tao, K.; Guo, J.; Wei, W. The emerging roles of protein homeostasis-governing pathways in Alzheimer’s disease. Aging Cell 2018, 17, e12801. [Google Scholar]
- Englander, S.W.; Mayne, L. The nature of protein folding pathways. Proc. Natl. Acad. Sci. USA 2014, 111, 15873. [Google Scholar]
- Vabulas, R.M.; Raychaudhuri, S.; Hayer-Hartl, M.; Hartl, F.U. Protein folding in the cytoplasm and the heat shock response. Cold Spring. Harb. Perspect Biol. 2010, 2, a004390. [Google Scholar]
- Skach, W.R. Cellular mechanisms of membrane protein folding. Nat. Struct Mol. Biol. 2009, 16, 606–612. [Google Scholar]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar]
- Mori, K. Signalling Pathways in the unfolded protein response: Development from yeast to mammals. J. Biochem. 2009, 146, 743–750. [Google Scholar]
- Chakrabarti, A.; Chen, A.W.; Varner, J.D. A review of the mammalian unfolded protein response. Biotechnol. Bioeng. 2011, 108, 2777–2793. [Google Scholar]
- Schröder, M.; Kaufman, R.J. ER stress and the unfolded protein response. Mutat. Res./Fundam. Mol. Mech. Mutagenesis 2005, 569, 29–63. [Google Scholar]
- Guerra-Moreno, A.; Ang, J.; Welsch, H.; Jochem, M.; Hanna, J. Regulation of the unfolded protein response in yeast by oxidative stress. FEBS Lett. 2019, 593, 1080–1088. [Google Scholar]
- Wu, H.; Ng, B.S.; Thibault, G. Endoplasmic reticulum stress response in yeast and humans. Biosci. Rep. 2014, 34, e00118. [Google Scholar]
- Okamura, K.; Kimata, Y.; Higashio, H.; Tsuru, A.; Kohno, K. Dissociation of Kar2p/BiP from an ER sensory molecule, Ire1p, triggers the unfolded protein response in yeast. Biochem. Biophys. Res. Commun. 2000, 279, 445–450. [Google Scholar] [PubMed]
- Sidrauski, C.; Walter, P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 1997, 90, 1031–1039. [Google Scholar]
- Tsvetanova, N.G.; Riordan, D.P.; Brown, P.O. The yeast rab GTPase Ypt1 modulates unfolded protein response dynamics by regulating the stability of HAC1 RNA. PLoS Genet. 2012, 8, e1002862. [Google Scholar]
- Mori, T.; Ogasawara, C.; Inada, T.; Englert, M.; Beier, H.; Takezawa, M.; Endo, T.; Yoshihisa, T. Dual functions of yeast tRNA ligase in the unfolded protein response: Unconventional cytoplasmic splicing of HAC1 pre-mRNA is not sufficient to release translational attenuation. Mol. Biol. Cell 2010, 21, 3722–3734. [Google Scholar] [PubMed]
- Ogawa, N.; Mori, K. Autoregulation of the HAC1 gene is required for sustained activation of the yeast unfolded protein response. Genes Cells 2004, 9, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Qi, L. Quality control in the endoplasmic reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef]
- Protter, D.S.W.; Parker, R. Principles and properties of stress granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef]
- Buchan, J.R.; Parker, R. Eukaryotic stress granules: The ins and outs of translation. Mol. Cell 2009, 36, 932–941. [Google Scholar] [CrossRef]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef]
- Plumb, R.; Zhang, Z.-R.; Appathurai, S.; Mariappan, M. A functional link between the co-translational protein translocation pathway and the UPR. eLife 2015, 4, e07426. [Google Scholar] [CrossRef]
- Adachi, Y.; Yamamoto, K.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct. Funct. 2008, 33, 75–89. [Google Scholar] [CrossRef]
- Galehdar, Z.; Swan, P.; Fuerth, B.; Callaghan, S.M.; Park, D.S.; Cregan, S.P. Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4–CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J. Neurosci. 2010, 30, 16938. [Google Scholar] [CrossRef] [PubMed]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Landau, G.; Kodali, V.K.; Malhotra, J.D.; Kaufman, R.J. Chapter Fourteen—Detection of oxidative damage in response to protein misfolding in the endoplasmic reticulum. In Methods in Enzymology; Cadenas, E., Packer, L., Eds.; Academic Press: Cambridge, MA, USA, 2013; Volume 526, pp. 231–250. [Google Scholar]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Rose, M.D.; Misra, L.M.; Vogel, J.P. KAR2, a karyogamy gene, is the yeast homolog of the mammalian BiP/GRP78 gene. Cell 1989, 57, 1211–1221. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, C.; Wang, A. Divergence and conservation of the major UPR branch IRE1-bZIP signaling pathway across eukaryotes. Sci. Rep. 2016, 6, 27362. [Google Scholar] [CrossRef]
- Kohno, K. Stress-sensing mechanisms in the unfolded protein response: Similarities and differences between yeast and mammals. J. Biochem. 2010, 147, 27–33. [Google Scholar] [CrossRef]
- Laurino, J.P.; Thompson, G.M.; Pacheco, E.; Castilho, B.A. The beta subunit of eukaryotic translation initiation factor 2 binds mRNA through the lysine repeats and a region comprising the C2-C2 motif. Mol. Cell. Biol. 1999, 19, 173–181. [Google Scholar] [CrossRef]
- Finley, D.; Ulrich, H.D.; Sommer, T.; Kaiser, P. The ubiquitin-proteasome system of Saccharomyces cerevisiae. Genetics 2012, 192, 319–360. [Google Scholar] [CrossRef]
- Vaden, J.H.; Tian, T.; Golf, S.; McLean, J.W.; Wilson, J.A.; Wilson, S.M. Chronic over-expression of ubiquitin impairs learning, reduces synaptic plasticity, and enhances GRIA receptor turnover in mice. J. Neurochem. 2019, 148, 386–399. [Google Scholar] [CrossRef]
- Rinetti, G.V.; Schweizer, F.E. Ubiquitination acutely regulates presynaptic neurotransmitter release in mammalian neurons. J. Neurosci. 2010, 30, 3157. [Google Scholar] [PubMed]
- Speese, S.D.; Trotta, N.; Rodesch, C.K.; Aravamudan, B.; Broadie, K. The ubiquitin proteasome system acutely regulates presynaptic protein turnover and synaptic efficacy. Curr. Biol. 2003, 13, 899–910. [Google Scholar]
- Sun, L.; Fan, G.; Shan, P.; Qiu, X.; Dong, S.; Liao, L.; Yu, C.; Wang, T.; Gu, X.; Li, Q.; et al. Regulation of energy homeostasis by the ubiquitin-independent REGγ proteasome. Nat. Commun. 2016, 7, 12497. [Google Scholar]
- Cheon, S.; Dean, M.; Chahrour, M. The ubiquitin proteasome pathway in neuropsychiatric disorders. Neurobiol. Learn. Mem. 2019, 165, 106791. [Google Scholar] [PubMed]
- Lambert-Smith, I.A.; Saunders, D.N.; Yerbury, J.J. The pivotal role of ubiquitin-activating enzyme E1 (UBA1) in neuronal health and neurodegeneration. Int. J. Biochem. Cell Biol. 2020, 123, 105746. [Google Scholar]
- Muratani, M.; Tansey, W.P. How the ubiquitin–proteasome system controls transcription. Nat. Rev. Mol. Cell Biol. 2003, 4, 192–201. [Google Scholar]
- Braten, O.; Livneh, I.; Ziv, T.; Admon, A.; Kehat, I.; Caspi, L.H.; Gonen, H.; Bercovich, B.; Godzik, A.; Jahandideh, S.; et al. Numerous proteins with unique characteristics are degraded by the 26S proteasome following monoubiquitination. Proc. Natl. Acad. Sci. USA 2016, 113, E4639. [Google Scholar]
- Ohtake, F.; Tsuchiya, H.; Saeki, Y.; Tanaka, K. K63 ubiquitylation triggers proteasomal degradation by seeding branched ubiquitin chains. Proc. Natl. Acad. Sci. USA 2018, 115, E1401. [Google Scholar]
- Ozkaynak, E.; Finley, D.; Solomon, M.J.; Varshavsky, A. The yeast ubiquitin genes: A family of natural gene fusions. EMBO J. 1987, 6, 1429–1439. [Google Scholar]
- McGrath, J.P.; Jentsch, S.; Varshavsky, A. UBA 1: An essential yeast gene encoding ubiquitin-activating enzyme. EMBO J. 1991, 10, 227–236. [Google Scholar]
- Seufert, W.; McGrath, J.P.; Jentsch, S. UBC1 encodes a novel member of an essential subfamily of yeast ubiquitin-conjugating enzymes involved in protein degradation. EMBO J. 1990, 9, 4535–4541. [Google Scholar] [CrossRef] [PubMed]
- Shieh, H.L.; Chen, Y.; Brown, C.R.; Chiang, H.L. Biochemical analysis of fructose-1,6-bisphosphatase import into vacuole import and degradation vesicles reveals a role for UBC1 in vesicle biogenesis. J. Biol. Chem. 2001, 276, 10398–10406. [Google Scholar] [PubMed]
- Preston, G.M.; Brodsky, J.L. The evolving role of ubiquitin modification in endoplasmic reticulum-associated degradation. Biochem. J. 2017, 474, 445–469. [Google Scholar] [CrossRef]
- Silva, G.M.; Finley, D.; Vogel, C. K63 polyubiquitination is a new modulator of the oxidative stress response. Nat. Struct Mol. Biol. 2015, 22, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Prakash, S.; Sung, P.; Prakash, L. DNA repair genes and proteins of Saccharomyces cerevisiae. Annu. Rev. Genet. 1993, 27, 33–70. [Google Scholar] [CrossRef] [PubMed]
- Craig, K.L.; Tyers, M. The F-box: A new motif for ubiquitin dependent proteolysis in cell cycle regulation and signal transduction. Prog. Biophys. Mol. Biol. 1999, 72, 299–328. [Google Scholar] [CrossRef]
- Tkach, J.M.; Yimit, A.; Lee, A.Y.; Riffle, M.; Costanzo, M.; Jaschob, D.; Hendry, J.A.; Ou, J.; Moffat, J.; Boone, C.; et al. Dissecting DNA damage response pathways by analysing protein localization and abundance changes during DNA replication stress. Nat. Cell Biol. 2012, 14, 966–976. [Google Scholar] [CrossRef] [PubMed]
- Parag, H.A.; Dimitrovsky, D.; Raboy, B.; Kulka, R.G. Selective ubiquitination of calmodulin by UBC4 and a putative ubiquitin protein ligase (E3) from Saccharomyces Cerevisiae. FEBS Lett. 1993, 325, 242–246. [Google Scholar] [CrossRef]
- Singh, R.K.; Kabbaj, M.H.; Paik, J.; Gunjan, A. Histone levels are regulated by phosphorylation and ubiquitylation-dependent proteolysis. Nat. Cell Biol. 2009, 11, 925–933. [Google Scholar] [CrossRef]
- Haworth, J.; Alver, R.C.; Anderson, M.; Bielinsky, A.K. Ubc4 and Not4 regulate steady-state levels of DNA polymerase-α to promote efficient and accurate DNA replication. Mol. Biol. Cell 2010, 21, 3205–3219. [Google Scholar] [CrossRef][Green Version]
- Xu, P.; Duong, D.M.; Seyfried, N.T.; Cheng, D.; Xie, Y.; Robert, J.; Rush, J.; Hochstrasser, M.; Finley, D.; Peng, J. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 2009, 137, 133–145. [Google Scholar] [CrossRef]
- Swanson, R.; Locher, M.; Hochstrasser, M. A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matα2 repressor degradation. Genes Dev. 2001, 15, 2660–2674. [Google Scholar] [CrossRef] [PubMed]
- Schüle, T.; Rose, M.; Entian, K.D.; Thumm, M.; Wolf, D.H. Ubc8p functions in catabolite degradation of fructose-1, 6-bisphosphatase in yeast. EMBO J. 2000, 19, 2161–2167. [Google Scholar] [CrossRef] [PubMed]
- Dieckhoff, P.; Bolte, M.; Sancak, Y.; Braus, G.H.; Irniger, S. Smt3/SUMO and Ubc9 are required for efficient APC/C-mediated proteolysis in budding yeast. Mol. Microbiol. 2004, 51, 1375–1387. [Google Scholar] [CrossRef]
- Eckert, J.H.; Johnsson, N. Pex10p links the ubiquitin conjugating enzyme Pex4p to the protein import machinery of the peroxisome. J. Cell Sci. 2003, 116, 3623–3634. [Google Scholar] [CrossRef] [PubMed]
- Brusky, J.; Zhu, Y.; Xiao, W. UBC13, a DNA-damage-inducible gene, is a member of the error-free postreplication repair pathway in Saccharomyces cerevisiae. Curr. Genet. 2000, 37, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Vanácová, S.; Wolf, J.; Martin, G.; Blank, D.; Dettwiler, S.; Friedlein, A.; Langen, H.; Keith, G.; Keller, W. A new yeast poly(A) polymerase complex involved in RNA quality control. PLoS Biol. 2005, 3, e189. [Google Scholar] [CrossRef] [PubMed]
- Kohlmann, S.; Schäfer, A.; Wolf, D.H. Ubiquitin ligase Hul5 is required for fragment-specific substrate degradation in endoplasmic reticulum-associated degradation. J. Biol. Chem. 2008, 283, 16374–16383. [Google Scholar] [CrossRef]
- Aviram, S.; Kornitzer, D. The ubiquitin ligase Hul5 promotes proteasomal processivity. Mol. Cell. Biol. 2010, 30, 985–994. [Google Scholar] [CrossRef]
- Fang, N.N.; Chan, G.T.; Zhu, M.; Comyn, S.A.; Persaud, A.; Deshaies, R.J.; Rotin, D.; Gsponer, J.; Mayor, T. Rsp5/Nedd4 is the main ubiquitin ligase that targets cytosolic misfolded proteins following heat stress. Nat. Cell Biol. 2014, 16, 1227–1237. [Google Scholar] [CrossRef]
- Kaida, D.; Toh-e, A.; Kikuchi, Y. Rsp5-Bul1/2 complex is necessary for the HSE-mediated gene expression in budding yeast. Biochem. Biophys. Res. Commun. 2003, 306, 1037–1041. [Google Scholar] [CrossRef]
- Hatakeyama, R.; Kamiya, M.; Takahara, T.; Maeda, T. Endocytosis of the aspartic acid/glutamic acid transporter Dip5 is triggered by substrate-dependent recruitment of the Rsp5 ubiquitin ligase via the arrestin-like protein Aly2. Mol. Cell. Biol. 2010, 30, 5598–5607. [Google Scholar] [CrossRef]
- Duncan, K.; Umen, J.G.; Guthrie, C. A putative ubiquitin ligase required for efficient mRNA export differentially affects hnRNP transport. Curr. Biol. Cb 2000, 10, 687–696. [Google Scholar] [CrossRef]
- Kim, D.H.; Zhang, W.; Koepp, D.M. The Hect domain E3 ligase Tom1 and the F-box protein Dia2 control Cdc6 degradation in G1 phase. J. Biol. Chem. 2012, 287, 44212–44220. [Google Scholar] [CrossRef] [PubMed]
- Daulny, A.; Geng, F.; Muratani, M.; Geisinger, J.M.; Salghetti, S.E.; Tansey, W.P. Modulation of RNA polymerase II subunit composition by ubiquitylation. Proc. Natl. Acad. Sci. USA 2008, 105, 19649–19654. [Google Scholar] [CrossRef]
- Zou, W.; Yan, J.; Zhao, N.; Niu, S.; Huang, X. A novel role for the alcohol sensitive ring/PHD finger protein Asr1p in regulating cell cycle mediated by septin-dependent assembly in yeast. Biochem. Biophys. Res. Commun. 2015, 458, 208–213. [Google Scholar] [CrossRef]
- Wood, A.; Krogan, N.J.; Dover, J.; Schneider, J.; Heidt, J.; Boateng, M.A.; Dean, K.; Golshani, A.; Zhang, Y.; Greenblatt, J.F.; et al. Bre1, an E3 ubiquitin ligase required for recruitment and substrate selection of Rad6 at a promoter. Mol. Cell 2003, 11, 267–274. [Google Scholar] [CrossRef]
- Wysocki, R.; Javaheri, A.; Allard, S.; Sha, F.; Côté, J.; Kron, S.J. Role of Dot1-dependent histone H3 methylation in G1 and S phase DNA damage checkpoint functions of Rad9. Mol. Cell. Biol. 2005, 25, 8430–8443. [Google Scholar] [CrossRef]
- Moehle, E.A.; Ryan, C.J.; Krogan, N.J.; Kress, T.L.; Guthrie, C. The yeast SR-like protein Npl3 links chromatin modification to mRNA processing. PLoS Genet. 2012, 8, e1003101. [Google Scholar] [CrossRef]
- Bieganowski, P.; Shilinski, K.; Tsichlis, P.N.; Brenner, C. Cdc123 and checkpoint forkhead associated with RING proteins control the cell cycle by controlling eIF2gamma abundance. J. Biol. Chem. 2004, 279, 44656–44666. [Google Scholar] [CrossRef]
- Singh, R.K.; Gonzalez, M.; Kabbaj, M.H.; Gunjan, A. Novel E3 ubiquitin ligases that regulate histone protein levels in the budding yeast Saccharomyces cerevisiae. PLoS ONE 2012, 7, e36295. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Ikeuchi, K.; Saeki, Y.; Iwasaki, S.; Schmidt, C.; Udagawa, T.; Sato, F.; Tsuchiya, H.; Becker, T.; Tanaka, K.; et al. Ubiquitination of stalled ribosome triggers ribosome-associated quality control. Nat. Commun. 2017, 8, 159. [Google Scholar] [CrossRef] [PubMed]
- Bordallo, J.; Plemper, R.K.; Finger, A.; Wolf, D.H. Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol. Biol. Cell 1998, 9, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Regelmann, J.; Schüle, T.; Josupeit, F.S.; Horak, J.; Rose, M.; Entian, K.D.; Thumm, M.; Wolf, D.H. Catabolite degradation of fructose-1,6-bisphosphatase in the yeast Saccharomyces cerevisiae: A genome-wide screen identifies eight novel GID genes and indicates the existence of two degradation pathways. Mol. Biol. Cell 2003, 14, 1652–1663. [Google Scholar] [CrossRef]
- Khoury, C.M.; Yang, Z.; Li, X.Y.; Vignali, M.; Fields, S.; Greenwood, M.T. A TSC22-like motif defines a novel antiapoptotic protein family. FEMS Yeast Res. 2008, 8, 540–563. [Google Scholar] [CrossRef]
- Menssen, R.; Schweiggert, J.; Schreiner, J.; Kusevic, D.; Reuther, J.; Braun, B.; Wolf, D.H. Exploring the topology of the Gid complex, the E3 ubiquitin ligase involved in catabolite-induced degradation of gluconeogenic enzymes. J. Biol. Chem. 2012, 287, 25602–25614. [Google Scholar] [CrossRef]
- Alvaro, D.; Lisby, M.; Rothstein, R. Genome-wide analysis of Rad52 foci reveals diverse mechanisms impacting recombination. PLoS Genet. 2007, 3, e228. [Google Scholar] [CrossRef]
- Panasenko, O.; Landrieux, E.; Feuermann, M.; Finka, A.; Paquet, N.; Collart, M.A. The yeast Ccr4-Not complex controls ubiquitination of the nascent-associated polypeptide (NAC-EGD) complex. J. Biol. Chem. 2006, 281, 31389–31398. [Google Scholar] [CrossRef]
- Denis, C.L.; Chiang, Y.C.; Cui, Y.; Chen, J. Genetic evidence supports a role for the yeast CCR4-NOT complex in transcriptional elongation. Genetics 2001, 158, 627–634. [Google Scholar]
- Srivastava, A.; Woolford, C.A.; Jones, E.W. Pep3p/Pep5p complex: A putative docking factor at multiple steps of vesicular transport to the vacuole of Saccharomyces cerevisiae. Genetics 2000, 156, 105–122. [Google Scholar]
- Sacksteder, K.A.; Gould, S.J. The genetics of peroxisome biogenesis. Annu. Rev. Genet. 2000, 34, 623–652. [Google Scholar] [PubMed]
- Williams, C.; van den Berg, M.; Geers, E.; Distel, B. Pex10p functions as an E3 ligase for the Ubc4p-dependent ubiquitination of Pex5p. Biochem. Biophys. Res. Commun. 2008, 374, 620–624. [Google Scholar]
- Platta, H.W.; El Magraoui, F.; Bäumer, B.E.; Schlee, D.; Girzalsky, W.; Erdmann, R. Pex2 and pex12 function as protein-ubiquitin ligases in peroxisomal protein import. Mol. Cell. Biol. 2009, 29, 5505–5516. [Google Scholar] [PubMed]
- Shin, M.E.; Ogburn, K.D.; Varban, O.A.; Gilbert, P.M.; Burd, C.G. FYVE domain targets Pib1p ubiquitin ligase to endosome and vacuolar membranes. J. Biol. Chem. 2001, 276, 41388–41393. [Google Scholar] [PubMed]
- Hewawasam, G.; Shivaraju, M.; Mattingly, M.; Venkatesh, S.; Martin-Brown, S.; Florens, L.; Workman, J.L.; Gerton, J.L. Psh1 is an E3 ubiquitin ligase that targets the centromeric histone variant Cse4. Mol. Cell 2010, 40, 444–454. [Google Scholar] [PubMed]
- Torres-Ramos, C.A.; Prakash, S.; Prakash, L. Requirement of RAD5 and MMS2 for postreplication repair of UV-damaged DNA in Saccharomyces cerevisiae. Mol. Cell. Biol. 2002, 22, 2419–2426. [Google Scholar] [PubMed]
- Blastyák, A.; Pintér, L.; Unk, I.; Prakash, L.; Prakash, S.; Haracska, L. Yeast Rad5 protein required for postreplication repair has a DNA helicase activity specific for replication fork regression. Mol. Cell 2007, 28, 167–175. [Google Scholar]
- Tateishi, S.; Sakuraba, Y.; Masuyama, S.; Inoue, H.; Yamaizumi, M. Dysfunction of human Rad18 results in defective postreplication repair and hypersensitivity to multiple mutagens. Proc. Natl. Acad. Sci. USA 2000, 97, 7927–7932. [Google Scholar]
- Ulrich, H.D. Regulating post-translational modifications of the eukaryotic replication clamp PCNA. Dna Repair 2009, 8, 461–469. [Google Scholar]
- Braun, M.A.; Costa, P.J.; Crisucci, E.M.; Arndt, K.M. Identification of Rkr1, a nuclear RING domain protein with functional connections to chromatin modification in Saccharomyces cerevisiae. Mol. Cell. Biol. 2007, 27, 2800–2811. [Google Scholar]
- Crowder, J.J.; Geigges, M.; Gibson, R.T.; Fults, E.S.; Buchanan, B.W.; Sachs, N.; Schink, A.; Kreft, S.G.; Rubenstein, E.M. Rkr1/Ltn1 ubiquitin ligase-mediated degradation of translationally stalled endoplasmic reticulum proteins. J. Biol. Chem. 2015, 290, 18454–18466. [Google Scholar] [CrossRef]
- Matsuda, R.; Ikeuchi, K.; Nomura, S.; Inada, T. Protein quality control systems associated with no-go and nonstop mRNA surveillance in yeast. Genes Cells Devoted Mol. Cell. Mech. 2014, 19, 1–12. [Google Scholar]
- Dasgupta, A.; Ramsey, K.L.; Smith, J.S.; Auble, D.T. Sir Antagonist 1 (San1) is a ubiquitin ligase. J. Biol. Chem. 2004, 279, 26830–26838. [Google Scholar] [CrossRef] [PubMed]
- Amm, I.; Wolf, D.H. Molecular mass as a determinant for nuclear San1-dependent targeting of misfolded cytosolic proteins to proteasomal degradation. FEBS Lett. 2016, 590, 1765–1775. [Google Scholar] [CrossRef]
- Ii, T.; Fung, J.; Mullen, J.R.; Brill, S.J. The yeast Slx5-Slx8 DNA integrity complex displays ubiquitin ligase activity. Cell Cycle (Georget. Tex.) 2007, 6, 2800–2809. [Google Scholar] [CrossRef][Green Version]
- Baker, L.A.; Ueberheide, B.M.; Dewell, S.; Chait, B.T.; Zheng, D.; Allis, C.D. The yeast Snt2 protein coordinates the transcriptional response to hydrogen peroxide-mediated oxidative stress. Mol. Cell. Biol. 2013, 33, 3735–3748. [Google Scholar] [PubMed]
- Hochstrasser, M. Ubiquitin-dependent protein degradation. Annu. Rev. Genet. 1996, 30, 405–439. [Google Scholar]
- Ohi, M.D.; Vander Kooi, C.W.; Rosenberg, J.A.; Ren, L.; Hirsch, J.P.; Chazin, W.J.; Walz, T.; Gould, K.L. Structural and functional analysis of essential pre-mRNA splicing factor Prp19p. Mol. Cell. Biol. 2005, 25, 451–460. [Google Scholar]
- Lu, X.; Legerski, R.J. The Prp19/Pso4 core complex undergoes ubiquitylation and structural alterations in response to DNA damage. Biochem Biophys Res Commun 2007, 354, 968–974. [Google Scholar]
- Hänzelmann, P.; Schäfer, A.; Völler, D.; Schindelin, H. Structural insights into functional modes of proteins involved in ubiquitin family pathways. Methods Mol. Biol. 2012, 832, 547–576. [Google Scholar]
- Richly, H.; Rape, M.; Braun, S.; Rumpf, S.; Hoege, C.; Jentsch, S. A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell 2005, 120, 73–84. [Google Scholar] [CrossRef]
- Urakov, V.N.; Valouev, I.A.; Lewitin, E.I.; Paushkin, S.V.; Kosorukov, V.S.; Kushnirov, V.V.; Smirnov, V.N.; Ter-Avanesyan, M.D. Itt1p, a novel protein inhibiting translation termination in Saccharomyces cerevisiae. BMC Mol. Biol. 2001, 2, 9. [Google Scholar] [CrossRef]
- Chiba, T.; Tanaka, K. Cullin-based ubiquitin ligase and its control by NEDD8-conjugating system. Curr. Protein Pept. Sci. 2004, 5, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Kamura, T.; Conaway, J.W.; Conaway, R.C. Roles of SCF and VHL ubiquitin ligases in regulation of cell growth. Prog. Mol. Subcell. Biol. 2002, 29, 1–15. [Google Scholar]
- Ribar, B.; Prakash, L.; Prakash, S. ELA1 and CUL3 are required along with ELC1 for RNA polymerase II polyubiquitylation and degradation in DNA-damaged yeast cells. Mol. Cell. Biol. 2007, 27, 3211–3216. [Google Scholar] [CrossRef]
- Fujii, K.; Kitabatake, M.; Sakata, T.; Miyata, A.; Ohno, M. A role for ubiquitin in the clearance of nonfunctional rRNAs. Genes Dev. 2009, 23, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Michel, J.J.; McCarville, J.F.; Xiong, Y. A role for Saccharomyces cerevisiae Cul8 ubiquitin ligase in proper anaphase progression. J. Biol. Chem. 2003, 278, 22828–22837. [Google Scholar] [CrossRef] [PubMed]
- Connelly, C.; Hieter, P. Budding yeast SKP1 encodes an evolutionarily conserved kinetochore protein required for cell cycle progression. Cell 1996, 86, 275–285. [Google Scholar] [CrossRef]
- Seol, J.H.; Shevchenko, A.; Shevchenko, A.; Deshaies, R.J. Skp1 forms multiple protein complexes, including RAVE, a regulator of V-ATPase assembly. Nat Cell Biol 2001, 3, 384–391. [Google Scholar] [CrossRef]
- Lejeune, D.; Chen, X.; Ruggiero, C.; Berryhill, S.; Ding, B.; Li, S. Yeast Elc1 plays an important role in global genomic repair but not in transcription coupled repair. Dna Repair 2009, 8, 40–50. [Google Scholar] [CrossRef]
- Seol, J.H.; Feldman, R.M.; Zachariae, W.; Shevchenko, A.; Correll, C.C.; Lyapina, S.; Chi, Y.; Galova, M.; Claypool, J.; Sandmeyer, S.; et al. Cdc53/cullin and the essential Hrt1 RING-H2 subunit of SCF define a ubiquitin ligase module that activates the E2 enzyme Cdc34. Genes Dev. 1999, 13, 1614–1626. [Google Scholar] [PubMed]
- Landry, B.D.; Doyle, J.P.; Toczyski, D.P.; Benanti, J.A. F-box protein specificity for G1 cyclins is dictated by subcellular localization. PLoS Genet 2012, 8, e1002851. [Google Scholar]
- Feldman, R.M.; Correll, C.C.; Kaplan, K.B.; Deshaies, R.J. A complex of Cdc4p, Skp1p, and Cdc53p/cullin catalyzes ubiquitination of the phosphorylated CDK inhibitor Sic1p. Cell 1997, 91, 221–230. [Google Scholar] [CrossRef]
- Smothers, D.B.; Kozubowski, L.; Dixon, C.; Goebl, M.G.; Mathias, N. The abundance of Met30p limits SCF(Met30p) complex activity and is regulated by methionine availability. Mol. Cell. Biol. 2000, 20, 7845–7852. [Google Scholar] [CrossRef]
- Yen, J.L.; Flick, K.; Papagiannis, C.V.; Mathur, R.; Tyrrell, A.; Ouni, I.; Kaake, R.M.; Huang, L.; Kaiser, P. Signal-induced disassembly of the SCF ubiquitin ligase complex by Cdc48/p97. Mol. Cell 2012, 48, 288–297. [Google Scholar] [CrossRef]
- Botuyan, M.V.; Koth, C.M.; Mer, G.; Chakrabartty, A.; Conaway, J.W.; Conaway, R.C.; Edwards, A.M.; Arrowsmith, C.H.; Chazin, W.J. Binding of elongin A or a von Hippel-Lindau peptide stabilizes the structure of yeast elongin C. Proc. Natl. Acad. Sci. USA 1999, 96, 9033–9038. [Google Scholar] [CrossRef]
- Gillette, T.G.; Yu, S.; Zhou, Z.; Waters, R.; Johnston, S.A.; Reed, S.H. Distinct functions of the ubiquitin-proteasome pathway influence nucleotide excision repair. EMBO J. 2006, 25, 2529–2538. [Google Scholar] [PubMed]
- Ho, Y.; Gruhler, A.; Heilbut, A.; Bader, G.D.; Moore, L.; Adams, S.L.; Millar, A.; Taylor, P.; Bennett, K.; Boutilier, K.; et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 2002, 415, 180–183. [Google Scholar] [CrossRef]
- Sipos, G.; Brickner, J.H.; Brace, E.J.; Chen, L.; Rambourg, A.; Kepes, F.; Fuller, R.S. Soi3p/Rav1p functions at the early endosome to regulate endocytic trafficking to the vacuole and localization of trans-Golgi network transmembrane proteins. Mol Biol Cell 2004, 15, 3196–3209. [Google Scholar] [CrossRef]
- Smardon, A.M.; Tarsio, M.; Kane, P.M. The RAVE complex is essential for stable assembly of the yeast V-ATPase. J. Biol. Chem. 2002, 277, 13831–13839. [Google Scholar]
- Escusa, S.; Camblong, J.; Galan, J.M.; Pinson, B.; Daignan-Fornier, B. Proteasome- and SCF-dependent degradation of yeast adenine deaminase upon transition from proliferation to quiescence requires a new F-box protein named Saf1p. Mol. Microbiol. 2006, 60, 1014–1025. [Google Scholar] [CrossRef] [PubMed]
- Topper, L.M.; Campbell, M.S.; Tugendreich, S.; Daum, J.R.; Burke, D.J.; Hieter, P.; Gorbsky, G.J. The dephosphorylated form of the anaphase-promoting complex protein Cdc27/Apc3 concentrates on kinetochores and chromosome arms in mitosis. Cell Cycle (Georget. Tex.) 2002, 1, 282–292. [Google Scholar] [CrossRef]
- Zachariae, W.; Nasmyth, K. Whose end is destruction: Cell division and the anaphase-promoting complex. Genes Dev. 1999, 13, 2039–2058. [Google Scholar] [CrossRef] [PubMed]
- Passmore, L.A.; McCormack, E.A.; Au, S.W.; Paul, A.; Willison, K.R.; Harper, J.W.; Barford, D. Doc1 mediates the activity of the anaphase-promoting complex by contributing to substrate recognition. EMBO J. 2003, 22, 786–796. [Google Scholar] [CrossRef]
- Zachariae, W.; Shevchenko, A.; Andrews, P.D.; Ciosk, R.; Galova, M.; Stark, M.J.; Mann, M.; Nasmyth, K. Mass spectrometric analysis of the anaphase-promoting complex from yeast: Identification of a subunit related to cullins. Science 1998, 279, 1216–1219. [Google Scholar] [CrossRef]
- Rousseau, A.; Bertolotti, A. Regulation of proteasome assembly and activity in health and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 697–712. [Google Scholar] [CrossRef]
- Bedford, L.; Paine, S.; Sheppard, P.W.; Mayer, R.J.; Roelofs, J. Assembly, structure, and function of the 26S proteasome. Trends Cell Biol. 2010, 20, 391–401. [Google Scholar] [CrossRef]
- Marshall, R.S.; Vierstra, R.D. Dynamic Regulation of the 26S Proteasome: From Synthesis to Degradation. Front. Mol. Biosci. 2019, 6, 40. [Google Scholar] [CrossRef] [PubMed]
- Livneh, I.; Cohen-Kaplan, V.; Cohen-Rosenzweig, C.; Avni, N.; Ciechanover, A. The life cycle of the 26S proteasome: From birth, through regulation and function, and onto its death. Cell Res. 2016, 26, 869–885. [Google Scholar] [CrossRef]
- Mannhaupt, G.; Schnall, R.; Karpov, V.; Vetter, I.; Feldmann, H. Rpn4p acts as a transcription factor by binding to PACE, a nonamer box found upstream of 26S proteasomal and other genes in yeast. FEBS Lett. 1999, 450, 27–34. [Google Scholar] [CrossRef]
- Ma, M.; Liu, Z.L. Comparative transcriptome profiling analyses during the lag phase uncover YAP1, PDR1, PDR3, RPN4, and HSF1 as key regulatory genes in genomic adaptation to the lignocellulose derived inhibitor HMF for Saccharomyces cerevisiae. BMC Genom. 2010, 11, 660. [Google Scholar] [CrossRef] [PubMed]
- Tobias, J.W.; Varshavsky, A. Cloning and functional analysis of the ubiquitin-specific protease gene UBP1 of Saccharomyces cerevisiae. J. Biol. Chem. 1991, 266, 12021–12028. [Google Scholar] [PubMed]
- Baker, R.T.; Tobias, J.W.; Varshavsky, A. Ubiquitin-specific proteases of Saccharomyces cerevisiae. Cloning of UBP2 and UBP3, and functional analysis of the UBP gene family. J. Biol. Chem. 1992, 267, 23364–23375. [Google Scholar] [PubMed]
- Cohen, M.; Stutz, F.; Belgareh, N.; Haguenauer-Tsapis, R.; Dargemont, C. Ubp3 requires a cofactor, Bre5, to specifically de-ubiquitinate the COPII protein, Sec23. Nat. Cell Biol. 2003, 5, 661–667. [Google Scholar] [CrossRef]
- Solé, C.; Nadal-Ribelles, M.; Kraft, C.; Peter, M.; Posas, F.; de Nadal, E. Control of Ubp3 ubiquitin protease activity by the Hog1 SAPK modulates transcription upon osmostress. EMBO J. 2011, 30, 3274–3284. [Google Scholar] [CrossRef]
- Amerik, A.Y.; Nowak, J.; Swaminathan, S.; Hochstrasser, M. The Doa4 deubiquitinating enzyme is functionally linked to the vacuolar protein-sorting and endocytic pathways. Mol. Biol. Cell 2000, 11, 3365–3380. [Google Scholar] [CrossRef]
- Swaminathan, S.; Amerik, A.Y.; Hochstrasser, M. The Doa4 deubiquitinating enzyme is required for ubiquitin homeostasis in yeast. Mol. Biol. Cell 1999, 10, 2583–2594. [Google Scholar] [CrossRef]
- Hanna, J.; Hathaway, N.A.; Tone, Y.; Crosas, B.; Elsasser, S.; Kirkpatrick, D.S.; Leggett, D.S.; Gygi, S.P.; King, R.W.; Finley, D. Deubiquitinating enzyme Ubp6 functions noncatalytically to delay proteasomal degradation. Cell 2006, 127, 99–111. [Google Scholar] [CrossRef]
- Böhm, S.; Szakal, B.; Herken, B.W.; Sullivan, M.R.; Mihalevic, M.J.; Kabbinavar, F.F.; Branzei, D.; Clark, N.L.; Bernstein, K.A. The budding yeast ubiquitin protease Ubp7 is a novel component involved in S phase progression. J. Biol. Chem. 2016, 291, 4442–4452. [Google Scholar] [CrossRef]
- Henry, K.W.; Wyce, A.; Lo, W.S.; Duggan, L.J.; Emre, N.C.; Kao, C.F.; Pillus, L.; Shilatifard, A.; Osley, M.A.; Berger, S.L. Transcriptional activation via sequential histone H2B ubiquitylation and deubiquitylation, mediated by SAGA-associated Ubp8. Genes Dev. 2003, 17, 2648–2663. [Google Scholar] [CrossRef]
- Gallego-Sánchez, A.; Andrés, S.; Conde, F.; San-Segundo, P.A.; Bueno, A. Reversal of PCNA ubiquitylation by Ubp10 in Saccharomyces cerevisiae. PLoS Genet. 2012, 8, e1002826. [Google Scholar]
- Huh, W.K.; Falvo, J.V.; Gerke, L.C.; Carroll, A.S.; Howson, R.W.; Weissman, J.S.; O’Shea, E.K. Global analysis of protein localization in budding yeast. Nature 2003, 425, 686–691. [Google Scholar]
- Debelyy, M.O.; Platta, H.W.; Saffian, D.; Hensel, A.; Thoms, S.; Meyer, H.E.; Warscheid, B.; Girzalsky, W.; Erdmann, R. Ubp15p, a ubiquitin hydrolase associated with the peroxisomal export machinery. J. Biol. Chem. 2011, 286, 28223–28234. [Google Scholar] [PubMed]
- Ostapenko, D.; Burton, J.L.; Solomon, M.J. The Ubp15 deubiquitinase promotes timely entry into S phase in Saccharomyces cerevisiae. Mol. Biol. Cell 2015, 26, 2205–2216. [Google Scholar] [PubMed]
- Kinner, A.; Kölling, R. The yeast deubiquitinating enzyme Ubp16 is anchored to the outer mitochondrial membrane. FEBS Lett. 2003, 549, 135–140. [Google Scholar] [PubMed]
- Verma, R.; Aravind, L.; Oania, R.; McDonald, W.H.; Yates, J.R., 3rd; Koonin, E.V.; Deshaies, R.J. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 2002, 298, 611–615. [Google Scholar]
- Guterman, A.; Glickman, M.H. Complementary roles for Rpn11 and Ubp6 in deubiquitination and proteolysis by the proteasome. J. Biol. Chem. 2004, 279, 1729–1738. [Google Scholar]
- Rumpf, S.; Jentsch, S. Functional division of substrate processing cofactors of the ubiquitin-selective Cdc48 chaperone. Mol. Cell 2006, 21, 261–269. [Google Scholar]
- Linghu, B.; Callis, J.; Goebl, M.G. Rub1p processing by Yuh1p is required for wild-type levels of Rub1p conjugation to Cdc53p. Eukaryot. Cell 2002, 1, 491–494. [Google Scholar]
- Groll, M.; Ditzel, L.; Löwe, J.; Stock, D.; Bochtler, M.; Bartunik, H.D.; Huber, R. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 1997, 386, 463–471. [Google Scholar]
- Heinemeyer, W.; Tröndle, N.; Albrecht, G.; Wolf, D.H. PRE5 and PRE6, the last missing genes encoding 20S proteasome subunits from yeast? Indication for a set of 14 different subunits in the eukaryotic proteasome core. Biochemistry 1994, 33, 12229–12237. [Google Scholar] [CrossRef]
- Georgatsou, E.; Georgakopoulos, T.; Thireos, G. Molecular cloning of an essential yeast gene encoding a proteasomal subunit. FEBS Lett. 1992, 299, 39–43. [Google Scholar] [CrossRef]
- Jäger, S.; Groll, M.; Huber, R.; Wolf, D.H.; Heinemeyer, W. Proteasome beta-type subunits: Unequal roles of propeptides in core particle maturation and a hierarchy of active site function. J. Mol. Biol. 1999, 291, 997–1013. [Google Scholar] [CrossRef] [PubMed]
- Arendt, C.S.; Hochstrasser, M. Identification of the yeast 20S proteasome catalytic centers and subunit interactions required for active-site formation. Proc. Natl. Acad. Sci. USA 1997, 94, 7156–7161. [Google Scholar] [CrossRef]
- Takeuchi, J.; Toh-e, A. Genetic evidence for interaction between components of the yeast 26S proteasome: Combination of a mutation in RPN12 (a lid component gene) with mutations in RPT1 (an ATPase gene) causes synthetic lethality. Mol. Gen. Genet. Mgg 1999, 262, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Rubin, D.M.; Fried, V.A.; Finley, D. The regulatory particle of the Saccharomyces cerevisiae proteasome. Mol. Cell. Biol. 1998, 18, 3149–3162. [Google Scholar] [CrossRef] [PubMed]
- McDonald, H.B.; Byers, B. A proteasome cap subunit required for spindle pole body duplication in yeast. J. Cell Biol. 1997, 137, 539–553. [Google Scholar] [CrossRef]
- Gonzalez, F.; Delahodde, A.; Kodadek, T.; Johnston, S.A. Recruitment of a 19S proteasome subcomplex to an activated promoter. Science 2002, 296, 548–550. [Google Scholar] [CrossRef]
- Glickman, M.H.; Rubin, D.M.; Coux, O.; Wefes, I.; Pfeifer, G.; Cjeka, Z.; Baumeister, W.; Fried, V.A.; Finley, D. A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and eIF3. Cell 1998, 94, 615–623. [Google Scholar] [CrossRef]
- Husnjak, K.; Elsasser, S.; Zhang, N.; Chen, X.; Randles, L.; Shi, Y.; Hofmann, K.; Walters, K.J.; Finley, D.; Dikic, I. Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 2008, 453, 481–488. [Google Scholar] [CrossRef]
- van Nocker, S.; Sadis, S.; Rubin, D.M.; Glickman, M.; Fu, H.; Coux, O.; Wefes, I.; Finley, D.; Vierstra, R.D. The multiubiquitin-chain-binding protein Mcb1 is a component of the 26S proteasome in Saccharomyces cerevisiae and plays a nonessential, substrate-specific role in protein turnover. Mol. Cell. Biol. 1996, 16, 6020–6028. [Google Scholar] [CrossRef]
- Bailly, E.; Reed, S.I. Functional characterization of Rpn3 uncovers a distinct 19S proteasomal subunit requirement for ubiquitin-dependent proteolysis of cell cycle regulatory proteins in budding yeast. Mol. Cell. Biol. 1999, 19, 6872–6890. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Santamaria, P.G.; Finley, D.; Ballesta, J.P.; Remacha, M. Rpn6p, a proteasome subunit from Saccharomyces cerevisiae, is essential for the assembly and activity of the 26 S proteasome. J. Biol. Chem. 2003, 278, 6687–6695. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a promoter of longevity: Insights from model organisms. Nat. Rev. Mol. Cell Biol. 2018, 19, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Su, T.; Li, X.; Yang, M.; Shao, Q.; Zhao, Y.; Ma, C.; Wang, P. Autophagy: An intracellular degradation pathway regulating plant survival and stress response. Front. Plant Sci. 2020, 11, 164. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Li, J.; Bao, J.K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. Cmls 2012, 69, 1125–1136. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef]
- Beese, C.J.; Brynjólfsdóttir, S.H.; Frankel, L.B. Selective autophagy of the protein homeostasis machinery: Ribophagy, proteaphagy and ER-phagy. Front. Cell Dev. Biol. 2020, 7, 373. [Google Scholar] [CrossRef]
- Kirkin, V. History of the selective autophagy research: How did it begin and where does it stand today? J. Mol. Biol. 2020, 432, 3–27. [Google Scholar] [PubMed]
- Farré, J.-C.; Subramani, S. Mechanistic insights into selective autophagy pathways: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 2016, 17, 537–552. [Google Scholar] [PubMed]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. AMPK/Snf1 signaling regulates histone acetylation: Impact on gene expression and epigenetic functions. Cell. Signal. 2016, 28, 887–895. [Google Scholar] [PubMed]
- Sacitharan, P.K.; Bou-Gharios, G.; Edwards, J.R. SIRT1 directly activates autophagy in human chondrocytes. Cell Death Discov. 2020, 6, 41. [Google Scholar]
- Hong, S.-P.; Leiper, F.C.; Woods, A.; Carling, D.; Carlson, M. Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proc. Natl. Acad. Sci. USA 2003, 100, 8839. [Google Scholar]
- Hedbacker, K.; Carlson, M. SNF1/AMPK pathways in yeast. Front Biosci 2008, 13, 2408–2420. [Google Scholar]
- Rodríguez-Colman, M.J.; Sorolla, M.A.; Vall-llaura, N.; Tamarit, J.; Ros, J.; Cabiscol, E. The FOX transcription factor Hcm1 regulates oxidative metabolism in response to early nutrient limitation in yeast. Role of Snf1 and Tor1/Sch9 kinases. Biochim. Et Biophys. Acta (Bba) Mol. Cell Res. 2013, 1833, 2004–2015. [Google Scholar]
- Ghavidel, A.; Baxi, K.; Prusinkiewicz, M.; Swan, C.; Belak, Z.R.; Eskiw, C.H.; Carvalho, C.E.; Harkness, T.A. Rapid nuclear exclusion of Hcm1 in aging Saccharomyces cerevisiae leads to vacuolar alkalization and replicative senescence. G3 (Bethesda) 2018, 8, 1579–1592. [Google Scholar]
- Rodriguez-Colman, M.J.; Reverter-Branchat, G.; Sorolla, M.A.; Tamarit, J.; Ros, J.; Cabiscol, E. The forkhead transcription factor Hcm1 promotes mitochondrial biogenesis and stress resistance in yeast. J. Biol. Chem. 2010, 285, 37092–37101. [Google Scholar]
- Linke, C.; Klipp, E.; Lehrach, H.; Barberis, M.; Krobitsch, S. Fkh1 and Fkh2 associate with Sir2 to control CLB2 transcription under normal and oxidative stress conditions. Front. Physiol. 2013, 4, 173. [Google Scholar]
- Murakami, H.; Aiba, H.; Nakanishi, M.; Murakami-Tonami, Y. Regulation of yeast forkhead transcription factors and FoxM1 by cyclin-dependent and polo-like kinases. Cell Cycle 2010, 9, 3253–3262. [Google Scholar] [CrossRef] [PubMed]
- Madia, F.; Wei, M.; Yuan, V.; Hu, J.; Gattazzo, C.; Pham, P.; Goodman, M.F.; Longo, V.D. Oncogene homologue Sch9 promotes age-dependent mutations by a superoxide and Rev1/Polzeta-dependent mechanism. J. Cell Biol. 2009, 186, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Urban, J.; Soulard, A.; Huber, A.; Lippman, S.; Mukhopadhyay, D.; Deloche, O.; Wanke, V.; Anrather, D.; Ammerer, G.; Riezman, H.; et al. Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol. Cell 2007, 26, 663–674. [Google Scholar] [CrossRef] [PubMed]
- McManus, E.J.; Alessi, D.R. TSC1–TSC2: A complex tale of PKB-mediated S6K regulation. Nat. Cell Biol. 2002, 4, E214–E216. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef] [PubMed]
- Kijanska, M.; Dohnal, I.; Reiter, W.; Kaspar, S.; Stoffel, I.; Ammerer, G.; Kraft, C.; Peter, M. Activation of Atg1 kinase in autophagy by regulated phosphorylation. Autophagy 2010, 6, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Raucci, S.; Jaquenoud, M.; Hatakeyama, R.; Stumpe, M.; Rohr, R.; Reggiori, F.; De Virgilio, C.; Dengjel, J. Multilayered control of protein turnover by TORC1 and Atg1. Cell Rep. 2019, 28, 3486–3496.e6. [Google Scholar] [CrossRef]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef]
- Vázquez-Ibarra, A.; Subirana, L.; Ongay-Larios, L.; Kawasaki, L.; Rojas-Ortega, E.; Rodríguez-González, M.; de Nadal, E.; Posas, F.; Coria, R. Activation of the Hog1 MAPK by the Ssk2/Ssk22 MAP3Ks, in the absence of the osmosensors, is not sufficient to trigger osmostress adaptation in Saccharomyces cerevisiae. FEBS J. 2018, 285, 1079–1096. [Google Scholar] [CrossRef]
- Murakami, Y.; Tatebayashi, K.; Saito, H. Two adjacent docking sites in the yeast Hog1 mitogen-activated protein (MAP) kinase differentially interact with the Pbs2 MAP kinase kinase and the Ptp2 protein tyrosine phosphatase. Mol. Cell Biol. 2008, 28, 2481–2494. [Google Scholar] [CrossRef]
- Manjithaya, R.; Jain, S.; Farré, J.-C.; Subramani, S. A yeast MAPK cascade regulates pexophagy but not other autophagy pathways. J. Cell Biol. 2010, 189, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Mao, K.; Wang, K.; Zhao, M.; Xu, T.; Klionsky, D.J. Two MAPK-signaling pathways are required for mitophagy in Saccharomyces cerevisiae. J. Cell Biol. 2011, 193, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Gualtieri, T.; Ragni, E.; Mizzi, L.; Fascio, U.; Popolo, L. The cell wall sensor Wsc1p is involved in reorganization of actin cytoskeleton in response to hypo-osmotic shock in Saccharomyces cerevisiae. Yeast (Chichesterengland) 2004, 21, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef]
- Nakatogawa, H. Mechanisms governing autophagosome biogenesis. Nat. Rev. Mol. Cell Biol. 2020, 21, 439–458. [Google Scholar] [CrossRef]
- Wang, S.; Li, Y.; Ma, C. Atg3 promotes Atg8 lipidation via altering lipid diffusion and rearrangement. Protein Sci. 2020, 29, 1511–1523. [Google Scholar] [CrossRef]
- Martens, S.; Fracchiolla, D. Activation and targeting of ATG8 protein lipidation. Cell Discov. 2020, 6, 23. [Google Scholar] [CrossRef]
- Hanada, T.; Noda, N.N.; Satomi, Y.; Ichimura, Y.; Fujioka, Y.; Takao, T.; Inagaki, F.; Ohsumi, Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem. 2007, 282, 37298–37302. [Google Scholar] [CrossRef]
- Kihara, A.; Noda, T.; Ishihara, N.; Ohsumi, Y. Two distinct Vps34 phosphatidylinositol 3–kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J. Cell Biol. 2001, 152, 519–530. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef]
- Abdollahzadeh, I.; Schwarten, M.; Gensch, T.; Willbold, D.; Weiergräber, O.H. The Atg8 family of proteins—Modulating shape and functionality of autophagic membranes. Front. Genet. 2017, 8, 109. [Google Scholar] [PubMed]
- Feng, Y.; Klionsky, D.J. Autophagic membrane delivery through ATG9. Cell Res. 2017, 27, 161–162. [Google Scholar] [CrossRef]
- Shintani, T.; Mizushima, N.; Ogawa, Y.; Matsuura, A.; Noda, T.; Ohsumi, Y. Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. EMBO J. 1999, 18, 5234–5241. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Kamada, Y.; Baba, M.; Takikawa, H.; Sasaki, M.; Ohsumi, Y. Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol. Biol. Cell 2005, 16, 2544–2553. [Google Scholar] [PubMed]
- Kametaka, S.; Okano, T.; Ohsumi, M.; Ohsumi, Y. Apg14p and Apg6/Vps30p form a protein complex essential for autophagy in the yeast, Saccharomyces cerevisiae. J. Biol. Chem. 1998, 273, 22284–22291. [Google Scholar] [CrossRef] [PubMed]
- Meijer, W.H.; van der Klei, I.J.; Veenhuis, M.; Kiel, J.A. ATG genes involved in non-selective autophagy are conserved from yeast to man, but the selective Cvt and pexophagy pathways also require organism-specific genes. Autophagy 2007, 3, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Slessareva, J.E.; Routt, S.M.; Temple, B.; Bankaitis, V.A.; Dohlman, H.G. Activation of the phosphatidylinositol 3-kinase Vps34 by a G protein alpha subunit at the endosome. Cell 2006, 126, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Stjepanovic, G.; Baskaran, S.; Lin, M.G.; Hurley, J.H. Vps34 kinase domain dynamics regulate the autophagic PI 3-kinase complex. Mol. Cell 2017, 67, 528–534.e3. [Google Scholar] [CrossRef]
- Kamada, Y.; Yoshino, K.; Kondo, C.; Kawamata, T.; Oshiro, N.; Yonezawa, K.; Ohsumi, Y. Tor directly controls the Atg1 kinase complex to regulate autophagy. Mol Cell Biol 2010, 30, 1049–1058. [Google Scholar]
- Fujioka, Y.; Suzuki, S.W.; Yamamoto, H.; Kondo-Kakuta, C.; Kimura, Y.; Hirano, H.; Akada, R.; Inagaki, F.; Ohsumi, Y.; Noda, N.N. Structural basis of starvation-induced assembly of the autophagy initiation complex. Nat. Struct. Mol. Biol. 2014, 21, 513–521. [Google Scholar]
- Reggiori, F.; Tucker, K.A.; Stromhaug, P.E.; Klionsky, D.J. The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev. Cell 2004, 6, 79–90. [Google Scholar] [CrossRef]
- Obara, K.; Sekito, T.; Niimi, K.; Ohsumi, Y. The Atg18-Atg2 complex is recruited to autophagic membranes via phosphatidylinositol 3-phosphate and exerts an essential function. J. Biol. Chem. 2008, 283, 23972–23980. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Sugita, H.; Yoshimori, T.; Ohsumi, Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 1998, 273, 33889–33892. [Google Scholar] [CrossRef]
- Wen, X.; Klionsky, D.J. An overview of macroautophagy in yeast. J. Mol. Biol. 2016, 428, 1681–1699. [Google Scholar] [CrossRef]
- de Marcos Lousa, C.; Denecke, J. Lysosomal and vacuolar sorting: Not so different after all! Biochem Soc Trans 2016, 44, 891–897. [Google Scholar] [CrossRef]
- Wang, C.-W.; Klionsky, D.J. The molecular mechanism of autophagy. Mol. Med. 2003, 9, 65–76. [Google Scholar] [CrossRef]
- Nair, U.; Jotwani, A.; Geng, J.; Gammoh, N.; Richerson, D.; Yen, W.-L.; Griffith, J.; Nag, S.; Wang, K.; Moss, T.; et al. SNARE proteins are required for macroautophagy. Cell 2011, 146, 290–302. [Google Scholar] [CrossRef]
- Li, S.C.; Kane, P.M. The yeast lysosome-like vacuole: Endpoint and crossroads. Biochim Biophys Acta 2009, 1793, 650–663. [Google Scholar] [CrossRef]
- Pamarthy, S.; Kulshrestha, A.; Katara, G.K.; Beaman, K.D. The curious case of vacuolar ATPase: Regulation of signaling pathways. Mol. Cancer 2018, 17, 41. [Google Scholar] [CrossRef]
- Colacurcio, D.J.; Nixon, R.A. Disorders of lysosomal acidification-The emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res Rev 2016, 32, 75–88. [Google Scholar] [CrossRef]
- Kane, P.M.; Smardon, A.M. Assembly and regulation of the yeast vacuolar H+-ATPase. J. Bioenerg. Biomembr. 2003, 35, 313–321. [Google Scholar] [CrossRef]
- Wilms, T.; Swinnen, E.; Eskes, E.; Dolz-Edo, L.; Uwineza, A.; Van Essche, R.; Rosseels, J.; Zabrocki, P.; Cameroni, E.; Franssens, V.; et al. The yeast protein kinase Sch9 adjusts V-ATPase assembly/disassembly to control pH homeostasis and longevity in response to glucose availability. PLoS Genet. 2017, 13, e1006835. [Google Scholar] [CrossRef] [PubMed]
- Hirata, R.; Ohsumk, Y.; Nakano, A.; Kawasaki, H.; Suzuki, K.; Anraku, Y. Molecular structure of a gene, VMA1, encoding the catalytic subunit of H(+)-translocating adenosine triphosphatase from vacuolar membranes of Saccharomyces cerevisiae. J. Biol. Chem. 1990, 265, 6726–6733. [Google Scholar] [PubMed]
- Ruckenstuhl, C.; Netzberger, C.; Entfellner, I.; Carmona-Gutierrez, D.; Kickenweiz, T.; Stekovic, S.; Gleixner, C.; Schmid, C.; Klug, L.; Sorgo, A.G.; et al. Lifespan extension by methionine restriction requires autophagy-dependent vacuolar acidification. PLoS Genet. 2014, 10, e1004347. [Google Scholar] [CrossRef] [PubMed]
- Forgac, M. Structure and properties of the vacuolar (H+)-ATPases. J. Biol. Chem. 1999, 274, 12951–12954. [Google Scholar]
- Vitavska, O.; Wieczorek, H.; Merzendorfer, H. A novel role for subunit C in mediating binding of the H+-V-ATPase to the actin cytoskeleton. J. Biol. Chem. 2003, 278, 18499–18505. [Google Scholar] [CrossRef]
- Graham, L.A.; Hill, K.J.; Stevens, T.H. VMA8 encodes a 32-kDa V1 subunit of the Saccharomyces cerevisiae vacuolar H(+)-ATPase required for function and assembly of the enzyme complex. J. Biol. Chem. 1995, 270, 15037–15044. [Google Scholar]
- Xu, T.; Forgac, M. Subunit D (Vma8p) of the yeast vacuolar H+-ATPase plays a role in coupling of proton transport and ATP hydrolysis. J. Biol. Chem. 2000, 275, 22075–22081. [Google Scholar] [CrossRef]
- Foury, F. The 31-kDa polypeptide is an essential subunit of the vacuolar ATPase in Saccharomyces cerevisiae. J. Biol. Chem. 1990, 265, 18554–18560. [Google Scholar]
- Graham, L.A.; Hill, K.J.; Stevens, T.H. VMA7 encodes a novel 14-kDa subunit of the Saccharomyces cerevisiae vacuolar H(+)-ATPase complex. J. Biol. Chem. 1994, 269, 25974–25977. [Google Scholar]
- Supeková, L.; Supek, F.; Nelson, N. The Saccharomyces cerevisiae VMA10 is an intron-containing gene encoding a novel 13-kDa subunit of vacuolar H(+)-ATPase. J. Biol. Chem. 1995, 270, 13726–13732. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.N.; Hirata, R.; Umemoto, N.; Ohya, Y.; Takatsuki, A.; Stevens, T.H.; Anraku, Y. VMA13 encodes a 54-kDa vacuolar H(+)-ATPase subunit required for activity but not assembly of the enzyme complex in Saccharomyces cerevisiae. J. Biol. Chem. 1993, 268, 18286–18292. [Google Scholar] [PubMed]
- Preston, R.A.; Murphy, R.F.; Jones, E.W. Assay of vacuolar pH in yeast and identification of acidification-defective mutants. Proc. Natl. Acad. Sci. USA 1989, 86, 7027–7031. [Google Scholar] [CrossRef] [PubMed]
- Manolson, M.F.; Wu, B.; Proteau, D.; Taillon, B.E.; Roberts, B.T.; Hoyt, M.A.; Jones, E.W. STV1 gene encodes functional homologue of 95-kDa yeast vacuolar H(+)-ATPase subunit Vph1p. J. Biol. Chem. 1994, 269, 14064–14074. [Google Scholar] [PubMed]
- Banerjee, S.; Clapp, K.; Tarsio, M.; Kane, P.M. Interaction of the late endo-lysosomal lipid PI(3,5)P2 with the Vph1 isoform of yeast V-ATPase increases its activity and cellular stress tolerance. J. Biol. Chem. 2019, 294, 9161–9171. [Google Scholar] [CrossRef]
- Umemoto, N.; Yoshihisa, T.; Hirata, R.; Anraku, Y. Roles of the VMA3 gene product, subunit c of the vacuolar membrane H(+)-ATPase on vacuolar acidification and protein transport. A study with VMA3-disrupted mutants of Saccharomyces cerevisiae. J. Biol. Chem. 1990, 265, 18447–18453. [Google Scholar]
- Rane, H.S.; Bernardo, S.M.; Raines, S.M.; Binder, J.L.; Parra, K.J.; Lee, S.A. Candida albicans VMA3 is necessary for V-ATPase assembly and function and contributes to secretion and filamentation. Eukaryot Cell 2013, 12, 1369–1382. [Google Scholar] [CrossRef][Green Version]
- Hirata, R.; Graham, L.A.; Takatsuki, A.; Stevens, T.H.; Anraku, Y. VMA11 and VMA16 encode second and third proteolipid subunits of the Saccharomyces cerevisiae vacuolar membrane H+-ATPase. J. Biol. Chem. 1997, 272, 4795–4803. [Google Scholar] [CrossRef]
- Flannery, A.R.; Graham, L.A.; Stevens, T.H. Topological characterization of the c, c’, and c” subunits of the vacuolar ATPase from the yeast Saccharomyces cerevisiae. J. Biol. Chem. 2004, 279, 39856–39862. [Google Scholar] [CrossRef]
- Bauerle, C.; Ho, M.N.; Lindorfer, M.A.; Stevens, T.H. The Saccharomyces cerevisiae VMA6 gene encodes the 36-kDa subunit of the vacuolar H(+)-ATPase membrane sector. J. Biol. Chem. 1993, 268, 12749–12757. [Google Scholar]
- Jia, C.; Zhang, K.; Zhang, D.; Yu, Q.; Zhao, Q.; Xiao, C.; Dong, Y.; Chu, M.; Li, M. Roles of VPH2 and VMA6 in localization of V-ATPase subunits, cell wall functions and filamentous development in Candida albicans. Fungal Genet. Biol. Fg B 2018, 114, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Compton, M.A.; Graham, L.A.; Stevens, T.H. Vma9p (subunit e) is an integral membrane V0 subunit of the yeast V-ATPase. J. Biol. Chem. 2006, 281, 15312–15319. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Kondo-Okamoto, N. Mitochondria and autophagy: Critical interplay between the two homeostats. Biochim. Et Biophys. Acta (Bba) Gen. Subj. 2012, 1820, 595–600. [Google Scholar] [CrossRef]
- Dokladny, K.; Myers, O.B.; Moseley, P.L. Heat shock response and autophagy—Cooperation and control. Autophagy 2015, 11, 200–213. [Google Scholar] [CrossRef]
- Russell, R.C.; Yuan, H.-X.; Guan, K.-L. Autophagy regulation by nutrient signaling. Cell Res. 2014, 24, 42–57. [Google Scholar] [CrossRef]
- Mao, K.; Klionsky, D.J. AMPK activates autophagy by phosphorylating ULK1. Circ. Res. 2011, 108, 787–788. [Google Scholar] [CrossRef]
- Carling, D.; Sanders, M.J.; Woods, A. The regulation of AMP-activated protein kinase by upstream kinases. Int. J. Obes. 2008, 32, S55–S59. [Google Scholar] [CrossRef]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 2008, 283, 27628–27635. [Google Scholar] [CrossRef]
- Satoh, A.; Brace, C.S.; Rensing, N.; Cliften, P.; Wozniak, D.F.; Herzog, E.D.; Yamada, K.A.; Imai, S.-I. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab 2013, 18, 416–430. [Google Scholar] [PubMed]
- Cantó, C.; Menzies, K.J.; Auwerx, J. NAD(+) metabolism and the control of energy homeostasis: A balancing act between mitochondria and the nucleus. Cell Metab 2015, 22, 31–53. [Google Scholar] [PubMed]
- Cerutti, R.; Pirinen, E.; Lamperti, C.; Marchet, S.; Sauve, A.A.; Li, W.; Leoni, V.; Schon, E.A.; Dantzer, F.; Auwerx, J.; et al. NAD(+)-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab 2014, 19, 1042–1049. [Google Scholar]
- White, E. Autophagy and p53. Cold Spring Harb. Perspect. Med. 2016, 6, a026120. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Woods, N.T.; Piluso, L.G.; Lee, H.H.; Chen, J.; Bhalla, K.N.; Monteiro, A.; Liu, X.; Hung, M.C.; Wang, H.G. p53 acetylation is crucial for its transcription-independent proapoptotic functions. J. Biol. Chem. 2009, 284, 11171–11183. [Google Scholar] [PubMed]
- Langley, E.; Pearson, M.; Faretta, M.; Bauer, U.-M.; Frye, R.A.; Minucci, S.; Pelicci, P.G.; Kouzarides, T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 2002, 21, 2383–2396. [Google Scholar] [PubMed]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar]
- Demers-Lamarche, J.; Guillebaud, G.; Tlili, M.; Todkar, K.; Bélanger, N.; Grondin, M.; Nguyen, A.P.; Michel, J.; Germain, M. Loss of mitochondrial function impairs lysosomes. J. Biol. Chem. 2016, 291, 10263–10276. [Google Scholar]
- Cheng, X.-T.; Xie, Y.-X.; Zhou, B.; Huang, N.; Farfel-Becker, T.; Sheng, Z.-H. Revisiting LAMP1 as a marker for degradative autophagy-lysosomal organelles in the nervous system. Autophagy 2018, 14, 1472–1474. [Google Scholar]
- Deus, C.M.; Yambire, K.F.; Oliveira, P.J.; Raimundo, N. Mitochondria–lysosome crosstalk: From physiology to neurodegeneration. Trends Mol. Med. 2020, 26, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.; Goodwin, J.G.; Chauhan, S.; Manyam, G.; Wang, J.; Kamat, A.M.; Boyd, D.D. ZKSCAN3 is a master transcriptional repressor of autophagy. Mol. Cell 2013, 50, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Guan, K.-L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, T.; Powers, S.; Cameron, S.; Fasano, O.; Goldfarb, M.; Broach, J.; Wigler, M. Functional homology of mammalian and yeast RAS genes. Cell 1985, 40, 19–26. [Google Scholar] [CrossRef]
- Cazzanelli, G.; Pereira, F.; Alves, S.; Francisco, R.; Azevedo, L.; Dias Carvalho, P.; Almeida, A.; Côrte-Real, M.; Oliveira, M.J.; Lucas, C.; et al. The yeast Saccharomyces cerevisiae as a model for understanding RAS proteins and their role in human tumorigenesis. Cells 2018, 7, 14. [Google Scholar] [CrossRef]
- Castellano, E.; Downward, J. RAS interaction with PI3K: More than just another effector pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef]
- Bhaskar, P.T.; Hay, N. The two TORCs and Akt. Dev. Cell 2007, 12, 487–502. [Google Scholar] [CrossRef]
- De Virgilio, C.; Loewith, R. The TOR signalling network from yeast to man. Int. J. Biochem. Cell Biol. 2006, 38, 1476–1481. [Google Scholar] [CrossRef]
- Díaz-Troya, S.; Pérez-Pérez, M.E.; Florencio, F.J.; Crespo, J.L. The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy 2008, 4, 851–865. [Google Scholar] [CrossRef]
- Sanz, P.; Viana, R.; Garcia-Gimeno, M.A. AMPK in yeast: The SNF1 (Sucrose Non-fermenting 1) protein kinase complex. EXS 2016, 107, 353–374. [Google Scholar]
- Peña-Llopis, S.; Vega-Rubin-de-Celis, S.; Schwartz, J.C.; Wolff, N.C.; Tran, T.A.T.; Zou, L.; Xie, X.-J.; Corey, D.R.; Brugarolas, J. Regulation of TFEB and V-ATPases by mTORC1. EMBO J. 2011, 30, 3242–3258. [Google Scholar] [PubMed]
- Roberts, D.J.; Tan-Sah, V.P.; Ding, E.Y.; Smith, J.M.; Miyamoto, S. Hexokinase-II positively regulates glucose starvation-induced autophagy through TORC1 inhibition. Mol. Cell 2014, 53, 521–533. [Google Scholar] [CrossRef]
- Bootman, M.D.; Chehab, T.; Bultynck, G.; Parys, J.B.; Rietdorf, K. The regulation of autophagy by calcium signals: Do we have a consensus? Cell Calcium 2018, 70, 32–46. [Google Scholar] [CrossRef]
- Milles, L.F.; Unterauer, E.M.; Nicolaus, T.; Gaub, H.E. Calcium stabilizes the strongest protein fold. Nat. Commun. 2018, 9, 4764. [Google Scholar] [CrossRef]
- Mekahli, D.; Bultynck, G.; Parys, J.B.; De Smedt, H.; Missiaen, L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb. Perspect Biol. 2011, 3, a004317. [Google Scholar] [CrossRef] [PubMed]
- Cakir, I.; Nillni, E.A. Endoplasmic reticulum stress, the hypothalamus, and energy balance. Trends Endocrinol. Metab. 2019, 30, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Ermak, G.; Davies, K.J.A. Calcium and oxidative stress: From cell signaling to cell death. Mol. Immunol. 2002, 38, 713–721. [Google Scholar] [CrossRef]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef]
- Ghislat, G.; Patron, M.; Rizzuto, R.; Knecht, E. Withdrawal of essential amino acids increases autophagy by a pathway involving Ca2+/calmodulin-dependent kinase kinase-β (CaMKK-β). J. Biol. Chem. 2012, 287, 38625–38636. [Google Scholar] [CrossRef]
- Zündorf, G.; Reiser, G. Calcium dysregulation and homeostasis of neural calcium in the molecular mechanisms of neurodegenerative diseases provide multiple targets for neuroprotection. Antioxid. Redox Signal. 2011, 14, 1275–1288. [Google Scholar] [CrossRef]
- Grotemeier, A.; Alers, S.; Pfisterer, S.G.; Paasch, F.; Daubrawa, M.; Dieterle, A.; Viollet, B.; Wesselborg, S.; Proikas-Cezanne, T.; Stork, B. AMPK-independent induction of autophagy by cytosolic Ca2+ increase. Cell Signal 2010, 22, 914–925. [Google Scholar] [CrossRef] [PubMed]
- Filippi-Chiela, E.C.; Viegas, M.S.; Thomé, M.P.; Buffon, A.; Wink, M.R.; Lenz, G. Modulation of autophagy by calcium signalosome in human disease. Mol. Pharmacol. 2016, 90, 371. [Google Scholar] [CrossRef] [PubMed]
- Coen, K.; Flannagan, R.S.; Baron, S.; Carraro-Lacroix, L.R.; Wang, D.; Vermeire, W.; Michiels, C.; Munck, S.; Baert, V.; Sugita, S.; et al. Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J. Cell Biol. 2012, 198, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Di Malta, C.; Cinque, L.; Settembre, C. Transcriptional regulation of autophagy: Mechanisms and diseases. Front. Cell Dev. Biol. 2019, 7, 114. [Google Scholar] [CrossRef]
- Tuteja, G.; Kaestner, K.H. Forkhead transcription factors II. Cell 2007, 131, 192. [Google Scholar] [CrossRef]
- Jiao, R.; Postnikoff, S.; Harkness, T.A.; Arnason, T.G. The SNF1 kinase ubiquitin-associated domain restrains its activation, activity, and the yeast life span. J. Biol. Chem. 2015, 290, 15393–15404. [Google Scholar] [CrossRef]
- Zhao, Y.; Yang, J.; Liao, W.; Liu, X.; Zhang, H.; Wang, S.; Wang, D.; Feng, J.; Yu, L.; Zhu, W.-G. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol. 2010, 12, 665–675. [Google Scholar] [CrossRef]
- Sanchez, A.M.J.; Csibi, A.; Raibon, A.; Cornille, K.; Gay, S.; Bernardi, H.; Candau, R. AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1. J. Cell. Biochem. 2012, 113, 695–710. [Google Scholar] [CrossRef]
- Füllgrabe, J.; Ghislat, G.; Cho, D.-H.; Rubinsztein, D.C. Transcriptional regulation of mammalian autophagy at a glance. J. Cell Sci. 2016, 129, 3059. [Google Scholar] [CrossRef]
- Hariharan, N.; Maejima, Y.; Nakae, J.; Paik, J.; DePinho Ronald, A.; Sadoshima, J. Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ. Res. 2010, 107, 1470–1482. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Et Biophys. Acta (Bba) Mol. Cell Res. 2011, 1813, 1978–1986. [Google Scholar] [CrossRef]
- Nakka, V.P.; Prakash-babu, P.; Vemuganti, R. Crosstalk between endoplasmic reticulum stress, oxidative stress, and autophagy: Potential therapeutic targets for acute CNS injuries. Mol. Neurobiol. 2016, 53, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Chiang, H.-H.; Louw, M.; Susanto, A.; Chen, D. Nutrient sensing and the oxidative stress response. Trends Endocrinol. Metab. 2017, 28, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. -Lung Cell. Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef] [PubMed]
- Fuse, Y.; Kobayashi, M. Conservation of the Keap1-Nrf2 system: An evolutionary journey through stressful space and time. Molecules 2017, 22, 436. [Google Scholar] [CrossRef]
- Vomund, S.; Schäfer, A.; Parnham, M.J.; Brüne, B.; von Knethen, A. Nrf2, the master regulator of anti-oxidative responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef]
- van der Laan, K.J.; Morita, A.; Perona-Martinez, F.P.; Schirhagl, R. Evaluation of the oxidative stress response of aging yeast cells in response to internalization of fluorescent nanodiamond biosensors. Nanomaterials 2020, 10, 372. [Google Scholar] [CrossRef] [PubMed]
- Norris, E.H.; Giasson, B.I. Role of oxidative damage in protein aggregation associated with Parkinson’s disease and related disorders. Antioxid. Redox Signal. 2005, 7, 672–684. [Google Scholar] [CrossRef]
- Rezatabar, S.; Karimian, A.; Rameshknia, V.; Parsian, H.; Majidinia, M.; Kopi, T.A.; Bishayee, A.; Sadeghinia, A.; Yousefi, M.; Monirialamdari, M.; et al. RAS/MAPK signaling functions in oxidative stress, DNA damage response and cancer progression. J. Cell. Physiol. 2019, 234, 14951–14965. [Google Scholar] [CrossRef]
- Morrison, D.K. MAP kinase pathways. Cold Spring Harb. Perspect Biol. 2012, 4, a011254. [Google Scholar] [CrossRef]
- Till, A.; Lakhani, R.; Burnett, S.F.; Subramani, S. Pexophagy: The selective degradation of peroxisomes. Int. J. Cell Biol. 2012, 2012, 512721. [Google Scholar] [PubMed]
- Courtial, L.; Picco, V.; Grover, R.; Cormerais, Y.; Rottier, C.; Labbe, A.; Pagès, G.; Ferrier-Pagès, C. The c-Jun N-terminal kinase prevents oxidative stress induced by UV and thermal stresses in corals and human cells. Sci. Rep. 2017, 7, 45713. [Google Scholar] [PubMed]
- Lorin, S.; Pierron, G.; Ryan, K.M.; Codogno, P.; Djavaheri-Mergny, M. Evidence for the interplay between JNK and p53-DRAM signaling pathways in the regulation of autophagy. Autophagy 2010, 6, 153–154. [Google Scholar] [CrossRef]
- Xu, P.; Das, M.; Reilly, J.; Davis, R.J. JNK regulates FoxO-dependent autophagy in neurons. Genes Dev. 2011, 25, 310–322. [Google Scholar] [CrossRef]
- Lin, X.; Wang, Y.-J.; Li, Q.; Hou, Y.-Y.; Hong, M.-H.; Cao, Y.-L.; Chi, Z.-Q.; Liu, J.-G. Chronic high-dose morphine treatment promotes SH-SY5Y cell apoptosis via c-Jun N-terminal kinase-mediated activation of mitochondria-dependent pathway. FEBS J. 2009, 276, 2022–2036. [Google Scholar]
- Kroemer, G.; Jäättelä, M. Lysosomes and autophagy in cell death control. Nat. Rev. Cancer 2005, 5, 886–897. [Google Scholar] [PubMed]
- Marshansky, V.; Futai, M. The V-type H+-ATPase in vesicular trafficking: Targeting, regulation and function. Curr. Opin. Cell Biol. 2008, 20, 415–426. [Google Scholar] [CrossRef]
- Parra, K.J.; Chan, C.-Y.; Chen, J. Saccharomyces cerevisiae vacuolar H+-ATPase regulation by disassembly and reassembly: One structure and multiple signals. Eukaryot Cell 2014, 13, 706–714. [Google Scholar]
- Collins, M.P.; Forgac, M. Regulation of V-ATPase assembly in nutrient sensing and function of V-ATPases in breast cancer metastasis. Front. Physiol. 2018, 9, 902. [Google Scholar]
- Bond, S.; Forgac, M. The Ras/cAMP/protein kinase A pathway regulates glucose-dependent assembly of the vacuolar (H+)-ATPase in yeast. J. Biol. Chem. 2008, 283, 36513–36521. [Google Scholar] [CrossRef]
- Smardon, A.M.; Diab, H.I.; Tarsio, M.; Diakov, T.T.; Nasab, N.D.; West, R.W.; Kane, P.M. The RAVE complex is an isoform-specific V-ATPase assembly factor in yeast. Mol. Biol. Cell 2014, 25, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Meo-Evoli, N.; Almacellas, E.; Massucci, F.A.; Gentilella, A.; Ambrosio, S.; Kozma, S.C.; Thomas, G.; Tauler, A. V-ATPase: A master effector of E2F1-mediated lysosomal trafficking, mTORC1 activation and autophagy. Oncotarget 2015, 6, 28057–28070. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-S.; Jiang, B.; Li, M.; Zhu, M.; Peng, Y.; Zhang, Y.-L.; Wu, Y.-Q.; Li, T.Y.; Liang, Y.; Lu, Z.; et al. The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 2014, 20, 526–540. [Google Scholar] [CrossRef]
- McGuire, C.M.; Forgac, M. Glucose starvation increases V-ATPase assembly and activity in mammalian cells through AMP kinase and phosphatidylinositide 3-kinase/Akt signaling. J. Biol. Chem. 2018, 293, 9113–9123. [Google Scholar] [CrossRef]
- Zhang, C.S.; Li, M.; Zong, Y.; Lin, S.C. Determining AMPK activation via the lysosomal v-ATPase-Ragulator-AXIN/LKB1 axis. Methods Mol. Biol. (Cliftonn. J.) 2018, 1732, 393–411. [Google Scholar]
- Mputhia, Z.; Hone, E.; Tripathi, T.; Sargeant, T.; Martins, R.; Bharadwaj, P. Autophagy modulation as a treatment of amyloid diseases. Molecules 2019, 24, 3372. [Google Scholar] [CrossRef] [PubMed]
- Ohno, M. Roles of eIF2α kinases in the pathogenesis of Alzheimer’s disease. Front Mol Neurosci 2014, 7, 22. [Google Scholar] [CrossRef]
- Li, J.-Q.; Yu, J.-T.; Jiang, T.; Tan, L. Endoplasmic reticulum dysfunction in Alzheimer’s disease. Mol. Neurobiol. 2015, 51, 383–395. [Google Scholar] [CrossRef]
- Lee, J.H.; Won, S.M.; Suh, J.; Son, S.J.; Moon, G.J.; Park, U.J.; Gwag, B.J. Induction of the unfolded protein response and cell death pathway in Alzheimer’s disease, but not in aged Tg2576 mice. Exp. Mol. Med. 2010, 42, 386–394. [Google Scholar] [CrossRef]
- Nijholt, D.A.T.; Nölle, A.; van Haastert, E.S.; Edelijn, H.; Toonen, R.F.; Hoozemans, J.J.M.; Scheper, W. Unfolded protein response activates glycogen synthase kinase-3 via selective lysosomal degradation. Neurobiol. Aging 2013, 34, 1759–1771. [Google Scholar] [CrossRef]
- LaFerla, F.M. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Halliwell, B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 2019, 20, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Vanderweyde, T.; Apicco, D.J.; Youmans-Kidder, K.; Ash, P.E.A.; Cook, C.; Lummertz da Rocha, E.; Jansen-West, K.; Frame, A.A.; Citro, A.; Leszyk, J.D.; et al. Interaction of tau with the RNA-binding protein TIA1 regulates tau pathophysiology and toxicity. Cell Rep. 2016, 15, 1455–1466. [Google Scholar] [CrossRef] [PubMed]
- Gadhave, K.; Bolshette, N.; Ahire, A.; Pardeshi, R.; Thakur, K.; Trandafir, C.; Istrate, A.; Ahmed, S.; Lahkar, M.; Muresanu, D.F.; et al. The ubiquitin proteasomal system: A potential target for the management of Alzheimer’s disease. J. Cell Mol. Med. 2016, 20, 1392–1407. [Google Scholar] [CrossRef] [PubMed]
- Al Mamun, A.; Uddin, M.S.; Kabir, M.T.; Khanum, S.; Sarwar, M.S.; Mathew, B.; Rauf, A.; Ahmed, M.; Ashraf, G.M. Exploring the promise of targeting ubiquitin-proteasome system to combat Alzheimer’s disease. Neurotox. Res. 2020, 38, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Gao, J.; Kim, J.; Hong, C.; Kim, J.; Tontonoz, P. The E3 ubiquitin ligase Idol controls brain LDL receptor expression, ApoE clearance, and Aβ amyloidosis. Sci. Transl. Med. 2015, 7, 314ra184. [Google Scholar] [CrossRef]
- Lee, M.J.; Lee, J.H.; Rubinsztein, D.C. Tau degradation: The ubiquitin–proteasome system versus the autophagy-lysosome system. Prog. Neurobiol. 2013, 105, 49–59. [Google Scholar] [CrossRef]
- Toral-Rios, D.; Pichardo-Rojas, P.S.; Alonso-Vanegas, M.; Campos-Peña, V. GSK3β and tau protein in Alzheimer’s Disease and epilepsy. Front. Cell. Neurosci. 2020, 14, 19. [Google Scholar] [CrossRef]
- Nair, S.; Traini, M.; Dawes, I.W.; Perrone, G.G. Genome-wide analysis of Saccharomyces cerevisiae identifies cellular processes affecting intracellular aggregation of Alzheimer’s amyloid-β42: Importance of lipid homeostasis. Mol. Biol. Cell 2014, 25, 2235–2249. [Google Scholar] [CrossRef]
- Nixon, R.A.; Yang, D.S. Autophagy failure in Alzheimer’s disease—Locating the primary defect. Neurobiol. Dis. 2011, 43, 38–45. [Google Scholar] [CrossRef]
- Piras, A.; Collin, L.; Grüninger, F.; Graff, C.; Rönnbäck, A. Autophagic and lysosomal defects in human tauopathies: Analysis of post-mortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol. Commun. 2016, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Rios, L.; Yan, Z.; Jasper, A.M.; Luera, D.; Luo, S.; Rao, H. Autophagy regulator Atg9 is degraded by the proteasome. Biochem. Biophys. Res. Commun. 2020, 522, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Aiello, A.; Accardi, G.; Candore, G.; Gambino, C.M.; Mirisola, M.; Taormina, G.; Virruso, C.; Caruso, C. Nutrient sensing pathways as therapeutic targets for healthy ageing. Expert Opin. Ther. Targets 2017, 21, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 2008, 28, 6926. [Google Scholar] [CrossRef]
- He, L.Q.; Lu, J.H.; Yue, Z.Y. Autophagy in ageing and ageing-associated diseases. Acta Pharmacol. Sin. 2013, 34, 605–611. [Google Scholar] [CrossRef]
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef]
- Cai, Z.; Zhou, Y.; Liu, Z.; Ke, Z.; Zhao, B. Autophagy dysfunction upregulates beta-amyloid peptides via enhancing the activity of γ-secretase complex. Neuropsychiatr Dis Treat 2015, 11, 2091–2099. [Google Scholar] [CrossRef]
- McDermott, J.R.; Gibson, A.M. Degradation of Alzheimer’s beta-amyloid protein by human cathepsin D. Neuroreport 1996, 7, 2163–2166. [Google Scholar]
- Papassotiropoulos, A.; Bagli, M.; Kurz, A.; Kornhuber, J.; Förstl, H.; Maier, W.; Pauls, J.; Lautenschlager, N.; Heun, R. A genetic variation of cathepsin D is a major risk factor for Alzheimer’s disease. Ann. Neurol. 2000, 47, 399–403. [Google Scholar] [CrossRef]
- Campanella, C.; Pace, A.; Caruso Bavisotto, C.; Marzullo, P.; Marino Gammazza, A.; Buscemi, S.; Palumbo Piccionello, A. Heat shock proteins in Alzheimer’s disease: Role and targeting. Int. J. Mol. Sci. 2018, 19, 2603. [Google Scholar] [CrossRef]
- Luo, G.R.; Le, W.D. Collective roles of molecular chaperones in protein degradation pathways associated with neurodegenerative diseases. Curr. Pharm. Biotechnol. 2010, 11, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Chang, P.T. Acidic pH promotes the formation of toxic fibrils from beta-amyloid peptide. Brain Res. 2001, 893, 287–291. [Google Scholar]
- Lanoiselée, H.-M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.-C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef] [PubMed]
- Veugelen, S.; Saito, T.; Saido, T.C.; Chávez-Gutiérrez, L.; De Strooper, B. Familial Alzheimer’s disease mutations in Presenilin generate amyloidogenic Aβ peptide seeds. Neuron 2016, 90, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Esselens, C.; Oorschot, V.; Baert, V.; Raemaekers, T.; Spittaels, K.; Serneels, L.; Zheng, H.; Saftig, P.; De Strooper, B.; Klumperman, J.; et al. Presenilin 1 mediates the turnover of telencephalin in hippocampal neurons via an autophagic degradative pathway. J. Cell Biol. 2004, 166, 1041–1054. [Google Scholar] [CrossRef] [PubMed]
- Katayama, T.; Imaizumi, K.; Sato, N.; Miyoshi, K.; Kudo, T.; Hitomi, J.; Morihara, T.; Yoneda, T.; Gomi, F.; Mori, Y.; et al. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat. Cell Biol. 1999, 1, 479–485. [Google Scholar] [CrossRef]
- Jung, T.; Bader, N.; Grune, T. Lipofuscin. Ann. N. Y. Acad. Sci. 2007, 1119, 97–111. [Google Scholar]
- Park, J.T.; Lee, Y.-S.; Cho, K.A.; Park, S.C. Adjustment of the lysosomal-mitochondrial axis for control of cellular senescence. Ageing. Res. Rev. 2018, 47, 176–182. [Google Scholar] [CrossRef]
- Moreno-García, A.; Kun, A.; Calero, O.; Medina, M.; Calero, M. An overview of the role of lipofuscin in age-related neurodegeneration. Front Neurosci 2018, 12, 464. [Google Scholar] [CrossRef]
- Moh, C.; Kubiak, J.Z.; Bajic, V.P.; Zhu, X.; Smith, M.A.; Lee, H.-g. Cell cycle deregulation in the neurons of Alzheimer’s disease. In Cell Cycle in Development; Kubiak, J.Z., Ed.; Springer Berlin Heidelberg: Berlin/Heidelberg, Germany, 2011; pp. 565–576. [Google Scholar]
- Höhn, A.; Jung, T.; Grimm, S.; Catalgol, B.; Weber, D.; Grune, T. Lipofuscin inhibits the proteasome by binding to surface motifs. Free Radic. Biol. Med. 2011, 50, 585–591. [Google Scholar] [CrossRef]
- Höhn, A.; Grune, T. Lipofuscin: Formation, effects and role of macroautophagy. Redox Biol. 2013, 1, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Pan, Q.; Zhang, W.; Xiang, J.; Jiang, X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. [Google Scholar] [CrossRef]
- Schedin-Weiss, S.; Inoue, M.; Hromadkova, L.; Teranishi, Y.; Yamamoto, N.G.; Wiehager, B.; Bogdanovic, N.; Winblad, B.; Sandebring-Matton, A.; Frykman, S.; et al. Monoamine oxidase B is elevated in Alzheimer disease neurons, is associated with γ-secretase and regulates neuronal amyloid β-peptide levels. Alzheimers Res. Ther. 2017, 9, 57. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shi, Y.; Wei, H. Calcium dysregulation in Alzheimer’s disease: A target for new drug development. J. Alzheimers Dis. Parkinsonism 2017, 7, 374. [Google Scholar] [CrossRef]
- Wong, S.Y.; Tang, B.L. SIRT1 as a therapeutic target for Alzheimer’s disease. Rev. Neurosci. 2016, 27, 813–825. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Kosik, K.S.; Kennedy, M.B.; Kowall, N.W. Hippocampal neurons predisposed to neurofibrillary tangle formation are enriched in type II calcium/calmodulin-dependent protein kinase. J. Neuropathol. Exp. Neurol. 1990, 49, 49–63. [Google Scholar] [CrossRef]
- Wang, Y.J.; Chen, G.H.; Hu, X.Y.; Lu, Y.P.; Zhou, J.N.; Liu, R.Y. The expression of calcium/calmodulin-dependent protein kinase II-alpha in the hippocampus of patients with Alzheimer’s disease and its links with AD-related pathology. Brain Res. 2005, 1031, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Beran, K. Budding of yeast cells, their scars and ageing. In Advances in Microbial Physiology; Rose, A.H., Wilkinson, J.F., Eds.; Academic Press: Cambridge, MA, USA, 1968; Volume 2, pp. 143–171. [Google Scholar]
- Sorokin, M.I.; Knorre, D.A.; Severin, F.F. Early manifestations of replicative aging in the yeast Saccharomyces cerevisiae. Microb Cell 2014, 1, 37–42. [Google Scholar] [CrossRef]
- Steinkraus, K.A.; Kaeberlein, M.; Kennedy, B.K. Replicative aging in yeast: The means to the end. Annu. Rev. Cell Dev. Biol. 2008, 24, 29–54. [Google Scholar] [CrossRef]
- Tzur, A.; Moore, J.K.; Jorgensen, P.; Shapiro, H.M.; Kirschner, M.W. Optimizing optical flow cytometry for cell volume-based sorting and analysis. PLoS ONE 2011, 6, e16053. [Google Scholar] [CrossRef]
- Zhang, H.; Komano, H.; Fuller, R.S.; Gandy, S.E.; Frail, D.E. Proteolytic processing and secretion of human beta-amyloid precursor protein in yeast. Evidence for a yeast secretase activity. J. Biol. Chem. 1994, 269, 27799–27802. [Google Scholar] [PubMed]
- Moosavi, B.; Mousavi, B.; Macreadie, I.G. Yeast model of Amyloid-β and tau aggregation in Alzheimer’s disease. J. Alzheimer’s Dis. Jad 2015, 47, 9–16. [Google Scholar] [CrossRef]
- Almshawit, H.; Macreadie, I. Fungicidal effect of thymoquinone involves generation of oxidative stress in Candida glabrata. Microbiol. Res. 2017, 195, 81–88. [Google Scholar] [CrossRef]
- Dhakal, S.; Subhan, M.; Fraser, J.M.; Gardiner, K.; Macreadie, I. Simvastatin efficiently reduces levels of Alzheimer’s amyloid beta in yeast. Int. J. Mol. Sci. 2019, 20, 3531. [Google Scholar] [CrossRef] [PubMed]
- Dhakal, S.; Macreadie, I. Tyramine and amyloid beta 42: A toxic synergy. Biomedicines 2020, 8, 145. [Google Scholar] [CrossRef]
- Luu, Y.N.; Macreadie, I. Development of convenient system for detecting yeast cell stress, including that of amyloid beta. Int. J. Mol. Sci. 2018, 19, 2136. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Jordan, J.J.; Ciribilli, Y.; Resnick, M.A.; Bisio, A.; Inga, A. Quantitative analysis of NF-κB transactivation specificity using a yeast-based functional assay. PLoS ONE 2015, 10, e0130170. [Google Scholar] [CrossRef][Green Version]








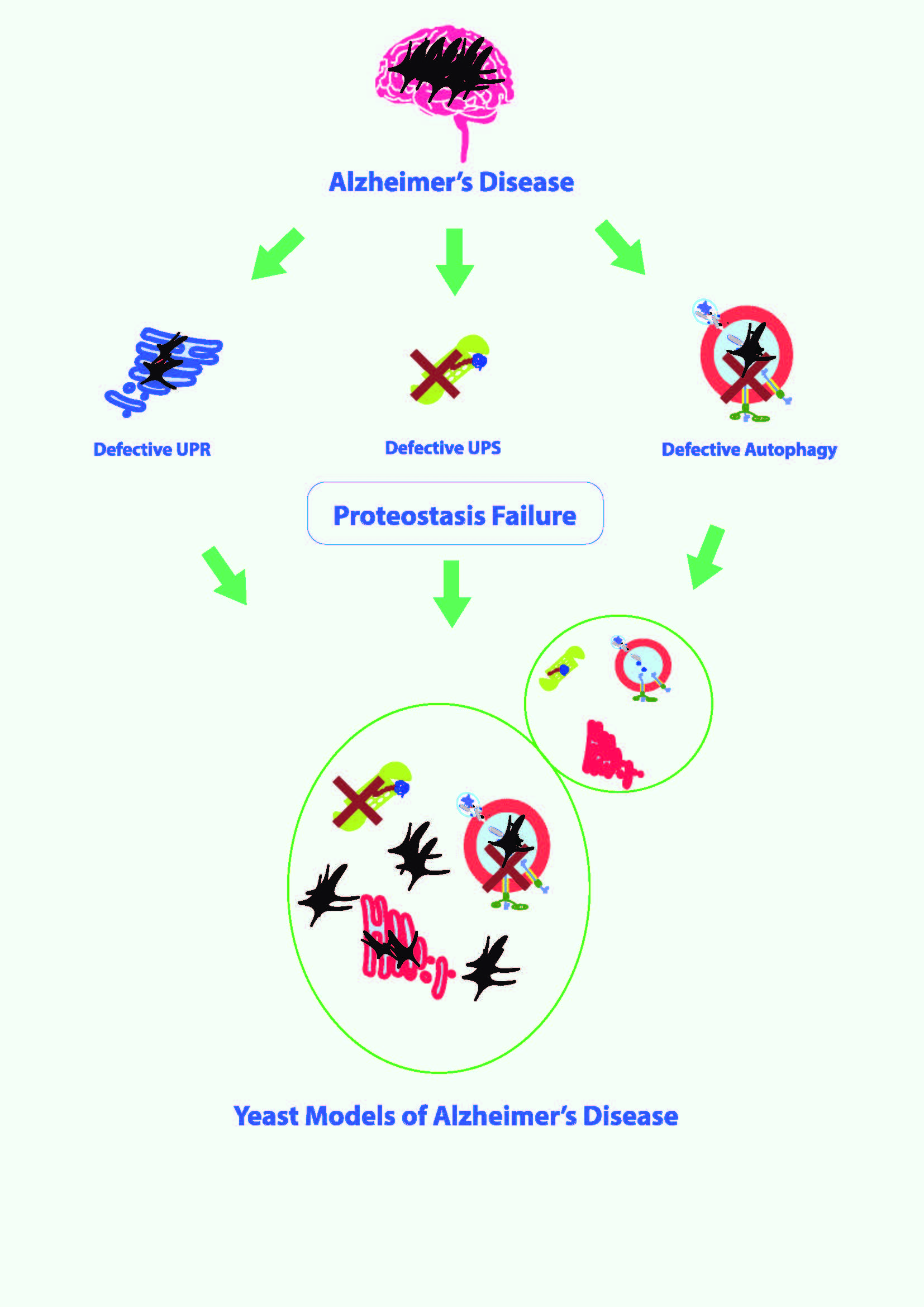{kind=link}
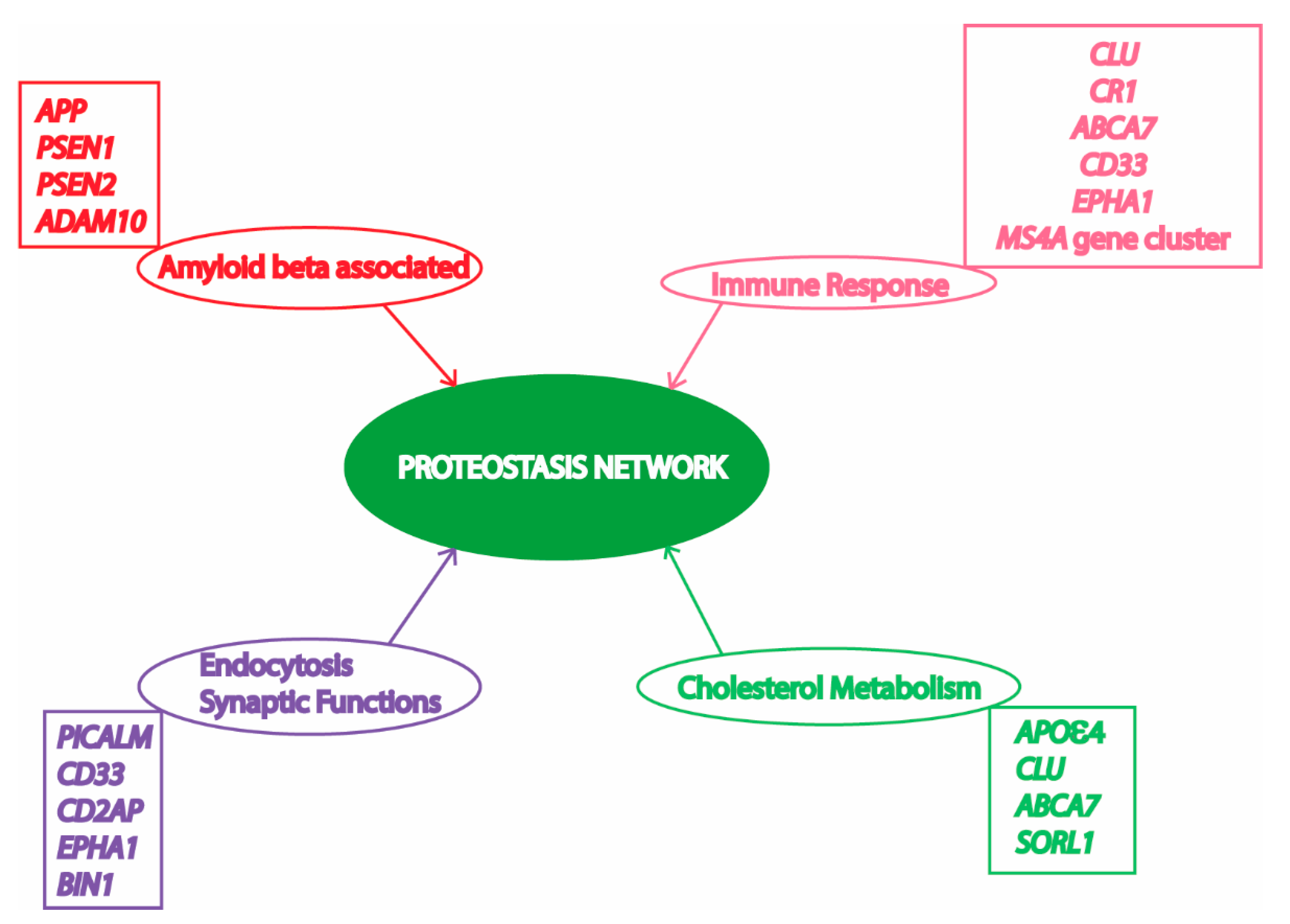{kind=link}
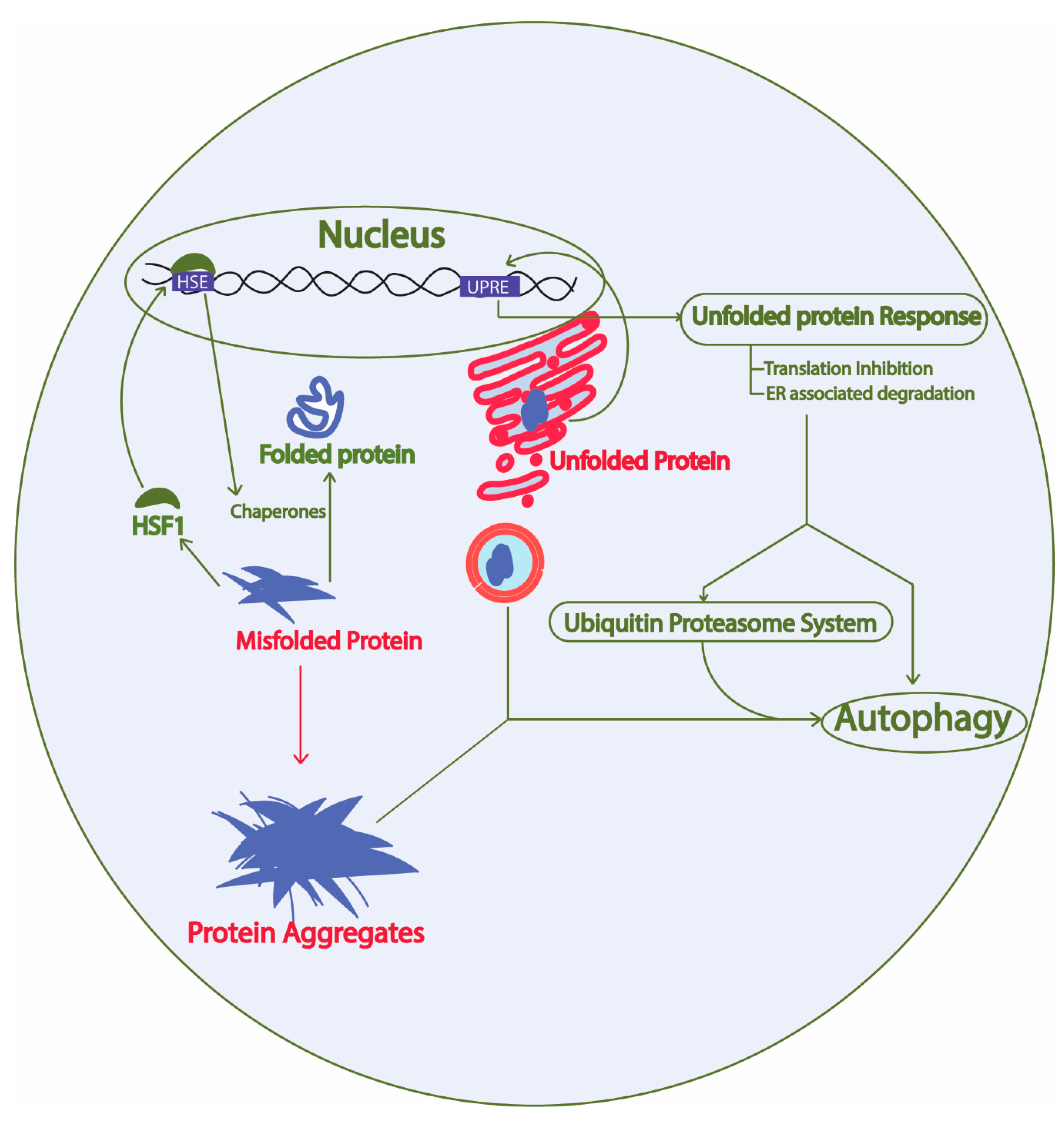{kind=link}
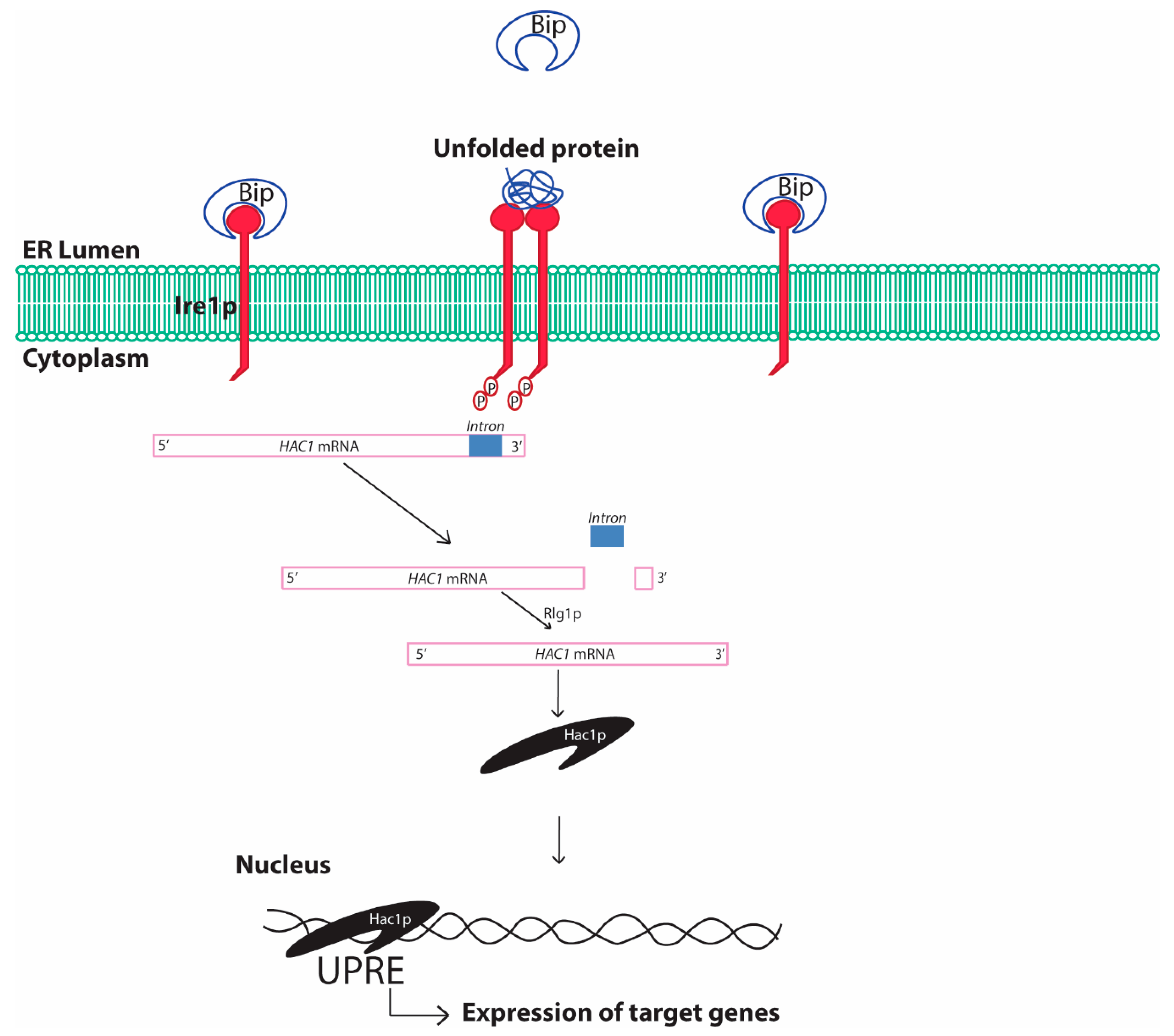{kind=link}
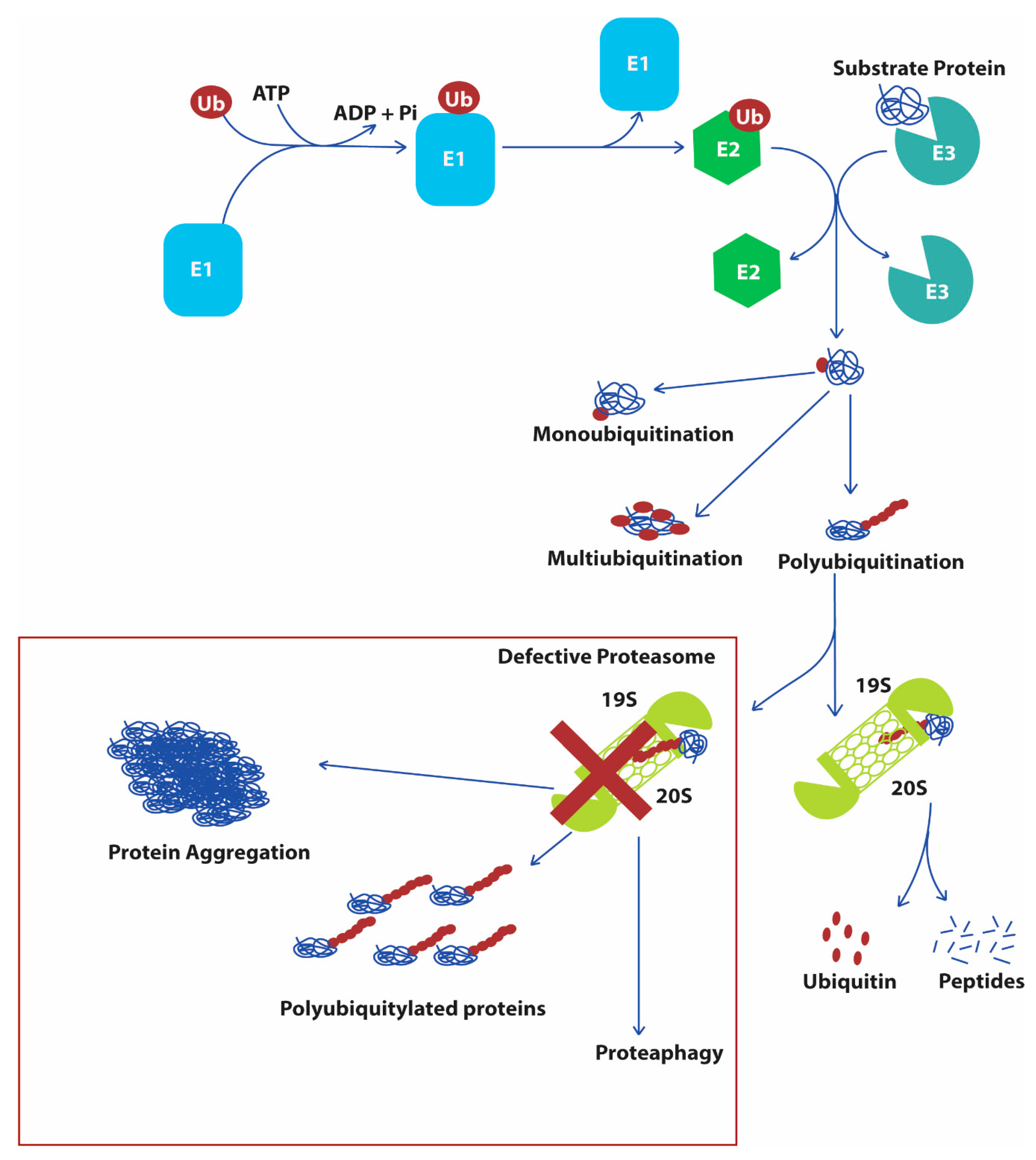{kind=link}
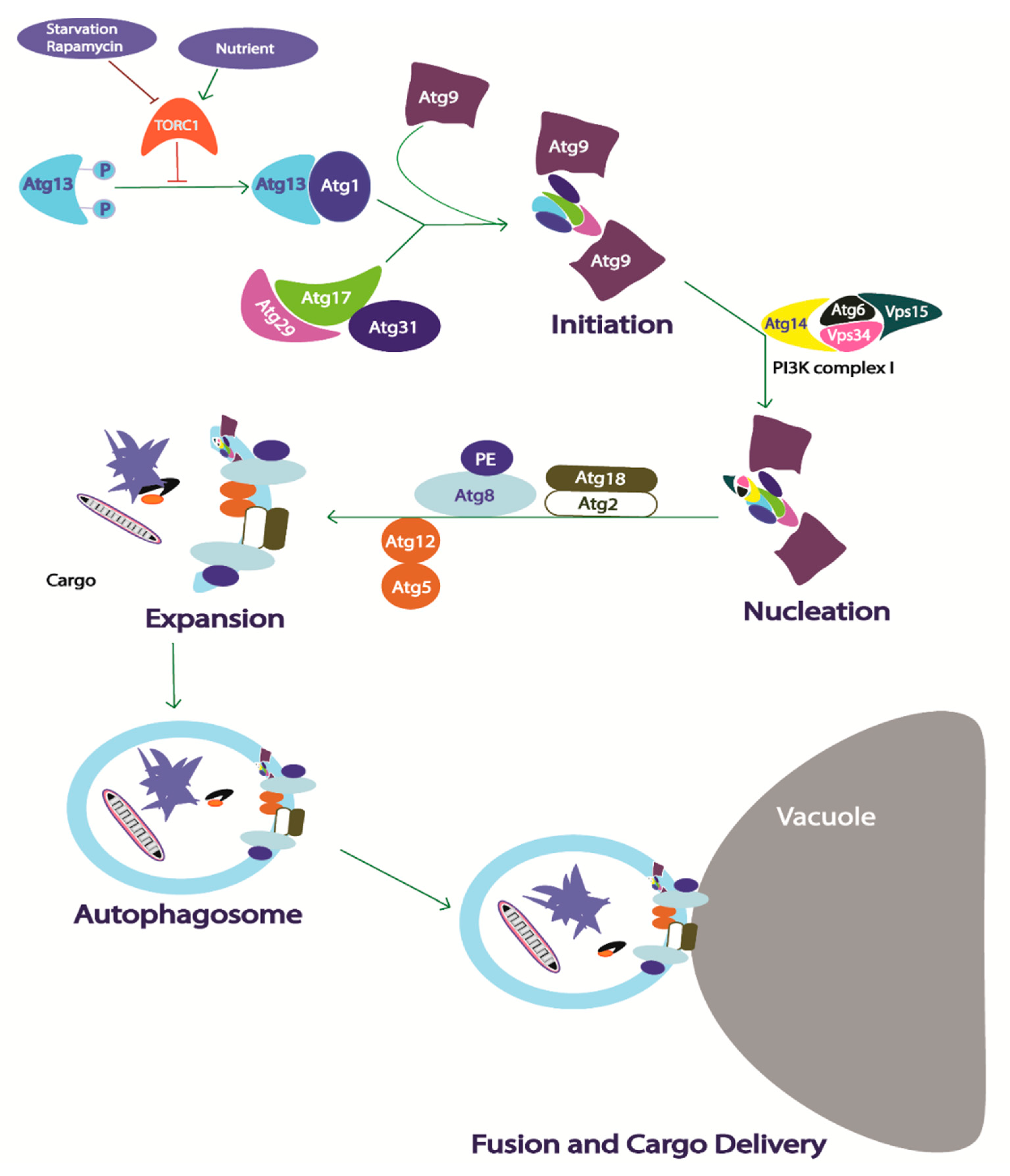{kind=link}
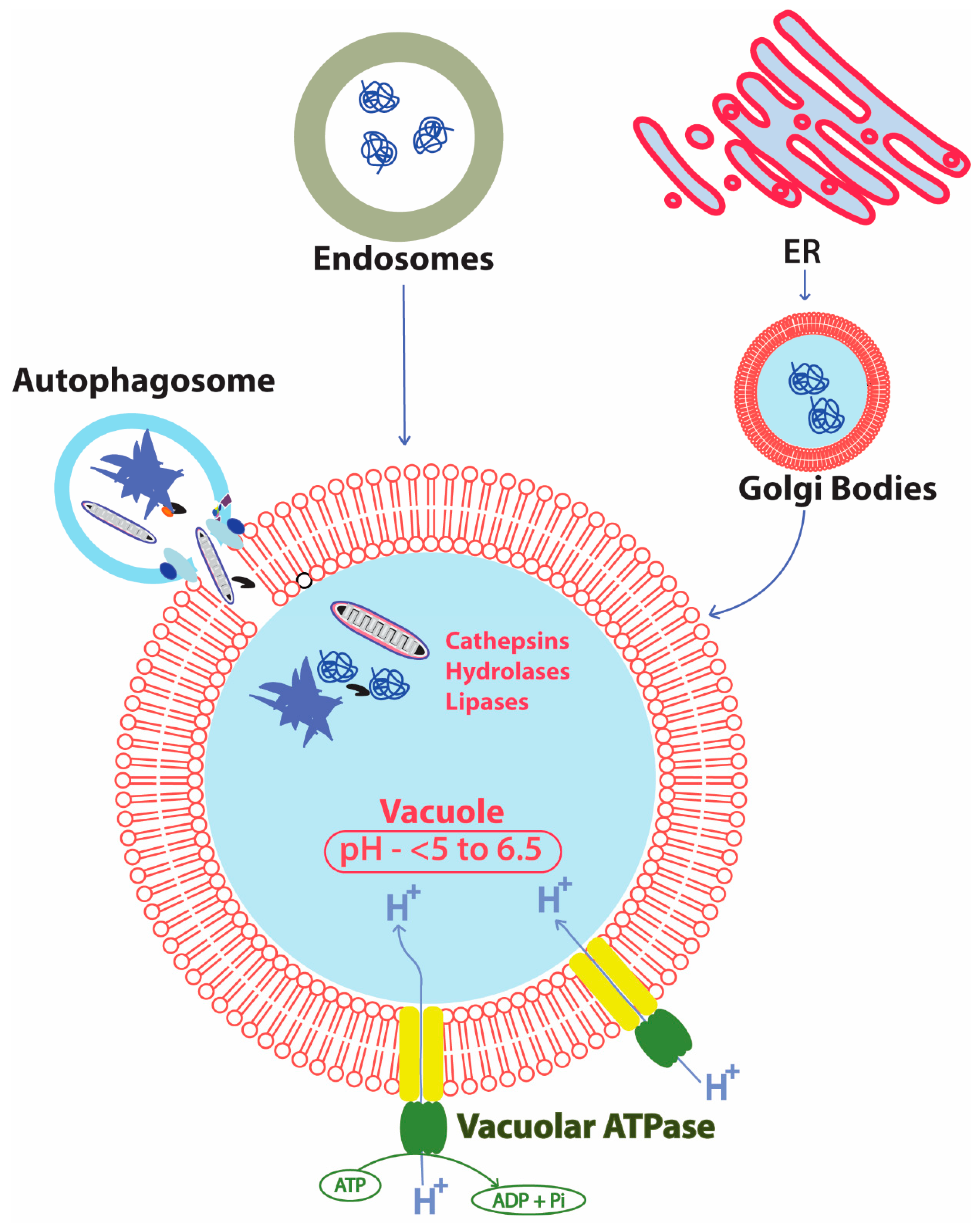{kind=link}
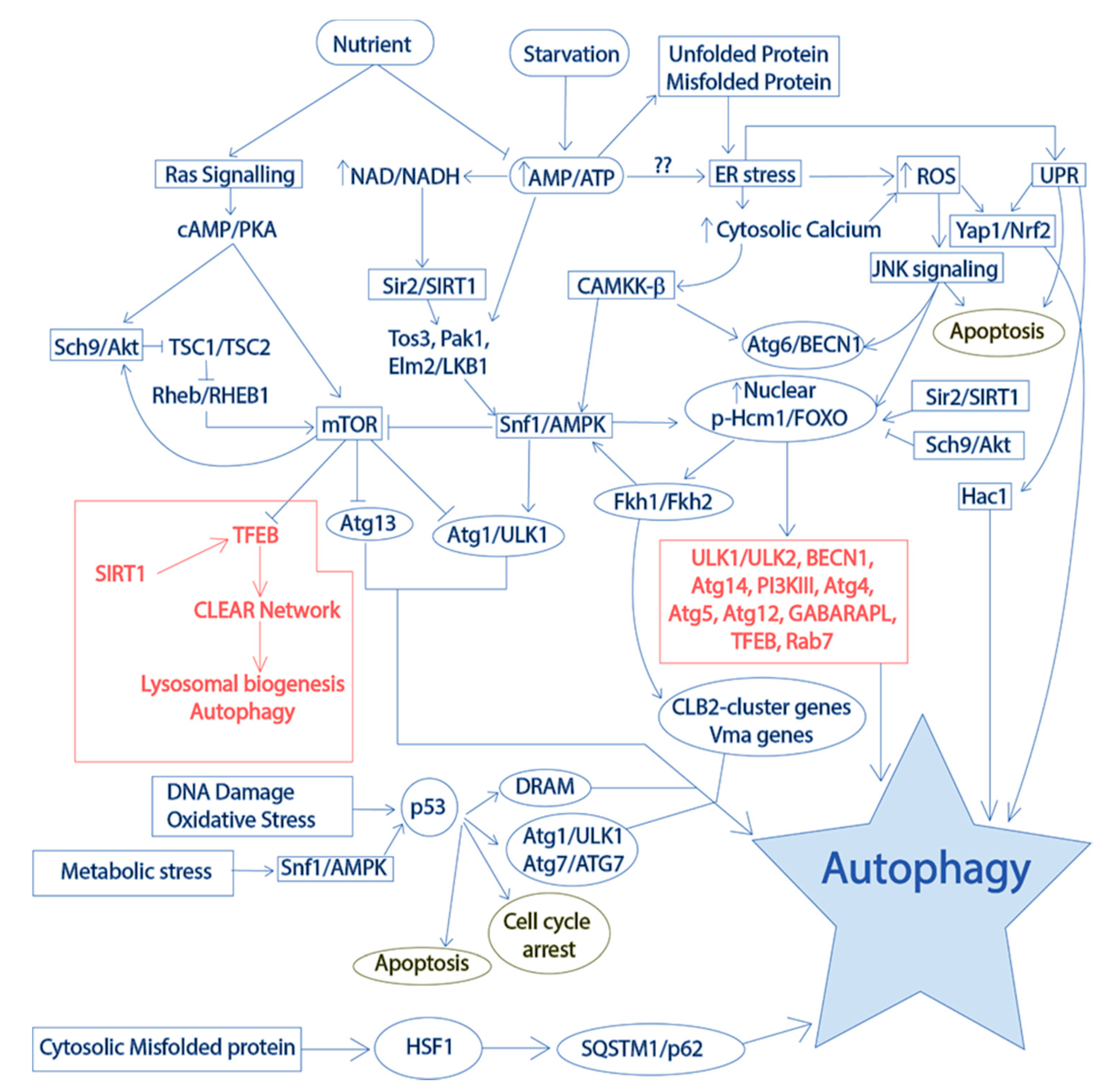{kind=link}
| Yeast Protein | Human Homolog | Function | References |
|---|---|---|---|
| Kar2p/Bip/Grp78p | HSPA5/BiP/GRP78 | Chaperone regulating activation of Ire1p | [42,57] |
| Ire1p | IRE1α IRE1β | Stress sensing and endonuclease activity | [58] |
| PERK | Stress sensing and protein kinase | [59] | |
| Hac1p | XBP1p | UPRE binding and expression of target genes | [59] |
| Sui2p | eIF2α | Eukaryotic translation initiation factor involved in protein translation regulation | [60] |
| Ypt1p | RAB1Ap | Regulates UPR by HAC1 mRNA decay | [44] |
| Yeast Protein | Human Homolog | Functions of Proteins in Yeast | Ref |
|---|---|---|---|
| Ubiquitin | |||
| Ubi1, Ubi2, Ubi3, Ubi4 | UBB, UBC | Cellular stress response and ubiquitination of proteins | [71] |
| Ubiquitin activating E1 enzyme | |||
| Uba1 | UBA1 | Ubiquitin-mediated protein degradation | [72] |
| Ubiquitin conjugating E2 enzyme | |||
| Ubc1 | UBE2K | Major E2 enzyme works together with Ubc4, degradation of short-lived and aberrant proteins, increases protein during DNA stress, cellular stress response, vesicle biogenesis, and ERAD | [73,74,75] |
| Ubc2 | UBE2A, UBE2B | K63 ubiquitination mediated oxidative stress response; turnover of Rpn4; DNA repair | [76,77] |
| Ubc3 | UB2R1, UB2R2 | Regulation of cell cycle progression; protein abundance during DNA stress | [78,79] |
| Ubc4, Ubc5 | UB2D1, UB2D2, UB2D3, UB2D4 | Major E2 enzyme works together with Ubc1; degradation of abnormal calmodulin and H3 histone; regulates levels of DNA polymerase | [80,81,82] |
| Ubc6 | UB2J2 | ERAD; ER membrane (Cytosolic) resident E2 enzyme | [83] |
| Ubc7 | UB2G1, UB2G2 | ERAD; inner nuclear membrane associated degradation; chromatin assembly | [84] |
| Ubc8 | UBE2H | Regulates gluconeogenesis | [85] |
| Ubc9 | UBC9 | Anaphase promoting complex cyclosome mediated proteolysis; Cell cycle progression | [86] |
| Ubc10 | UB2D1, UB2D2 | Peroxisome biogenesis | [87] |
| Ubc13 | UBE2N | DNA damage repair, DNA stress response | [88] |
| Yeast Protein | Human Homolog | Functions of Proteins in Yeast | Ref |
|---|---|---|---|
| Ubiquitin ligase E3 enzyme | |||
| Hul4 | HECTD2 | HECT ubiquitin ligase | [89] |
| Hul5 | UBE3C | HECT ubiquitin ligase; essential for elongation of polyubiquitin chains in the misfolded proteins during heat shock response; ERAD | [90,91,92] |
| Rsp5 | NEDD4L | NEDD4 family E3 ubiquitin ligase; regulates heat shock response; endocytosis and vesicle trafficking. | [92,93,94] |
| Tom1 | HUWE1 | HECT ubiquitin ligase; export of mRNA to cytosol; balancing histones; cell cycle regulation. | [81,95,96] |
| Ufd4 | TRIP12 | E3 ubiquitin ligase; involved in DNA stress. | [79] |
| Asr1 | RNF165 | Ubiquitin E3 ligase; regulates RNA Polymerase II subunits; actin cytoskeleton organization | [97,98] |
| Bre1 | RNF40 | E3 ubiquitin ligase conjugates with Rad6p and regulates histone methylation; oxidative stress response mediated proteolysis; double stranded break repair; pre-mRNA splicing. | [76,99,100,101] |
| Dma1, Dma2 | RNF8 | E3 ubiquitin ligase; regulates level of eIF2 | [102] |
| Ssm4/Doa10 | MARCHF6 | RING-CH domain E3 ligase associated with ER and nuclear membrane; involved in ERAD, | [84] |
| Hel2 | ZNF598 | RING finger E3 ubiquitin ligase; excess histone clearance; involved in ribosome associated quality control | [103,104] |
| Hrd1 | SYVN1 | H2 ring finger E3 ubiquitin ligase; involved in UPR and ERAD | [105] |
| Fyv10 | MAEA | Ring finger ubiquitin ligase; inhibits gluconeogenesis; anti-apoptotic; part of glucose-induced degradation-deficient (GID) complex | [106,107,108] |
| Irc20 | SHPRH | Ring finger E3 ubiquitin ligase; involved in non-homologous end joining DNA repair pathway | [109] |
| Nam7 | UPF1 | RING-related E3 ubiquitin ligase; nonsense mutation mediated mRNA decay | [79] |
| Mot2 | CNOT4 | Ubiquitin ligase of CCR4-NOT complex; regulates transcription, post transcriptional modifications and mRNA degradation | [110,111] |
| Pep5 | VPS11 | Histone E3 ligase that enhances degradation of excess histones; vacuole biogenesis. | [103,112] |
| Pex2 | PEX2 | RING finger peroxisomal E3 ubiquitin ligase | [113] |
| Pex10 | PEX10 | Peroxisomal membrane associated E3 ubiquitin ligase | [114] |
| Pex12 | PEX12 | RING finger peroxisomal E3 ubiquitin ligase | [115] |
| Pib1 | FYCO1 | RING finger ubiquitin ligase of endosome and vacuole membranes | [116] |
| Psh1 | TRIM25 | E3 ubiquitin ligase for degradation of centromere binding protein | [117] |
| Rad5 | HLTF | Ubiquitin ligase or DNA helicase involved in DNA damage stress response | [118,119] |
| Rad18 | RAD18 | E3 ubiquitin ligase complexes with PCNA; involved in post replication repair of DNA | [120,121] |
| Rmd5 | RMND5A | RING finger E3 ubiquitin ligase; inhibits gluconeogenesis; part of GID complex | [106] |
| Rkr1 | LTN1 | RING domain E3 ubiquitin ligase; part of ribosome quality control complex; clearance of defective mRNA | [122,123,124] |
| San1 | RNF13 | E3 ubiquitin ligase; involved in selective clearance of defective aggregate prone proteins | [125,126] |
| Slx8 | RNF10 | RING domain ubiquitin ligase; required for genomic integrity | [127] |
| Snt2 | PHF14 | RING finger E3 ubiquitin ligase; oxidative stress response; degradation of excess histones. | [103,128] |
| Ubr2 | UBR2 | Cytoplasmic E3 ubiquitin ligase | [129] |
| Prp19 | PRPF19 | E3 ubiquitin ligase; DNA damage response; pre-mRNA splicing | [130,131] |
| Ufd2 | UBE4B | Polyubiquitylation of substrate protein; stress response; ERAD of misfolded proteins | [132,133] |
| Hel1 | ARIH1 | RING finger E3 ubiquitin ligase; clearance of excess histones | [103] |
| Itt1 | RNF14 | E3 ubiquitin ligase; modulates termination of translation | [134] |
| Cdc53 | CUL1 | Cullin family E3 ubiquitin ligase; cell cycle progression | [135,136] |
| Cul3 | CUL3 | Cullin family E3 ubiquitin ligase; degradation of RNA Polymerase II | [137] |
| Rtt101 | CUL4 | Cullin subunit of a E3 ubiquitin ligase complex; cell cycle progression; double stranded break repair; rRNA decay | [138,139] |
| Skp1 | SKP1 | Major component of SCF (Skp1-Cullin-F-box) E3 ubiquitin ligase complex; V-ATPase assembly; cell cycle progression; DNA replication stress response | [79,140,141] |
| Elc1 | ELOC | E3 ubiquitin ligase; genomic repair; degradation of RNA polymerase II | [137,142] |
| Hrt1 | RBX1 | Part of various ubiquitin ligase SCF complex | [143] |
| Cdc4 | FBXW7 | F-box protein; part of SCF-Cdc4 ubiquitin ligase complex; ubiquitination of cyclin-dependent kinases | [144,145] |
| Met30 | FBXW7 | Essential protein; F-box protein; part of SCF complex; heavy metal stress response; Cell cycle | [146,147] |
| Ela1 | ELOA | F-box protein heterodimerizes with Elc1; subunit of Elongin-Cullin-Socs ubiquitin ligase complex | [137,148,149] |
| Hrt3 | FBX9 | F-box protein of SCF ubiquitin ligase complex | [150] |
| Rav1 | DMXL1 | Part of RAVE complex; V-ATPase assembly; role in endocytosis | [151,152] |
| Saf1 | HERC2 | F-box protein involved in cell quiescence | [153] |
| Cdc27 | CDC27 | Part of anaphase promoting complex (APC)/cyclosome, a ubiquitin ligase complex involved in the degradation of anaphase inhibitors | [154] |
| Cdc16 | CDC16 | Part of APC/C, a ubiquitin ligase complex involved in the degradation of anaphase inhibitors | [155] |
| Cdc23 | CDC23 | Part of APC/C, a ubiquitin ligase complex involved in the degradation of anaphase inhibitors | [155] |
| Doc1/Apc10 | ANAPC10 | Subunit of APC; Substrate ubiquitination | [156,157] |
| Yeast Protein | Human Homolog | Functions of Yeast Proteins | Ref |
|---|---|---|---|
| Deubiquitinating enzymes (DUBs) | |||
| Ubp1 | USP19 | Ubiquitin specific protease—carboxy terminal hydrolase activity | [164] |
| Ubp2 | USP28 | Ubiquitin specific protease—deubiquitinates proteins, controls K63 during oxidative stress | [76] |
| Ubp3 | USP10 | Regulate transport between ER and Golgi; regulates osmotic stress response; deubiquitinates COPI and COPII proteins; increases protein during DNA stress | [165,166,167] |
| Ubp4/Ubp5 | USP8 | Deubiquitinates proteins inside endosomes that are bound to be delivered to vacuole for degradation; increases intracellular free ubiquitin. | [168,169] |
| Ubp6 | USP14 | Negatively regulates branched polyubiquitinated proteins from proteasomal degradation | [170] |
| Ubp7/Ubp11 | USP21 | Deubiquitinating proteins involved in cell cycle progression | [171] |
| Ubp8 | USP22 | SAGA-mediated histone 2B ubiquitination | [172] |
| Ubp9/Ubp13 | USP46 | Deubiquitinating ubiquitin-protein fusions | [129] |
| Ubp10 | USP20 | Ribosome biogenesis and rRNA formation; deubiquitinates several cellular proteins. | [173] |
| Ubp12 | USP15 | Deubiquitinates proteins | [174] |
| Ubp14 | USP5 | Regulates gluconeogenesis | [106] |
| Ubp15 | USP7 | Peroxisome biogenesis and cell cycle progression | [175,176] |
| Ubp16 | USP16 | Mitochondria associated deubiquitinating enzyme | [177] |
| Rpn11 | PSMD14 | Metalloprotease present in 26S proteasome; helps in deubiquitylation and proteolytic cleavage of substrates | [178,179] |
| Otu1 | YOD1 | Deubiquitylation of proteins ubiquitinated by Ufd2 | [180] |
| Otu2 | OTUD6B | Increases abundance of protein during DNA stress | [79] |
| Yuh1 | UCHL3 | Hydrolyses C-terminal of ubiquitin peptide bonds and release monomeric ubiquitin | [181] |
| Proteasomal Core (20S subunit) | |||
| α-subunit (α1-7) | |||
| Scl1 | PSMA6 | Essential for survival, alpha-1 subunit of 20S core machinery, degradation of protein substrates | [182] |
| Pre8 | PSMA2 | Alpha-2 subunit of 20S proteasome | [183] |
| Pre9 | PSMA4 | Alpha-3 subunit of 20S proteasome | [182] |
| Pre6 | PSMA7 | Alpha-4 subunit of 20S proteasome | [183] |
| Pup2 | PSMA5 | Alpha-5 subunit of 20S proteasome | [184] |
| Pre5 | PSMA1 | Alpha-6 subunit of 20S proteasome | [183] |
| Pre10 | PSMA3 | Alpha-7 subunit of 20S proteasome | [79] |
| β-subunit (β1-7) | |||
| Pre3 | PSMB6 | Beta-1 subunit of 20S proteasome | [182] |
| Pup1 | PSMB7 | Beta-2 subunit of 20S proteasome | [185] |
| Pup3 | PSMB3 | Beta-3 subunit of 20S proteasome | [186] |
| Pre1 | PSMB2 | Beta-4 subunit of 20S proteasome | [182] |
| Pre2 | PSMB5 | Beta-5 subunit of 20S proteasome | [182] |
| Pre7 | PSMB1 | Beta-6 subunit of 20S proteasome | [182] |
| Pre4 | PSMB4 | Beta-7 subunit of 20S proteasome | [185] |
| Regulatory Particle (19S subunit) | |||
| RP base | |||
| Rpt1 | PSMC2 | ATPase of 19S RP | [187] |
| Rpt2 | PSMC1 | ATPase of 19S RP | [188] |
| Rpt3 | PSMC4 | ATPase of 19S RP | [188] |
| Rpt4 | PSMC6 | ATPase of 19S RP; spindle pole duplication; ERAD | [188,189] |
| Rpt5 | PSMC3 | ATPase of 19S RP | [190] |
| Rpt6 | PSMC5 | ATPase of 19S RP | [79] |
| Rpn1 | PSMD2 | Protein–protein interaction; proteasomal ligand recognition and non-ATPase subunit of 19S RP | [191] |
| Rpn2 | PSMD1 | Non-ATPase subunit of 19S RP | [79] |
| Rpn13 | ADRM1 | Ubiquitin receptor of proteasome | [192] |
| Rpn10 | PSMD4 | Assembly of regulatory particle; non-ATPase subunit of 19S RP | [193] |
| RP Lid | |||
| Rpn3 | PSMD3 | Essential non-ATPase subunit of 19S RP lid | [194] |
| Rpn5 | PSMD12 | Essential non-ATPase subunit of 19S RP lid | [188] |
| Rpn6 | PSMD11 | Essential non-ATPase subunit of 19S RP lid; assembly and activity of proteasome | [195] |
| Rpn7 | PSMD6 | Essential non-ATPase subunit of 19S RP lid | [188] |
| Rpn8 | PSMD7 | Essential non-ATPase subunit of 19S RP lid | [188] |
| Rpn11 | PSMD14 | Essential non-ATPase subunit of 19S RP lid, degradation and deubiquitylation of substrate proteins, mitochondrial and peroxisomal fission. | [178] |
| Rpn12 | PSMD8 | Essential non-ATPase subunit of 19S RP lid | [61] |
| Proteins Involved in Yeast Autophagy | Human Homologs | Function | Ref |
|---|---|---|---|
| Sir2 | SIRT1 | Sirtuin family NAD+-dependent histone deacetylase that activates upstream kinases that activate Snf1/AMPK and activates TFEB and autophagy in mammalian system. | [205,206] |
| Tos3, Sak1, Elm1 | LKB1, CAMKK | Upstream protein kinases that activates AMPK/Snf1 | [207,208] |
| Hcm1 | FOXO3 | Expression of other fork head box transcription factor proteins and expression of various proteins involved in autophagy; nuclear localization and expression of target genes essential for vacuolar acidification; mitochondrial biogenesis and stress resistance | [209,210,211] |
| Fkh1, Fkh2 | FOXM | Expression required for stress resistance, autophagy, and cell cycle progression | [212,213] |
| Sch9 | AKT/PKB | Regulates autophagy negatively works together with TORC1 | [214,215] |
| Tsc1, Tsc2 | TSC1, TSC2 | Inhibits TORC1 activity and activates autophagy | [216] |
| Rhb1 | Rheb | Promotes TORC1 activity and inhibits autophagy | [217] |
| Snf1 | AMPK | Activates Atg1; nutrient sensing; inactivates TORC1 complex and positively regulate autophagy | [208,218] |
| Torc1 | TORC1 | Blocks interaction of Atg1 and Atg13; nutrient sensing, negatively regulates autophagy | [219] |
| Gtr1 | RAGA, RAGB | Part of Rag GTPase complex; regulates TORC1 activity | [220] |
| Gtr2 | RAGC, RAGD | Part of Rag GTPase complex; regulates TORC1 activity | [220] |
| Ssk2, Ssk22 | MAP3K4/MEKK4 | MAPK kinase kinase for Hog1 | [221] |
| Pbs2 | MAP2K2/MEK2 | MAPK kinase for Hog1 | [222] |
| Hog1 | MAPK14/p38 | Involved in mitophagy and pexophagy | [223,224] |
| Slg1/Wsc1 | WSCD2 | Important for mitophagy; stress response | [224,225] |
| Pkc1 | PRKCA, PRKCB, PRKCD, PRKCE, PRKCG, PRKCH, PRKCI, PRKCQ | Activation of Bck2 in turn activating Slt2 involved in pexophagy and mitophagy | [224] |
| Slt2 | MAPK7/ERK5 | MAPK involved in pexophagy and mitophagy | [224] |
| Atg1 | ULK1, ULK2 | Serine/threonine protein kinase activity; conjugates with Atg13; autophagy initiation regulation | [226] |
| Atg2 | ATG2A, ATG2B | Conjugates with Atg18; forms Atg2-Atg18 complex; and involved in autophagosome expansion | [227] |
| Atg3 | ATG3 | E2-like enzyme for formation of Atg8 lipidation complex | [228] |
| Atg4 | ATG4A, ATG4B, ATG4C, ATG4D | Cysteine protease that processes the inactive Atg-8 at carboxy terminus to give mature Atg8 | [229] |
| Atg5 | ATG5 | Conjugates with Atg12 | [230] |
| Atg6/Vps30 | BECN1 | Important part of PI3K complexes | [231] |
| Atg7 | ATG7 | E1 like enzyme involved in both Atg12-Atg5 and Atg8-PE complex formation | [232] |
| Atg8 | LC3A, LC3B, LC3B2, LC3C, GABARAP, GATE16 | Conjugates with PE to form lipidation complex; important part of autophagosome membrane; interacts with SNARE proteins to deliver the autophagosomal cargo; determines the size of autophagosome | [233] |
| Atg9 | ATG9 | The only transmembrane protein of autophagosome required for phagophore expansion; interacts with Atg2 and regulates autophagy | [234] |
| Atg10 | ATG10 | E2-like enzyme conjugating Atg5-Atg12 | [235] |
| Atg12 | ATG12 | Forms Atg5-Atg12 complex involved in phagophore expansion | [230] |
| Atg13 | ATG13 | Activated by dephosphorylation during starvation, conjugates with Atg1 to initiate formation of phagophore assembly site | [236] |
| Atg14 | ATG14 | PI3K complex I component | [237] |
| Atg17 | FIP200/RB1CC1 | Atg1 kinase complex component involved in initiation of autophagy | [236] |
| Atg18 | WIPI1, WIPI2 | Conjugates with Atg2; forms Atg2-Atg18 complex; binds with PI3P; and involved in elongation of phagophore. | [238] |
| Vps15 | PIK3R4/VPS15/p150 | Protein kinase required for activation of Vps34 | [239] |
| Vps34 | PIK3C3/VPS34 | PI3K catalytic component | [240] |
| Vacuolar ATPase Subunits | Yeast Proteins | Human Homologs | Function of the Yeast Protein | Ref |
|---|---|---|---|---|
| V1 Subunit | ||||
| A | Vma1 | ATP6V1A | Site specific endonuclease activity; methionine restriction regulation of life span; stress response | [256,257] |
| B | Vma2 | ATP6V1B1, ATP6V1B2 | Proton pump of endomembrane; protein abundance during DNA replication stress | [79,258] |
| C | Vma5 | ATP6V1C1, ATP6V1C2 | Part of proton pump; required for assembly of V-ATPase subunits at vacuolar membrane | [259] |
| D | Vma8 | ATP6V1D | Role in proton pumping and ATP hydrolysis | [260,261] |
| E | Vma4 | ATP6V1E1, ATP6V1E2 | Part of V-ATPase; protein content increases during DNA replication stress | [79,262] |
| F | Vma7 | ATP6V1F | Part of proton pump; required for V-ATPase subunits assembly at vacuolar membrane | [258,263] |
| G | Vma10 | ATP6V1G1, ATP6V1G2, ATP6V1G3 | Part of proton pump and role in vacuole acidification | [264] |
| H | Vma13 | ATP6V1H | Part of proton pump; activates and stabilizes v-ATPase | [258,265] |
| V0 Subunit | ||||
| a | Vph1, Stv1 | ATP6V0A1, ATP6V0A2, ATP6V0A3, ATP6V0A4 | Part of V-ATPase complex; regulates V-ATPase activity; present in vacuoles, Golgi bodies and endosomes | [266,267,268] |
| c | Vma3 | ATP6V0C | Proteolipid subunit; Vacuole acidification; Copper and Iron homeostasis | [269,270] |
| c’ | Vma11 | ATP6V0B, ATP6V0C | Integral hydrophobic membrane proteolipid; required for proton pump | [271,272] |
| c” | Vma16 | ATP6V0B, ATP6V0C | Part of V-ATPase | [271] |
| d | Vma6 | ATP6V0D1, ATP6V0D2 | Part of proton pump; required for V1 assembly at vacuole membrane | [273,274] |
| e | Vma9 | - | V0 biogenesis; vacuole acidification; part of V0 subunit. | [275] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dhakal, S.; Macreadie, I. Protein Homeostasis Networks and the Use of Yeast to Guide Interventions in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 8014. https://doi.org/10.3390/ijms21218014
Dhakal S, Macreadie I. Protein Homeostasis Networks and the Use of Yeast to Guide Interventions in Alzheimer’s Disease. International Journal of Molecular Sciences. 2020; 21(21):8014. https://doi.org/10.3390/ijms21218014
Chicago/Turabian StyleDhakal, Sudip, and Ian Macreadie. 2020. "Protein Homeostasis Networks and the Use of Yeast to Guide Interventions in Alzheimer’s Disease" International Journal of Molecular Sciences 21, no. 21: 8014. https://doi.org/10.3390/ijms21218014
APA StyleDhakal, S., & Macreadie, I. (2020). Protein Homeostasis Networks and the Use of Yeast to Guide Interventions in Alzheimer’s Disease. International Journal of Molecular Sciences, 21(21), 8014. https://doi.org/10.3390/ijms21218014