Interdependence between Chromogranin-A, Alternatively Activated Macrophages, Tight Junction Proteins and the Epithelial Functions. A Human and In-Vivo/In-Vitro Descriptive Study

 , ,
, , 
Abstract
:1. Introduction
2. Results
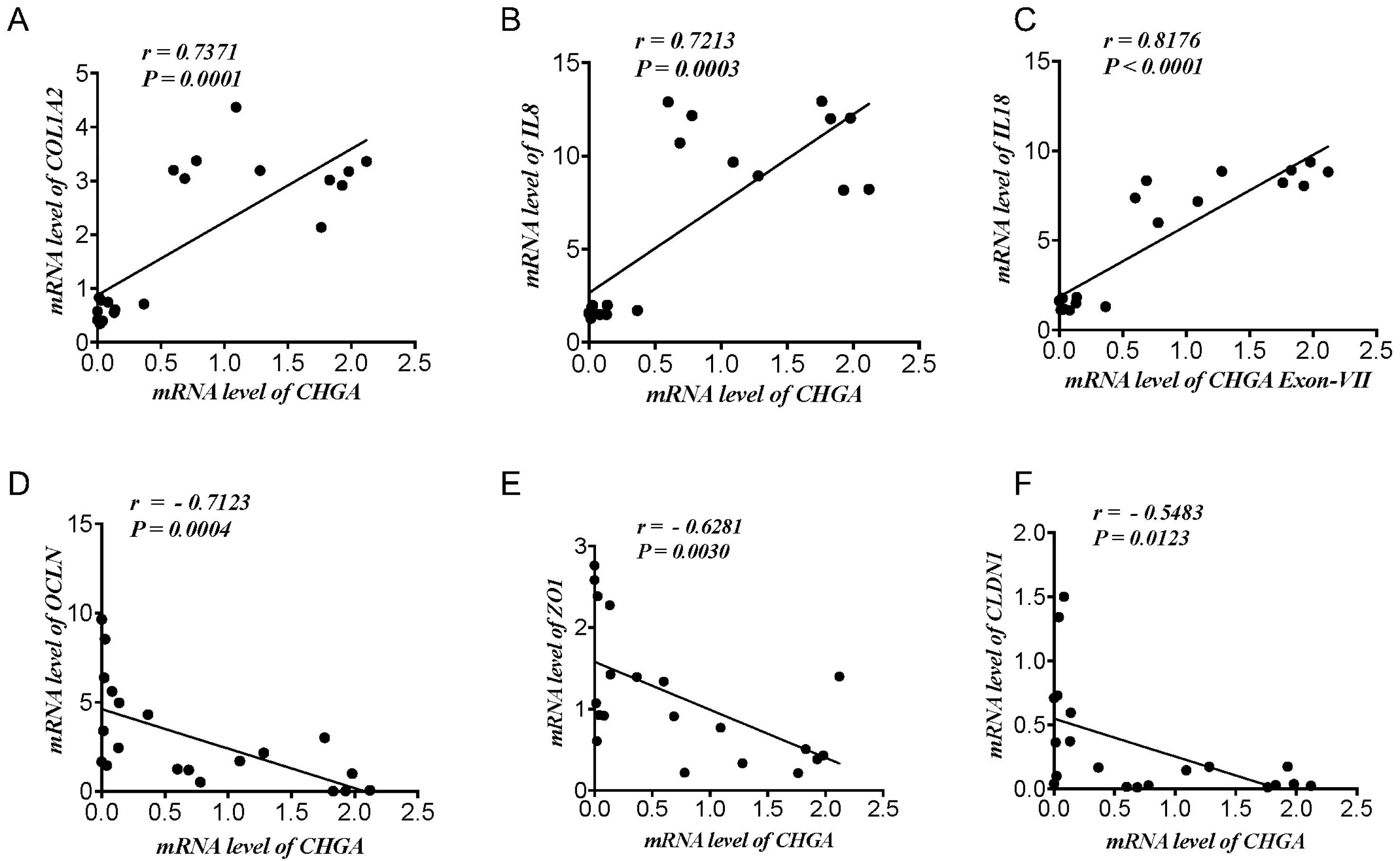
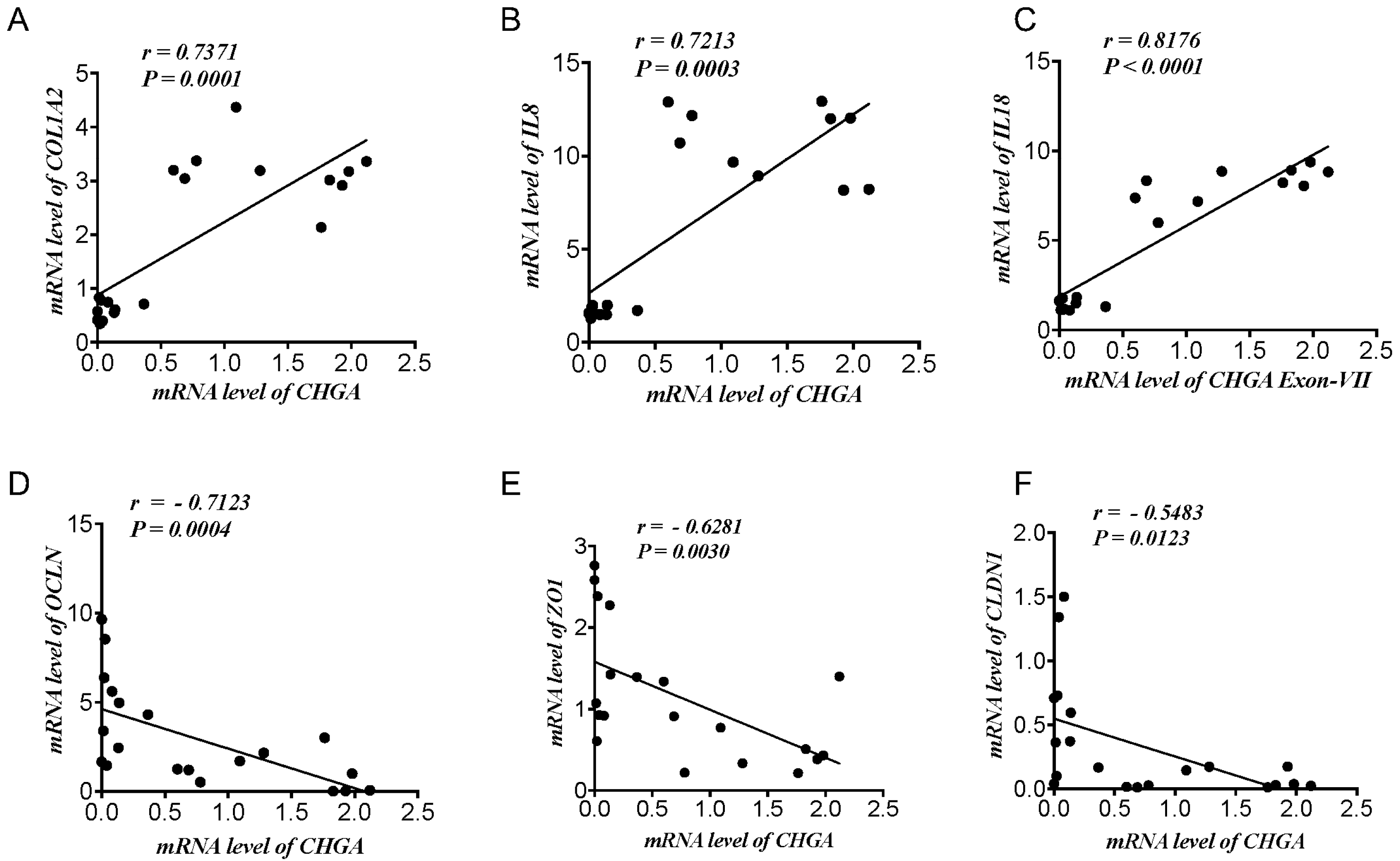
2.1. CHGA mRNA Expression Correlates Positively with Epithelial-Associated Cytokines & Collagen mRNA Expression and Negatively with TJ Proteins mRNA Expression Markers in UC Patients and Healthy Individuals
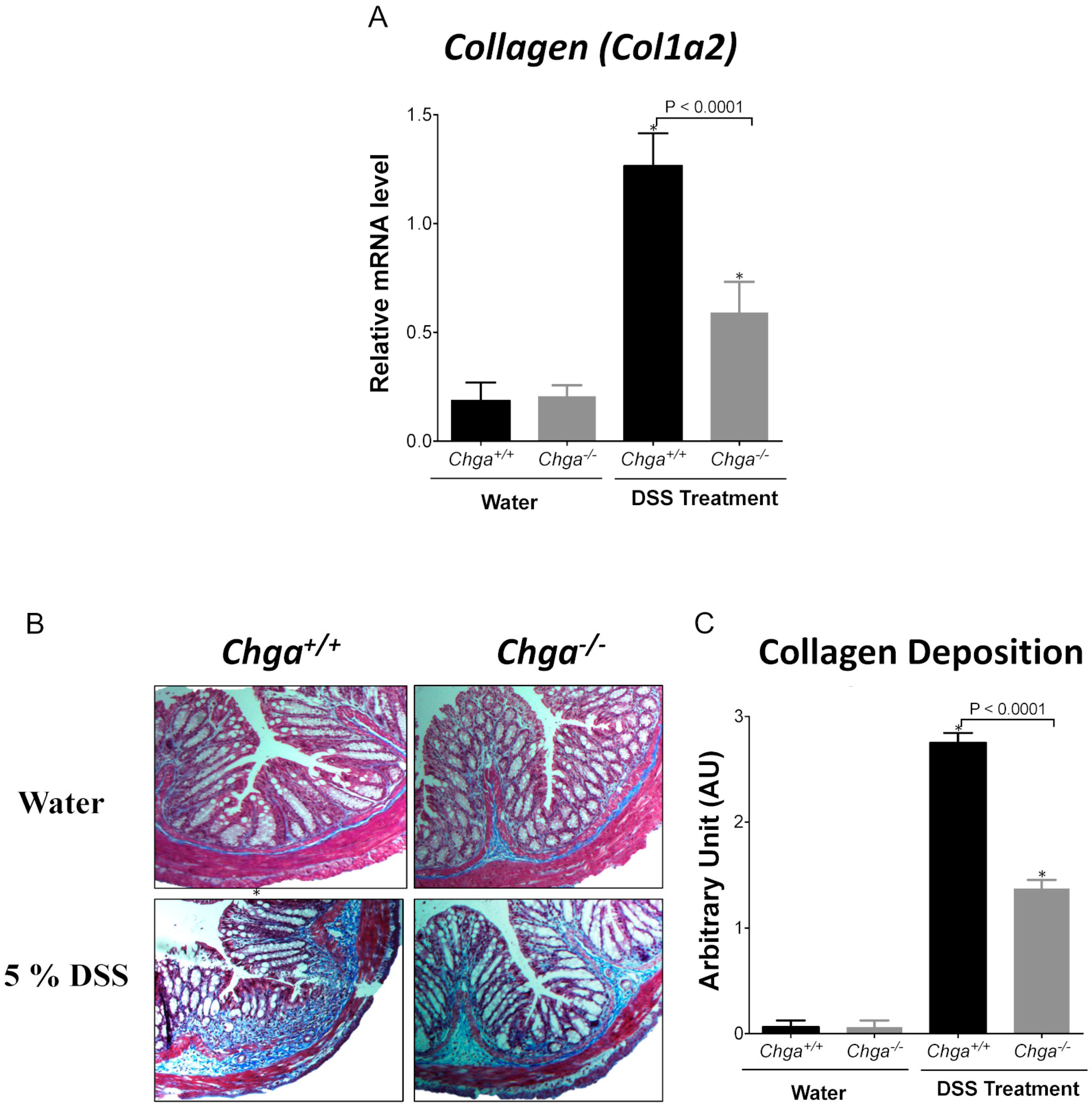
2.2. Deletion of Chga Attenuates Collagen Expression and Deposition in Acute DSS-Induced Colitis
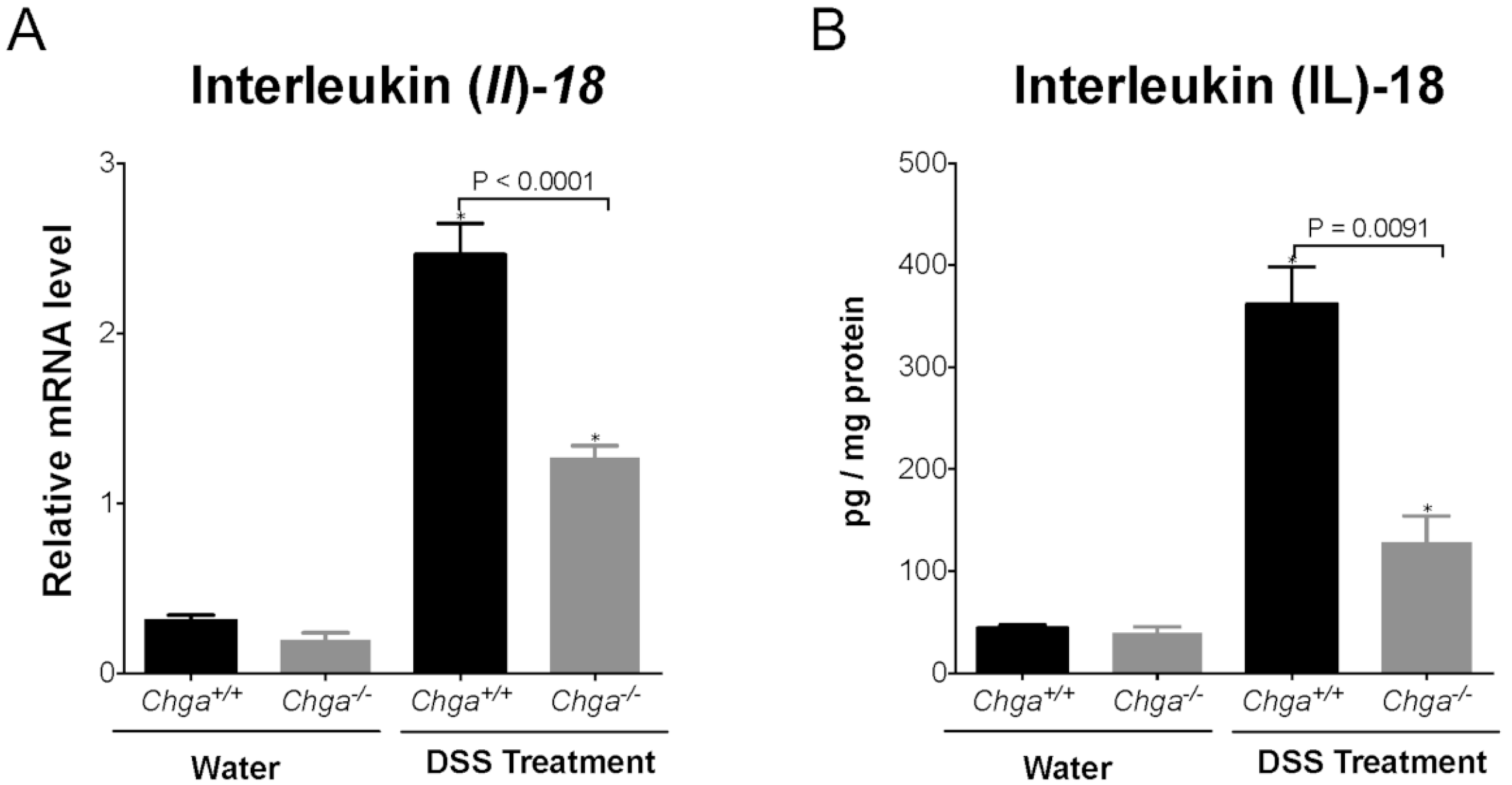
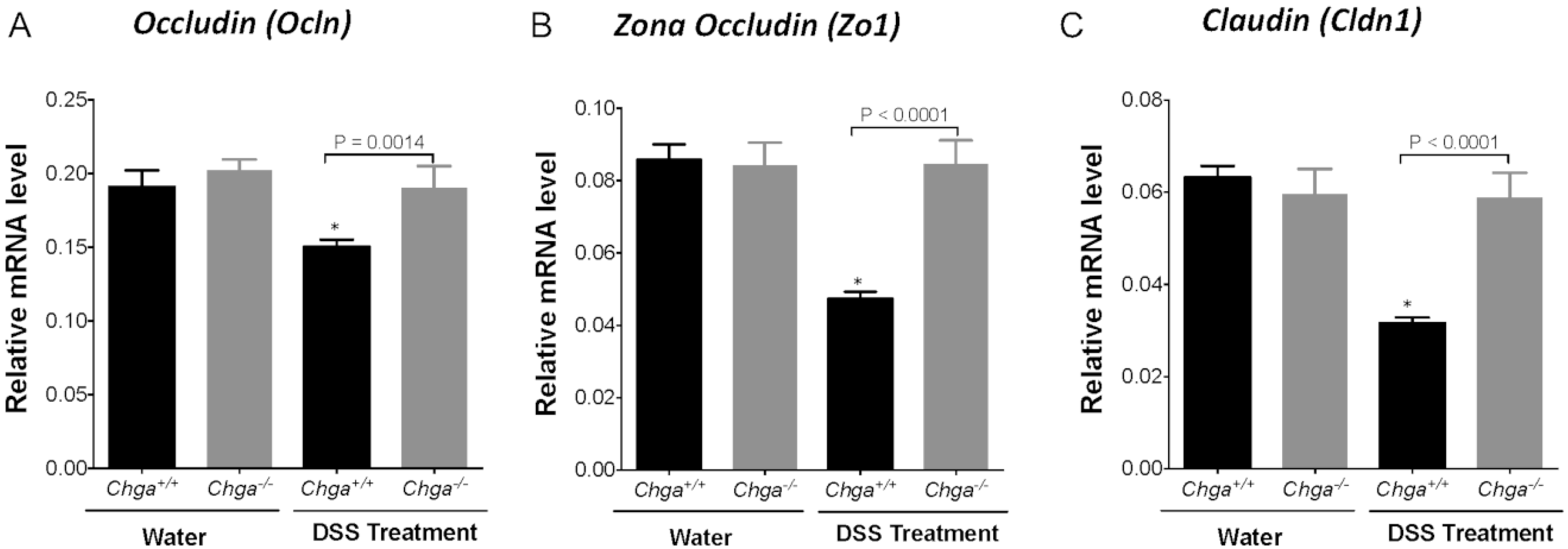
2.3. Deletion of Chga Maintains TJ Protein mRNA Expression and Decreases Colonic IL-18 Expression and Release in Acute DSS-Induced Colitis
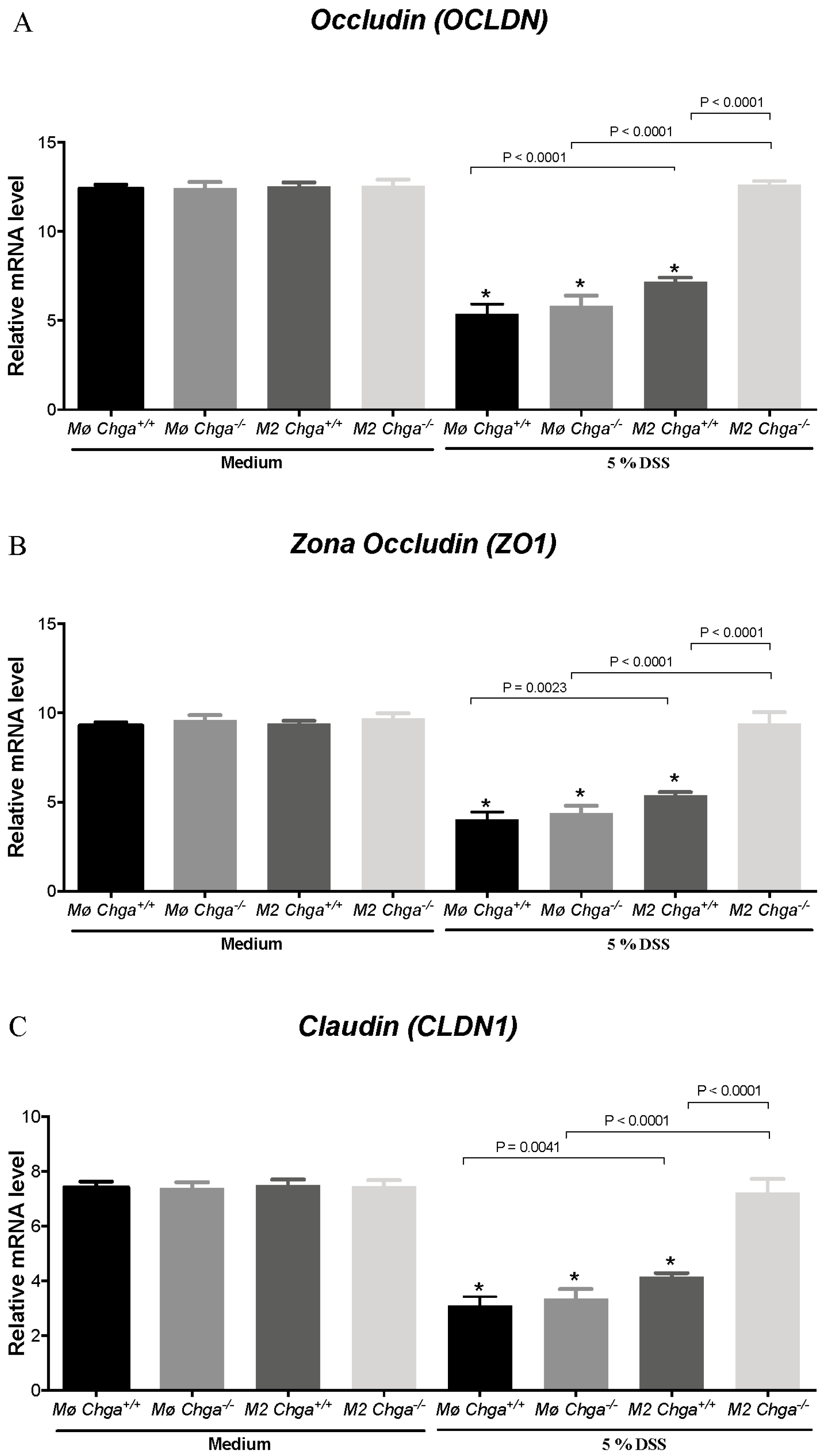
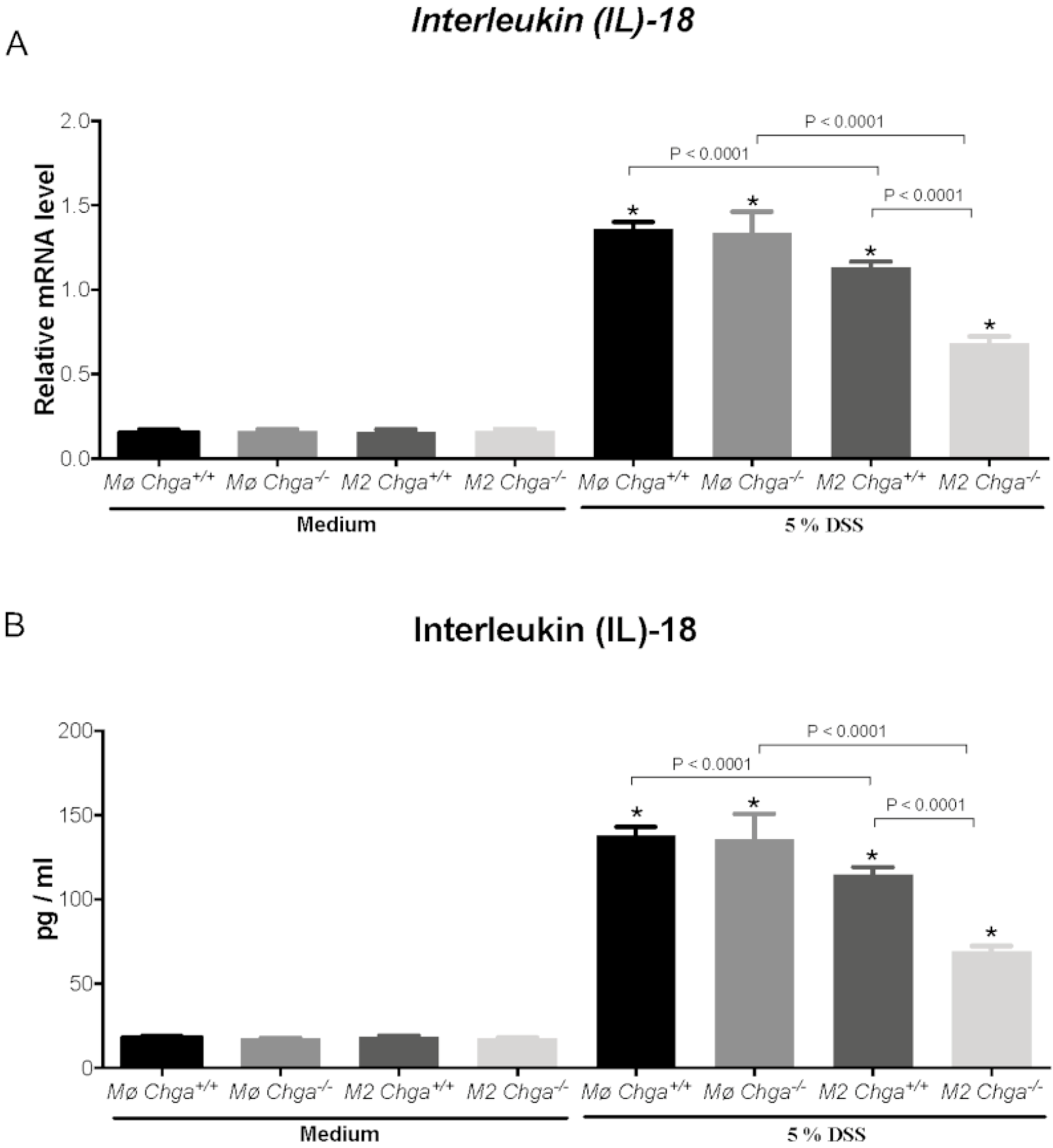
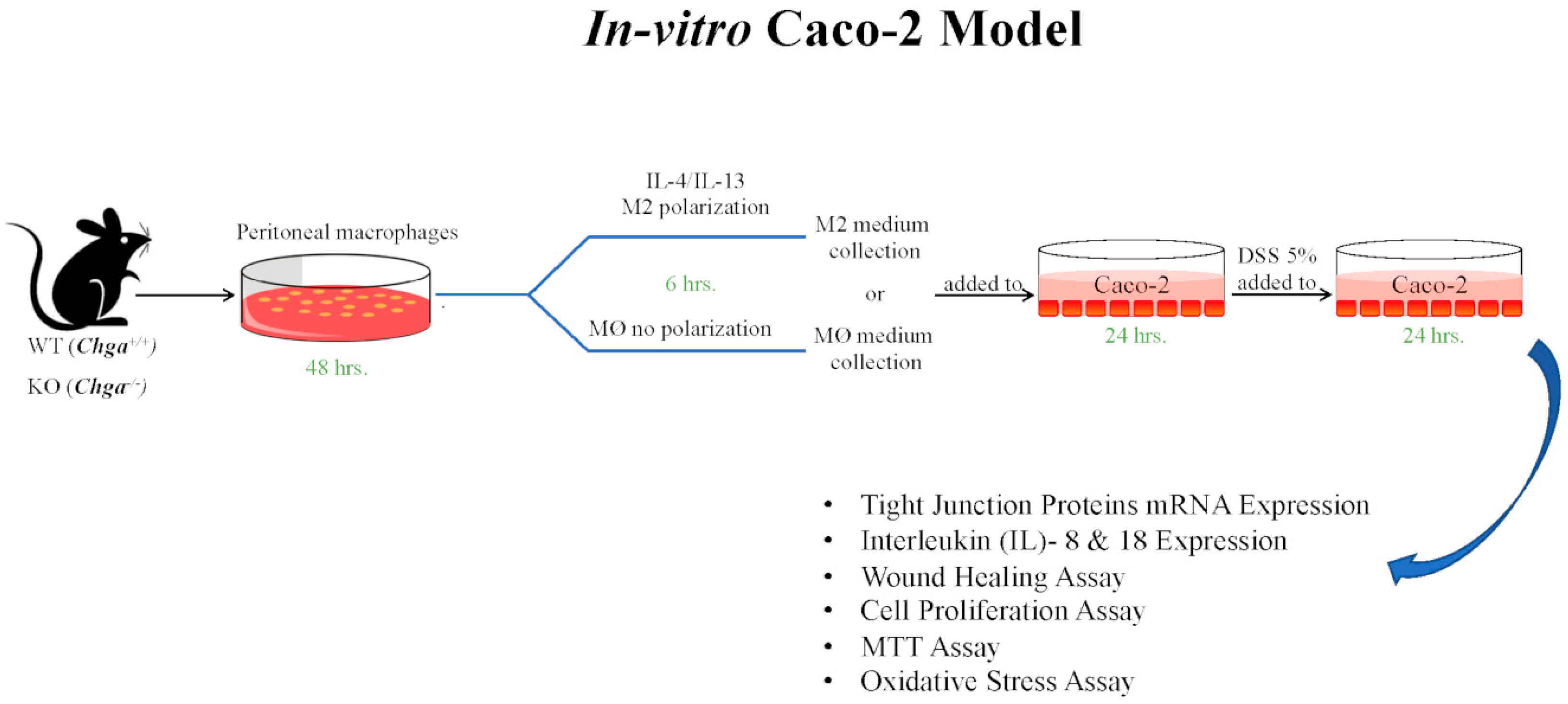
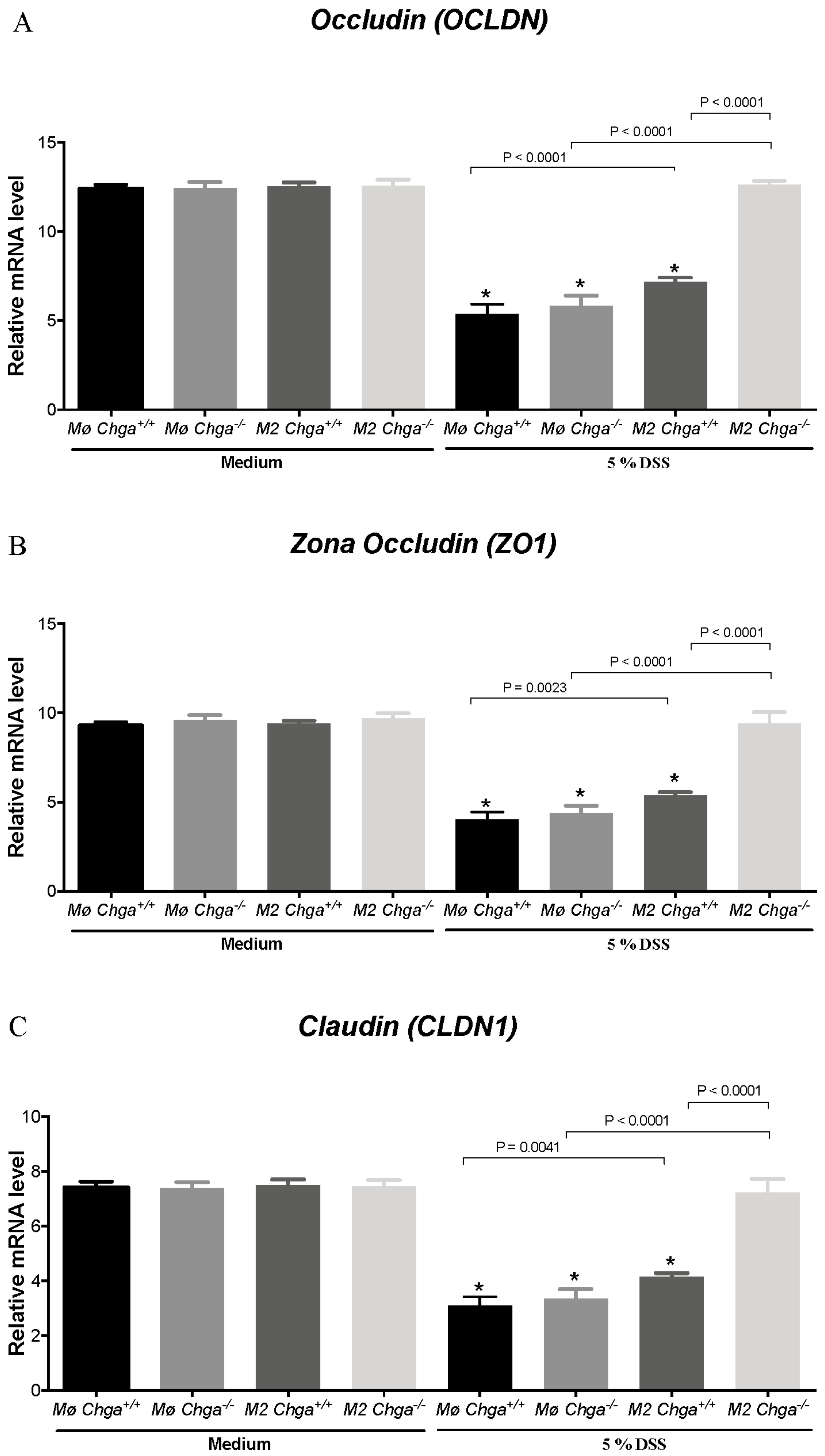
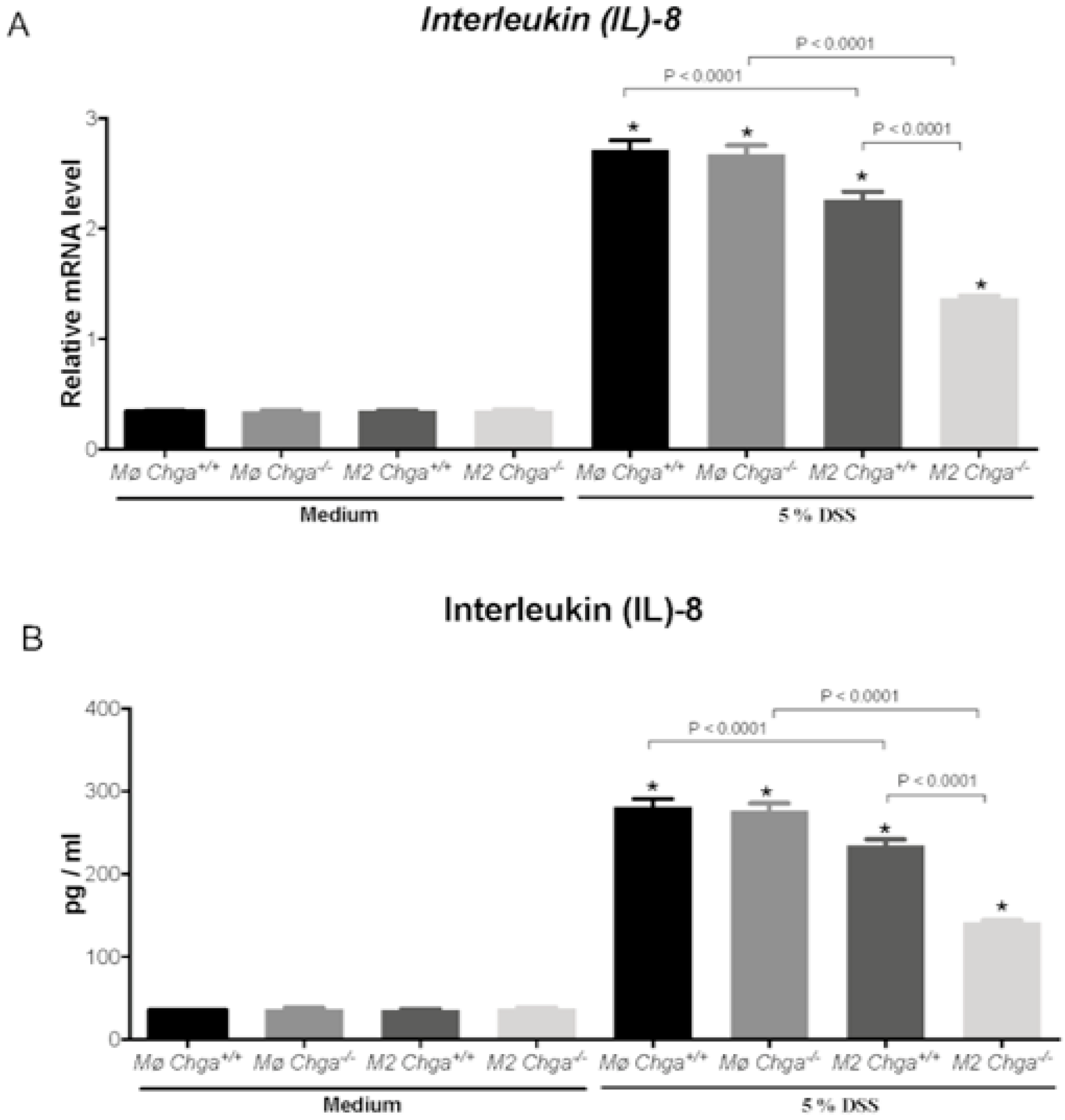
2.4. Chga-Deficient M2-Conditioned Supernatant Maintains TJ Protein mRNA Expression and Decreases IL-8 and IL-18 Expression and Release in DSS-Stimulated Human Caco-2 Epithelial Cells
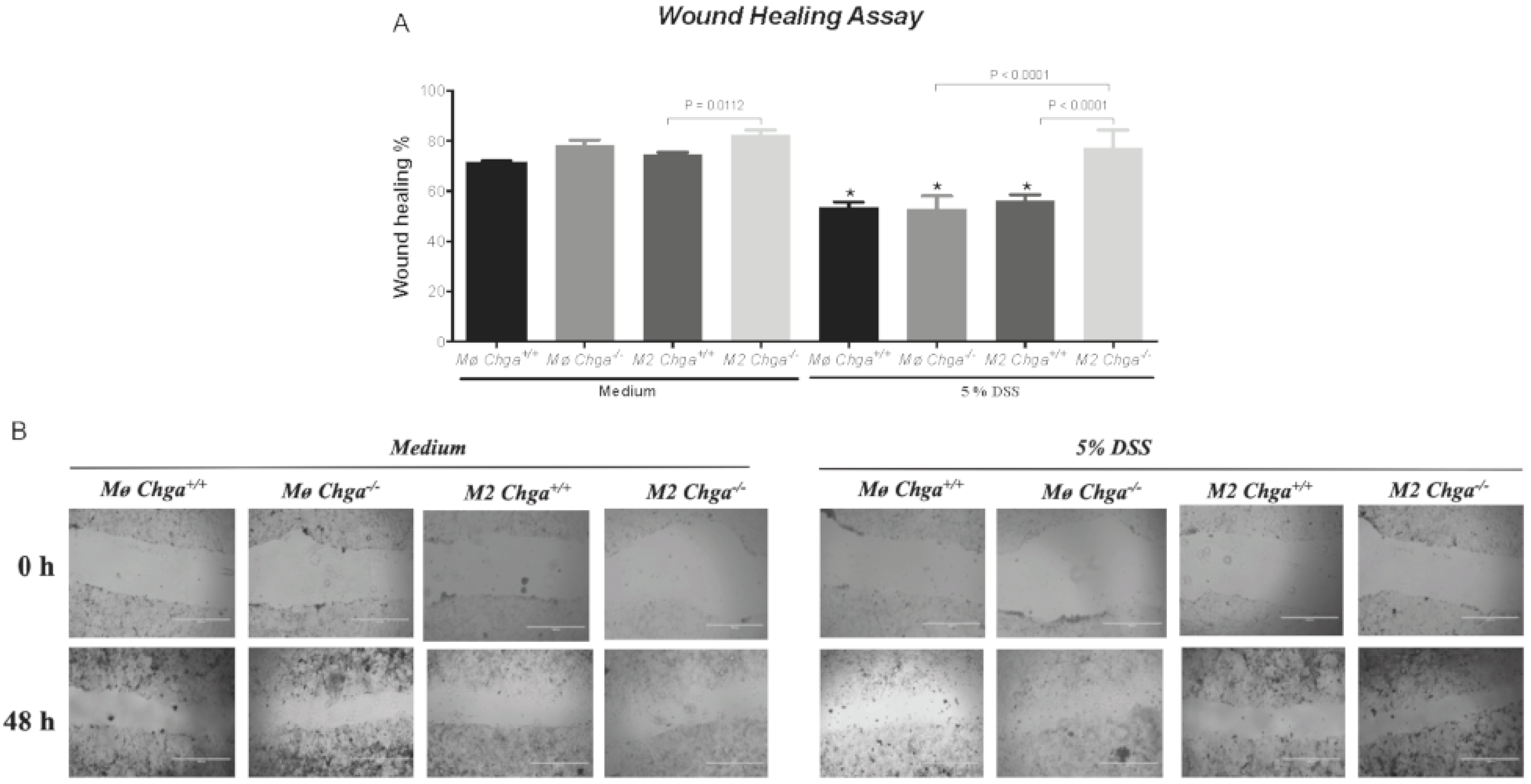
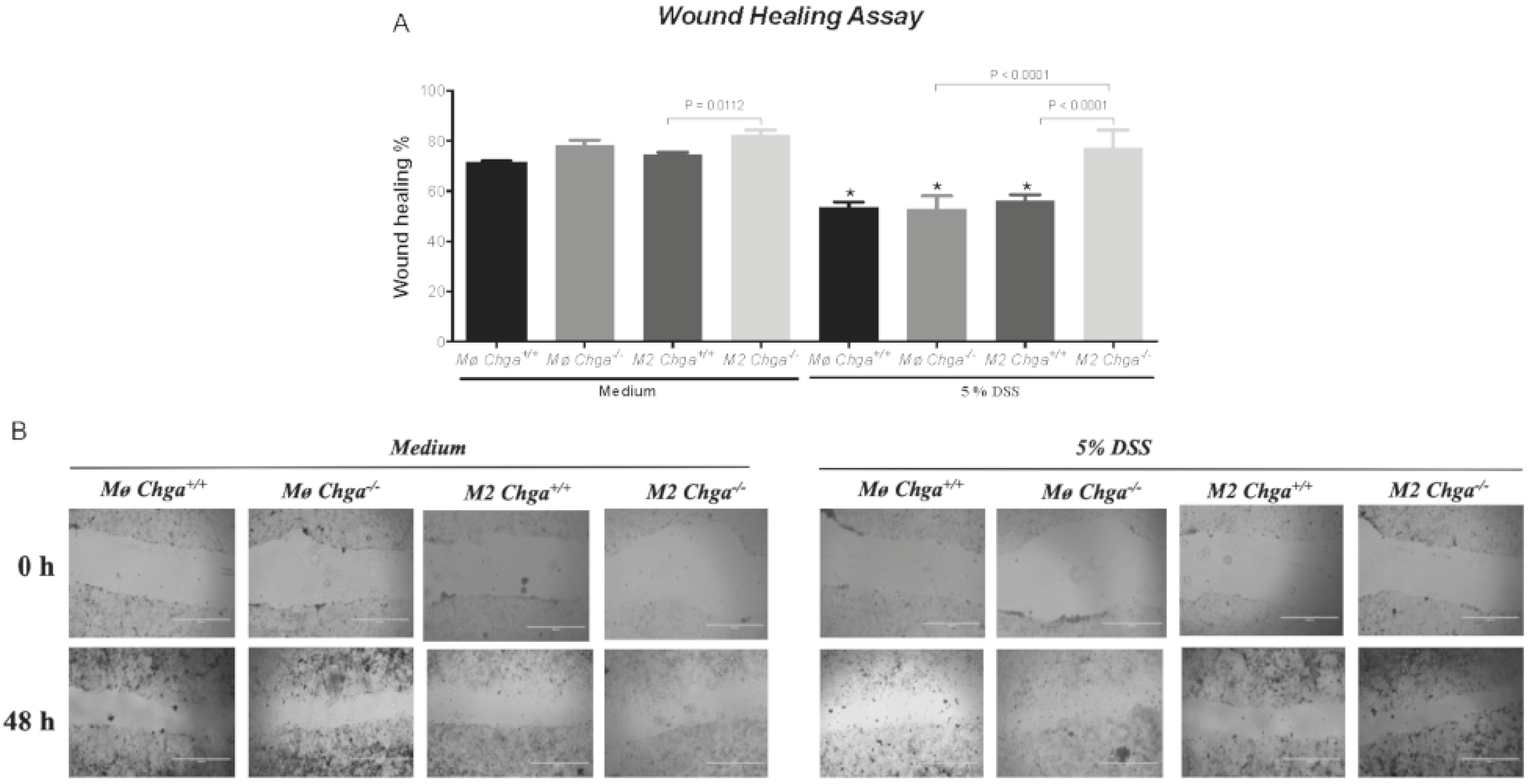
2.5. Chga-Deficient M2-Conditioned Supernatant Enhances the Functional Abilities of Human Caco-2 Epithelial Cells
3. Discussion
4. Material and Methods
4.1. Active Ulcerative Colitis & Control Subjects
4.2. Chga-Knockout Mice
4.3. DSS Injury Model
4.4. Assessment of Colitis Severity and the Collagen Deposition
4.5. Protein Assay
4.6. Intraperitoneal Macrophages Cell Culture
4.7. Cell Line Culture
4.8. Gene Expression
4.9. Data Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Turner, J.R. Molecular basis of epithelial barrier regulation: From basic mechanisms to clinical application. Am. J. Pathol. 2006, 169, 1901–1909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eissa, N.; Hussein, H.; Hendy, G.N.; Bernstein, C.N.; Ghia, J.-E. Chromogranin-A and its derived peptides and their pharmacological effects during intestinal inflammation. Biochem. Pharmacol. 2018, 152, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Eissa, N.; Mujawar, Q.; Abdul-Salam, T.; Zohni, S.; El-Matary, W. The Immune-Sleep Crosstalk in Inflammatory Bowel Disease. Sleep Med. 2020, 73, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-Z.; Li, Y.-Y. Inflammatory bowel disease: Pathogenesis. World J. Gastroenterol. WJG 2014, 20, 91. [Google Scholar] [CrossRef] [PubMed]
- Eissa, N.; Ghia, J. Immunomodulatory effect of ghrelin in the intestinal mucosa. Neurogastroenterol. Motil. 2015, 27, 1519–1527. [Google Scholar] [CrossRef]
- Eissa, N.; Kermarrec, L.; Metz-Boutigue, M.-H.; Hendy, G.N.; Bernstein, C.N.; Ghia, J.-E. Chromofungin treatment promotes alternatively activated macrophages, suppresses classically activated macrophages and improves epithelial cell functions during colitis. Gastroenterology 2017, 152, S143. [Google Scholar] [CrossRef]
- Na, Y.R.; Stakenborg, M.; Seok, S.H.; Matteoli, G. Macrophages in intestinal inflammation and resolution: A potential therapeutic target in IBD. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 531–543. [Google Scholar] [CrossRef]
- Quiros, M.; Nishio, H.; Neumann, P.A.; Siuda, D.; Brazil, J.C.; Azcutia, V.; Hilgarth, R.; O’Leary, M.N.; Garcia-Hernandez, V.; Leoni, G. Macrophage-derived IL-10 mediates mucosal repair by epithelial WISP-1 signaling. J. Clin. Investig. 2017, 127, 3510–3520. [Google Scholar] [CrossRef]
- Seyedizade, S.S.; Afshari, K.; Bayat, S.; Rahmani, F.; Momtaz, S.; Rezaei, N.; Abdolghaffari, A.H. Current Status of M1 and M2 Macrophages Pathway as Drug Targets for Inflammatory Bowel Disease. Arch. Immunol. Ther. Exp. 2020, 68, 10. [Google Scholar] [CrossRef]
- Eissa, N.; Hussein, H.; Ghia, J.-E. A Gene Expression Analysis of M1 and M2 Polarized Macrophages. In Immunometabolism; Springer: Berlin/Heidelberg, Germany, 2020; pp. 131–144. [Google Scholar]
- D’amico, M.A.; Ghinassi, B.; Izzicupo, P.; Manzoli, L.; Di Baldassarre, A. Biological function and clinical relevance of chromogranin A and derived peptides. Endocr. Connect. 2014, 3, R45–R54. [Google Scholar] [CrossRef] [Green Version]
- El-Salhy, M.; Danielsson, Å.; Stenling, R.; Grimelius, L. Colonic endocrine cells in inflammatory bowel disease. J. Intern. Med. 1997, 242, 413–419. [Google Scholar] [CrossRef]
- Sciola, V.; Massironi, S.; Conte, D.; Caprioli, F.; Ferrero, S.; Ciafardini, C.; Peracchi, M.; Bardella, M.T.; Piodi, L. Plasma chromogranin a in patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2009, 15, 867–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidhu, R.; Drew, K.; McAlindon, M.E.; Lobo, A.J.; Sanders, D.S. Elevated serum chromogranin A in irritable bowel syndrome (IBS) and inflammatory bowel disease (IBD): A shared model for pathogenesis? Inflamm. Bowel Dis. 2010, 16, 361. [Google Scholar] [CrossRef] [PubMed]
- Eissa, N.; Hussein, H.; Kermarrec, L.; Ali, A.Y.; Marshall, A.; Metz-Boutigue, M.-H.; Hendy, G.N.; Bernstein, C.N.; Ghia, J.-E. Chromogranin-A Regulates Macrophage Function and the Apoptotic Pathway in Murine DSS colitis. J. Mol. Med. 2018, 96, 183–198. [Google Scholar] [CrossRef]
- Rabbi, M.F.; Munyaka, P.M.; Eissa, N.; Metz-Boutigue, M.-H.; Khafipour, E.; Ghia, J.E. Human catestatin alters gut microbiota composition in mice. Front. Microbiol. 2017, 7, 2151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eissa, N.; Hussein, H.; Mesgna, R.; Bonin, S.; Hendy, G.N.; Metz-Boutigue, M.-H.; Bernstein, C.N.; Ghia, J.-E. Catestatin regulates epithelial cell dynamics to improve intestinal inflammation. Vaccines 2018, 6, 67. [Google Scholar] [CrossRef] [Green Version]
- Eissa, N.; Hussein, H.; Kermarrec, L.; Elgazzar, O.; Metz-Boutigue, M.-H.; Bernstein, C.N.; Ghia, J.-E. Chromofungin (CHR: CHGA47–66) is downregulated in persons with active ulcerative colitis and suppresses pro-inflammatory macrophage function through the inhibition of NF-κB signaling. Biochem. Pharmacol. 2017, 145, 102–113. [Google Scholar] [CrossRef]
- Eissa, N.; Hussein, H.; Kermarrec, L.; Grover, J.; Metz-Boutigue, M.-H.E.; Bernstein, C.N.; Ghia, J.-E. chromofungin ameliorates the Progression of colitis by regulating alternatively activated Macrophages. Front. Immunol. 2017, 8, 1131. [Google Scholar] [CrossRef] [Green Version]
- Rabbi, M.F.; Labis, B.; Metz-Boutigue, M.-H.; Bernstein, C.N.; Ghia, J.-E. Catestatin decreases macrophage function in two mouse models of experimental colitis. Biochem. Pharmacol. 2014, 89, 386–398. [Google Scholar] [CrossRef]
- Gleeson, J.P.; Estrada, H.Q.; Yamashita, M.; Svendsen, C.N.; Targan, S.R.; Barrett, R.J. Development of Physiologically Responsive Human iPSC-Derived Intestinal Epithelium to Study Barrier Dysfunction in IBD. Int. J. Mol. Sci. 2020, 21, 1438. [Google Scholar] [CrossRef] [Green Version]
- Mortensen, J.; Lindholm, M.; Langholm, L.; Kjeldsen, J.; Bay-Jensen, A.; Karsdal, M.; Manon-Jensen, T. The intestinal tissue homeostasis–the role of extracellular matrix remodeling in inflammatory bowel disease. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 977–993. [Google Scholar] [CrossRef] [PubMed]
- Massironi, S.; Zilli, A.; Cavalcoli, F.; Conte, D.; Peracchi, M. Chromogranin A and other enteroendocrine markers in inflammatory bowel disease. Neuropeptides 2016, 58, 127–134. [Google Scholar] [CrossRef]
- Wagner, M.; Peterson, C.G.; Ridefelt, P.; Sangfelt, P.; Carlson, M. Fecal markers of inflammation used as surrogate markers for treatment outcome in relapsing inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 5584–5589. [Google Scholar] [CrossRef]
- Zissimopoulos, A.; Vradelis, S.; Konialis, M.; Chadolias, D.; Bampali, A.; Constantinidis, T.; Efremidou, E.; Kouklakis, G. Chromogranin A as a biomarker of disease activity and biologic therapy in inflammatory bowel disease: A prospective observational study. Scand. J. Gastroenterol. 2014, 49, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Rumio, C.; Dusio, G.F.; Colombo, B.; Gasparri, A.; Cardani, D.; Marcucci, F.; Corti, A. The N-terminal fragment of chromogranin A, vasostatin-1 protects mice from acute or chronic colitis upon oral administration. Dig. Dis. Sci. 2012, 57, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.-H.; Lee, S.-H.; Park, J.-W.; Go, D.-M.; Kim, D.-Y. Osteopontin protects colonic mucosa from dextran sodium sulfate-induced acute colitis in mice by regulating junctional distribution of occludin. Dig. Dis. Sci. 2019, 64, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Poritz, L.S.; Garver, K.I.; Green, C.; Fitzpatrick, L.; Ruggiero, F.; Koltun, W.A. Loss of the tight junction protein ZO-1 in dextran sulfate sodium induced colitis. J. Surg. Res. 2007, 140, 12–19. [Google Scholar] [CrossRef]
- Nowarski, R.; Jackson, R.; Gagliani, N.; de Zoete, M.R.; Palm, N.W.; Bailis, W.; Low, J.S.; Harman, C.C.; Graham, M.; Elinav, E. Epithelial IL-18 equilibrium controls barrier function in colitis. Cell 2015, 163, 1444–1456. [Google Scholar] [CrossRef] [Green Version]
- Nighot, P.; Ma, T. Endocytosis of Intestinal Tight Junction Proteins: In Time and Space. Inflamm. Bowel Dis. 2020. [CrossRef]
- Pastorelli, L.; De Salvo, C.; Mercado, J.R.; Vecchi, M.; Pizarro, T.T. Central role of the gut epithelial barrier in the pathogenesis of chronic intestinal inflammation: Lessons learned from animal models and human genetics. Front. Immunol. 2013, 4, 280. [Google Scholar] [CrossRef] [Green Version]
- Lichtenberger, G.S.; Flavell, R.A.; Alexopoulou, L. Innate immunity and apoptosis in IBD. Inflamm. Bowel Dis. 2004, 10, S58–S62. [Google Scholar] [CrossRef]
- Steinbach, E.C.; Plevy, S.E. The role of macrophages and dendritic cells in the initiation of inflammation in IBD. Inflamm. Bowel Dis. 2014, 20, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.-Z.; Winston, J.H.; Sarna, S.K. Differential immune and genetic responses in rat models of Crohn’s colitis and ulcerative colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G41–G51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piechota-Polanczyk, A.; Fichna, J. Review article: The role of oxidative stress in pathogenesis and treatment of inflammatory bowel diseases. Naunyn-Schmiedebergs Arch. Pharmacol. 2014, 387, 605–620. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Yang, Q.; Rogers, C.J.; Du, M.; Zhu, M.-J. AMPK improves gut epithelial differentiation and barrier function via regulating Cdx2 expression. Cell Death Differ. 2017, 24, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Iacomino, G.; Aufiero, V.R.; Iannaccone, N.; Melina, R.; Giardullo, N.; De Chiara, G.; Venezia, A.; Taccone, F.S.; Iaquinto, G.; Mazzarella, G. IBD: Role of intestinal compartments in the mucosal immune response. Immunobiology 2020, 225, 151849. [Google Scholar] [CrossRef] [PubMed]
- Marafini, I.; Sedda, S.; Dinallo, V.; Monteleone, G. Inflammatory cytokines: From discoveries to therapies in IBD. Expert Opin. Biol. Ther. 2019, 19, 1207–1217. [Google Scholar] [CrossRef]
- Kermarrec, L.; Eissa, N.; Wang, H.; Kapoor, K.; Diarra, A.; Gounni, A.S.; Bernstein, C.N.; Ghia, J.E. Semaphorin-3E attenuates intestinal inflammation through the regulation of the communication between splenic CD11C+ and CD4+ CD25− T-cells. Br. J. Pharmacol. 2019, 176, 1235–1250. [Google Scholar] [CrossRef]
- Hendy, G.N.; Li, T.; Girard, M.; Feldstein, R.C.; Mulay, S.; Desjardins, R.; Day, R.; Karaplis, A.C.; Tremblay, M.L.; Canaff, L. Targeted ablation of the chromogranin a (Chga) gene: Normal neuroendocrine dense-core secretory granules and increased expression of other granins. Mol. Endocrinol. 2006, 20, 1935–1947. [Google Scholar] [CrossRef]
- Okayasu, I.; Hatakeyama, S.; Yamada, M.; Ohkusa, T.; Inagaki, Y.; Nakaya, R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990, 98, 694–702. [Google Scholar] [CrossRef]
- Eissa, N.; Hussein, H.; Diarra, A.; Elgazzar, O.; Gounni, A.S.; Bernstein, C.N.; Ghia, J.-E. Semaphorin 3E regulates apoptosis in the intestinal epithelium during the development of colitis. Biochem. Pharmacol. 2019, 166, 264–273. [Google Scholar] [CrossRef]
- Mosser, D.M.; Zhang, X. Activation of Murine Macrophages. Curr. Protoc. Immunol. 2008. [Google Scholar] [CrossRef] [PubMed]
- Eissa, N.; Hussein, H.; Wang, H.; Rabbi, M.F.; Bernstein, C.N.; Ghia, J.-E. Stability of Reference Genes for Messenger RNA Quantification by Real-Time PCR in Mouse Dextran Sodium Sulfate Experimental Colitis. PLoS ONE 2016, 11, e0156289. [Google Scholar] [CrossRef] [PubMed]
- Eissa, N.; Kermarrec, L.; Hussein, H.; Bernstein, C.N.; Ghia, J.-E. Appropriateness of reference genes for normalizing messenger RNA in mouse 2, 4-dinitrobenzene sulfonic acid (DNBS)-induced colitis using quantitative real time PCR. Sci. Rep. 2017, 7, 42427. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward | Reverse |
|---|---|---|
| CLDN1 | AGGTGCTATCTGTTCAGTGATG | TGGCTGACTTTCCTTGTGTAG |
| COL1A2 | GAGCGGTAACAAGGGTGAGC | CTTCCCCATTAGGGCCTCTC |
| IL18 | GCGTCACTACACTCAGCTAAT | GCGTCACTACACTCAGCTAAT |
| IL8 | ACTGAGAGTGATTGAGAGTGGAC | AACCCTCTGCACCCAGTTTTC |
| OCLDN | ACAAGCGGTTTTATCCAGAGTC | GTCATCCACAGGCGAAGTTAAT |
| TBP | CCCGAAACGCCGAATATAATCC | AATCAGTGCCGTGGTTCGTG |
| ZO1 | CCAGCCTGCTAAACCTACTAAA | ATCTCTTGCTGCCAAACTATCT |
| Gene | Forward | Reverse |
|---|---|---|
| Cldn1 | GGGGACAACATCGTGACCG | AGGAGTCGAAGACTTTGCACT |
| Col1a2 | GGTGAGCCTGGTCAAACGG | ACTGTGTCCTTTCACGCCTTT |
| Eef2 | TGTCAGTCATCGCCCATGTG | CATCCTTGCGAGTGTCAGTGA |
| Il18 | GACTCTTGCGTCAACTTCAAGG | CAGGCTGTCTTTTGTCAACGA |
| Ocldn | TTGAAAGTCCACCTCCTTACAGA | CCGGATAAAAAGAGTACGCTGG |
| Zo1 | GCCGCTAAGAGCACAGCAA | TCCCCACTCTGAAAATGAGGA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eissa, N.; Hussein, H.; Tshikudi, D.M.; Hendy, G.N.; Bernstein, C.N.; Ghia, J.-E. Interdependence between Chromogranin-A, Alternatively Activated Macrophages, Tight Junction Proteins and the Epithelial Functions. A Human and In-Vivo/In-Vitro Descriptive Study. Int. J. Mol. Sci. 2020, 21, 7976. https://doi.org/10.3390/ijms21217976
Eissa N, Hussein H, Tshikudi DM, Hendy GN, Bernstein CN, Ghia J-E. Interdependence between Chromogranin-A, Alternatively Activated Macrophages, Tight Junction Proteins and the Epithelial Functions. A Human and In-Vivo/In-Vitro Descriptive Study. International Journal of Molecular Sciences. 2020; 21(21):7976. https://doi.org/10.3390/ijms21217976
Chicago/Turabian StyleEissa, Nour, Hayam Hussein, Diane M. Tshikudi, Geoffrey N. Hendy, Charles N. Bernstein, and Jean-Eric Ghia. 2020. "Interdependence between Chromogranin-A, Alternatively Activated Macrophages, Tight Junction Proteins and the Epithelial Functions. A Human and In-Vivo/In-Vitro Descriptive Study" International Journal of Molecular Sciences 21, no. 21: 7976. https://doi.org/10.3390/ijms21217976
APA StyleEissa, N., Hussein, H., Tshikudi, D. M., Hendy, G. N., Bernstein, C. N., & Ghia, J.-E. (2020). Interdependence between Chromogranin-A, Alternatively Activated Macrophages, Tight Junction Proteins and the Epithelial Functions. A Human and In-Vivo/In-Vitro Descriptive Study. International Journal of Molecular Sciences, 21(21), 7976. https://doi.org/10.3390/ijms21217976