Factors Affecting the Formation and Treatment of Thrombosis by Natural and Synthetic Compounds


Abstract
:1. Introduction
2. The Basic Mechanism of Blood Clotting
3. Causes of Vein Thrombosis
3.1. Hypoxia and Thrombus Formation
3.2. Role of Patogens and Oxidtive Stress
4. Regulation of Cells Activation in Development of VTE
4.1. Platelets Activation
4.2. Endothelial Cells Activation
5. ROS and Oxidative Stress
6. Advances in Treatment of Thromboembolism
6.1. Endovascular Treatment of DVT
6.2. Anticoagulants
6.2.1. Inhibitors of FX
6.2.2. Thrombin Inhibitors
6.2.3. Other Compounds
6.3. Antiplatelet Therapy
6.4. Emerging Drugs
6.4.1. RUC-1, RUC-2, and RUC-4
6.4.2. PAR-1 and PAR-4 Antagonist
6.4.3. Inhibitors of PDI and PI3Kβ
6.4.4. Other Compounds
7. Natural Compounds
7.1. Flavonoids
7.1.1. Antithrombotic Properties of Flavonoids
7.1.2. Isoflavonoids
7.1.3. Traditional Korean and Chinese Medicine
7.2. Saponins
7.2.1. Plant to the Genus Panax
7.2.2. Liriope muscari
7.2.3. Steroidal Sapogenin
7.2.4. Other Saponins
7.3. Stevioside and Derivertes
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AA | Arachidonic acid |
| ACE | Angiotensin-converting enzyme |
| ADP | Adenosine diphosphate |
| AGEs | Advanced glycation end products |
| Akt | Protein kinase B |
| AMI | Acute myocardial infarction |
| aPTT | Activated partial thromboplastin time |
| apoAI | Apoplipoprotein A-I |
| APS | Antiphospholipid syndrome |
| ASA | Acetylsalicylic acid |
| ASO | Antisense oligonucleotides |
| AT | Antithrombin |
| ATP | Adenosine triphosphate |
| cAMP | Cyclic adenosine monophosphate |
| CAT | Catalase |
| CCL | Chemokine (C-C motif) ligand |
| CD | Cluster of differentiation |
| CDT | Catheter-directed thrombolysis |
| CEP | Centrosome-associated protein |
| CHCL1 | Chemokine induces the infiltration of neutrophils |
| CLEC | C-type lectin-like receptor |
| COX | Cyclooxygenase |
| CT | Computed tomography |
| CX3CL1 | Fractalkine |
| CX3CR1 | CX3C chemokine receptor 1 |
| CXCL | Chemokine CXC ligand-1 |
| CYP | Cytochrome P450 |
| CypA | Cyclophilin A |
| DNA | Deoxyribonucleic acid |
| DVT | Deep vein thrombosis |
| eATP | Extracellular ATP |
| ECE | Endothelin converting enzyme |
| ECs | Endothelial cells |
| ED | Endothelial dysfunction |
| EDCF | Endothelium-derived contracting factor |
| EDGF | Endothelium-derived growth factor |
| EDHF | Endothelium-derived hyperpolarizing factor |
| EGF | Epidermal growth factor |
| EMP | Microparticles of endothelial cells |
| EMT | Epithelial-mesenchymal transition |
| eNOS | Endothelial nitric oxide synthase |
| ERK | Extracellular signal-regulated kinase |
| ETC | Electron transport chain |
| FGF | Fibroblast growth factor |
| GP | Glycoprotein |
| GSK | Glycogen synthase kinase |
| H2O | Water |
| H2O2 | Hydrogen peroxide |
| Hb | Haemoglobin |
| HClO | Hypochlorous acid |
| HIF | Hypoxia inducible factor |
| HNE | 4-hydroxy-2-nonenal |
| HO• | Hydroxyl radical |
| HO2• | Hydroperoxyl radical |
| Hp | Haptoglobin |
| HRE | Hormone response element |
| HsCRP | High-sensitivity C-reactive protein |
| 5HT2A | Serotonin receptor 2A |
| HUVEC | Human umbilical vein endothelial cells |
| Hx | Hemopexin |
| IAV | Influenza A virus |
| ICAM | Intercellular adhesion molecule |
| iCypA | Extracellular cyclophilin A |
| IDUs | Intravenous drug users |
| IFN | Interferon |
| IGF | Insulin-like growth factor |
| IL | Interleukin |
| iNOS | Induced nitric oxide synthase |
| INR | International normalized ratio |
| IVC | Inferior vena cava |
| IκBα | Inhibitor of kappa B |
| JAK-2 | Janus kinase 2 |
| JNK | C-Jun N-terminal kinase |
| LMWH | Low molecular weight heparin |
| LOX | Lipoxygenase |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinases |
| MCP | Monocyte chemoattractant protein |
| metHb | Methaemoglobin |
| MHC | Major histocompatibility complex |
| MIDAS | Metal ion-dependent adhesion site |
| MIP-2 | Macrophage inflammatory protein 2 |
| MKP-1 | Mitogen-activated protein kinases phosphatase 1 |
| MMP | Matrix metalloproteinase |
| MPO | Myeloperoxidase |
| MPV | Mean platelets volume |
| mPTP | Mitochondrial permeability transition pore |
| MRI | Magnetic resonance imaging |
| NAD+ | Nicotinamide-adenine dinucleotide phosphate (oxidised form) |
| NADPH | Nicotinamide-adenine dinucleotide phosphate (reduced form) |
| NAPc2 | Nematode anticoagulant protein c2 |
| NETs | Neutrophil extracellular traps |
| NF-κB | Nuclear factor kappa B |
| NLRP3 | Inflammasome is component of the innate immune system |
| NMMHC IIA | Non-muscle myosin heavy chain IIa |
| NO• | Nitric oxide |
| NOX | NADPH oxidase |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| NSCLC | Non-small-cell lung carcinoma |
| O2 | Oxygen |
| 1O2 | Singlet oxygen |
| O2•− | Superoxide anion radical |
| ONOO− | Peroxynitrite |
| ox-L | Oxidised lipids |
| ox-P | Oxidised proteins |
| oxyHb | Oxyhaemoglobin |
| PAF | Platelet activating factor |
| PAI-I | Plasminogen activator inhibitor-1 |
| PAR | Protease-activated receptor |
| PAT | Percutaneous aspiration thrombectomy |
| PCDT | Catheter-directed pharmacomechanical thrombolysis |
| PCI | Percutaneous coronary intervention |
| PD-L1 | Programmed death-ligand 1 |
| PDE3 | Phosphodiesterase type 3 |
| PDGF | Platelet-derived growth factor |
| PDI | Protein disulfide-isomerase |
| PE | Pulmonary embolism |
| PF | Platelet factor |
| PGE | Prostaglandin E |
| PGG | Prostaglandin G |
| PGH | Prostaglandin H |
| PGI | Prostacycline |
| PI3Kβ | Phosphatidylinositol 3 kinase β |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| PLa | Activated platelets |
| PLA | Phospholipase A |
| PLC | Phospholipase C |
| PLCγ2 | Phospholipase Cγ2 |
| PMN | Polymorphonuclear leukocyte |
| PMQ | Pentamethyl quercetin |
| PNS | Saponins Panax notoginseng |
| PPAR-γ | Peroxisome proliferator-activated receptor γ |
| PSGL-1 | P-selectin glycoprotein ligand-1 |
| PT | Prothrombin time |
| PTS | Polygala fallax Hesml root saponin extract |
| RBCs | Red blood cells |
| RNA | Ribonucleic acid |
| ROS | Reactive oxygen species |
| SBSI | Simultaneous decrease in prostacyclin synthase |
| SLO | Streptolysin O |
| SLS | Streptolysin S |
| SOD | Superoxide dismutase |
| STAT3 | Signal transducer and activator of transcription 3 |
| STEC | Shigatoxigenic Escherichia coli |
| TF | Tissue factor |
| TGF-β | Transforming growth factor beta |
| TLR | Toll-like receptor |
| TNF-α | Tumour necrosis factor alfa |
| t-PA | Tissue plasminogen activator |
| TRAP | Thrombin receptor activator peptide |
| TTP | Thrombocytopenic purpura |
| TXA2 | Tromboxan A2 |
| UFH | Unfractionated heparin |
| UV | Ultraviolet |
| VCAM | Vascular cell adhesion molecule |
| VEGF | Vascular endothelial growth factor |
| VKORC1 | Vitamin K epoxide reductase complex |
| VSMCs | Vascular smooth muscle cells |
| VTE | Venous thromboembolism |
| vWF | Von Willebrand factor |
References
- Weitz, J.I.; Chan, N.C. Advances in Antithrombotic Therapy. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 7–12. [Google Scholar] [CrossRef] [Green Version]
- Essien, E.-O.; Rali, P.; Mathai, S.C. Pulmonary Embolism. Med Clin. N. Am. 2019, 103, 549–564. [Google Scholar] [CrossRef] [PubMed]
- Flumignan, C.D.; Flumignan, R.L.; Baptista-Silva, J.C. Antiplatelet agents for the treatment of deep venous thrombosis. Cochrane Database Syst. Rev. 2016, 2016, CD012369. [Google Scholar] [CrossRef]
- Wattanakit, K.; Lutsey, P.L.; Bell, E.J.; Gornik, H.; Cushman, M.; Heckbert, S.R.; Rosamond, W.D.; Folsom, A.R. Association between cardiovascular disease risk factors and occurrence of venous thromboembolism. Thromb. Haemost. 2012, 108, 508–515. [Google Scholar] [PubMed] [Green Version]
- Stone, J.; Hangge, P.; Albadawi, H.; Wallace, A.; Shamoun, F.; Knuttien, M.G.; Naidu, S.; Oklu, R. Deep vein thrombosis: Pathogenesis, diagnosis, and medical management. Cardiovasc. Diagn. Ther. 2017, 7 (Suppl. 3), 276–284. [Google Scholar] [CrossRef] [PubMed]
- Criqui, M.H.; Denenberg, J.O.; Bergan, J.; Langer, R.D.; Fronek, A. Risk factors for chronic venous disease: The San Diego Population Study. J. Vasc. Surg. 2007, 46, 331–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwiatkowska, W.; Knysz, B.; Gąsiorowski, J.; Witkiewicz, W. Deep vein thrombosis of the lower limbs in intravenous drug users. Postępy Hig. Med. Doświadczalnej 2015, 69, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.D. 1 Endothelial cell function and thrombosis. Baillière’s Clin. Haematol. 1994, 7, 441–452. [Google Scholar] [CrossRef]
- Van Hinsbergh, V.W.M. Endothelium—Role in regulation of coagulation and inflammation. Semin. Immunopathol. 2012, 34, 93–106. [Google Scholar] [CrossRef] [Green Version]
- Wolberg, A.S.; Campbell, R.A. Thrombin generation, fibrin clot formation and hemostasis. Transfus. Apher. Sci. 2008, 38, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Weisel, J.W.; Litvinov, R.I. Fibrin Formation, Structure and Properties. Subcell Biochem. 2017, 82, 405–456. [Google Scholar]
- Petrillo, G.; Cirillo, P.; Luana D’Ascoli, G.-; Maresca, F.; Ziviello, F.; Chiariello, M. Tissue Factor/Factor FVII Complex Inhibitors in Cardiovascular Disease. Are Things Going Well? Curr. Cardiol. Rev. 2010, 6, 325–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budnik, I.; Brill, A. Immune Factors in Deep Vein Thrombosis Initiation. Trends Immunol. 2018, 39, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Kehrel, B.E.; Corkrey, H.A.; Freedman, J.E. Thrombosis and platelets: An update. Eur. Heart J. 2016, 38, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.A.; Key, N.S.; Levy, J.H. Blood Coagulation: Hemostasis and Thrombin Regulation. Anesth. Analg. 2009, 108, 1433–1446. [Google Scholar] [CrossRef] [PubMed]
- MCKenzie, P.J. Deep venous thrombosis and anaesthesia. Br. J. Anaesth. 1991, 67, 128. [Google Scholar] [CrossRef] [Green Version]
- Branchford, B.R.; Carpenter, S.L. The Role of Inflammation in Venous Thromboembolism. Front. Pediatrics 2018, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Levy, J.H. Inflammation and thrombosis: Roles of neutrophils, platelets and endothelial cells and their interactions in thrombus formation during sepsis. J. Thromb. Haemost. 2018, 16, 231–241. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Apta, B.H.R.; Bonna, A.M.; Harper, M.T. Platelet P-selectin triggers rapid surface exposure of tissue factor in monocytes. Sci. Rep. 2019, 9, 13397. [Google Scholar] [CrossRef]
- Waldron, B.; Moll, S. A patient’s guide to recovery after deep vein thrombosis or pulmonary embolism. Circulation 2014, 129, 477–479. [Google Scholar] [CrossRef] [Green Version]
- Swystun, L.L.; Liaw, P.C. The role of leukocytes in thrombosis. Blood 2016, 128, 753–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brill, A.; Suidan, G.L.; Wagner, D.D. Hypoxia, such as encountered at high altitude, promotes deep vein thrombosis in mice. J. Thromb. Haemost. 2013, 11, 1773–1775. [Google Scholar] [CrossRef]
- Gupta, N.; Sahu, A.; Prabhakar, A.; Chatterjee, T.; Tyagi, T.; Kumari, B.; Khan, N.; Nair, V.; Bajaj, N.; Sharma, M.; et al. Activation of NLRP3 inflammasome complex potentiates venous thrombosis in response to hypoxia. Proc. Natl. Acad. Sci. USA 2017, 114, 4763–4768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malone, P.C. The aetiology of deep venous thrombosis. QJM 2006, 99, 581–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esmon, C.T. Basic mechanisms and pathogenesis of venous thrombosis. Blood Rev. 2009, 23, 225–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Jackson, R.M. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am. J. Physiol. Cell Physiol. 2002, 282, 227–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinsky, D.J.; Naka, Y.; Liao, H.; Oz, M.C.; Wagner, D.D.; Mayadas, T.N.; Johnson, R.C.; Hynes, R.O.; Heath, M.; Lawson, C.A.; et al. Hypoxia-induced exocytosis of endothelial cell weibel-palade bodies: A mechanism for rapid neutrophil recruitment after cardiac preservation. J. Clin. Investig. 1996, 97, 493–500. [Google Scholar] [CrossRef]
- Michiels, C.; Arnould, T.; Remacle, J. Endothelial cell responses to hypoxia: Initiation of a cascade of cellular interactions. Biochim. Et Biophys. Acta BbaMol. Cell Res. 2000, 1497, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Liu, K.; Li, B.; Li, Y.; Ye, K.; Qi, J.; Wang, Y. Macrophages Aggravate Hypoxia-Induced Cardiac Microvascular Endothelial Cell Injury via Peroxynitrite: Protection by Tongxinluo. Cell Commun. Adhes. 2015, 22, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Thompson, L.F.; Eltzschig, H.K.; Ibla, J.C.; Van De Wiele, C.J.; Resta, R.; Morote-Garcia, J.C.; Colgan, S.P. Crucial Role for Ecto-5′-Nucleotidase (CD73) in Vascular Leakage during Hypoxia. J. Exp. Med. 2004, 200, 1395–1405. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Abdulla, P.; Hoffman, E.; Hamilton, K.E.; Daniels, D.; Schönfeld, C.; Löffler, M.; Reyes, G.; Duszenko, M.; Karhausen, J.; et al. HIF-1–dependent repression of equilibrative nucleoside transporter (ENT) in hypoxia. J. Exp. Med. 2005, 202, 1493–1505. [Google Scholar] [CrossRef] [PubMed]
- Eckle, T.; Faigle, M.; Grenz, A.; Laucher, S.; Thompson, L.F.; Eltzschig, H.K. A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood 2008, 111, 2024–2035. [Google Scholar] [CrossRef] [Green Version]
- Blouin, C.C.; Pagé, E.L.; Soucy, G.M.; Richard, D.E. Hypoxic gene activation by lipopolysaccharide in macrophages: Implication of hypoxia-inducible factor 1α. Blood 2004, 103, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Kruger, B.; Krick, S.; Dhillon, N.; Lerner, S.M.; Ames, S.; Bromberg, J.S.; Lin, M.; Walsh, L.; Vella, J.; Fischereder, M.; et al. Donor Toll-like receptor 4 contributes to ischemia and reperfusion injury following human kidney transplantation. Proc. Natl. Acad. Sci. USA 2009, 106, 3390–3395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldea, I.; Teacoe, I.; Olteanu, D.E.; Vaida-Voievod, C.; Clichici, A.; Sirbu, A.; Filip, G.A.; Clichici, S. Effects of different hypoxia degrees on endothelial cell cultures—Time course study. Mech. Ageing Dev. 2018, 172, 45–50. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Karin, M. Regulation and Function of NF-κB Transcription Factors in the Immune System. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef] [PubMed]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Mold, C.; Morris, C.A. Complement activation by apoptotic endothelial cells following hypoxia/reoxygenation. Immunology 2001, 102, 359–364. [Google Scholar] [CrossRef] [PubMed]
- HART, M. Initiation of complement activation following oxidative stress.In vitro and in vivo observations. Mol. Immunol. 2004, 41, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Poyton, R.O.; Ball, K.A.; Castello, P.R. Mitochondrial generation of free radicals and hypoxic signaling. Trends Endocrinol. Metab. 2009, 20, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Castello, P.R.; David, P.S.; McClure, T.; Crook, Z.; Poyton, R.O. Mitochondrial cytochrome oxidase produces nitric oxide under hypoxic conditions: Implications for oxygen sensing and hypoxic signaling in eukaryotes. Cell Metab. 2006, 3, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Palacios-Callender, M.; Hollis, V.; Mitchison, M.; Frakich, N.; Unitt, D.; Moncada, S. Cytochrome c oxidase regulates endogenous nitric oxide availability in respiring cells: A possible explanation for hypoxic vasodilation. Proc. Natl. Acad. Sci. USA 2007, 104, 18508–18513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishino, T.; Okamoto, K.; Eger, B.T.; Pai, E.F.; Nishino, T. Mammalian xanthine oxidoreductase—Mechanism of transition from xanthine dehydrogenase to xanthine oxidase. FEBS J. 2008, 275, 3278–3289. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, J.O.; Weitzberg, E.; Gladwin, M.T. The nitrate-nitrite-nitric oxide pathway in physiology and therapeutics. Nat. Rev. Drug Discov. 2008, 7, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spooner, R.; Yilmaz, Ö. The Role of Reactive-Oxygen-Species in Microbial Persistence and Inflammation. Int. J. Mol. Sci. 2011, 12, 334–352. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, G. Gram-positive and gram-negative bacterial toxins in sepsis. Virulence 2014, 5, 213–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hynes, W.; Sloan, M. Secreted Extracellular Virulence Factors. In Streptococcus Pyogenes: Basic Biology to Clinical Manifestations [Internet]; Ferretti, J.J., Stevens, D.L., Fischetti, V.A., Eds.; University of Oklahoma Health Sciences Center: Oklahoma City, OK, USA, 2016. [Google Scholar]
- Jürgens, D. Production and characterization of Escherichia coli enterohemolysin and its effects on the structure of erythrocyte membranes. Cell Biol. Int. 2002, 26, 175–186. [Google Scholar] [CrossRef]
- Krause, M.; Barth, H.; Schmidt, H. Toxins of Locus of Enterocyte Effacement-Negative Shiga Toxin-Producing Escherichia coli. Toxins 2018, 10, 241. [Google Scholar] [CrossRef] [Green Version]
- Jiang, N.; Tan, N.S.; Ho, B.; Ding, J.L. Respiratory protein–generated reactive oxygen species as an antimicrobial strategy. Nat. Immunol. 2007, 8, 1114–1122. [Google Scholar] [CrossRef]
- Alayash, A.I.; Patel, R.P.; Cashon, R.E. Redox Reactions of Hemoglobin and Myoglobin: Biological and Toxicological Implications. Antioxid. Redox Signal. 2001, 3, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Coates, C.J.; Decker, H. Immunological properties of oxygen-transport proteins: Hemoglobin, hemocyanin and hemerythrin. Cell. Mol. Life Sci. 2017, 74, 293–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahl, N.; Winarsih, I.; Tucker-Kellogg, L.; Ding, J.L. Extracellular haemoglobin upregulates and binds to tissue factor on macrophages: Implications for coagulation and oxidative stress. Thromb. Haemost. 2014, 111, 67–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Tang, H. Aberrant coagulation causes a hyper-inflammatory response in severe influenza pneumonia. Cell. Mol. Immunol. 2016, 13, 432–442. [Google Scholar] [CrossRef]
- Barnes, M.; Heywood, A.E.; Mahimbo, A.; Rahman, B.; Newall, A.T.; Macintyre, C.R. Acute myocardial infarction and influenza: A meta-analysis of case–control studies. Heart 2015, 101, 1738–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, J.J.; Voora, D.; Cyr, D.D.; Lucas, J.E.; Zaas, A.K.; Woods, C.W.; Newby, L.K.; Kraus, W.E.; Ginsburg, G.S. Gene Expression Profiles Link Respiratory Viral Infection, Platelet Response to Aspirin, and Acute Myocardial Infarction. PLoS ONE 2015, 10, e0132259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauguel-Moreau, M.; El Hajjam, M.; De Baynast, Q.; Vieillard-Baron, A.; Lot, A.-S.; Chinet, T.; Mustafic, H.; Bégué, C.; Carlier, R.Y.; Geri, G.; et al. Occurrence of pulmonary embolism related to COVID-19. J. Thromb. Thrombolysis 2020. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Zawilska, K. Fizjologia hemostazy. In Podstawy Hematologii; Dmoszynska, A., Robak, T., Eds.; Wydawnictwo Czelej: Lublin, Poland, 2015; ISBN 978-83-7563-202-6. [Google Scholar]
- Korzonek-Szlacheta, I.; Zubelewicz-Szkodzińska, B.; Gąsior, M. Płytki krwi—Ogniwo łączące zakrzepicę ze stanem zapalnym. Folia Cardiol. 2018, 13, 303–308. [Google Scholar] [CrossRef]
- Violi, F.; Pignatelli, P. Platelet Oxidative Stress and Thrombosis. Thromb. Res. 2012, 129, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Seno, T.; Inoue, N.; Gao, D.; Okuda, M.; Sumi, Y.; Matsui, K.; Yamada, S.; Hirata, K.; Kawashima, S.; Tawa, R.; et al. Involvement of NADH/NADPH Oxidase in Human Platelet ROS Production. Thromb. Res. 2001, 103, 399–409. [Google Scholar] [CrossRef]
- Qiao, J.; Arthur, J.F.; Gardiner, E.E.; Andrews, R.K.; Zeng, L.; Xu, K. Regulation of platelet activation and thrombus formation by reactive oxygen species. Redox Biol. 2018, 14, 126–130. [Google Scholar] [CrossRef]
- Stirban, A.; Gawlowski, T.; Roden, M. Vascular effects of advanced glycation endproducts: Clinical effects and molecular mechanisms. Mol. Metab. 2014, 3, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Beristain-Covarrubias, N.; Perez-Toledo, M.; Thomas, M.R.; Henderson, I.R.; Watson, S.P.; Cunningham, A.F. Understanding Infection-Induced Thrombosis: Lessons Learned From Animal Models. Front. Immunol. 2019, 10, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Nguyen, K.-A.; Potempa, J. Dichotomy of gingipains action as virulence factors: From cleaving substrates with the precision of a surgeon’s knife to a meat chopper-like brutal degradation of proteins. Periodontology 2000 2010, 54, 15–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proulx, F.; Seidman, E.G.; Karpman, D. Pathogenesis of Shiga Toxin-Associated Hemolytic Uremic Syndrome. Pediatric Res. 2001, 50, 163–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldsmith, H.L.; Turitto, V.T. Rheological aspects of thrombosis and haemostasis: Basic principles and applications. ICTH-Report--Subcommittee on Rheology of the International Committee on Thrombosis and Haemostasis. Thromb. Haemost. 1986, 55, 415–435. [Google Scholar] [CrossRef] [PubMed]
- Rana, A.; Westein, E.; Niego, B.; Hagemeyer, C.E. Shear-Dependent Platelet Aggregation: Mechanisms and Therapeutic Opportunities. Front. Cardiovasc. Med. 2019, 6, 141. [Google Scholar] [CrossRef]
- Ruggeri, Z.M.; Orje, J.N.; Habermann, R.; Federici, A.B.; Reininger, A.J. Activation-independent platelet adhesion and aggregation under elevated shear stress. Blood 2006, 108, 1903–1910. [Google Scholar] [CrossRef]
- Spiel, A.O.; Gilbert, J.C.; Jilma, B. Von Willebrand Factor in Cardiovascular Disease. Circulation 2008, 117, 1449–1459. [Google Scholar] [CrossRef] [Green Version]
- Gragnano, F.; Sperlongano, S.; Golia, E.; Natale, F.; Bianchi, R.; Crisci, M.; Fimiani, F.; Pariggiano, I.; Diana, V.; Carbone, A.; et al. The Role of von Willebrand Factor in Vascular Inflammation: From Pathogenesis to Targeted Therapy. Mediat. Inflamm. 2017, 2017, 5620314. [Google Scholar] [CrossRef] [PubMed]
- Gorog, D.A. Coronary angioplasty enhances platelet reactivity through von Willebrand factor release. Heart 2003, 89, 329–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, T.A.; Moore, S.F.; Williams, C.M.; Poole, A.W.; Vanhaesebroeck, B.; Hers, I. Phosphoinositide 3-Kinases p110α and p110β Have Differential Roles in Insulin-Like Growth Factor-1–Mediated Akt Phosphorylation and Platelet Priming. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1681–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hathcock, J.J. Flow Effects on Coagulation and Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1729–1737. [Google Scholar] [CrossRef]
- Fogelson, A.L.; Neeves, K.B. Fluid Mechanics of Blood Clot Formation. Annu. Rev. Fluid Mech. 2015, 47, 377–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebas, H.; Yahiaoui, K.; Martos, R.; Boulaftali, Y. Platelets Are at the Nexus of Vascular Diseases. Front. Cardiovasc. Med. 2019, 6, 132. [Google Scholar] [CrossRef]
- Montoro-García, S.; Schindewolf, M.; Stanford, S.; Larsen, O.; Thiele, T. The Role of Platelets in Venous Thromboembolism. Semin. Thromb. Hemost. 2016, 42, 242–251. [Google Scholar] [CrossRef]
- Widlansky, M.E.; Gokce, N.; Keaney, J.F.; Vita, J.A. The clinical implications of endothelial dysfunction. J. Am. Coll. Cardiol. 2003, 42, 1149–1160. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.M.; Douglas, G.; Bendall, J.K.; McNeill, E.; Crabtree, M.J.; Hale, A.B.; Mai, A.; Li, J.-M.; McAteer, M.A.; Schneider, J.E.; et al. Endothelial Cell–Specific Reactive Oxygen Species Production Increases Susceptibility to Aortic Dissection. Circulation 2014, 129, 2661–2672. [Google Scholar] [CrossRef] [Green Version]
- Castro-Ferreira, R.; Cardoso, R.; Leite-Moreira, A.; Mansilha, A. The Role of Endothelial Dysfunction and Inflammation in Chronic Venous Disease. J. Vasc. Surg. Venous Lymphat. Disord. 2018, 6, 552–553. [Google Scholar] [CrossRef]
- Echeverría, C.; Montorfano, I.; Hermosilla, T.; Armisén, R.; Velásquez, L.A.; Cabello-Verrugio, C.; Varela, D.; Simon, F. Endotoxin Induces Fibrosis in Vascular Endothelial Cells through a Mechanism Dependent on Transient Receptor Protein Melastatin 7 Activity. PLoS ONE 2014, 9, e94146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deswal, A.; Petersen, N.J.; Feldman, A.M.; Young, J.B.; White, B.G.; Mann, D.L. Cytokines and Cytokine Receptors in Advanced Heart Failure. Circulation 2001, 103, 2055–2059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cines, D.B.; Pollak, E.S.; Buck, C.A.; Loscalzo, J.; Zimmerman, G.A.; McEver, R.P.; Pober, J.S.; Wick, T.M.; Konkle, B.A.; Schwartz, B.S.; et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood 1998, 91, 3527–3561. [Google Scholar] [PubMed]
- Fioranelli, M.; Bottaccioli, A.G.; Bottaccioli, F.; Bianchi, M.; Rovesti, M.; Roccia, M.G. Stress and Inflammation in Coronary Artery Disease: A Review Psychoneuroendocrineimmunology-Based. Front. Immunol. 2018, 9, 2031. [Google Scholar] [CrossRef] [PubMed]
- Wheway, J.; Latham, S.L.; Combes, V.; Grau, G.E.R. Endothelial Microparticles Interact with and Support the Proliferation of T Cells. J. Immunol. 2014, 193, 3378–3387. [Google Scholar] [CrossRef] [Green Version]
- Allen, R.C.; Stephens, J.T. Myeloperoxidase Selectively Binds and Selectively Kills Microbes. Infect. Immun. 2011, 79, 474–485. [Google Scholar] [CrossRef] [Green Version]
- Case, A. On the Origin of Superoxide Dismutase: An Evolutionary Perspective of Superoxide-Mediated Redox Signaling. Antioxidants 2017, 6, 82. [Google Scholar] [CrossRef] [Green Version]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free Radicals: Properties, Sources, Targets, and Their Implication in Various Diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Bielski, B.H.J.; Cabelli, D.E. Superoxide and Hydroxyl Radical Chemistry in Aqueous Solution. In Active Oxygen in Chemistry; Springer: Dordrecht, The Netherlands, 1995; pp. 66–104. [Google Scholar]
- Li, Y.; Zhu, H.; Kuppusamy, P.; Zweier, J.; Trush, M. Mitochondrial Electron Transport Chain-Derived Superoxide Exits Macrophages: Implications for Mononuclear Cell-Mediated Pathophysiological Processes. React. Oxyg. Species 2016, 1, 81–98. [Google Scholar] [CrossRef] [Green Version]
- Babior, B.M. NADPH oxidase. Curr. Opin. Immunol. 2004, 16, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G.P.; Schjoerring, J.K.; Jahn, T.P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta Bba Biomembr. 2006, 1758, 994–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, M.T.; Gauss, K.A. Structure and regulation of the neutrophil respiratory burst oxidase: Comparison with nonphagocyte oxidases. J. Leukoc. Biol. 2004, 76, 760–781. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.D.; Hsu, A.T.; Ellson, C.D.; Y. Miyazawa, B.; Kong, Y.-W.; Greenwood, J.D.; Dhara, S.; Neal, M.D.; Sperry, J.L.; Park, M.S.; et al. Blood clotting and traumatic injury with shock mediates complement-dependent neutrophil priming for extracellular ROS, ROS-dependent organ injury and coagulopathy. Clin. Exp. Immunol. 2018, 194, 103–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, W.N.; Deng, J.; Ruan, X.Z.; Xu, Q. Reactive Oxygen Species Generation and Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Galley, J.C.; Straub, A.C. Redox Control of Vascular Function. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 178–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldosari, S.; Awad, M.; Harrington, E.; Sellke, F.; Abid, M. Subcellular Reactive Oxygen Species (ROS) in Cardiovascular Pathophysiology. Antioxidants 2018, 7, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreml, S.; Szeimies, R.M.; Prantl, L.; Karrer, S.; Landthaler, M.; Babilas, P. Oxygen in acute and chronic wound healing. Br. J. Dermatol. 2010, 163, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Arvieux, J.; Regnault, V.; Hachulla, E.; Darnige, L.; Berthou, F.; Youinou, P. Oxidation of beta2-glycoprotein I (beta2GPI) by the hydroxyl radical alters phospholipid binding and modulates recognition by anti-beta2GPI autoantibodies. Arthritis Res. Ther. 2001, 3, P082. [Google Scholar] [CrossRef] [Green Version]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Lambeth, J.D. Nox enzymes, ROS, and chronic disease: An example of antagonistic pleiotropy. Free Radic. Biol. Med. 2007, 43, 332–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassègue, B.; Griendling, K.K. NADPH Oxidases: Functions and Pathologies in the Vasculature. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular Mechanisms of Angiotensin II–Mediated Mitochondrial Dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Haigh, S.; Barman, S.; Fulton, D.J.R. From form to function: The role of Nox4 in the cardiovascular system. Front. Physiol. 2012, 3, 412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervantes Gracia, K.; Llanas-Cornejo, D.; Husi, H. CVD and Oxidative Stress. J. Clin. Med. 2017, 6, 22. [Google Scholar] [CrossRef] [Green Version]
- Weyemi, U.; Dupuy, C. The emerging role of ROS-generating NADPH oxidase NOX4 in DNA-damage responses. Mutat. Res. Rev. Mutat. Res. 2012, 751, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-M.; Kim, S.-J.; Tatsunami, R.; Yamamura, H.; Fukai, T.; Ushio-Fukai, M. ROS-induced ROS release orchestrated by Nox4, Nox2, and mitochondria in VEGF signaling and angiogenesis. Am. J. Physiol. Cell Physiol. 2017, 312, 749–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini, R.; Singh, S. Inducible nitric oxide synthase: An asset to neutrophils. J. Leukoc. Biol. 2019, 105, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.K.; Joshi, M.B.; Philippova, M.; Erne, P.; Hasler, P.; Hahn, S.; Resink, T.J. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 2010, 584, 3193–3197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikshit, M.; Kumari, R.; Srimal, R.C. Pulmonary thromboembolism-induced alterations in nitric oxide release from rat circulating neutrophils. J. Pharmacol. Exp. Ther. 1993, 265, 1369–1373. [Google Scholar] [PubMed]
- Mitsuke, Y.; Lee, J.-D.; Shimizu, H.; Uzui, H.; Iwasaki, H.; Ueda, T. Nitric oxide synthase activity in peripheral polymorphonuclear leukocytes in patients with chronic congestive heart failure. Am. J. Cardiol. 2001, 87, 183–187. [Google Scholar] [CrossRef]
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front. Cell. Infect. Microbiol. 2017, 7, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, T.; Möller, S.; Klinger, M.; Solbach, W.; Laskay, T.; Behnen, M. The Impact of Various Reactive Oxygen Species on the Formation of Neutrophil Extracellular Traps. Mediat. Inflamm. 2012, 2012, 849136. [Google Scholar] [CrossRef] [PubMed]
- Paiva, C.N.; Bozza, M.T. Are Reactive Oxygen Species Always Detrimental to Pathogens? Antioxid. Redox Signal. 2014, 20, 1000–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Benna, J.; Hurtado-Nedelec, M.; Marzaioli, V.; Marie, J.-C.; Gougerot-Pocidalo, M.-A.; Dang, P.M.-C. Priming of the neutrophil respiratory burst: Role in host defense and inflammation. Immunol. Rev. 2016, 273, 180–193. [Google Scholar] [CrossRef]
- Sheshachalam, A.; Srivastava, N.; Mitchell, T.; Lacy, P.; Eitzen, G. Granule Protein Processing and Regulated Secretion in Neutrophils. Front. Immunol. 2014, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Lassègue, B.; Sorescu, D.; Szöcs, K.; Yin, Q.Q.; Akers, M.; Zhang, Y.; Grant, S.L.; Lambeth, J.D.; Griendling, K.K. Novel gp91phox homologues in vascular smooth muscle cells: Nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ. Res. 2001, 88, 888–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- San José, G.; Bidegain, J.; Robador, P.A.; Díez, J.; Fortuño, A.; Zalba, G. Insulin-induced NADPH oxidase activation promotes proliferation and matrix metalloproteinase activation in monocytes/macrophages. Free Radic. Biol. Med. 2009, 46, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Xue, C.; Sowden, M.P.; Berk, B.C. Extracellular and Intracellular Cyclophilin A, Native and Post-Translationally Modified, Show Diverse and Specific Pathological Roles in Diseases. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 986–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C. The role of inflammatory cytokines in endothelial dysfunction. Basic Res. Cardiol. 2008, 103, 398–406. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Hu, X.; Wang, M. Pterostilbene inhibited advanced glycation end products (AGEs)-induced oxidative stress and inflammation by regulation of RAGE/MAPK/NF-κB in RAW264.7 cells. J. Funct. Foods 2018, 40, 272–279. [Google Scholar] [CrossRef]
- Duque, G.A.; Descoteaux, A. Macrophage Cytokines: Involvement in Immunity and Infectious Diseases. Front. Immunol. 2014, 5, 1–14. [Google Scholar]
- Alvarez, B.; Radi, R. Peroxynitrite reactivity with amino acids and proteins. Amino Acids 2003, 25, 295–311. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, N.M. Toxicity of nitrogen dioxide: An introduction. Toxicology 1994, 89, 161–174. [Google Scholar] [CrossRef]
- Persinger, R.L.; Poynter, M.E.; Ckless, K.; Janssen-Heininger, Y.M.W. Molecular mechanisms of nitrogen dioxide induced epithelial injury in the lung. Mol. Cell. Biochem. 2002, 234, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Gray, B.; Carmichael, A.J. Kinetics of superoxide scavenging by dismutase enzymes and manganese mimics determined by electron spin resonance. Biochem. J. 1992, 281, 795–802. [Google Scholar] [CrossRef]
- Crow, J.P.; Beckman, J.S. Reactions between Nitric Oxide, Superoxide, and Peroxynitrite: Footprints of Peroxynitrite in Vivo. Adv. Pharmacol. 1995, 34, 17–43. [Google Scholar]
- Nauser, T.; Koppenol, W.H. The Rate Constant of the Reaction of Superoxide with Nitrogen Monoxide: Approaching the Diffusion Limit. J. Phys. Chem. A 2002, 106, 4084–4086. [Google Scholar] [CrossRef]
- Münzel, T.; Sinning, C.; Post, F.; Warnholtz, A.; Schulz, E. Pathophysiology, diagnosis and prognostic implications of endothelial dysfunction. Ann. Med. 2008, 40, 180–196. [Google Scholar] [CrossRef] [PubMed]
- Gow, A.J.; Luchsinger, B.P.; Pawloski, J.R.; Singel, D.J.; Stamler, J.S. The oxyhemoglobin reaction of nitric oxide. Proc. Natl. Acad. Sci. USA 1999, 96, 9027–9032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auten, R.L.; Davis, J.M. Oxygen Toxicity and Reactive Oxygen Species: The Devil Is in the Details. Pediatric Res. 2009, 66, 121–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brzeszczynska, J.; Gwozdzinski, K. Nitric oxide induced oxidative changes in erythrocyte membrane components. Cell Biol. Int. 2008, 32, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Ansari, S.A.; Pendurthi, U.R.; Rao, L.V.M. The lipid peroxidation product 4-hydroxy-2-nonenal induces tissue factor decryption via ROS generation and the thioredoxin system. Blood Adv. 2017, 1, 2399–2413. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, C.; Siow, R.; Gwozdz, A.M.; Saha, P.; Smith, A. Reactive Oxygen Species in Venous Thrombosis. Int. J. Mol. Sci. 2020, 21, 1918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golino, P.; Ragni, M.; Cirillo, P.; Avvdimento, V.E.; Feliciello, A.; Esposito, N.; Scognamiglio, A.; Trimarco, B.; Iaccarino, G.; Condorelli, M.; et al. Effects of tissue factor induced by oxygen free radicals on coronary flow during reperfusion. Nat. Med. 1996, 2, 35–40. [Google Scholar] [CrossRef]
- Cadroy, Y.; Dupouy, D.; Boneu, B.; Plaisancié, H. Polymorphonuclear Leukocytes Modulate Tissue Factor Production by Mononuclear Cells: Role of Reactive Oxygen Species. J. Immunol. 2000, 164, 3822–3828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herkert, O.; Diebold, I.; Brandes, R.P.; Hess, J.; Busse, R.; Görlach, A. NADPH Oxidase Mediates Tissue Factor—Dependent Surface Procoagulant Activity by Thrombin in Human Vascular Smooth Muscle Cells. Circulation 2002, 105, 2030–2036. [Google Scholar] [CrossRef] [Green Version]
- Mackman, N. Triggers, targets and treatments for thrombosis. Nature 2008, 451, 914–918. [Google Scholar] [CrossRef]
- Lichota, A.; Gwozdzinski, L.; Gwozdzinski, K. Therapeutic potential of natural compounds in inflammation and chronic venous insufficiency. Eur. J. Med. Chem. 2019, 176, 68–91. [Google Scholar] [CrossRef]
- Kovacs, M.J.; Kahn, S.R.; Wells, P.S.; Anderson, D.A.; Chagnon, I.; Le Gal, G.; Solymoss, S.; Crowther, M.; Perrier, A.; Ramsay, T.; et al. Patients with a first symptomatic unprovoked deep vein thrombosis are at higher risk of recurrent venous thromboembolism than patients with a first unprovoked pulmonary embolism. J. Thromb. Haemost. 2010, 8, 1926–1932. [Google Scholar] [CrossRef] [PubMed]
- Goktay, A.Y.; Senturk, C. Endovascular Treatment of Thrombosis and Embolism. Adv. Exp. Med. Biol. 2017, 906, 195–213. [Google Scholar] [PubMed]
- Shatila, W.; Almanfi, A.; Massumi, M.; Dougherty, K.G.; Parekh, D.R.; Strickman, N.E. Endovascular Treatment of Superior Vena Cava Syndrome via Balloon-in-Balloon Catheter Technique with a Palmaz Stent. Tex. Heart Inst. J. 2016, 43, 520–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ringleb, P.A. Thrombolytics, Anticoagulants, and Antiplatelet Agents. Stroke 2006, 37, 312–313. [Google Scholar] [CrossRef] [Green Version]
- Sen, C.K.; Roy, S. Wound healing. In Plastic Surgery; Rodriguez, E., Losee, J.E., Neligan, P.C., Eds.; Saunders: Philadelphia, PA, USA; pp. 240–266. ISBN 978-14-557-4047-5. 2012. [Google Scholar]
- Kearon, C.; Akl, E.A.; Ornelas, J.; Blaivas, A.; Jimenez, D.; Bounameaux, H.; Huisman, M.; King, C.S.; Morris, T.A.; Sood, N.; et al. Antithrombotic Therapy for VTE Disease. Chest 2016, 149, 315–352. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Lee, H.; Kim, M.-A.; Choi, J.; Kim, K.-M.; Hwang, J.S.; Na, M.; Bae, J.-S. Evaluation of novel factor Xa inhibitors from Oxya chinensis sinuosa with anti-platelet aggregation activity. Sci. Rep. 2017, 7, 7934. [Google Scholar] [CrossRef] [Green Version]
- Fischer, P.M. Design of Small-Molecule Active-Site Inhibitors of the S1A Family Proteases as Procoagulant and Anticoagulant Drugs. J. Med. Chem. 2018, 61, 3799–3822. [Google Scholar] [CrossRef]
- Zacconi, F.C. FXa Direct Synthetic Inhibitors. In Anticoagulant Drugs; InTech: London, UK, 2018. [Google Scholar]
- Byon, W.; Garonzik, S.; Boyd, R.A.; Frost, C.E. Apixaban: A Clinical Pharmacokinetic and Pharmacodynamic Review. Clin. Pharmacokinet. 2019, 58, 1265–1279. [Google Scholar] [CrossRef] [Green Version]
- Bratsos, S. Pharmacokinetic Properties of Rivaroxaban in Healthy Human Subjects. Cureus 2019, 11, e5484. [Google Scholar] [CrossRef] [Green Version]
- Rao, P.S.S.; Burkart, T. Advances in oral anticoagulation therapy—What’s in the pipeline? Blood Rev. 2017, 31, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Outes, A.; Terleira-Fernández, A.I.; Calvo-Rojas, G.; Suárez-Gea, M.L.; Vargas-Castrillón, E. Dabigatran, Rivaroxaban, or Apixaban versus Warfarin in Patients with Nonvalvular Atrial Fibrillation: A Systematic Review and Meta-Analysis of Subgroups. Thrombosis 2013, 2013, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Bushnell, C.; McCullough, L.D.; Awad, I.A.; Chireau, M.V.; Fedder, W.N.; Furie, K.L.; Howard, V.J.; Lichtman, J.H.; Lisabeth, L.D.; Piña, I.L.; et al. Guidelines for the Prevention of Stroke in Women. Stroke 2014, 45, 1545–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yee, J.; Kaide, C. Emergency Reversal of Anticoagulation. West. J. Emerg. Med. 2019, 20, 770–783. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Meng, J.; Guo, T.; Zhao, J.; Wang, Y.; Wang, J.; Qiu, Y.; Ding, H. Comparison of Apixaban and Low Molecular Weight Heparin in Preventing Deep Venous Thrombosis after Total Knee Arthroplasty in Older Adults. Yonsei Med. J. 2019, 60, 626. [Google Scholar] [CrossRef] [PubMed]
- Skelley, J.W.; Thomason, A.R.; Nolen, J.C. Betrixaban (Bevyxxa): A Direct-Acting Oral Anticoagulant Factor Xa Inhibitor. Pharm. Ther. 2018, 43, 85–88. [Google Scholar]
- Iwatsuki, Y.; Sato, T.; Moritani, Y.; Shigenaga, T.; Suzuki, M.; Kawasaki, T.; Funatsu, T.; Kaku, S. Biochemical and pharmacological profile of darexaban, an oral direct factor Xa inhibitor. Eur. J. Pharmacol. 2011, 673, 49–55. [Google Scholar] [CrossRef]
- Shiraga, T.; Yajima, K.; Suzuki, K.; Suzuki, K.; Hashimoto, T.; Iwatsubo, T.; Miyashita, A.; Usui, T. Identification of UDP-Glucuronosyltransferases Responsible for the Glucuronidation of Darexaban, an Oral Factor Xa Inhibitor, in Human Liver and Intestine. Drug Metab. Dispos. 2012, 40, 276–282. [Google Scholar] [CrossRef] [Green Version]
- Hirayama, F.; Koshio, H.; Ishihara, T.; Hachiya, S.; Sugasawa, K.; Koga, Y.; Seki, N.; Shiraki, R.; Shigenaga, T.; Iwatsuki, Y.; et al. Discovery of N-[2-Hydroxy-6-(4-methoxybenzamido)phenyl]-4- (4-methyl-1,4-diazepan-1-yl)benzamide (Darexaban, YM150) as a Potent and Orally Available Factor Xa Inhibitor. J. Med. Chem. 2011, 54, 8051–8065. [Google Scholar] [CrossRef]
- Clayville, L.R.; Vogel Anderson, K.; Miller, S.A.; Onge, E.L.S. New Options in Anticoagulation for the Prevention Of Venous Thromboembolism and Stroke. PT 2011, 36, 86–99. [Google Scholar]
- Shirley, M.; Dhillon, S. Edoxaban: A Review in Deep Vein Thrombosis and Pulmonary Embolism. Drugs 2015, 75, 2025–2034. [Google Scholar] [CrossRef]
- Brown, D.G.; Wilkerson, E.C.; Love, W.E. A review of traditional and novel oral anticoagulant and antiplatelet therapy for dermatologists and dermatologic surgeons. J. Am. Acad. Dermatol. 2015, 72, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Székely, O.; Miyazawa, K.; Lip, G.Y.H. Current and emerging pharmacotherapy for ischemic stroke prevention in patients with atrial fibrillation. Expert Opin. Pharmacother. 2018, 19, 1999–2009. [Google Scholar] [CrossRef]
- Feng, W.; Wu, K.; Liu, Z.; Kong, G.; Deng, Z.; Chen, S.; Wu, Y.; Chen, M.; Liu, S.; Wang, H. Oral direct factor Xa inhibitor versus enoxaparin for thromboprophylaxis after hip or knee arthroplasty: Systemic review, traditional meta-analysis, dose–response meta-analysis and network meta-analysis. Thromb. Res. 2015, 136, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.M.; Weitz, J.I. New anticoagulants: Beyond heparin, low-molecular-weight heparin and warfarin. Br. J. Pharmacol. 2005, 144, 1017–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamikubo, Y.; Mendolicchio, G.L.; Zampolli, A.; Marchese, P.; Rothmeier, A.S.; Orje, J.N.; Gale, A.J.; Krishnaswamy, S.; Gruber, A.; Østergaard, H.; et al. Selective factor VIII activation by the tissue factor–factor VIIa–factor Xa complex. Blood 2017, 130, 1661–1670. [Google Scholar] [CrossRef] [PubMed]
- Markwardt, F. Hirudin As Alternative Anticoagulant- A Historical Review. Semin. Thromb. Hemost. 2002, 28, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Ren, J.-X.; Wang, J.-J.; Ding, L.-S.; Zhao, J.-J.; Liu, S.-Y.; Gao, H.-M. Chinese Medicinal Leech: Ethnopharmacology, Phytochemistry, and Pharmacological Activities. Evid. Based Complementary Altern. Med. 2016, 2016, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Li, Z.; Li, J.; Liu, L. Effects of Natural Hirudin and Low Molecular Weight Heparin in Preventing Deep Venous Thrombosis in Aged Patients with Intertrochanteric Fracture. Sci. Rep. 2018, 8, 8847. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.L. Bivalirudin as an Alternative to Heparin for Anticoagulation in Infants and Children. J. Pediatric Pharmacol. Ther. 2015, 20, 408–417. [Google Scholar] [CrossRef] [Green Version]
- Kimmelstiel, C.; Zhang, P.; Kapur, N.K.; Weintraub, A.; Krishnamurthy, B.; Castaneda, V.; Covic, L.; Kuliopulos, A. Bivalirudin Is a Dual Inhibitor of Thrombin and Collagen-Dependent Platelet Activation in Patients Undergoing Percutaneous Coronary Intervention. Circ. Cardiovasc. Interv. 2011, 4, 171–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qureshi, W.; Ali, Z.; Amjad, W.; Alirhayim, Z.; Farooq, H.; Qadir, S.; Khalid, F.; Al-Mallah, M.H. Venous Thromboembolism in Cancer: An Update of Treatment and Prevention in the Era of Newer Anticoagulants. Front. Cardiovasc. Med. 2016, 3, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, A.W.; Schiering, N.; Melkko, S.; Ewert, S.; Salter, J.; Zhang, Y.; McCormack, P.; Yu, J.; Huang, X.; Chiu, Y.-H.; et al. MAA868, a novel FXI antibody with a unique binding mode, shows durable effects on markers of anticoagulation in humans. Blood 2019, 133, 1507–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Minno, A.; Frigerio, B.; Spadarella, G.; Ravani, A.; Sansaro, D.; Amato, M.; Kitzmiller, J.P.; Pepi, M.; Tremoli, E.; Baldassarre, D. Old and new oral anticoagulants: Food, herbal medicines and drug interactions. Blood Rev. 2017, 31, 193–203. [Google Scholar] [CrossRef]
- Samuelson, B.T.; Cuker, A. Measurement and reversal of the direct oral anticoagulants. Blood Rev. 2017, 31, 77–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesarman-Maus, G.; Ruiz-Argüelles, G.J. News in the Indications of Direct Oral Anticoagulants According to the American College of Chest Physicians 2016 Guidelines. Curr. Drug Metab. 2017, 18, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Ansell, J.E.; Bakhru, S.H.; Laulicht, B.E.; Steiner, S.S.; Grosso, M.A.; Brown, K.; Dishy, V.; Lanz, H.J.; Mercuri, M.F.; Noveck, R.J.; et al. Single-dose ciraparantag safely and completely reverses anticoagulant effects of edoxaban. Thromb. Haemost. 2017, 117, 238–245. [Google Scholar] [CrossRef]
- Okumura, K.; Lip, G.Y.H.; Akao, M.; Tanizawa, K.; Fukuzawa, M.; Abe, K.; Akishita, M.; Yamashita, T. Edoxaban for the management of elderly Japanese patients with atrial fibrillation ineligible for standard oral anticoagulant therapies: Rationale and design of the ELDERCARE-AF study. Am. Heart J. 2017, 194, 99–106. [Google Scholar] [CrossRef]
- Thomson, P.; Brocklebank, C.; Semchuk, W. Treatment Dosing of Low-Molecular-Weight Heparins and the Dose Cap Dilemma: Considerations for Patients in Canada. Can. J. Hosp. Pharm. 2009, 62, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Nadar, S.K.; Goyal, D.; Shantsila, E.; Banerjee, P.; Lip, G.Y. Fondaparinux: An overview. Expert Rev. Cardiovasc. Ther. 2009, 7, 577–585. [Google Scholar] [CrossRef]
- Eriksson, B.; Agnelli, G.; Gallus, A.; Lassen, M.; Prins, M.; Renfurm, R.; Kashiwa, M.; Turpie, A. Darexaban (YM150) versus enoxaparin for the prevention of venous thromboembolism after total hip arthroplasty: A randomised phase IIb dose confirmation study (ONYX-3). Thromb. Haemost. 2014, 111, 213–225. [Google Scholar] [PubMed]
- Dimitropoulos, G.; Rahim, S.M.Z.; Moss, A.S.; Lip, G.Y.H. New anticoagulants for venous thromboembolism and atrial fibrillation: What the future holds. Expert Opin. Investig. Drugs 2018, 27, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Choi, S.; Kim, Y.; Lim, H. Population Pharmacokinetic and Pharmacodynamic Modeling Analysis of GCC-4401C, a Novel Direct Factor Xa Inhibitor, in Healthy Volunteers. CPT Pharmacomet. Syst. Pharmacol. 2016, 5, 532–543. [Google Scholar] [CrossRef] [PubMed]
- Roffel, A.; van Marle, S.; Choi, S.; Lee, H.; Dueker, S.; Tiessen, R. Metabolism and excretion of GCC-4401C, a factor Xa inhibitor, in humans. Int. J. Pharmacokinet. 2017, 2, 93–104. [Google Scholar] [CrossRef] [Green Version]
- Smythe, M.A.; Priziola, J.; Dobesh, P.P.; Wirth, D.; Cuker, A.; Wittkowsky, A.K. Guidance for the practical management of the heparin anticoagulants in the treatment of venous thromboembolism. J. Thromb. Thrombolysis 2016, 41, 165–186. [Google Scholar] [CrossRef] [Green Version]
- Lippi, G.; Gosselin, R.; Favaloro, E.J. Current and Emerging Direct Oral Anticoagulants: State-of-the-Art. Semin. Thromb. Hemost. 2019, 45, 490–501. [Google Scholar] [CrossRef]
- Capodanno, D.; Ferreiro, J.L.; Angiolillo, D.J. Antiplatelet therapy: New pharmacological agents and changing paradigms. J. Thromb. Haemost. 2013, 11, 316–329. [Google Scholar] [CrossRef] [Green Version]
- McFadyen, J.D.; Schaff, M.; Peter, K. Current and future antiplatelet therapies: Emphasis on preserving haemostasis. Nat. Rev. Cardiol. 2018, 15, 181–191. [Google Scholar] [CrossRef]
- Harter, K.; Levine, M.; Henderson, S. Anticoagulation Drug Therapy: A Review. West. J. Emerg. Med. 2015, 16, 11–17. [Google Scholar] [CrossRef]
- Rubboli, A.; Patti, G. What is the Role for Glycoprotein IIB/IIIA Inhibitor Use in the Catheterization Laboratory in the Current Era? Curr. Vasc. Pharmacol. 2018, 16, 451–458. [Google Scholar] [CrossRef]
- Eisert, W.G. Dipyridamole in antithrombotic treatment. Adv. Interv. Cardiol. 2012, 47, 78–86. [Google Scholar]
- Kim, H.-H.; Liao, J.K. Translational Therapeutics of Dipyridamole. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 39–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, S. Cilostazol: Potential mechanism of action for antithrombotic effects accompanied by a low rate of bleeding. Atheroscler. Suppl. 2005, 6, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Niu, P.-P.; Guo, Z.-N.; Jin, H.; Xing, Y.-Q.; Yang, Y. Antiplatelet regimens in the long-term secondary prevention of transient ischaemic attack and ischaemic stroke: An updated network meta-analysis. BMJ Open 2016, 6, e009013. [Google Scholar] [CrossRef]
- Steinhubl, S.R.; Badimon, J.J.; Bhatt, D.L.; Herbert, J.-M.; Lüscher, T.F. Clinical evidence for anti-inflammatory effects of antiplatelet therapy in patients with atherothrombotic disease. Vasc. Med. 2007, 12, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Quinn, M.J.; Fitzgerald, D.J. Ticlopidine and Clopidogrel. Circulation 1999, 100, 1667–1672. [Google Scholar] [CrossRef]
- Jiang, M.; You, J.H. Review of pharmacoeconomic evaluation of genotype-guided antiplatelet therapy. Expert Opin. Pharmacother. 2015, 16, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Majithia, A.; Bhatt, D.L. Novel Antiplatelet Therapies for Atherothrombotic Diseases. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 546–557. [Google Scholar] [CrossRef] [Green Version]
- Tscharre, M.; Michelson, A.D.; Gremmel, T. Novel Antiplatelet Agents in Cardiovascular Disease. J. Cardiovasc. Pharmacol. Ther. 2020, 25, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.; Crain, E.; Watson, C.; Yang, W.; Wexler, R.; Lam, P.; Rehfuss, R.; Schumacher, W. Differential effects of P2Y1 versus P2Y12 receptor antagonism on thrombosis and bleeding in rabbits. Eur. Heart J. 2013, 34, P1431. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Wang, Y.; Lai, A.; Qiao, J.X.; Wang, T.C.; Hua, J.; Price, L.A.; Shen, H.; Chen, X.; Wong, P.; et al. Discovery of 4-Aryl-7-Hydroxyindoline-Based P2Y 1 Antagonists as Novel Antiplatelet Agents. J. Med. Chem. 2014, 57, 6150–6164. [Google Scholar] [CrossRef] [PubMed]
- Kolandaivelu, K.; Bhatt, D.L. Novel Antiplatelet Therapies. In Platelets; Elsevier: Amsterdam, The Netherland, 2019; pp. 991–1015. [Google Scholar]
- Bach, P.; Antonsson, T.; Bylund, R.; Björkman, J.-A.; Österlund, K.; Giordanetto, F.; van Giezen, J.J.J.; Andersen, S.M.; Zachrisson, H.; Zetterberg, F. Lead Optimization of Ethyl 6-Aminonicotinate Acyl Sulfonamides as Antagonists of the P2Y 12 Receptor. Separation of the Antithrombotic Effect and Bleeding for Candidate Drug AZD1283. J. Med. Chem. 2013, 56, 7015–7024. [Google Scholar] [CrossRef] [PubMed]
- Hashemzadeh, M.; Furukawa, M.; Goldsberry, S.; Movahed, M.R. Chemical structures and mode of action of intravenous glycoprotein IIb/IIIa receptor blockers: A review. Exp. Clin. Cardiol. 2008, 13, 192–197. [Google Scholar]
- Stoffer, K.; Shah, S. Abciximab; StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Bansal, A.B.; Sattar, Y.; Jamil, R.T. Eptifibatide. Drugs 1999, 57, 439–462. [Google Scholar]
- Zhao, C.; Cheng, G.; He, R.; Guo, H.; Li, Y.; Lu, X.; Zhang, Y.; Qiu, C. Effects of different routes of tirofiban injection on the left ventricular function and prognosis of patients with myocardial infarction treated with percutaneous coronary intervention. Exp. Ther. Med. 2015, 9, 2401–2405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beavers, C.J.; Naqvi, I.A. Clopidogrel. Drugs 1997, 54, 745–750. [Google Scholar]
- Hermanides, R.S.; Kilic, S.; van ’t Hof, A.W.J. Optimal pharmacological therapy in ST-elevation myocardial infarction—a review. Neth. Heart J. 2018, 26, 296–310. [Google Scholar] [CrossRef] [Green Version]
- Layne, K.; Ferro, A. Antiplatelet Therapy in Acute Coronary Syndrome. Eur. Cardiol. Rev. 2017, 12, 33. [Google Scholar] [CrossRef]
- Balinski, A.M.; Preuss, C.V. Cilostazol. Drugs Aging 1999, 14, 63–71. [Google Scholar]
- Royston, D. Anticoagulant and Antiplatelet Therapy. In Pharmacology and Physiology for Anesthesia; Elsevier: Amsterdam, The Netherlands, 2019; pp. 870–894. [Google Scholar]
- Desager, J.-P. Clinical Pharmacokinetics of Ticlopidine. Clin. Pharmacokinet. 1994, 26, 347–355. [Google Scholar] [CrossRef]
- Juif, P.; Boehler, M.; Dobrow, M.; Ufer, M.; Dingemanse, J. Clinical Pharmacology of the Reversible and Potent P2Y 12 Receptor Antagonist ACT-246475 After Single Subcutaneous Administration in Healthy Male Subjects. J. Clin. Pharmacol. 2019, 59, 123–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storey, R.F.; Gurbel, P.A.; ten Berg, J.; Bernaud, C.; Dangas, G.D.; Frenoux, J.-M.; Gorog, D.A.; Hmissi, A.; Kunadian, V.; James, S.K.; et al. Pharmacodynamics, pharmacokinetics, and safety of single-dose subcutaneous administration of selatogrel, a novel P2Y12 receptor antagonist, in patients with chronic coronary syndromes. Eur. Heart J. 2020, 41, 3132–3140. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Choi, W.-S.; McCoy, J.G.; Negri, A.; Zhu, J.; Naini, S.; Li, J.; Shen, M.; Huang, W.; Bougie, D.; et al. Structure-Guided Design of a High-Affinity Platelet Integrin IIb 3 Receptor Antagonist That Disrupts Mg2+ Binding to the MIDAS. Sci. Transl. Med. 2012, 4, 125ra32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Vootukuri, S.; Shang, Y.; Negri, A.; Jiang, J.; Nedelman, M.; Diacovo, T.G.; Filizola, M.; Thomas, C.J.; Coller, B.S. RUC-4. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2321–2329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vootukuri, S.; Li, J.; Nedelman, M.; Thomas, C.; Jiang, J.-K.; Babayeva, M.; Coller, B.S. Preclinical studies of RUC-4, a novel platelet αIIbβ3 antagonist, in non-human primates and with human platelets. J. Clin. Transl. Sci. 2019, 3, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Heuberger, D.M.; Schuepbach, R.A. Protease-activated receptors (PARs): Mechanisms of action and potential therapeutic modulators in PAR-driven inflammatory diseases. Thromb. J. 2019, 17, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gryka, R.J.; Buckley, L.F.; Anderson, S.M. Vorapaxar: The Current Role and Future Directions of a Novel Protease-Activated Receptor Antagonist for Risk Reduction in Atherosclerotic Disease. Drugs Rd 2017, 17, 65–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, S.J.; Ismat, F.A.; Wang, Z.; Cerra, M.; Narayan, H.; Raftis, J.; Gray, T.J.; Connell, S.; Garonzik, S.; Ma, X.; et al. PAR4 (Protease-Activated Receptor 4) Antagonism With BMS-986120 Inhibits Human Ex Vivo Thrombus Formation. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 448–456. [Google Scholar] [CrossRef] [Green Version]
- Dumas, M.; Nadal-Wollbold, F.; Gaussem, P.; Perez, M.; Mirault, T.; Létienne, R.; Bourbon, T.; Grelac, F.; Le Grand, B.; Bachelot-Loza, C. Antiplatelet and antithrombotic effect of F 16618, a new thrombin proteinase-activated receptor-1 (PAR1) antagonist. Br. J. Pharmacol. 2012, 165, 1827–1835. [Google Scholar] [CrossRef] [Green Version]
- Chieng-Yane, P.; Bocquet, A.; Létienne, R.; Bourbon, T.; Sablayrolles, S.; Perez, M.; Hatem, S.N.; Lompré, A.-M.; Le Grand, B.; David-Dufilho, M. Protease-Activated Receptor-1 Antagonist F 16618 Reduces Arterial Restenosis by Down-Regulation of Tumor Necrosis Factor α and Matrix Metalloproteinase 7 Expression, Migration, and Proliferation of Vascular Smooth Muscle Cells. J. Pharmacol. Exp. Ther. 2011, 336, 643–651. [Google Scholar] [CrossRef]
- Di Serio, C.; Pellerito, S.; Duarte, M.; Massi, D.; Naldini, A.; Cirino, G.; Prudovsky, I.; Santucci, M.; Geppetti, P.; Marchionni, N.; et al. Protease-Activated Receptor 1-Selective Antagonist SCH79797 Inhibits Cell Proliferation and Induces Apoptosis by a Protease-Activated Receptor 1-Independent Mechanism. Basic Clin. Pharmacol. Toxicol. 2007, 101, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, K.; Ohnishi, T.; Miura, N.; Sameshima, H.; Koide, T.; Tanaka, K.A.; Maruyama, I. Antithrombotic effects of PAR1 and PAR4 antagonists evaluated under flow and static conditions. Thromb. Res. 2014, 133, 66–72. [Google Scholar] [CrossRef]
- Chen, Z.; Li, T.; Kareem, K.; Tran, D.; Griffith, B.P.; Wu, Z.J. The role of PI3K/Akt signaling pathway in non-physiological shear stress-induced platelet activation. Artif. Organs 2019, 43, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Sturgeon, S.A.; Jones, C.; Angus, J.A.; Wright, C.E. Advantages of a selective β-isoform phosphoinositide 3-kinase antagonist, an anti-thrombotic agent devoid of other cardiovascular actions in the rat. Eur. J. Pharmacol. 2008, 587, 209–215. [Google Scholar] [CrossRef]
- Laurent, P.-A.; Séverin, S.; Hechler, B.; Vanhaesebroeck, B.; Payrastre, B.; Gratacap, M.-P. Platelet PI3Kβ and GSK3 regulate thrombus stability at a high shear rate. Blood 2015, 125, 881–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durrant, T.N.; Hers, I. PI3K inhibitors in thrombosis and cardiovascular disease. Clin. Transl. Med. 2020, 9, 8. [Google Scholar] [CrossRef] [PubMed]
- Nylander, S.; Kull, B.; Björkman, J.A.; Ulvinge, J.C.; Oakes, N.; Emanuelsson, B.M.; Andersson, M.; Skärby, T.; Inghardt, T.; Fjellström, O. Human target validation of phosphoinositide 3-kinase (PI3K)β: Effects on platelets and insulin sensitivity, using AZD6482 a novel PI3Kβ inhibitor. J. Thromb. Haemost. 2012, 10, 2127–2136. [Google Scholar] [CrossRef]
- Xu, P.-F.; Yang, J.-A.; Liu, J.-H.; Yang, X.; Liao, J.-M.; Yuan, F.-E.; Liu, B.-H.; Chen, Q.-X. PI3Kβ inhibitor AZD6482 exerts antiproliferative activity and induces apoptosis in human glioblastoma cells. Oncol. Rep. 2019, 41, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Luci, D.; Jameson, J.B.; Yasgar, A.; Diaz, G.; Joshi, N.; Kantz, A.; Markham, K.; Perry, S.; Kuhn, N.; Yeung, J.; et al. Discovery of ML355, a Potent and Selective Inhibitor of Human 12-Lipoxygenase; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010. [Google Scholar]
- Adili, R.; Tourdot, B.E.; Mast, K.; Yeung, J.; Freedman, J.C.; Green, A.; Luci, D.K.; Jadhav, A.; Simeonov, A.; Maloney, D.J.; et al. First Selective 12-LOX Inhibitor, ML355, Impairs Thrombus Formation and Vessel Occlusion In Vivo With Minimal Effects on Hemostasis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1828–1839. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, J.C.; DeFeo-Fraulini, T.; Hutabarat, R.M.; Horvath, C.J.; Merlino, P.G.; Marsh, H.N.; Healy, J.M.; BouFakhreddine, S.; Holohan, T.V.; Schaub, R.G. First-in-Human Evaluation of Anti–von Willebrand Factor Therapeutic Aptamer ARC1779 in Healthy Volunteers. Circulation 2007, 116, 2678–2686. [Google Scholar] [CrossRef]
- Huang, R.-H.; Fremont, D.H.; Diener, J.L.; Schaub, R.G.; Sadler, J.E. A Structural Explanation for the Antithrombotic Activity of ARC1172, a DNA Aptamer that Binds von Willebrand Factor Domain A1. Structure 2009, 17, 1476–1484. [Google Scholar] [CrossRef] [Green Version]
- Spiel, A.O.; Mayr, F.B.; Ladani, N.; Wagner, P.G.; Schaub, R.G.; Gilbert, J.C.; Jilma, B. The aptamer ARC1779 is a potent and specific inhibitor of von willebrand factor mediated ex vivo platelet function in acute myocardial infarction. Platelets 2009, 20, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Jilma-Stohlawetz, P.; Gilbert, J.; Gorczyca, M.; Knöbl, P.; Jilma, B. A dose ranging phase I/II trial of the von Willebrand factor inhibiting aptamer ARC1779 in patients with congenital thrombotic thrombo—Cytopenic purpura. Thromb. Haemost. 2011, 106, 391–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kageyama, S.; Yamamoto, H.; Nagano, M.; Arisaka, H.; Kayahara, T.; Yoshimoto, R. Anti-thrombotic effects and bleeding risk of AJvW-2, a monoclonal antibody against human von Willebrand factor. Br. J. Pharmacol. 1997, 122, 165–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siller-Matula, J.M.; Merhi, Y.; Tanguay, J.-F.; Duerschmied, D.; Wagner, D.D.; McGinness, K.E.; Pendergrast, P.S.; Chung, J.-K.; Tian, X.; Schaub, R.G.; et al. ARC15105 Is a Potent Antagonist of Von Willebrand Factor Mediated Platelet Activation and Adhesion. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 902–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanlon, A.; Metjian, A. Caplacizumab in adult patients with acquired thrombotic thrombocytopenic purpura. Ther. Adv. Hematol. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Bartunek, J.; Barbato, E.; Heyndrickx, G.; Vanderheyden, M.; Wijns, W.; Holz, J.-B. Novel Antiplatelet Agents: ALX-0081, a Nanobody Directed towards von Willebrand Factor. J. Cardiovasc. Transl. Res. 2013, 6, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Mao, Y.; Abdelgawwad, M.S.; Kocher, N.K.; Li, M.; Dai, X.; Li, B.; Zheng, X.L. Therapeutic efficacy of the platelet glycoprotein Ib antagonist anfibatide in murine models of thrombotic thrombocytopenic purpura. Blood Adv. 2016, 1, 75–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, Y.; Huo, J.; Xu, W.; Xiong, J.; Li, Y.; Wu, W. A novel anti-platelet aggregation tripeptide from Agkistrodon acutus venom: Isolation and characterization. Toxicon 2009, 54, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Reheman, A.; Hou, Y.; Zhou, H.; Wang, Y.; Marshall, A.; Liang, C.; Dai, X.; Li, B.X.; Vanhoorelbeke, K.; et al. Anfibatide, a novel GPIb complex antagonist, inhibits platelet adhesion and thrombus formation in vitro and in vivo in murine models of thrombosis. Thromb. Haemost. 2014, 111, 279–289. [Google Scholar]
- Li, T.-T.; Fan, M.-L.; Hou, S.-X.; Li, X.-Y.; Barry, D.M.; Jin, H.; Luo, S.-Y.; Kong, F.; Lau, L.-F.; Dai, X.-R.; et al. A novel snake venom-derived GPIb antagonist, anfibatide, protects mice from acute experimental ischaemic stroke and reperfusion injury. Br. J. Pharmacol. 2015, 172, 3904–3916. [Google Scholar] [CrossRef] [PubMed]
- Voors-Pette, C.; Lebozec, K.; Dogterom, P.; Jullien, L.; Billiald, P.; Ferlan, P.; Renaud, L.; Favre-Bulle, O.; Avenard, G.; Machacek, M.; et al. Safety and Tolerability, Pharmacokinetics, and Pharmacodynamics of ACT017, an Antiplatelet GPVI (Glycoprotein VI) Fab. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, T.; Yoshida, H.; Satoh, K. Regulation of CX3CL1/Fractalkine Expression in Endothelial Cells. J. Atheroscler. Thromb. 2004, 11, 15–21. [Google Scholar] [CrossRef] [Green Version]
- Stolla, M.; Pelisek, J.; von Brühl, M.-L.; Schäfer, A.; Barocke, V.; Heider, P.; Lorenz, M.; Tirniceriu, A.; Steinhart, A.; Bauersachs, J.; et al. Fractalkine Is Expressed in Early and Advanced Atherosclerotic Lesions and Supports Monocyte Recruitment via CX3CR1. PLoS ONE 2012, 7, e43572. [Google Scholar] [CrossRef] [PubMed]
- Riopel, M.; Vassallo, M.; Ehinger, E.; Pattison, J.; Bowden, K.; Winkels, H.; Wilson, M.; de Jong, R.; Patel, S.; Balakrishna, D.; et al. CX3CL1-Fc treatment prevents atherosclerosis in Ldlr KO mice. Mol. Metab. 2019, 20, 89–101. [Google Scholar] [CrossRef]
- Gailani, D.; Gruber, A. Factor XI as a Therapeutic Target. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1316–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallisch, M.; Lorentz, C.U.; Lakshmanan, H.H.S.; Johnson, J.; Carris, M.R.; Puy, C.; Gailani, D.; Hinds, M.T.; McCarty, O.J.T.; Gruber, A.; et al. Antibody inhibition of contact factor XII reduces platelet deposition in a model of extracorporeal membrane oxygenator perfusion in nonhuman primates. Res. Pract. Thromb. Haemost. 2020, 4, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Büller, H.R.; Bethune, C.; Bhanot, S.; Gailani, D.; Monia, B.P.; Raskob, G.E.; Segers, A.; Verhamme, P.; Weitz, J.I. Factor XI Antisense Oligonucleotide for Prevention of Venous Thrombosis. N. Engl. J. Med. 2015, 372, 232–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, D.; Thelen, K.; Kraff, S.; Schwers, S.; Schiffer, S.; Unger, S.; Yassen, A.; Boxnick, S. BAY 1213790, a fully human IgG1 antibody targeting coagulation factor XIa: First evaluation of safety, pharmacodynamics, and pharmacokinetics. Res. Pract. Thromb. Haemost. 2019, 3, 242–253. [Google Scholar] [CrossRef] [Green Version]
- Woodruff, R.S.; Ivanov, I.; Verhamme, I.M.; Sun, M.-F.; Gailani, D.; Sullenger, B.A. Generation and characterization of aptamers targeting factor XIa. Thromb. Res. 2017, 156, 134–141. [Google Scholar] [CrossRef]
- Hayward, N.J.; Goldberg, D.I.; Morrel, E.M.; Friden, P.M.; Bokesch, P.M. Phase 1a/1b study of EP-7041: A novel, potent, selective, small molecule FXIa inhibitor. Circulation 2017, 136, A13747. [Google Scholar]
- Phang, M.; Lazarus, S.; Wood, L.; Garg, M. Diet and Thrombosis Risk: Nutrients for Prevention of Thrombotic Disease. Semin. Thromb. Hemost. 2011, 37, 199–208. [Google Scholar] [CrossRef]
- Vazhappilly, C.G.; Ansari, S.A.; Al-Jaleeli, R.; Al-Azawi, A.M.; Ramadan, W.S.; Menon, V.; Hodeify, R.; Siddiqui, S.S.; Merheb, M.; Matar, R.; et al. Role of flavonoids in thrombotic, cardiovascular, and inflammatory diseases. Inflammopharmacology 2019, 27, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.E. Oxidative Stress and Platelets. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Faggio, C.; Sureda, A.; Morabito, S.; Sanches-Silva, A.; Mocan, A.; Nabavi, S.F.; Nabavi, S.M. Flavonoids and platelet aggregation: A brief review. Eur. J. Pharmacol. 2017, 807, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Maiti, S.; Nazmeen, A.; Medda, N.; Patra, R.; Ghosh, T.K. Flavonoids green tea against oxidant stress and inflammation with related human diseases. Clin. Nutr. Exp. 2019, 24, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Kosakowska, O. Experimental Paper. Intrapopulation variability of flavonoid content in roots of Baikal skullcap (Scutellaria baicalensis Georgi). Herba Pol. 2017, 63, 20–31. [Google Scholar] [CrossRef]
- Ryu, R.; Jung, U.J.; Seo, Y.-R.; Kim, H.-J.; Moon, B.S.; Bae, J.-S.; Lee, D.G.; Choi, M.-S. Beneficial effect of persimmon leaves and bioactive compounds on thrombosis. Food Sci. Biotechnol. 2015, 24, 233–240. [Google Scholar] [CrossRef]
- Khan, M.M.; Quoc Tran, B.; Jang, Y.-J.; Park, S.-H.; Fondrie, W.E.; Chowdhury, K.; Hwan Yoon, S.; Goodlett, D.R.; Chae, S.-W.; Chae, H.-J.; et al. Assessment of the Therapeutic Potential of Persimmon Leaf Extract on Prediabetic Subjects. Mol. Cells 2017, 40, 466–475. [Google Scholar]
- Zhang, Y.-X.; Yang, T.-T.; Xia, L.; Zhang, W.-F.; Wang, J.-F.; Wu, Y.-P. Inhibitory Effect of Propolis on Platelet Aggregation In Vitro. J. Healthc. Eng. 2017, 2017, 3050895. [Google Scholar] [CrossRef]
- Lu, W.-J.; Lin, K.-C.; Liu, C.-P.; Lin, C.-Y.; Wu, H.-C.; Chou, D.-S.; Geraldine, P.; Huang, S.-Y.; Hsieh, C.-Y.; Sheu, J.-R. Prevention of arterial thrombosis by nobiletin: In vitro and in vivo studies. J. Nutr. Biochem. 2016, 28, 1–8. [Google Scholar] [CrossRef]
- Ku, S.-K.; Bae, J.-S. Antithrombotic activities of wogonin and wogonoside via inhibiting platelet aggregation. Fitoterapia 2014, 98, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.-L.; Da, X.-W.; He, A.-D.; Yao, G.-Q.; Xie, W.; Liu, G.; Xiang, J.-Z.; Ming, Z.-Y. Pentamethylquercetin (PMQ) reduces thrombus formation by inhibiting platelet function. Sci. Rep. 2015, 5, 11142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.; Zhang, H.; Li, Y.; Zhou, Y.; Lu, W.; Yao, L. The effect of Diosmin on the blood proteome in a rat model of venous thrombosis. Int. J. Biol. Macromol. 2017, 104, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Hsia, C.-W.; Wu, M.-P.; Velusamy, M.; Hsia, C.-H.; Chou, D.-S.; Tsai, C.-L.; Hsu, C.-Y.; Jayakumar, T.; Chung, C.-L.; Sheu, J.-R. Novel Therapeutic Agent against Platelet Activation In Vitro and Arterial Thrombosis In Vivo by Morin Hydrate. Int. J. Mol. Sci. 2018, 19, 2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.H.; Son, Y.K.; Han, Y.N. Tissue factor inhibitory flavonoids from the fruits ofChaenomeles sinensis. Arch. Pharmacal Res. 2002, 25, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Karlíčková, J.; Říha, M.; Filipský, T.; Macáková, K.; Hrdina, R.; Mladěnka, P. Antiplatelet Effects of Flavonoids Mediated by Inhibition of Arachidonic Acid Based Pathway. Planta Med. 2015, 82, 76–83. [Google Scholar] [CrossRef]
- Dasgupta, A. Antiinflammatory Herbal Supplements. In Translational Inflammation; Elsevier: Amsterdam, The Netherlands, 2019; pp. 69–91. [Google Scholar]
- Liakopoulou, C.; Kazazis, C.; Vallianou, N.G. Silimarin and Cancer. Anti-Cancer Agents Med. Chem. 2019, 18, 1970–1974. [Google Scholar] [CrossRef] [PubMed]
- Bijak, M.; Dziedzic, A.; Saluk-Bijak, J. Flavonolignans reduce the response of blood platelet to collagen. Int. J. Biol. Macromol. 2018, 106, 878–884. [Google Scholar] [CrossRef]
- Bijak, M.; Szelenberger, R.; Dziedzic, A.; Saluk-Bijak, J. Inhibitory Effect of Flavonolignans on the P2Y12 Pathway in Blood Platelets. Molecules 2018, 23, 374. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Lee, J.; Kang, S.; Moon, H.; Chung, K.H.; Kim, K.R. Antiplatelet and Antithrombotic Effects of the Extract of Lindera obtusiloba Leaves. Biomol. Ther. 2016, 24, 659–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, W.; Liu, L.; Li, J.; Qu, L.; Pang, X.; Yu, H.; Zhang, Y.; Wang, T. Bioactive flavonoids from Flos Sophorae. J. Nat. Med. 2017, 71, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Yang, F.-Q.; Zhang, Q.; Wang, F.-Q.; Hu, Y.-J.; Xia, Z.-N. Natural Products for Antithrombosis. Evid. Based Complementary Altern. Med. 2015, 2015, 876426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kregiel, D.; Berlowska, J.; Witonska, I.; Antolak, H.; Proestos, C.; Babic, M.; Babic, L.; Zhang, B. Saponin-Based, Biological-Active Surfactants from Plants. In Application and Characterization of Surfactants; InTech: London, UK, 2017. [Google Scholar]
- Singh, D.; Chaudhuri, P.K. Structural characteristics, bioavailability and cardioprotective potential of saponins. Integr. Med. Res. 2018, 7, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.-H.; Kwon, H.-W.; Cho, H.-J.; Rhee, M.H.; Park, H.-J. Vasodilator-stimulated phosphoprotein-phosphorylation by ginsenoside Ro inhibits fibrinogen binding to αIIb/β3 in thrombin-induced human platelets. J. Ginseng Res. 2016, 40, 359–365. [Google Scholar] [CrossRef] [Green Version]
- Shen, Q.; Li, J.; Zhang, C.; Wang, P.; Mohammed, A.; Ni, S.; Tang, Z. Panax notoginseng saponins reduce high-risk factors for thrombosis through peroxisome proliferator-activated receptor -γ pathway. Biomed. Pharmacother. 2017, 96, 1163–1169. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Meng, X.; Zhai, Y.; Zhou, P.; Ye, T.; Wang, Z.; Sun, G.; Sun, X. Panax Notoginseng Saponins: A Review of Its Mechanisms of Antidepressant or Anxiolytic Effects and Network Analysis on Phytochemistry and Pharmacology. Molecules 2018, 23, 940. [Google Scholar] [CrossRef] [Green Version]
- Thom, V.; Tung, N.; Van Diep, D.; Thuy, D.; Hue, N.; Long, D.; Tung, B.; Huyen, P.; Ly Huong, D. Antithrombotic activity and saponin composition of the roots of Panax bipinnatifidus Seem. growing in Vietnam. Pharmacogn. Res. 2018, 10, 333. [Google Scholar]
- Zhai, K.; Zheng, J.; Tang, Y.; Li, F.; Lv, Y.; Zhang, Y.; Gao, Z.; Qi, J.; Yu, B.; Kou, J. The saponin D39 blocks dissociation of non-muscular myosin heavy chain IIA from TNF receptor 2, suppressing tissue factor expression and venous thrombosis. Br. J. Pharmacol. 2017, 174, 2818–2831. [Google Scholar] [CrossRef]
- Yu, B.; Tian, Y.; Ma, S.; Lin, B.; Kou, J. Anti-thrombotic activity of DT-13, a saponin isolated from the root tuber of Liriope muscari. Indian J. Pharmacol. 2013, 45, 283. [Google Scholar] [CrossRef]
- Kou, J.; Yu, B.; Xu, Q. Inhibitory effects of ethanol extract from Radix Ophiopogon japonicus on venous thrombosis linked with its endothelium-protective and anti-adhesive activities. Vasc. Pharmacol. 2005, 43, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Kou, J.; Tian, Y.; Tang, Y.; Yan, J.; Yu, B. Antithrombotic Activities of Aqueous Extract from Radix Ophiopogon japonicus and Its Two Constituents. Biol. Pharm. Bull. 2006, 29, 1267–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Huang, Y. Chinese Herbal Medicine on Cardiovascular Diseases and the Mechanisms of Action. Front. Pharmacol. 2016, 7, 469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Huang, B.; Du, D.; Guo, X.; Xin, G.; Xing, Z.; Liang, Y.; Chen, Y.; Chen, Q.; He, Y.; et al. Anti-thrombosis effect of diosgenyl saponins in vitro and in vivo. Steroids 2013, 78, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Kou, J.P.; Li, J.F.; Yan, J.; Zhu, D.N. Effect of total saponin from root of Polygala fallax Hesml. (PTS) on coagulation and thrombosis. J. China Pharm. Univ. 2003, 34, 63–65. [Google Scholar]
- Yoshikawa, M.; Matsuda, H. Chemical and pharmacological studies on triterpene saponins, escins, from horse chestnut seeds. In Saponins in Food, Feedstuffs and Medicinal Plants; Oleszek, W., Marston, A., Eds.; Springer: Dordrecht, The Netherlands, 2000; pp. 189–203. ISBN 978-90-481-5341-1. [Google Scholar]
- Wang, M.; Li, H.; Xu, F.; Gao, X.; Li, J.; Xu, S.; Zhang, D.; Wu, X.; Xu, J.; Hua, H.; et al. Diterpenoid lead stevioside and its hydrolysis products steviol and isosteviol: Biological activity and structural modification. Eur. J. Med. Chem. 2018, 156, 885–906. [Google Scholar] [CrossRef]
- Geuns, J.M. Stevioside. Phytochemistry 2003, 64, 913–921. [Google Scholar] [CrossRef]
- Carrera-Lanestosa, A.; Moguel-Ordóñez, Y.; Segura-Campos, M. Stevia rebaudiana Bertoni: A Natural Alternative for Treating Diseases Associated with Metabolic Syndrome. J. Med. Food 2017, 20, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ruiz, J.C.; Moguel-Ordoñez, Y.B.; Segura-Campos, M.R. Biological activity of Stevia rebaudiana Bertoni and their relationship to health. Crit. Rev. Food Sci. Nutr. 2017, 57, 2680–2690. [Google Scholar] [CrossRef]
- Salehi, B.; López, M.D.; Martínez-López, S.; Victoriano, M.; Sharifi-Rad, J.; Martorell, M.; Rodrigues, C.F.; Martins, N. Stevia rebaudiana Bertoni bioactive effects: From in vivo to clinical trials towards future therapeutic approaches. Phytother. Res. 2019, 33, 2904–2917. [Google Scholar] [CrossRef]
- KKak, S. Nrf2: A key regulator of redox signaling in liver diseases. In Liver Pathophysiology; Muriel, P., Ed.; Academic Press: Waltham, MA, USA, 2017; pp. 472–479. ISBN 978-0-12-804274-8. [Google Scholar]
- Wang, Z.; Zhao, Y. Gut microbiota derived metabolites in cardiovascular health and disease. Protein Cell 2018, 9, 416–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boonkaewwan, C.; Burodom, A. Anti-inflammatory and immunomodulatory activities of stevioside and steviol on colonic epithelial cells. J. Sci. Food Agric. 2013, 93, 3820–3825. [Google Scholar] [CrossRef] [PubMed]
- Yingkun, N.; Zhenyu, W.; Jing, L.; Xiuyun, L.; Huimin, Y. Stevioside Protects LPS-Induced Acute Lung Injury in Mice. Inflammation 2013, 36, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Boonkaewwan, C.; Ao, M.; Toskulkao, C.; Rao, M.C. Specific Immunomodulatory and Secretory Activities of Stevioside and Steviol in Intestinal Cells. J. Agric. Food Chem. 2008, 56, 3777–3784. [Google Scholar] [CrossRef]
- Bridel, M.; Lavieille, R. The sweet principle in Kaa-he-e (Stevia rebaudiana. Bertoni). II. Hydrolysis of stevioside by enzymes. III. Steviol by enzymic hydrolysis and isosteviol by acid hydrolysis. Bull. Soc. Chim. Biol. 1931, 13, 781–796. [Google Scholar]
- Ullah, A.; Munir, S.; Mabkhot, Y.; Badshah, S. Bioactivity Profile of the Diterpene Isosteviol and its Derivatives. Molecules 2019, 24, 678. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Zhang, D.; Li, M.; Wu, Q.; Lam, Y.P.Y.; Guo, Y.; Chen, C.; Bai, N.; Malhotra, S.; Li, W.; et al. Discovery of novel, potent, isosteviol-based antithrombotic agents. Eur. J. Med. Chem. 2019, 183, 111722. [Google Scholar] [CrossRef] [PubMed]
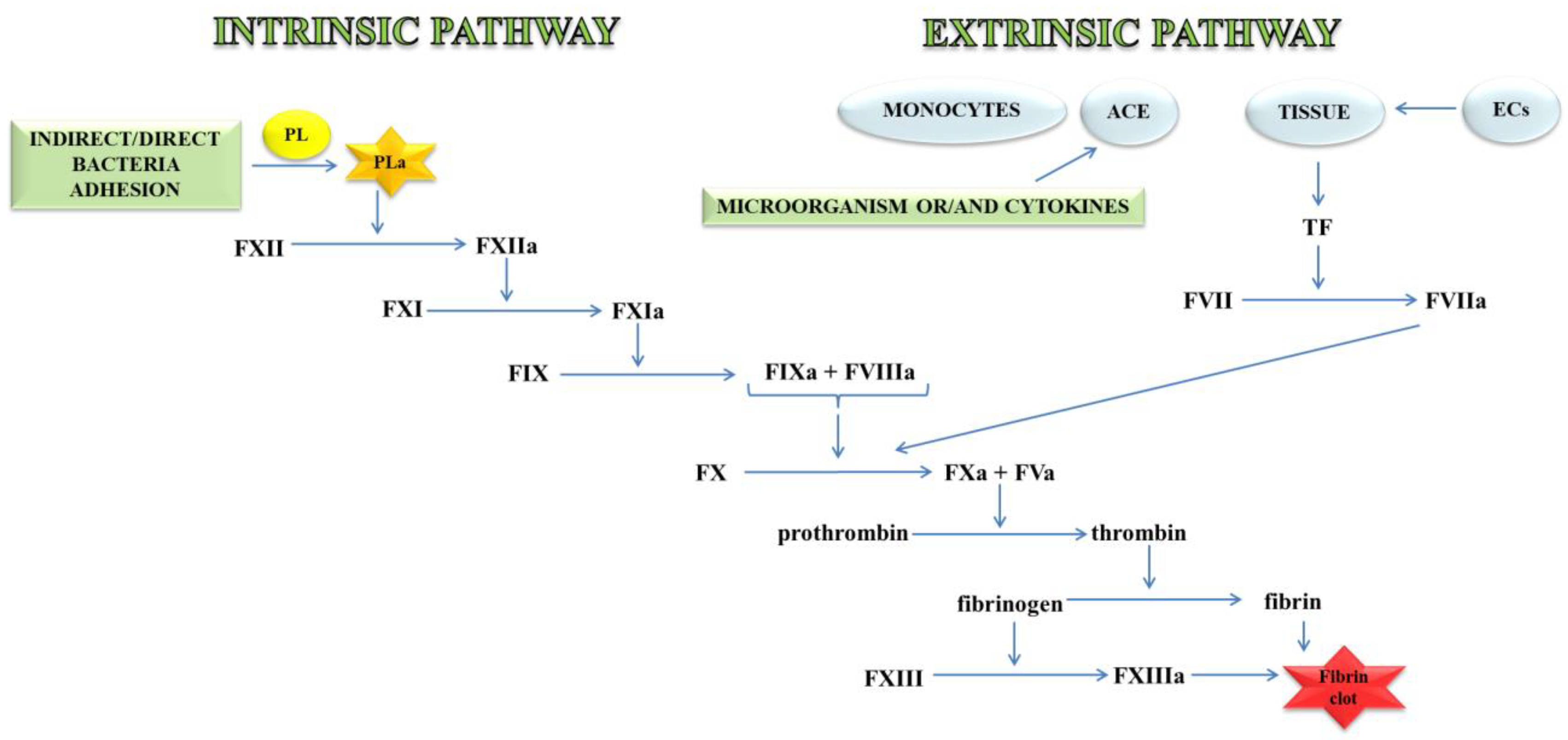
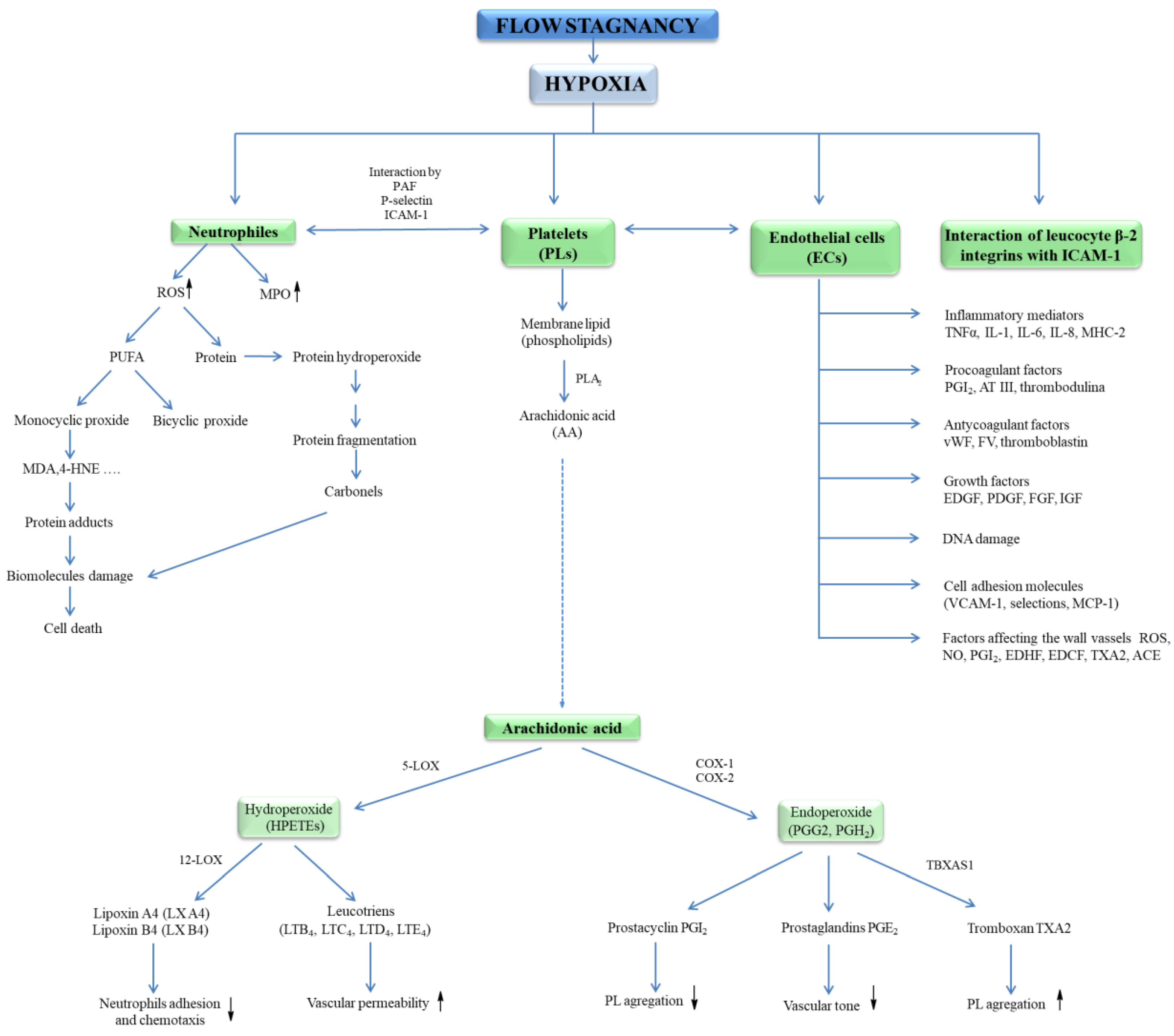
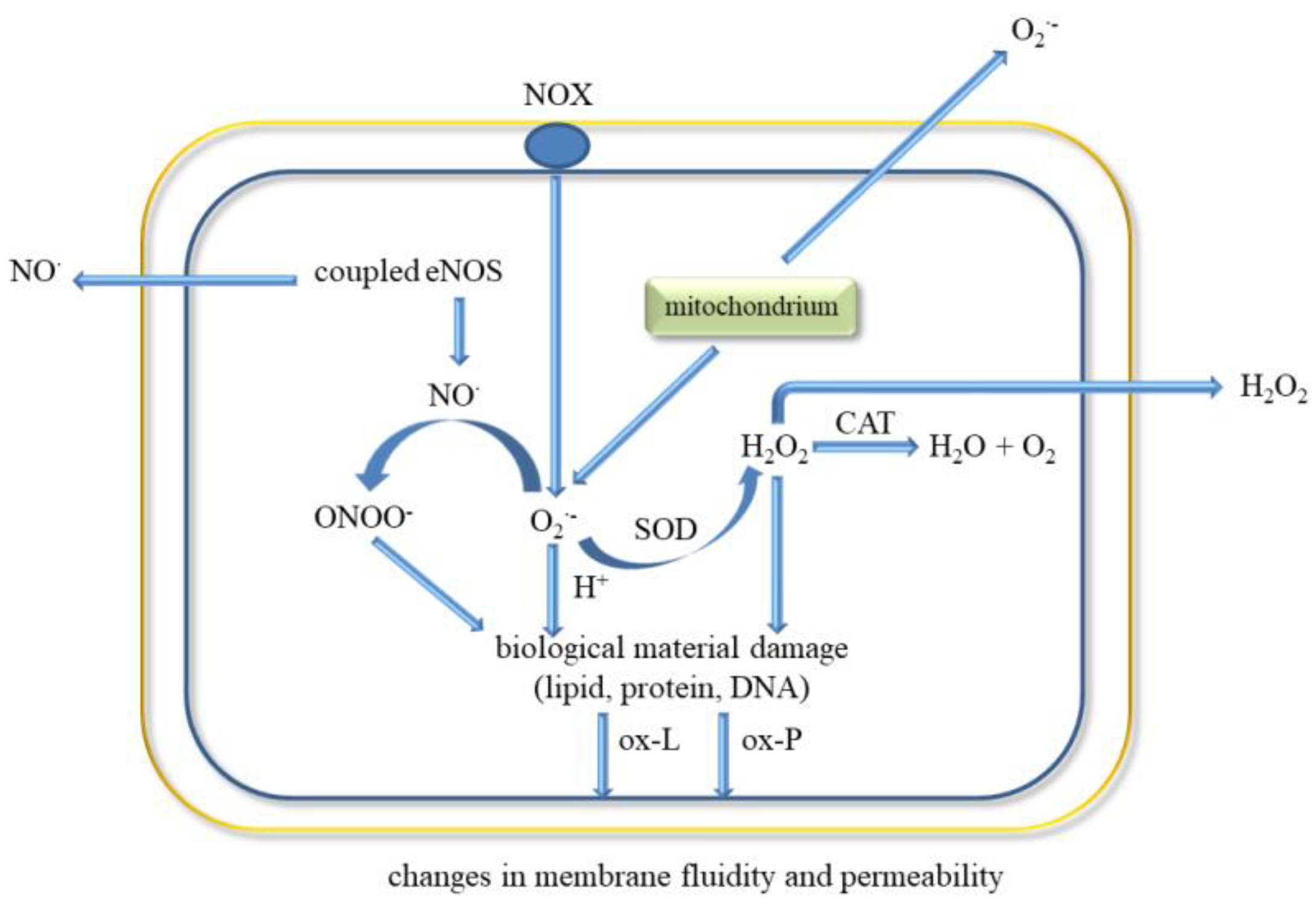

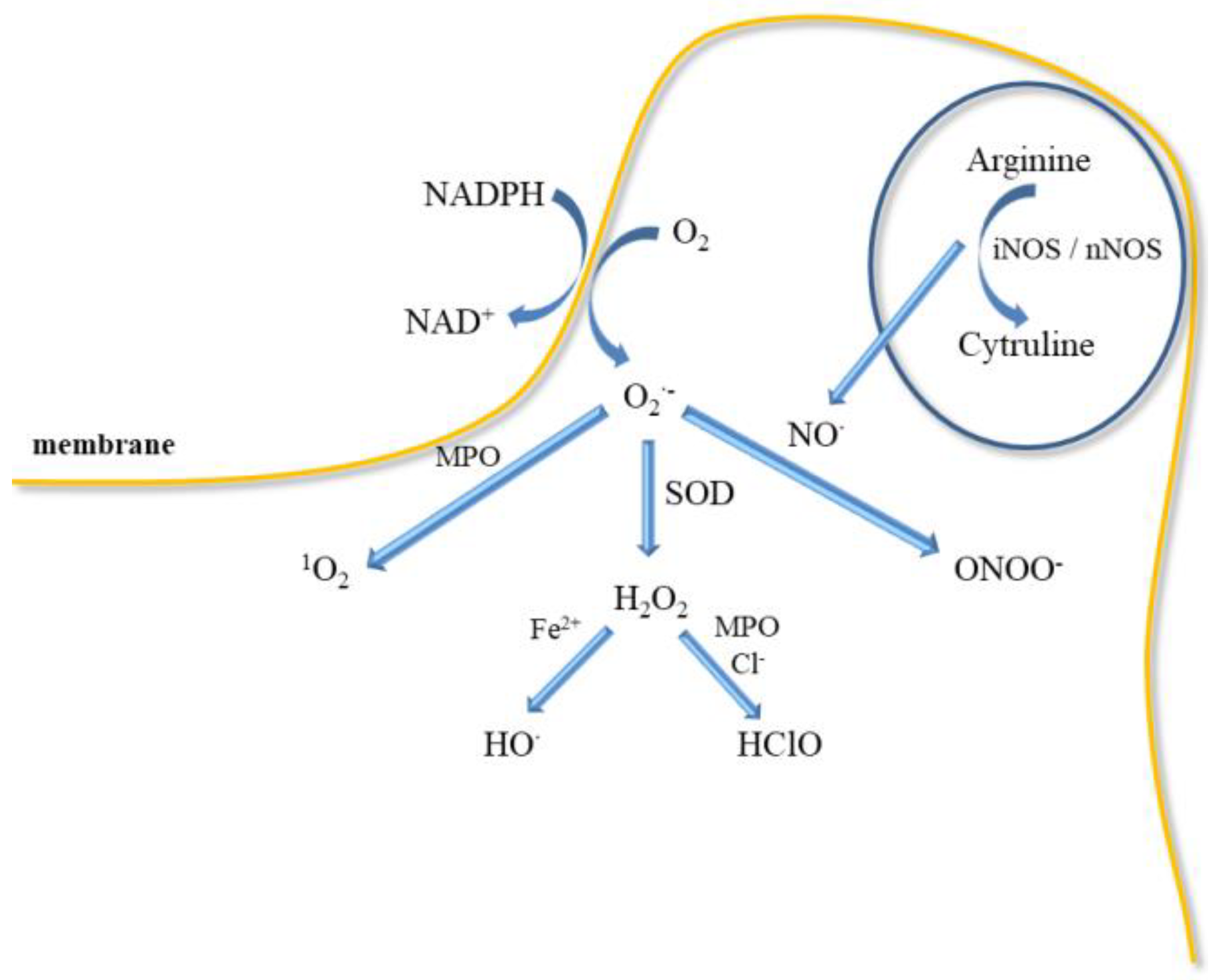
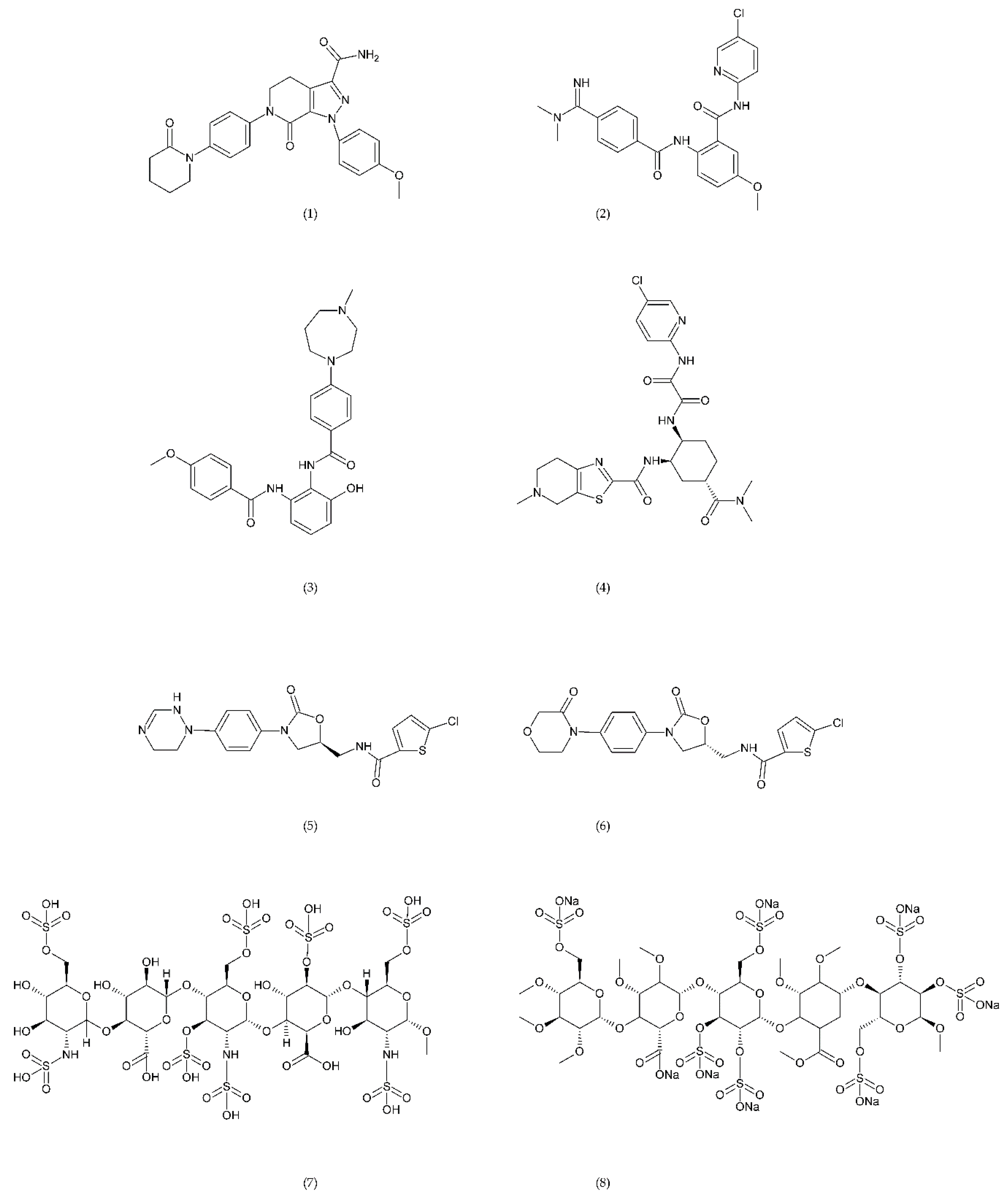
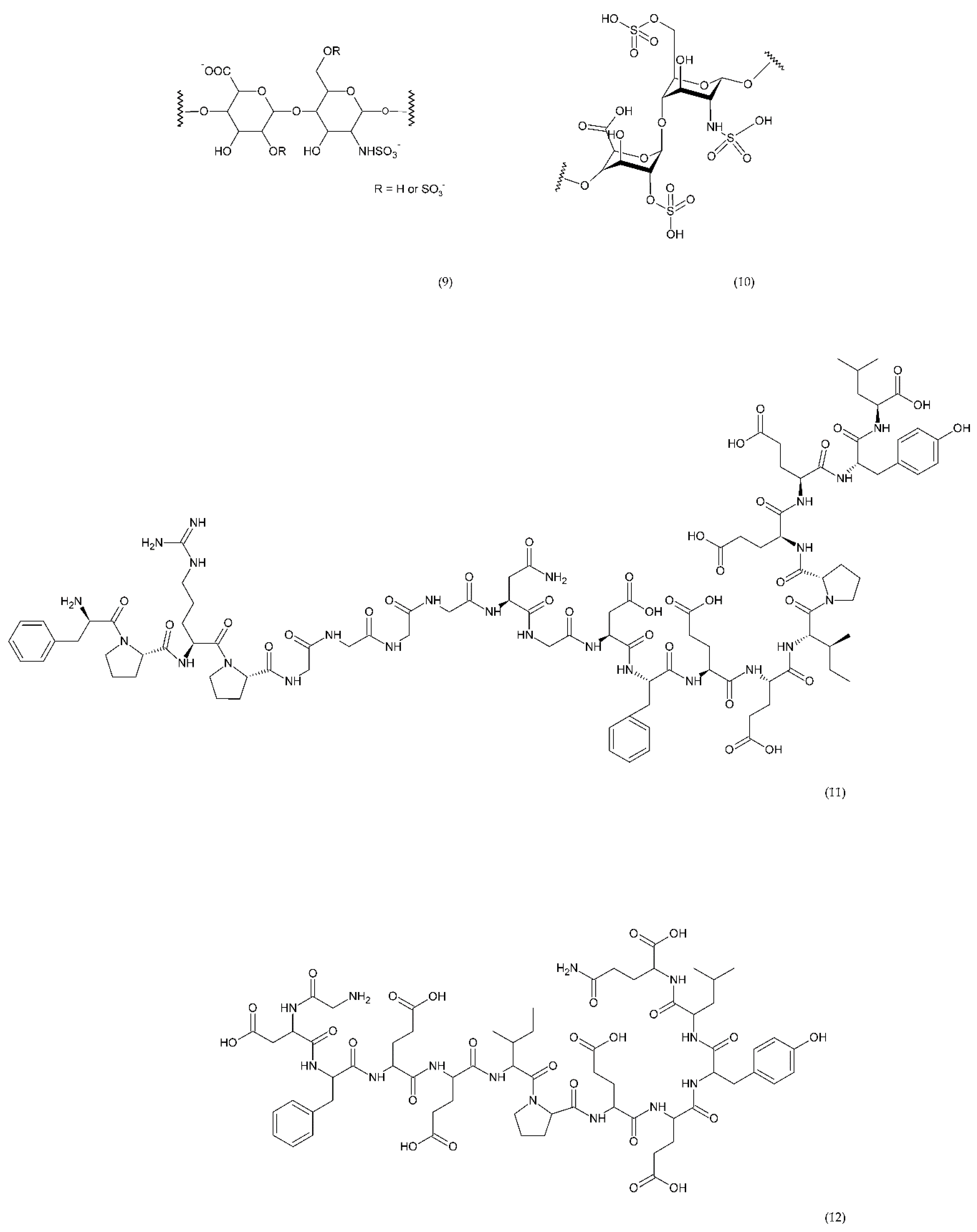
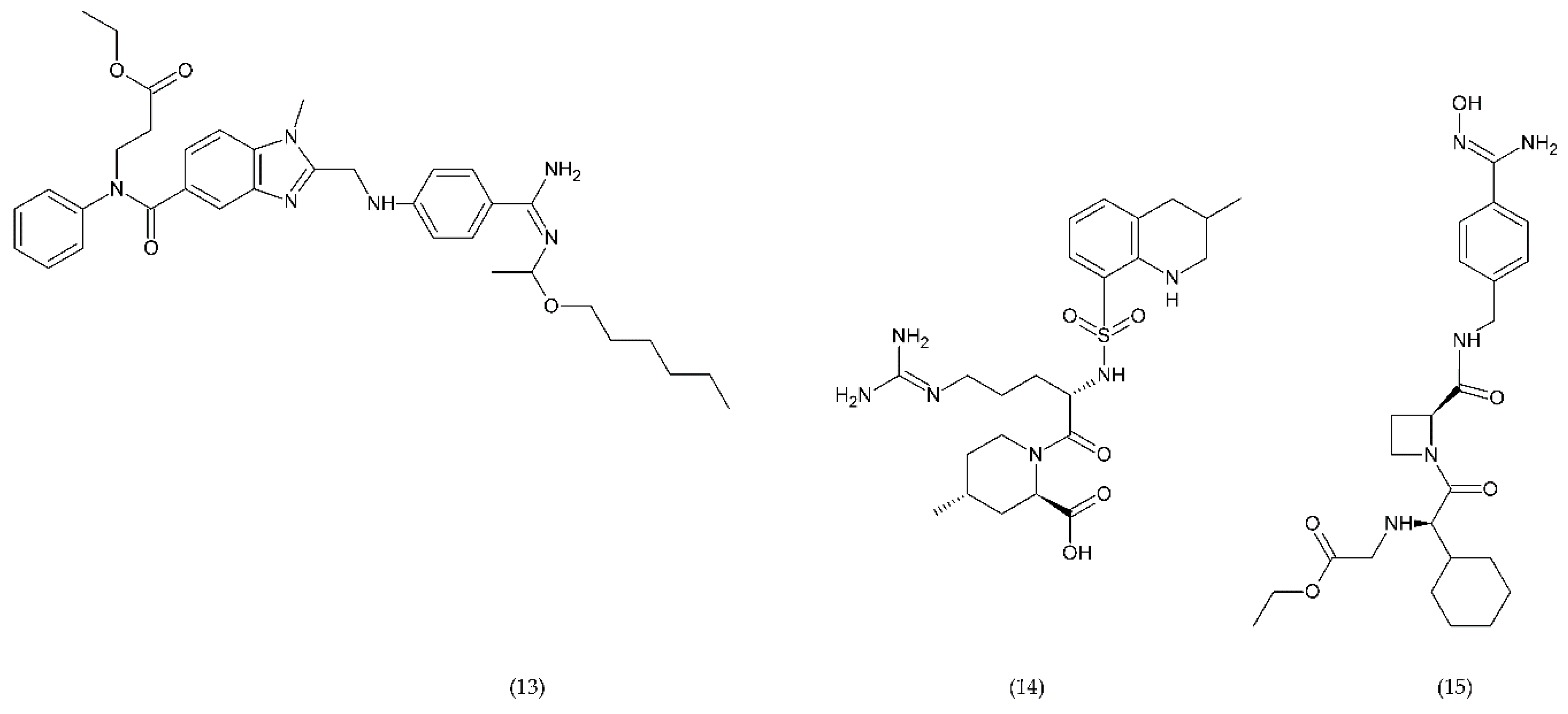
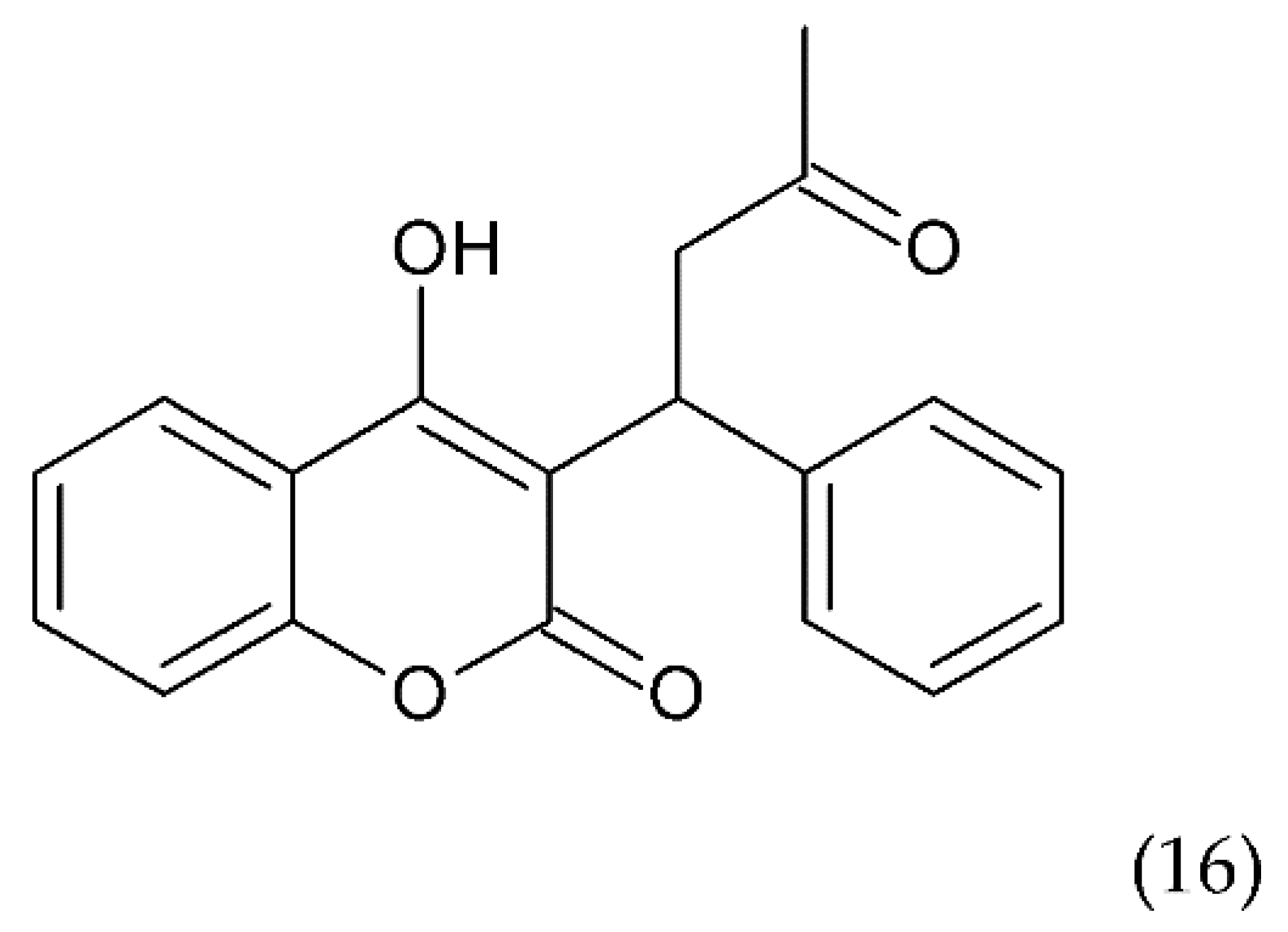

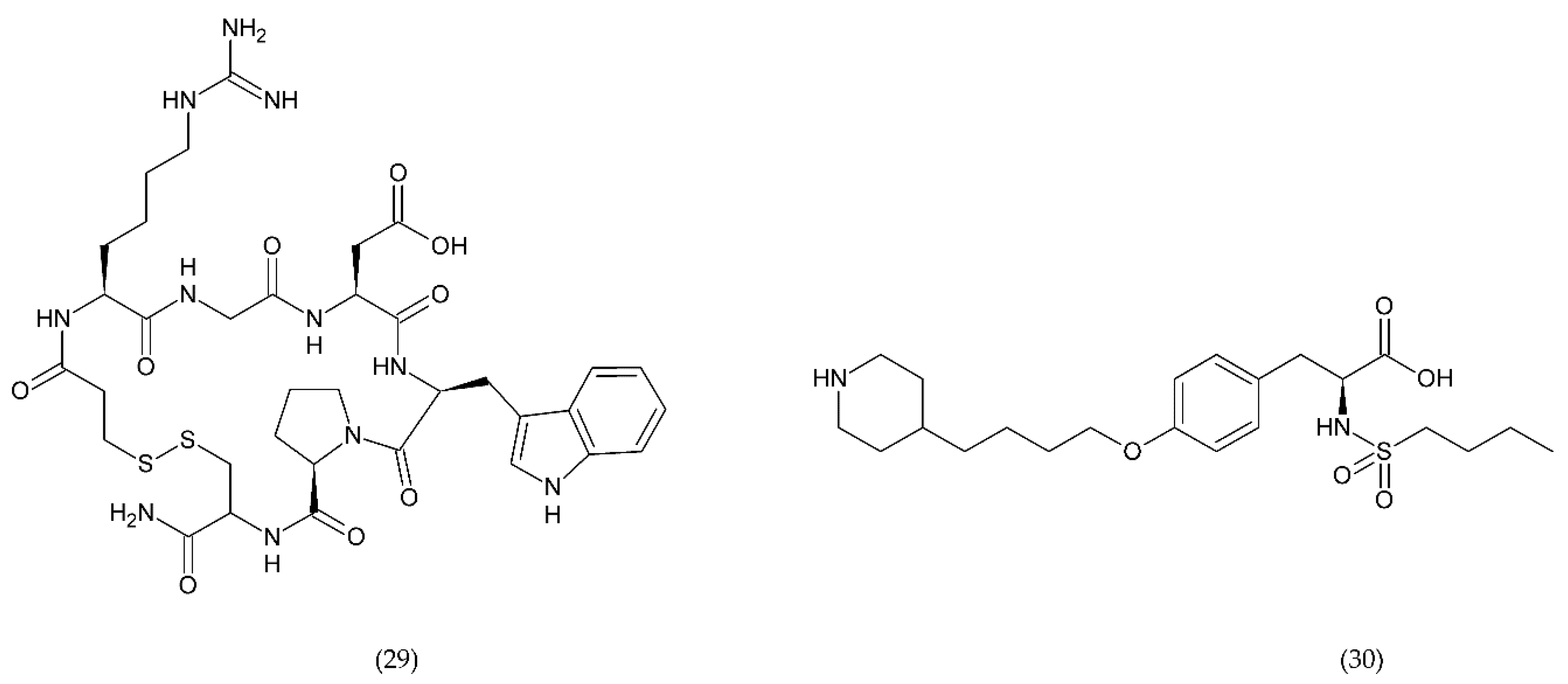
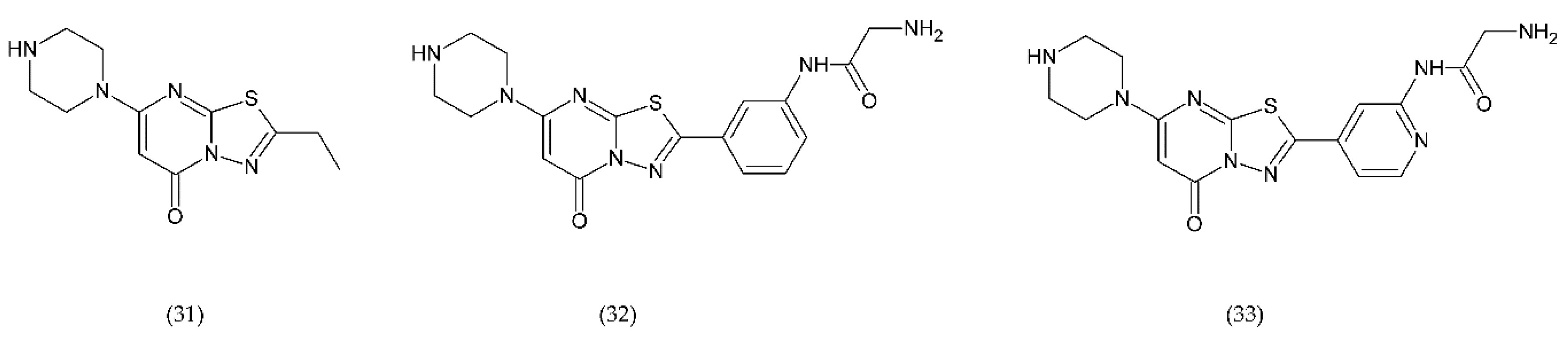
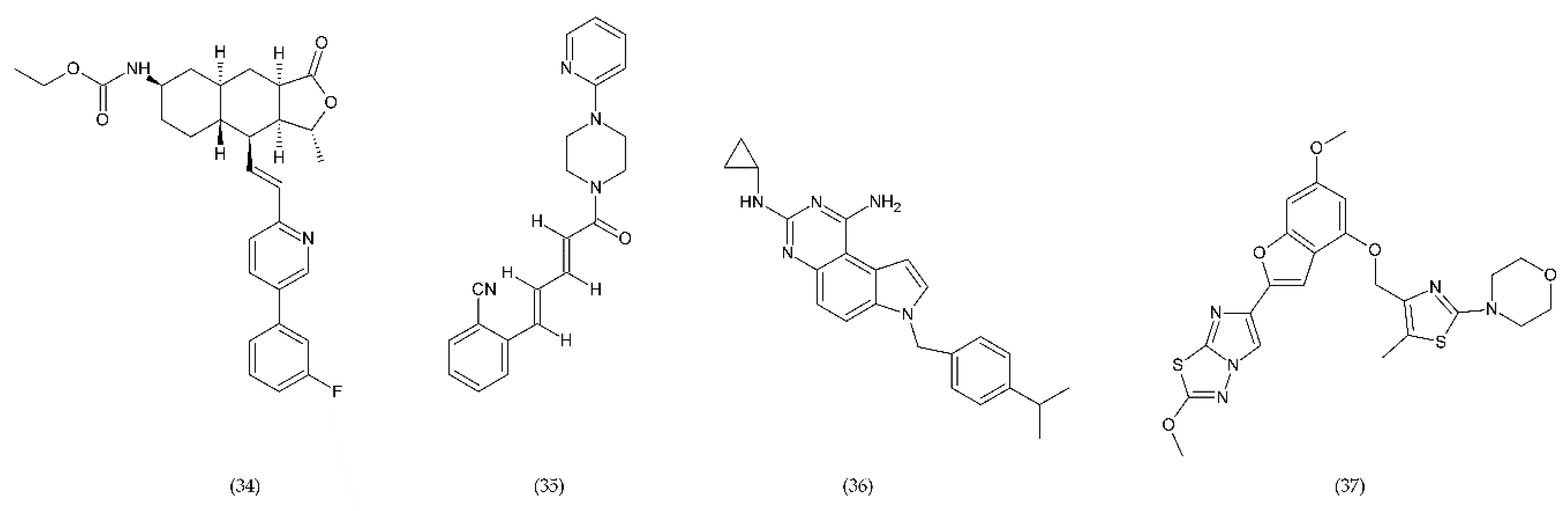
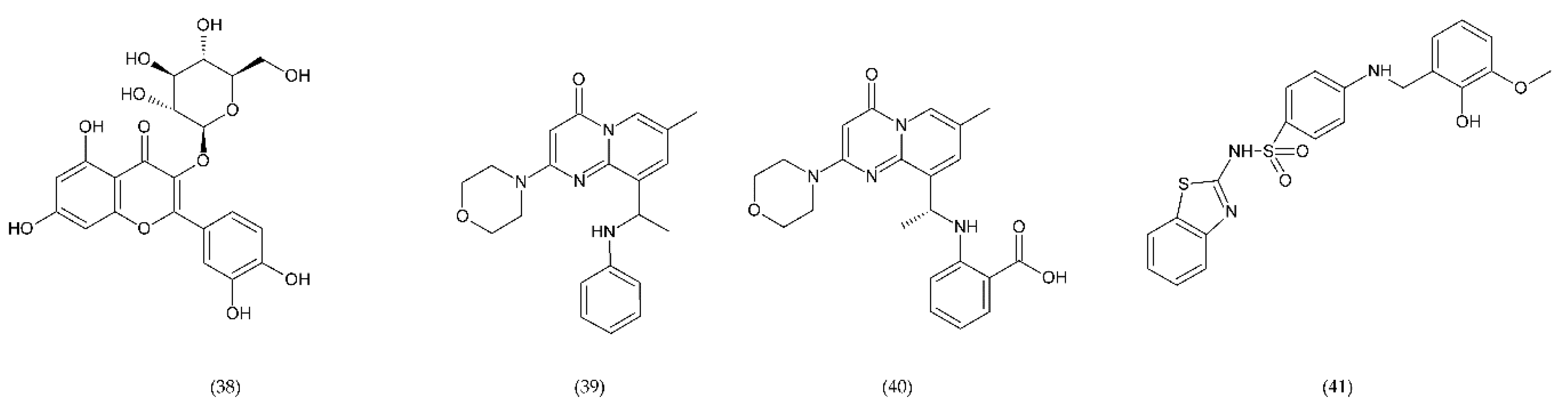
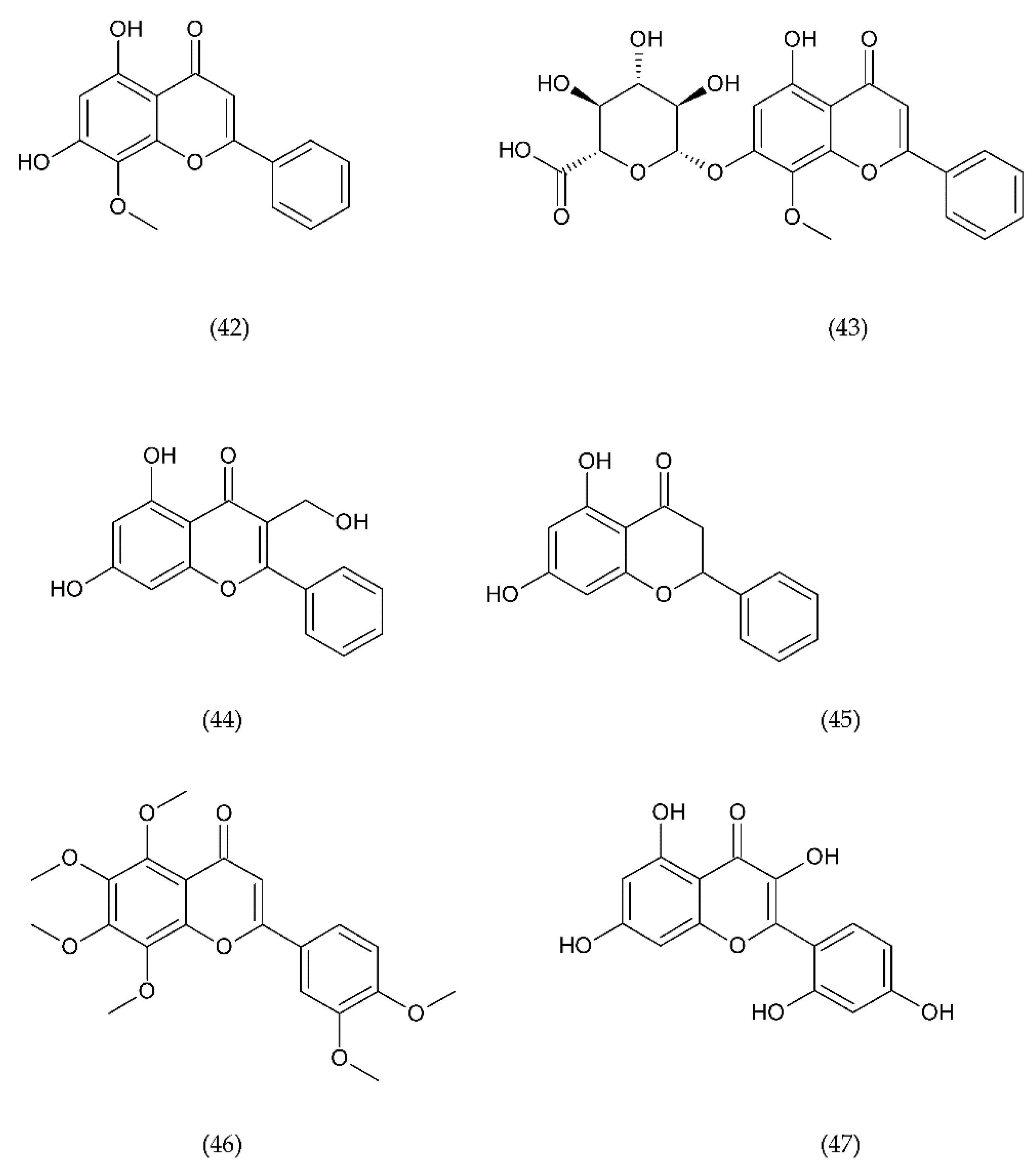
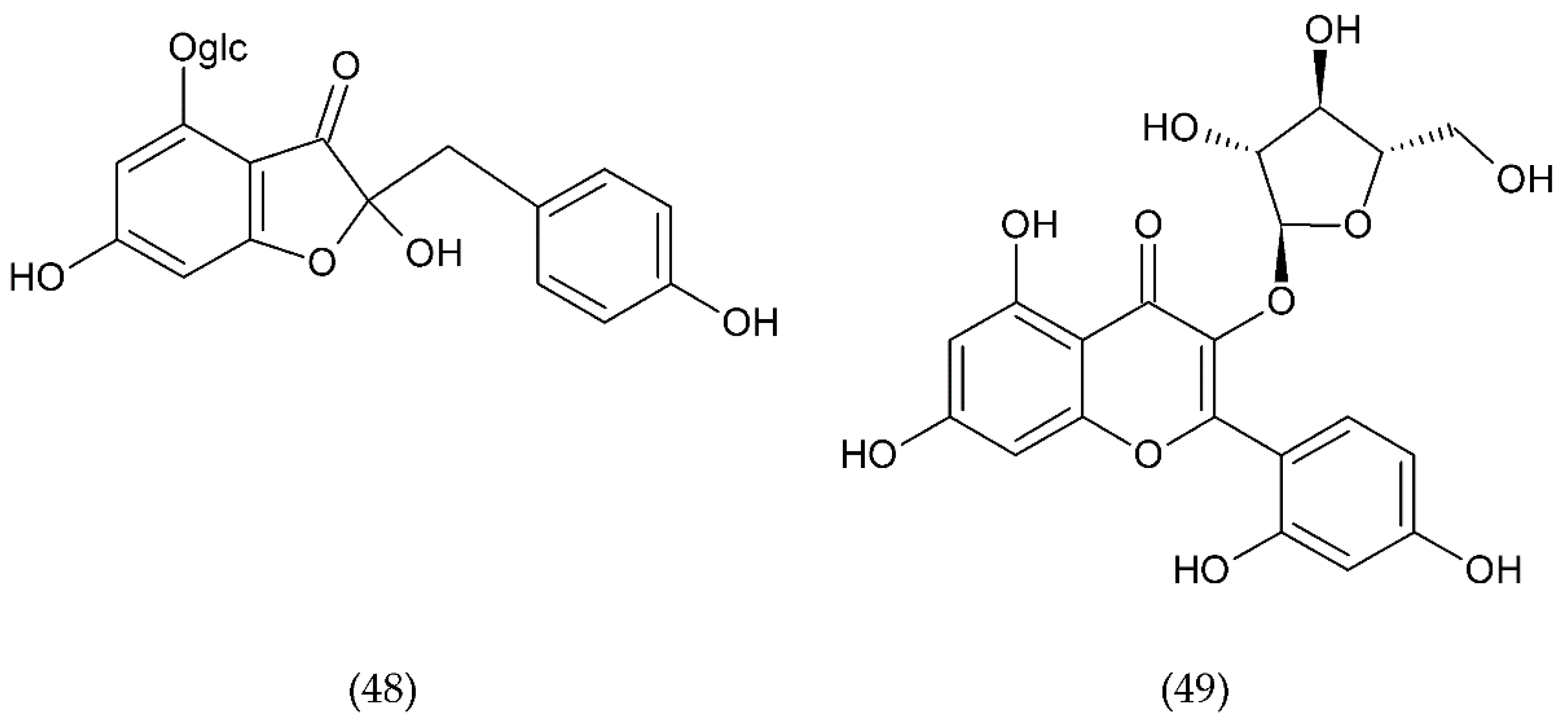
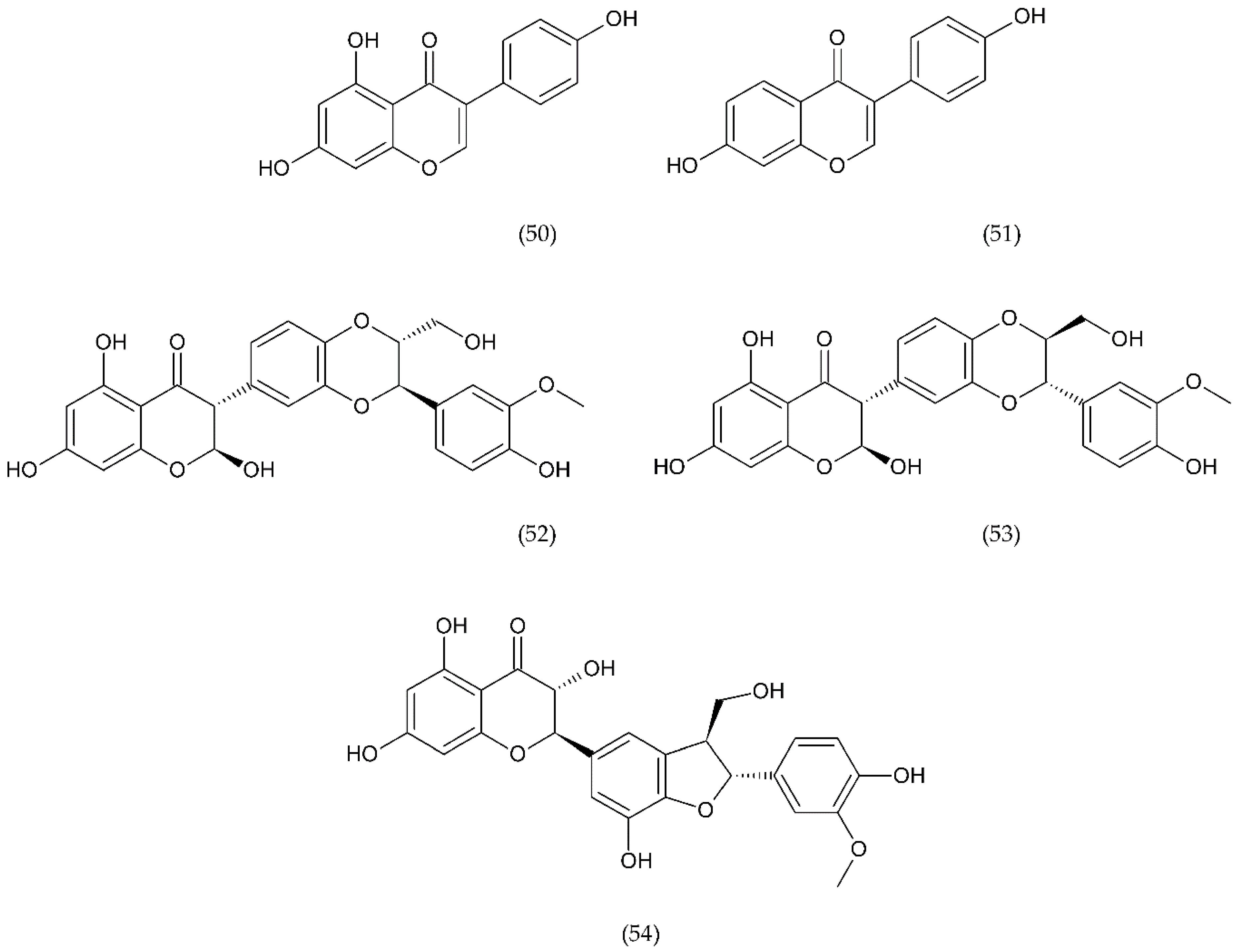
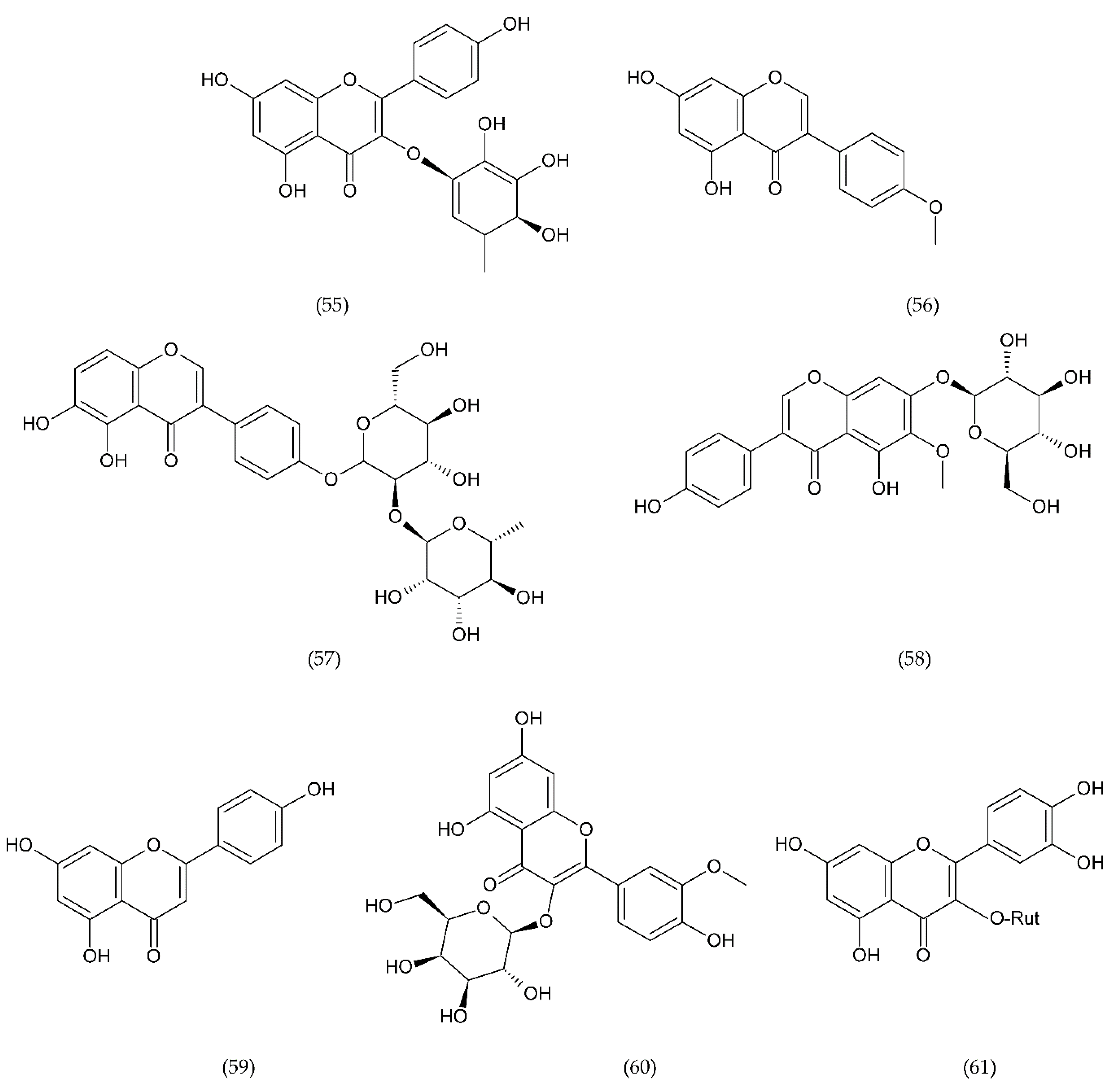
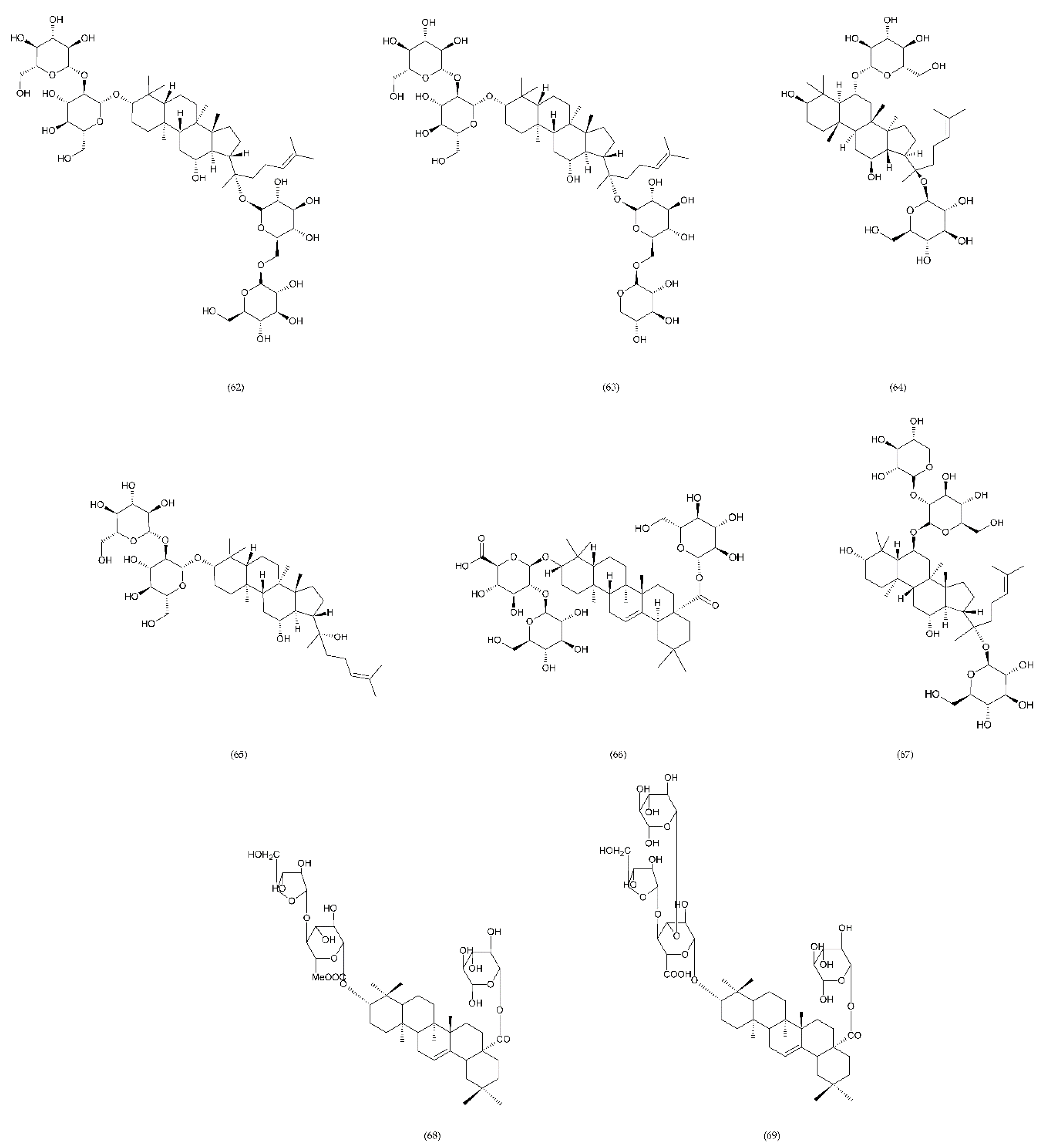
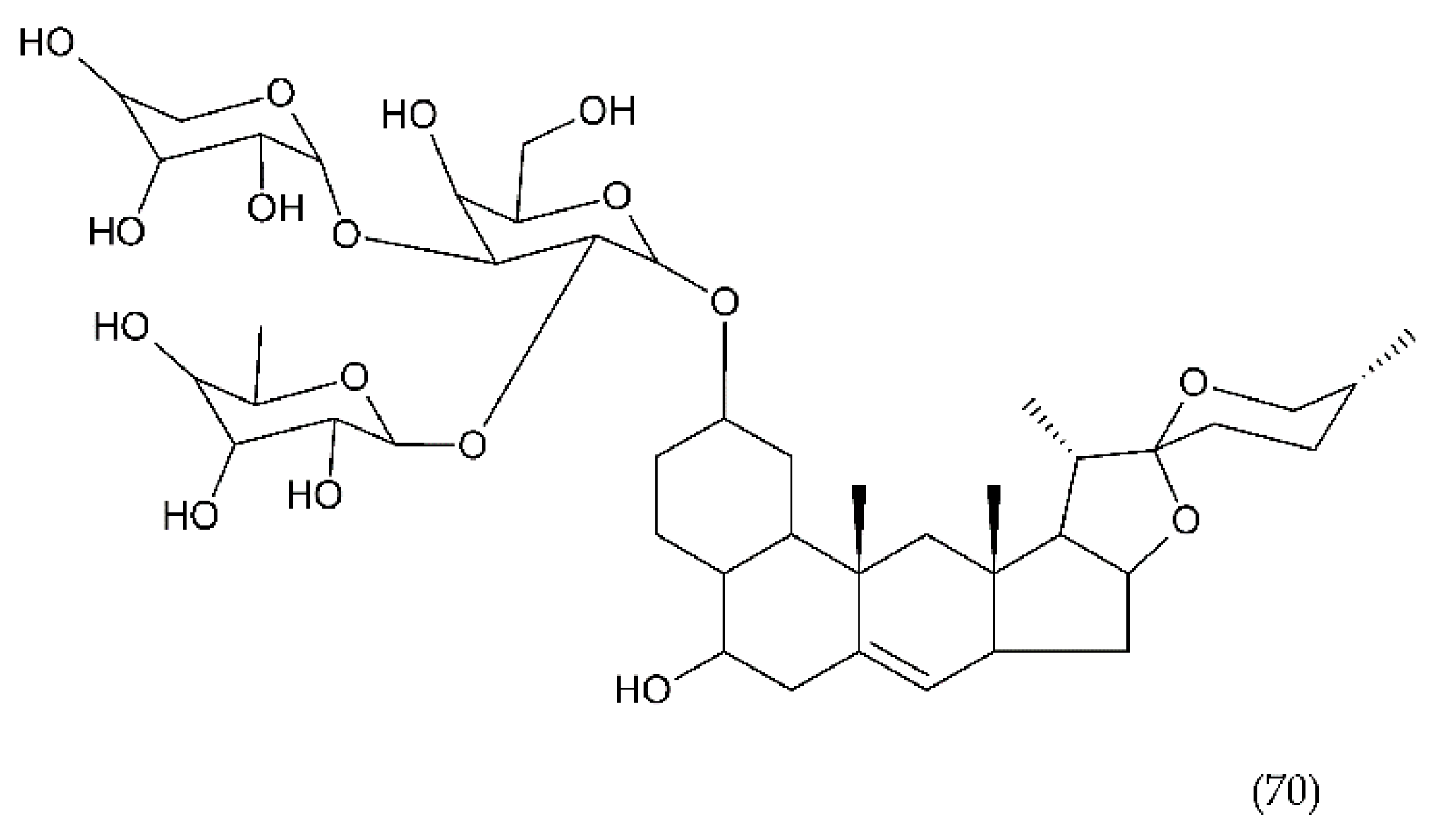
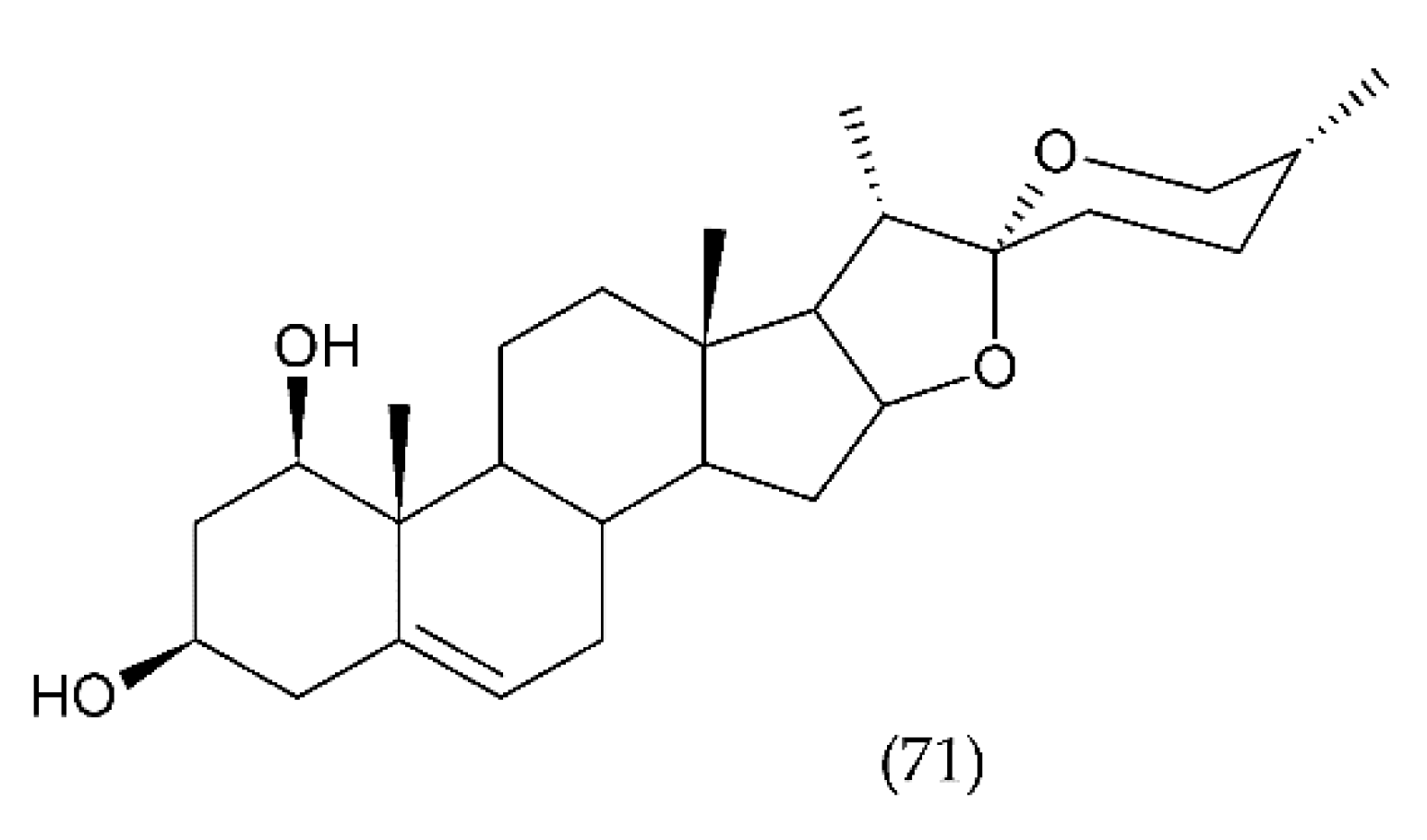
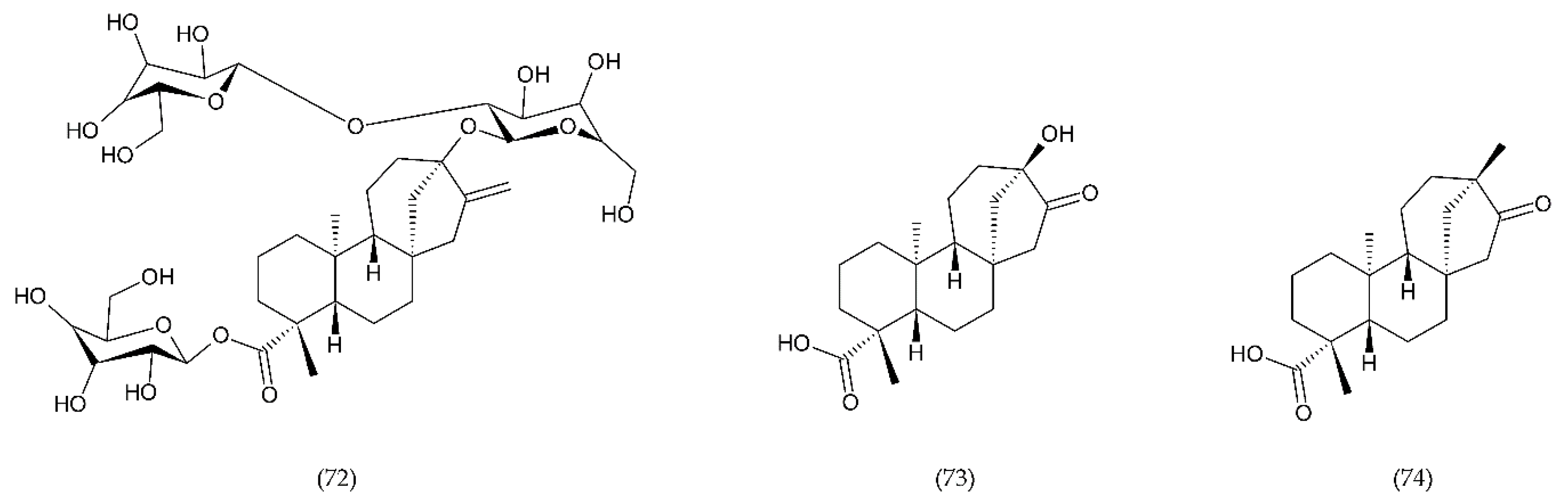

{kind=link}
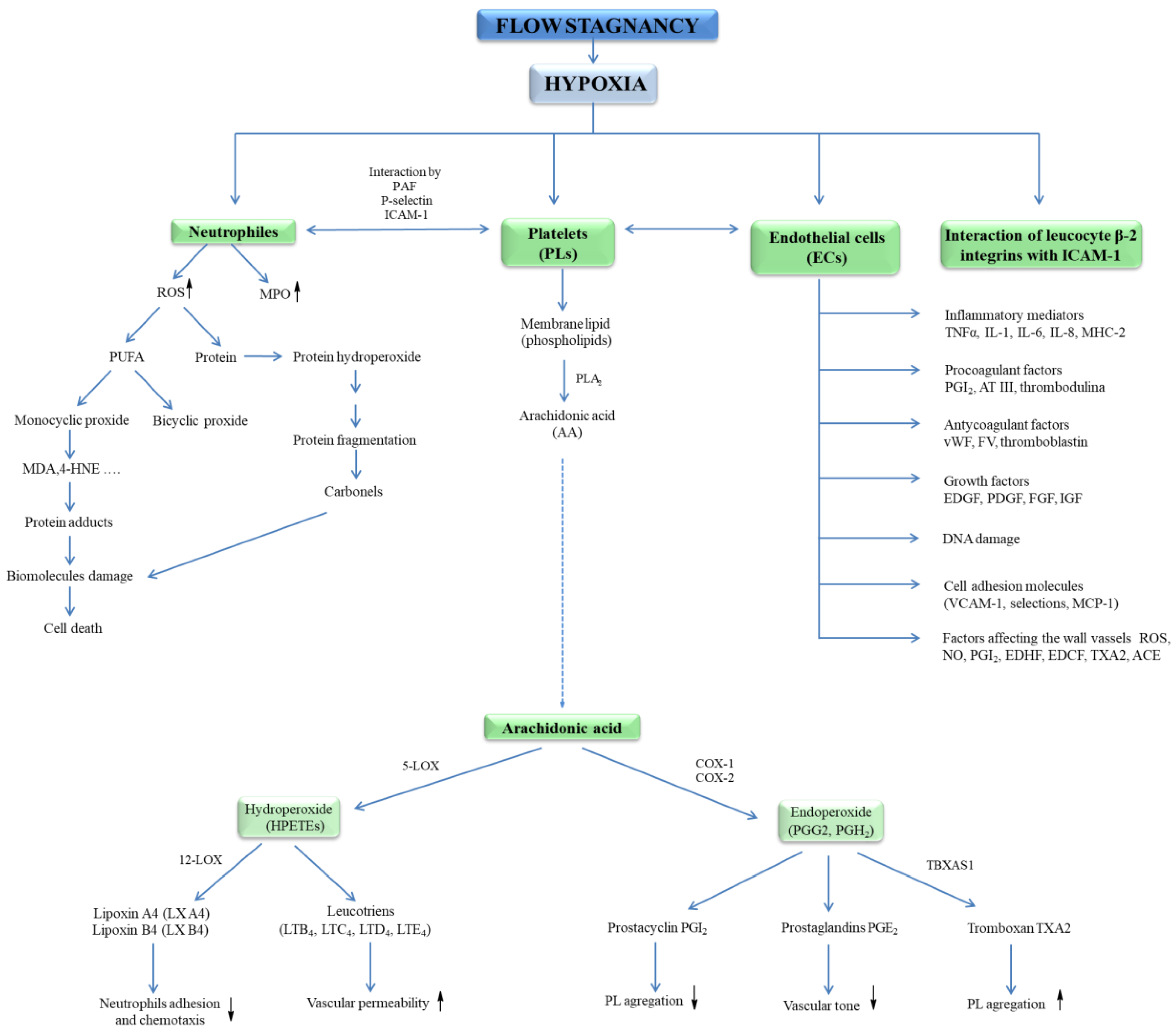{kind=link}
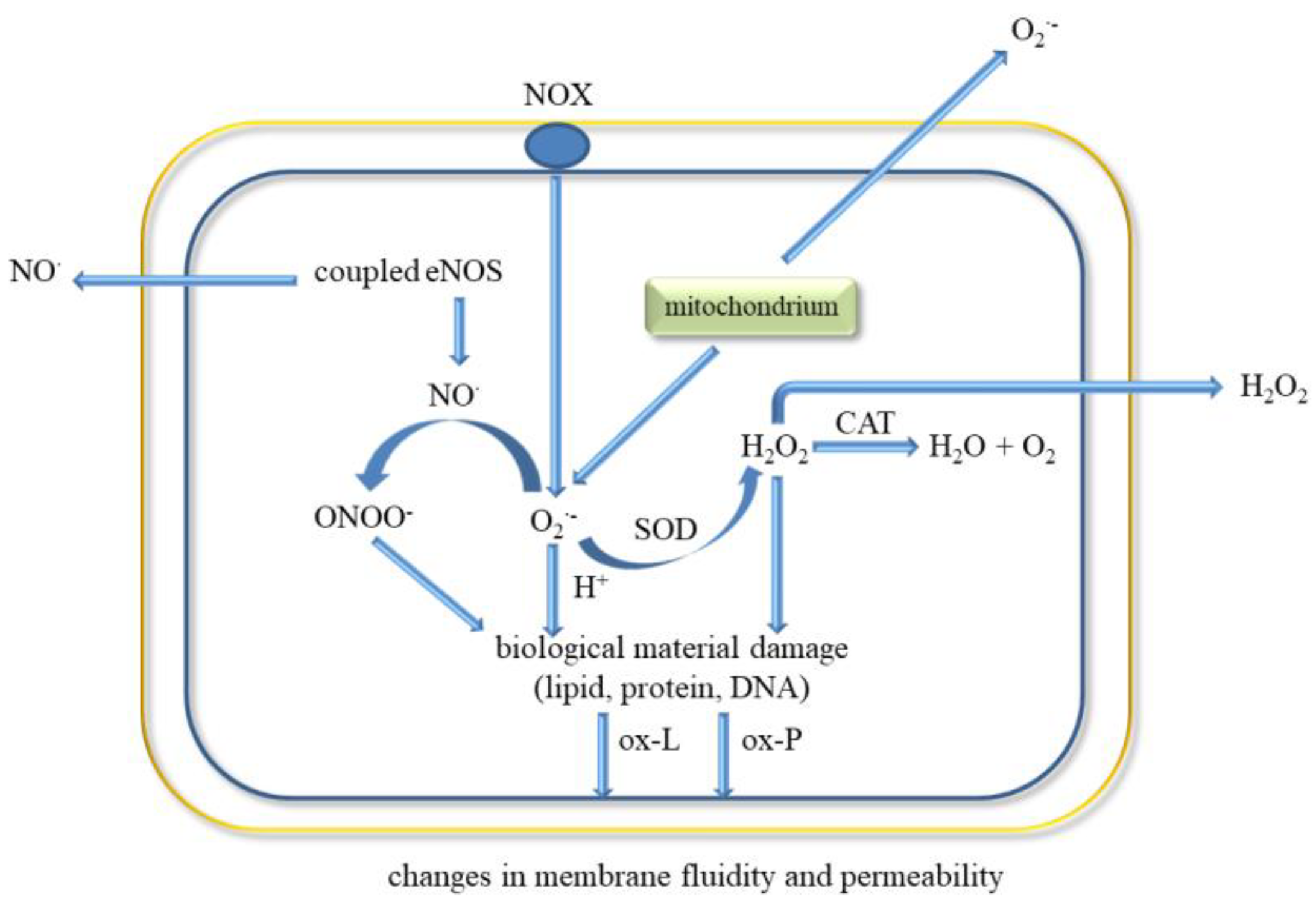{kind=link}
{kind=link}
{kind=link}
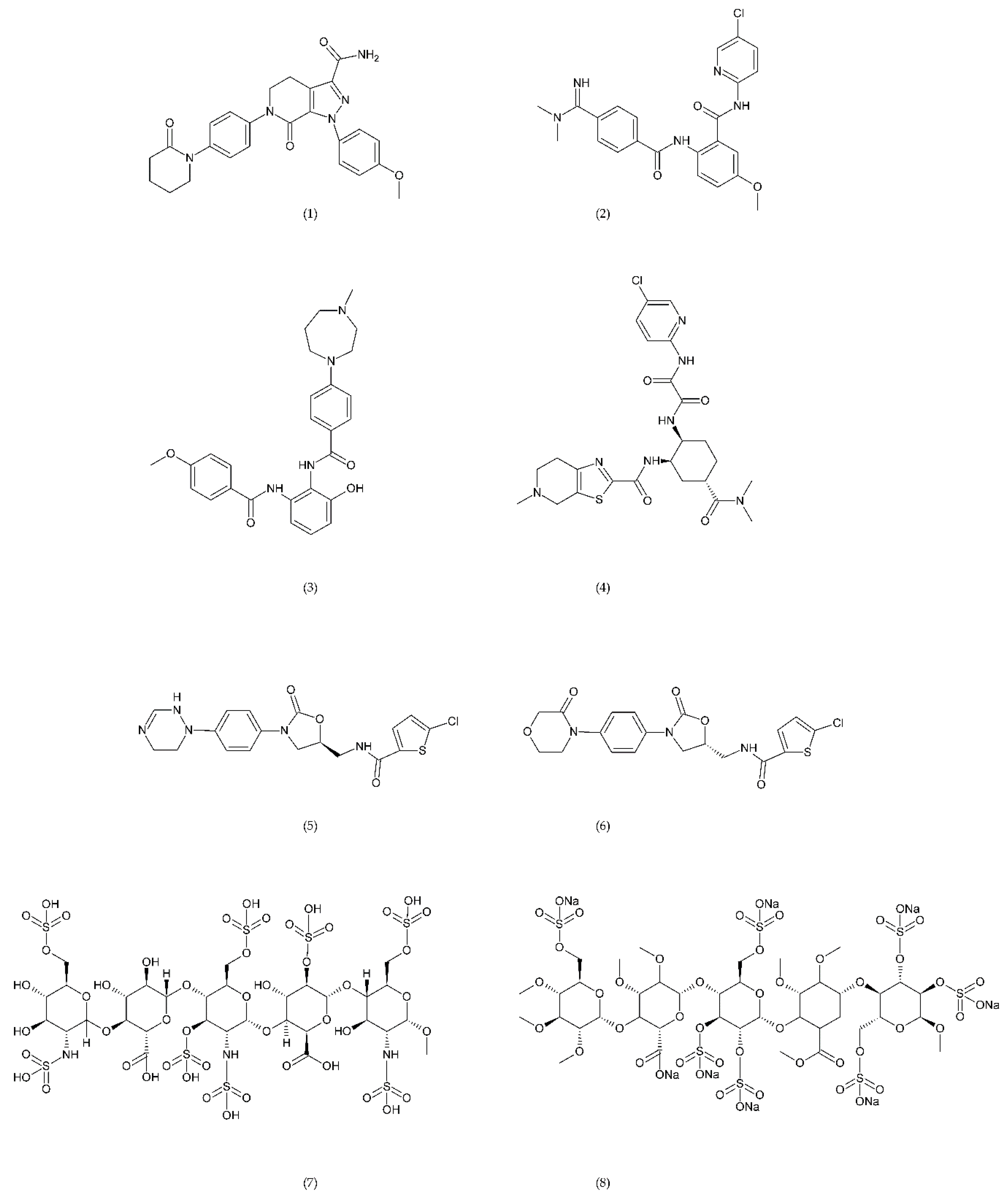{kind=link}
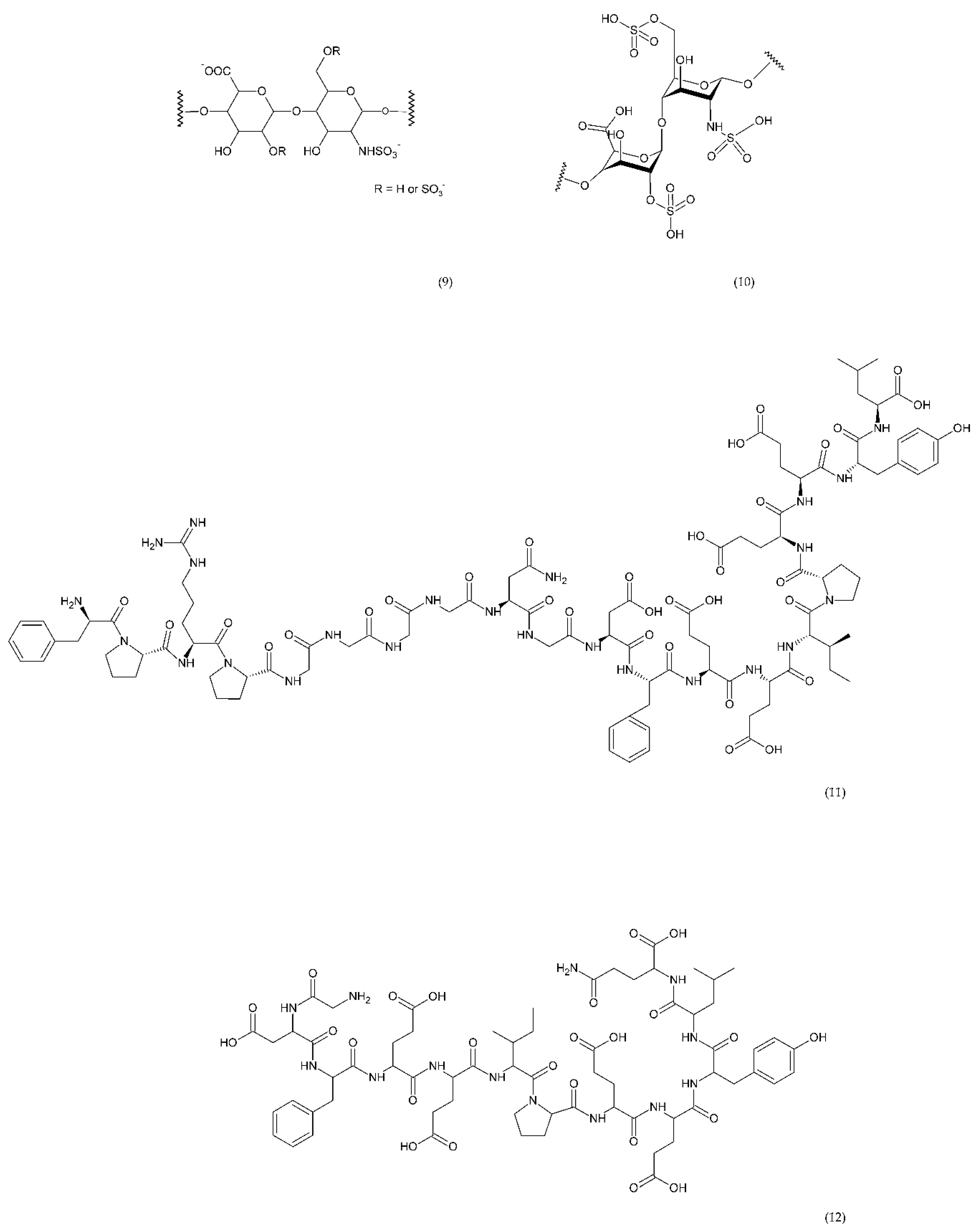{kind=link}
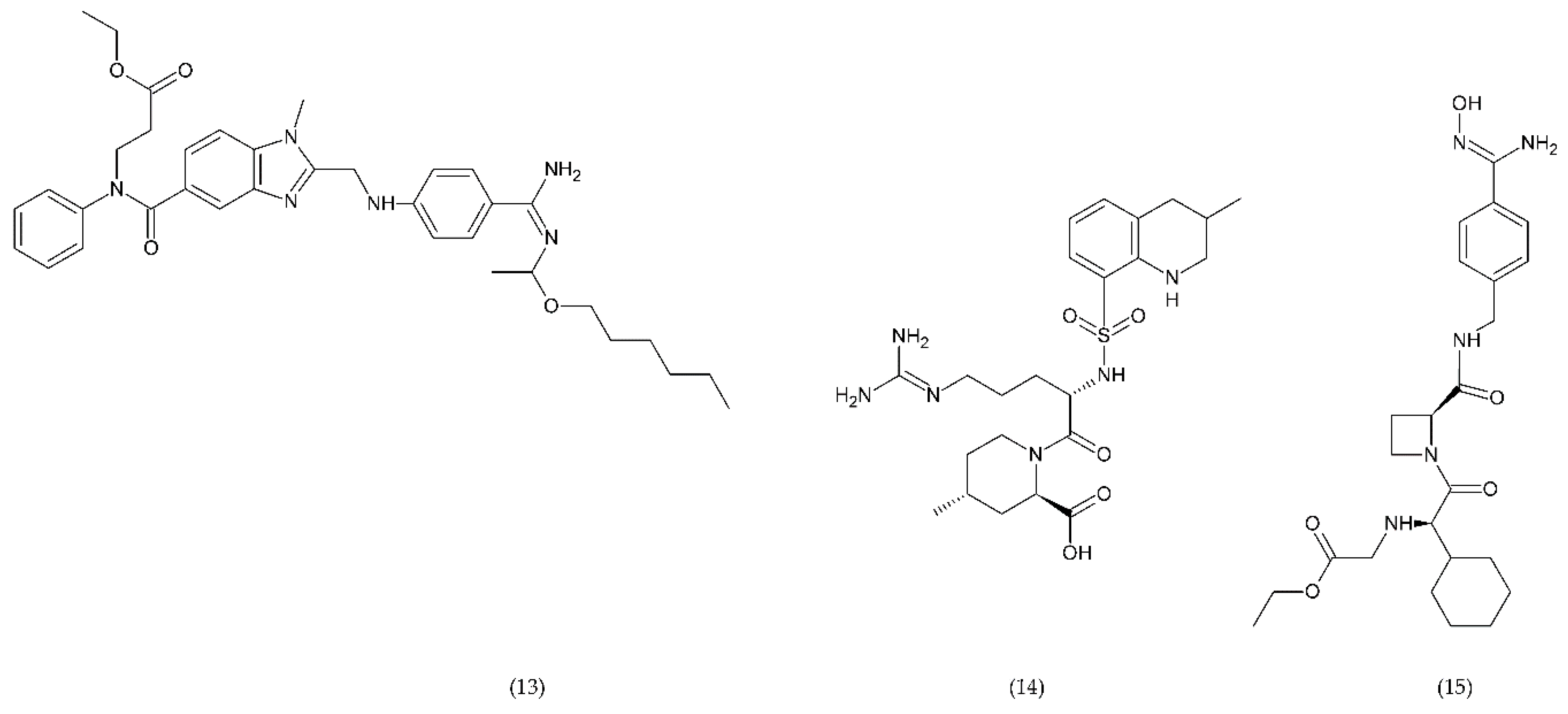{kind=link}
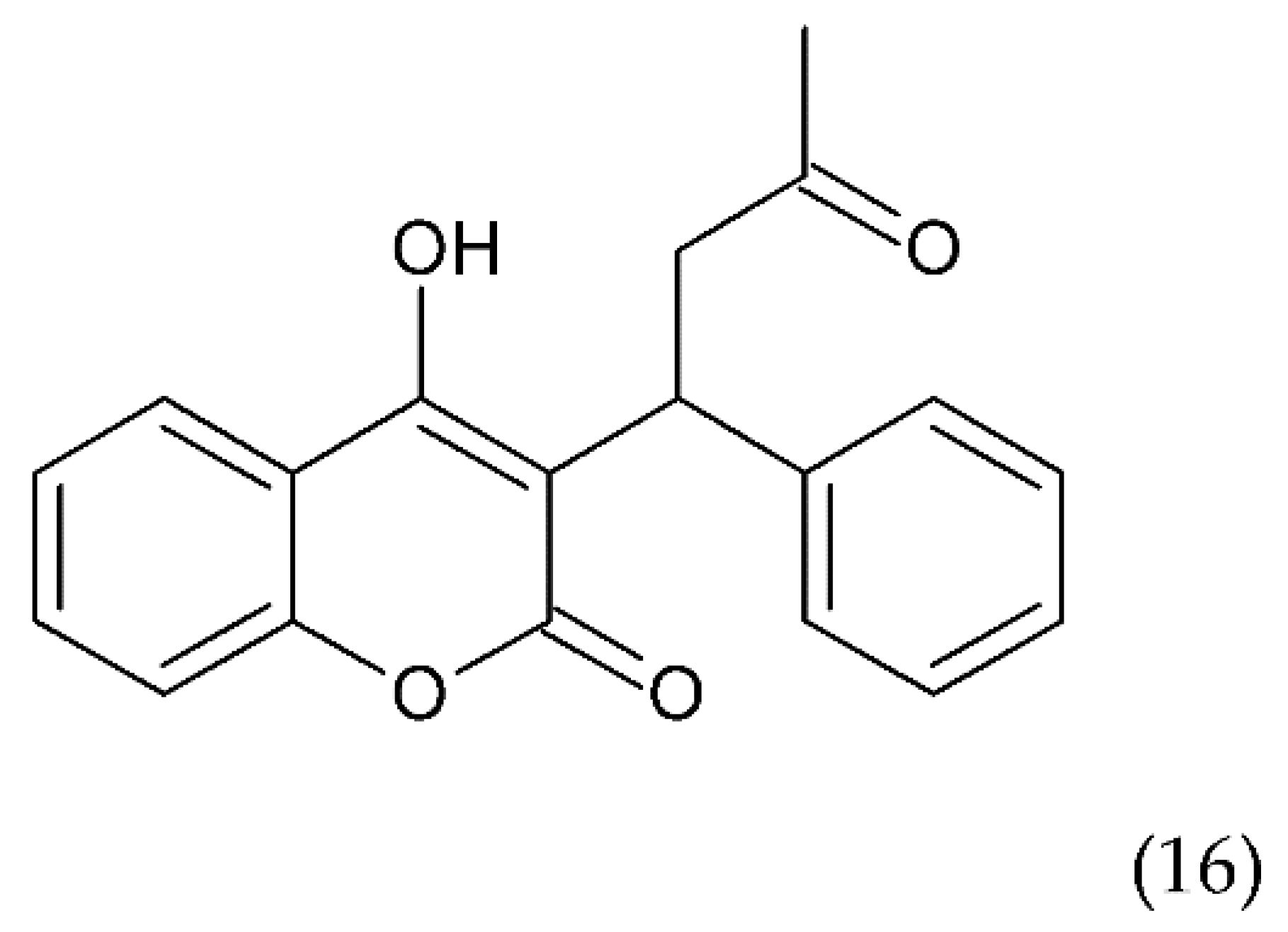{kind=link}
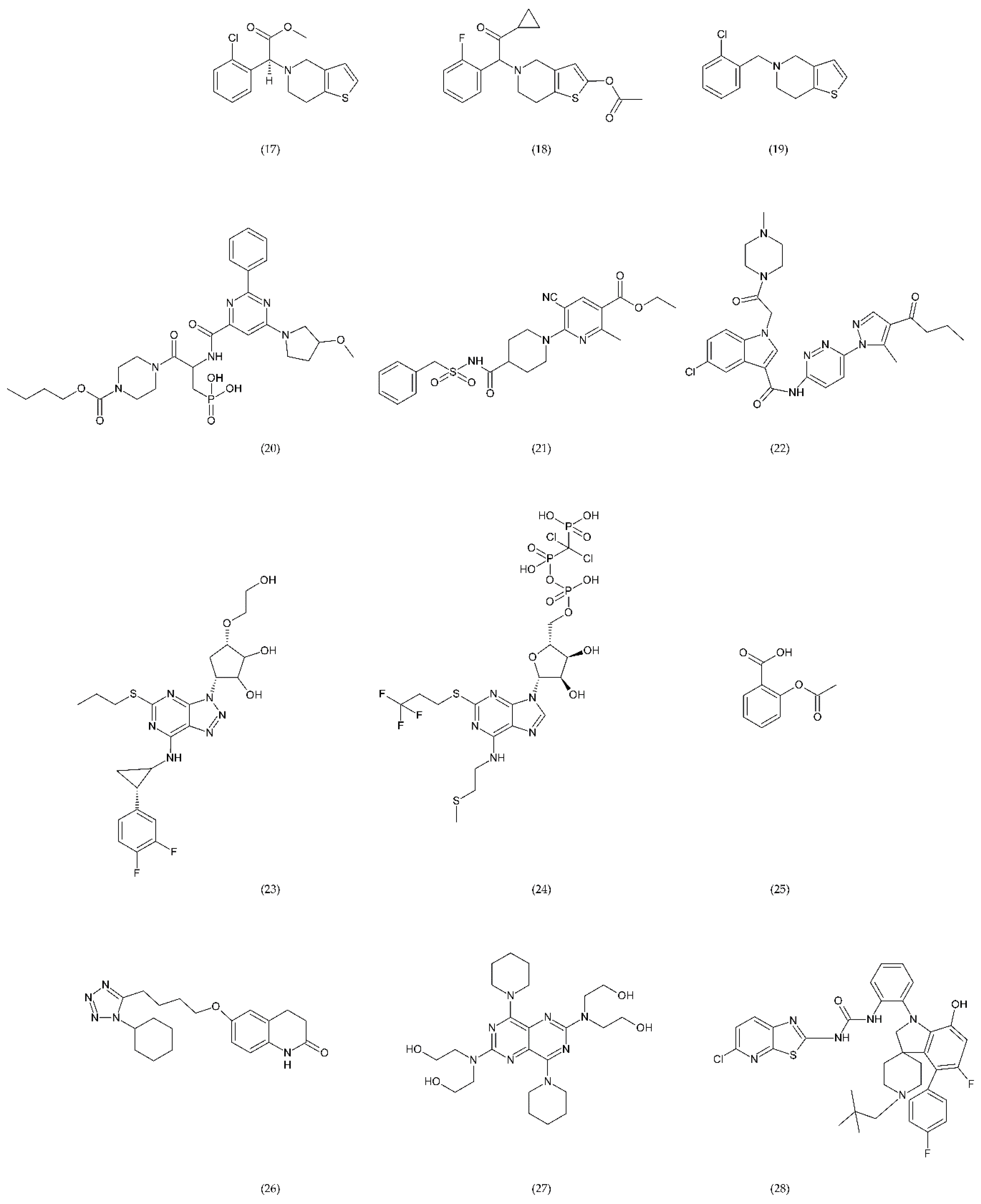{kind=link}
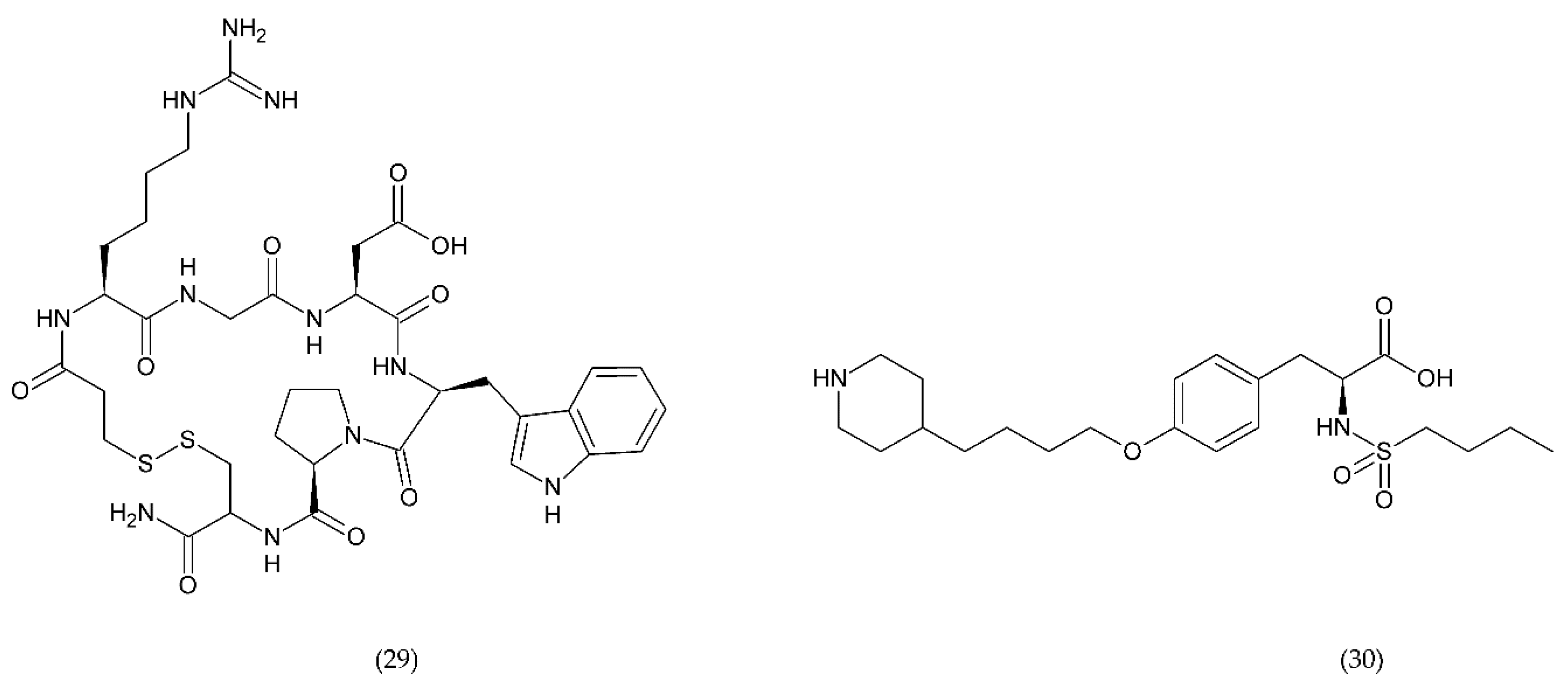{kind=link}
{kind=link}
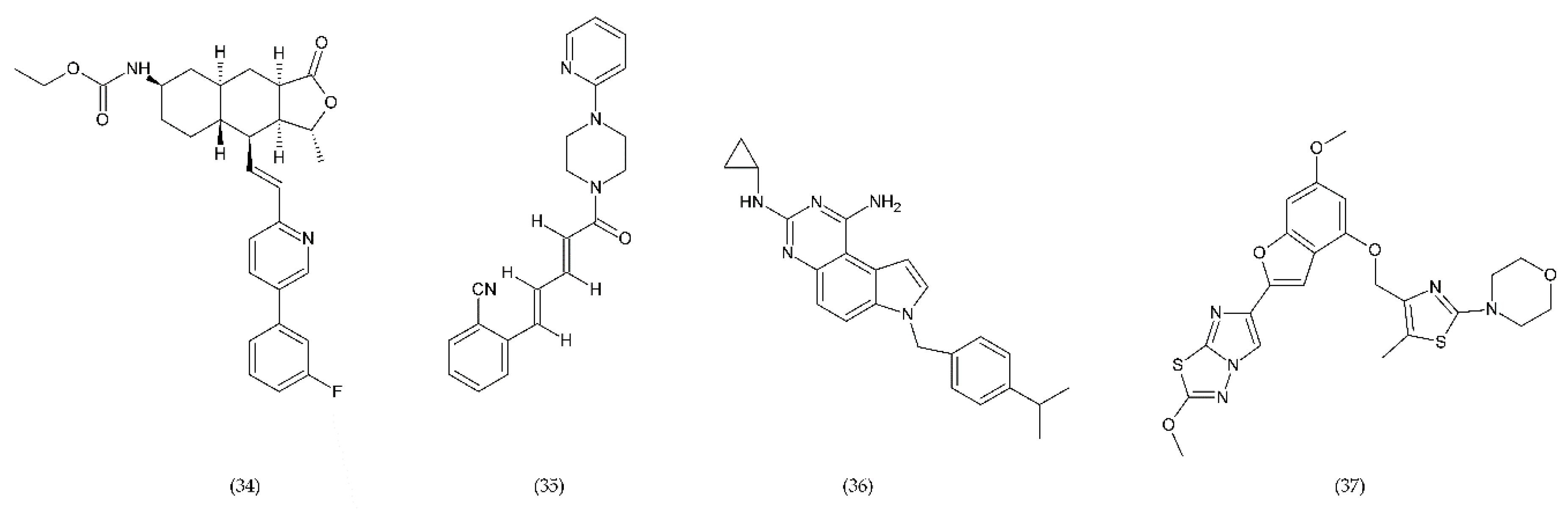{kind=link}
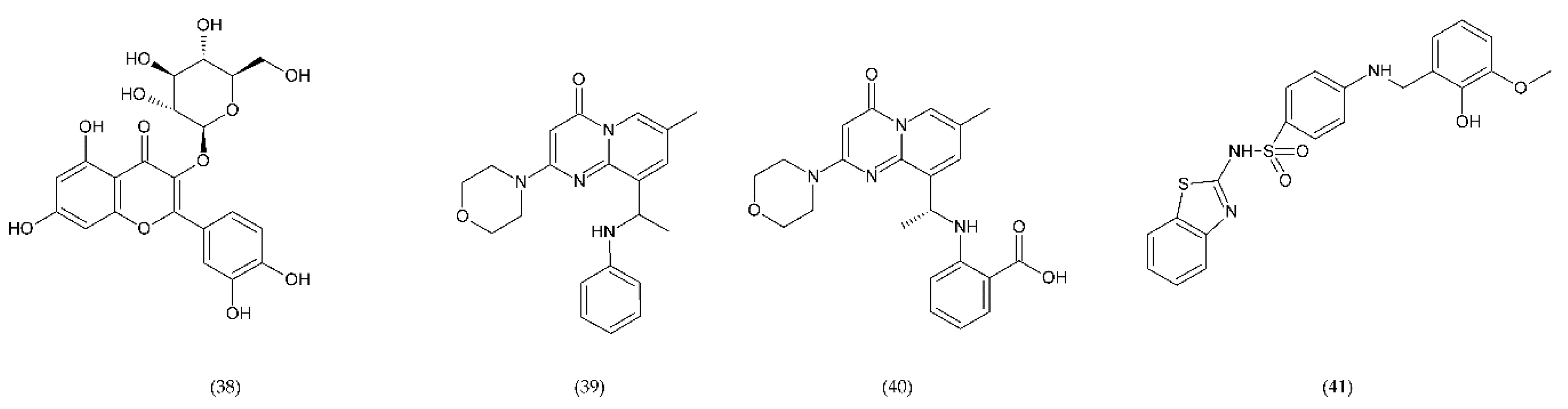{kind=link}
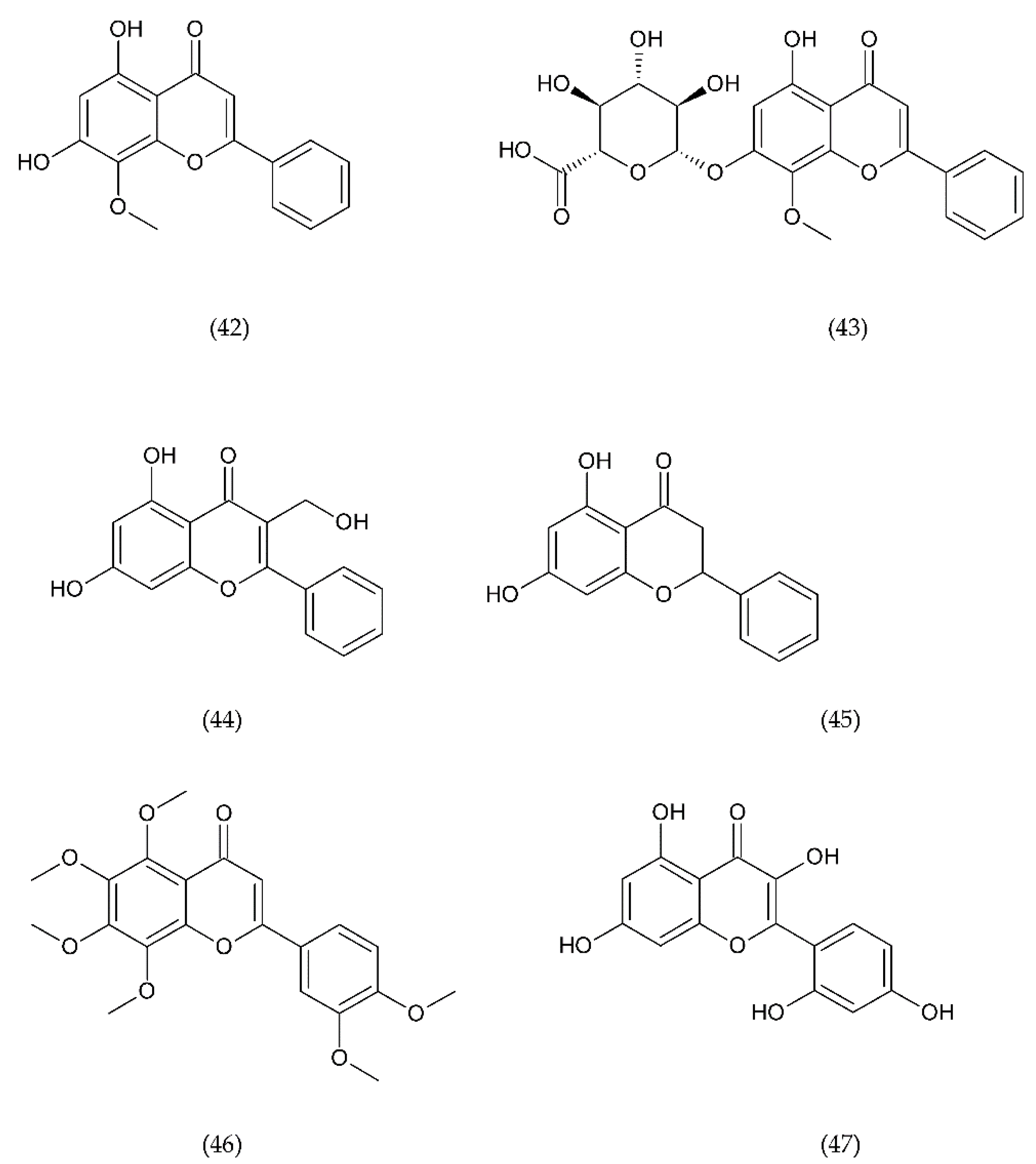{kind=link}
{kind=link}
{kind=link}
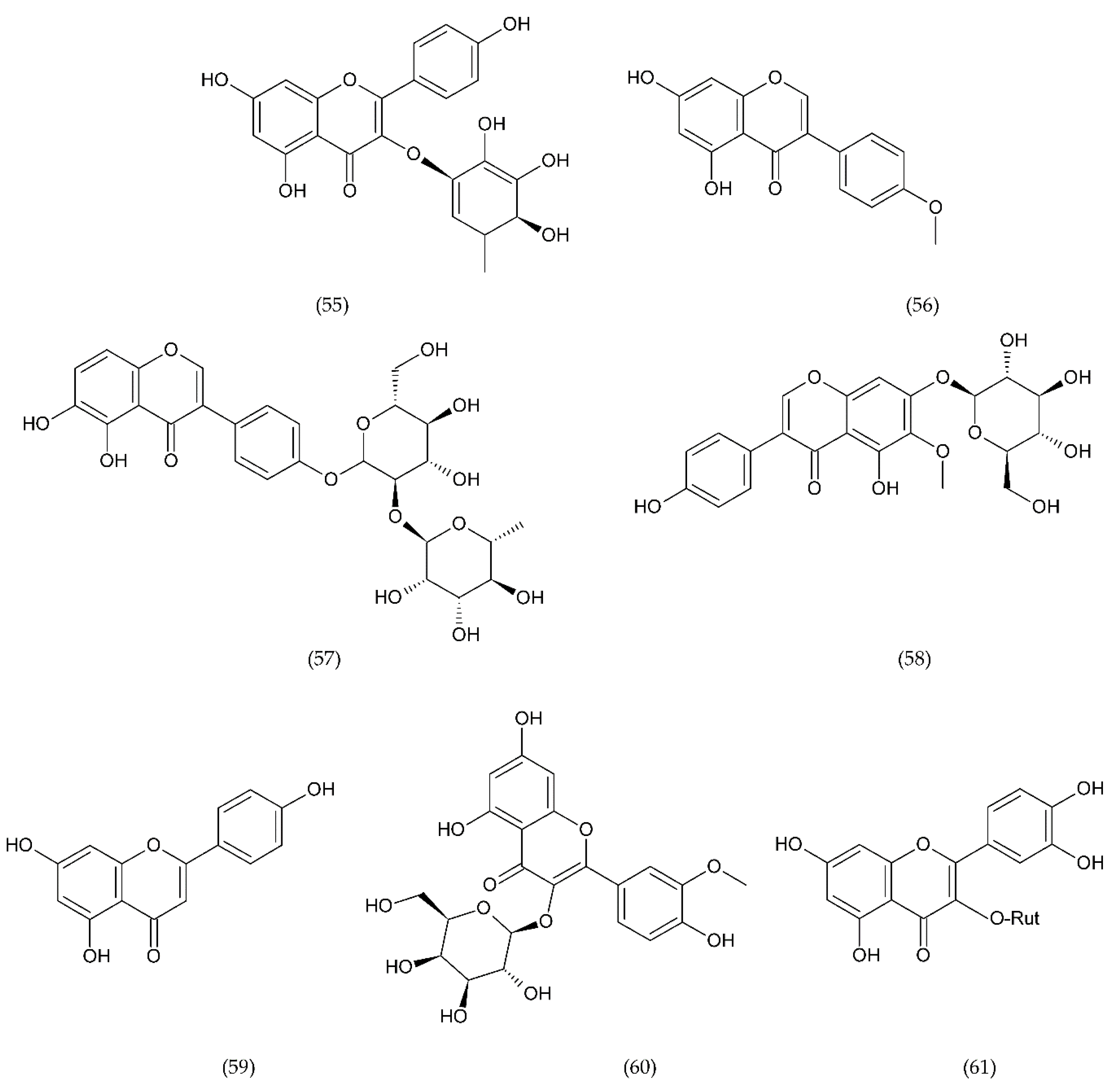{kind=link}
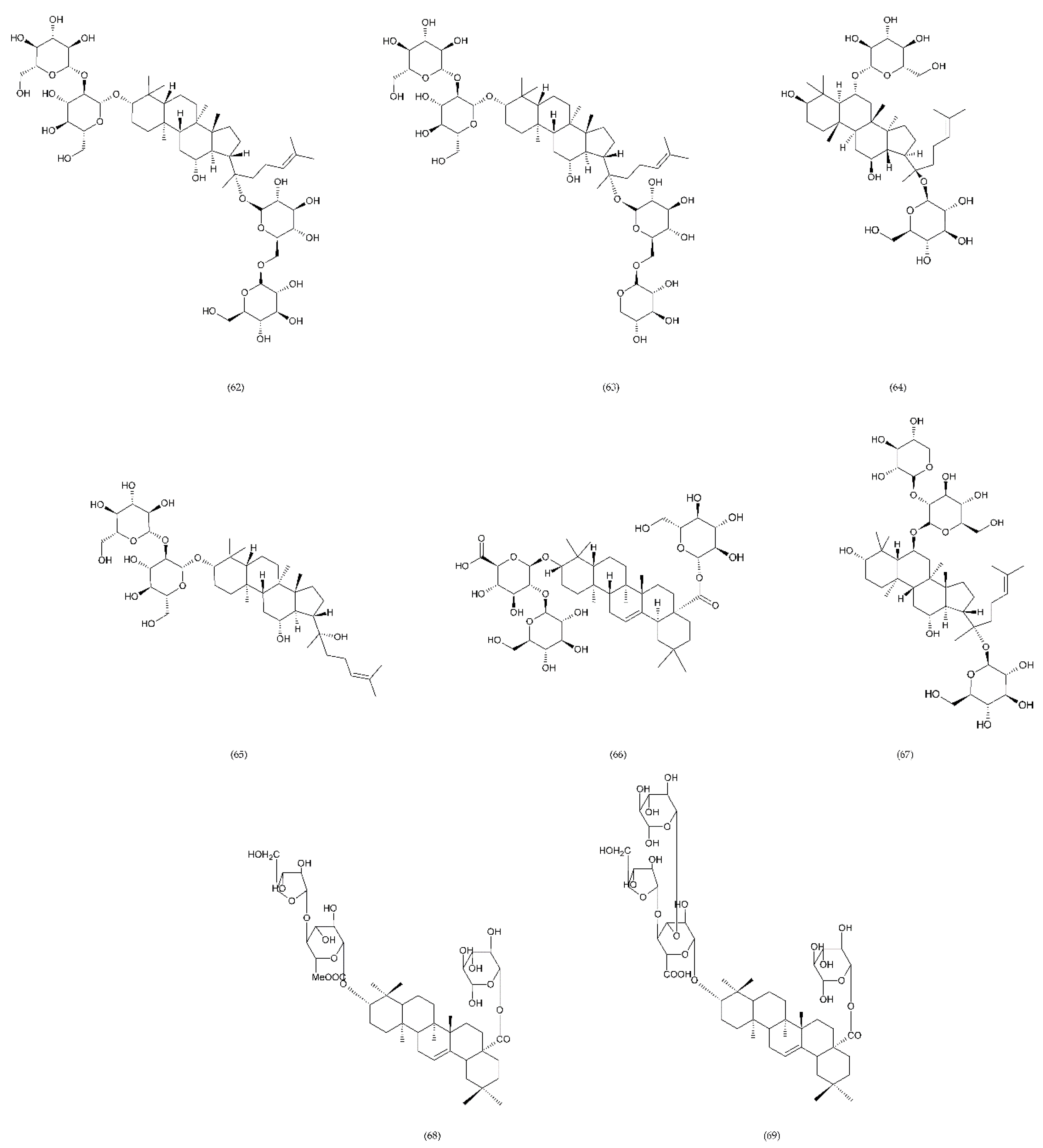{kind=link}
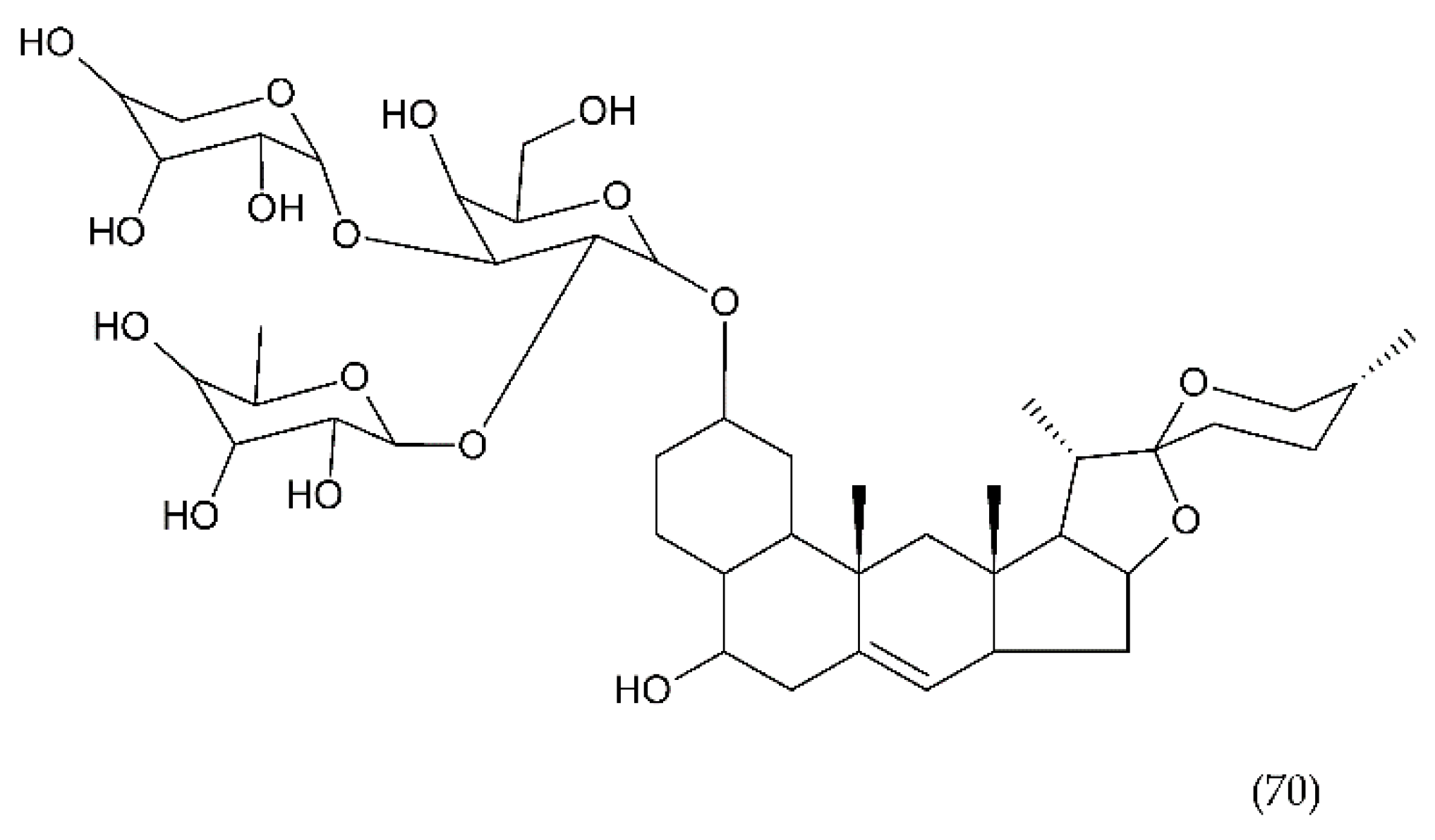{kind=link}
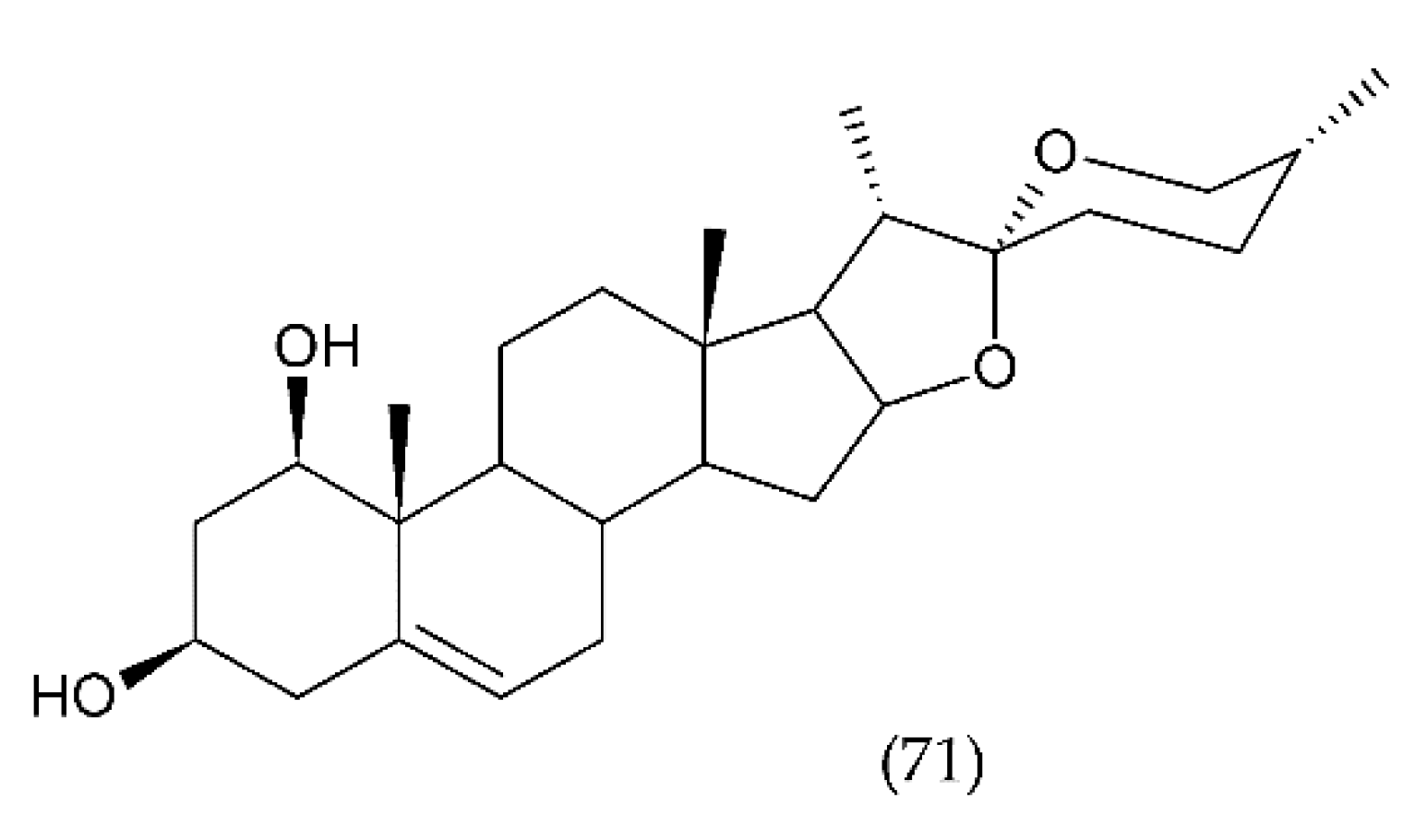{kind=link}
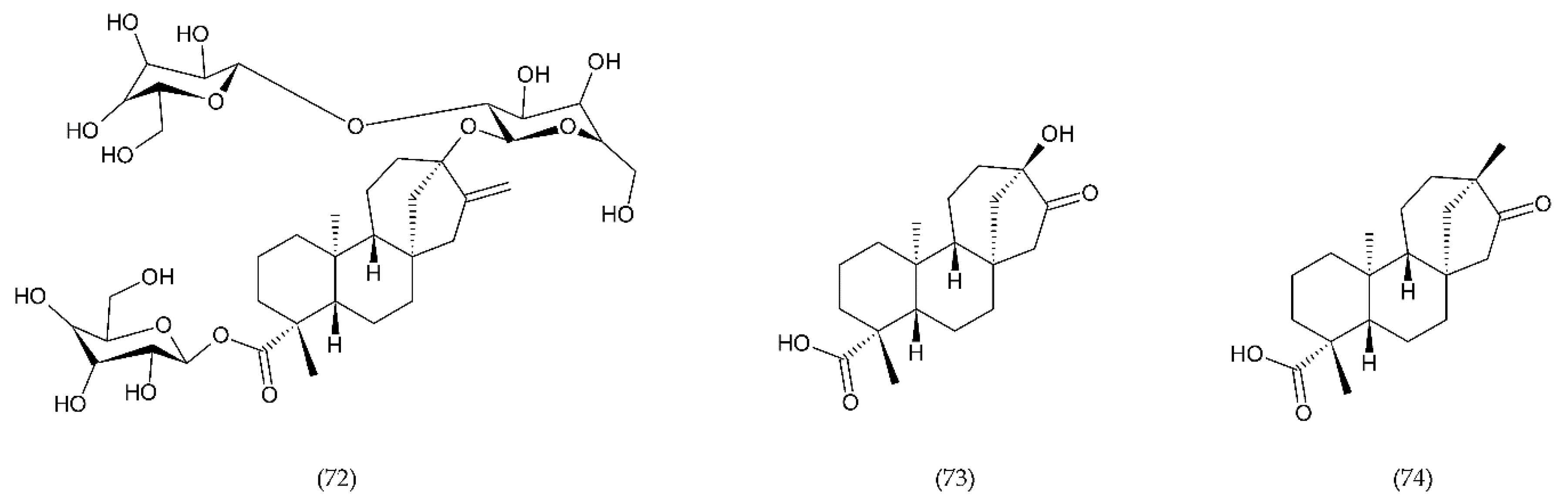{kind=link}
| Anticoagulants | ||||
|---|---|---|---|---|
| Drug | Formulation | Mechanisms of Action | Route of Administration | References |
| Apixaban BMS-562247 | 10 mg twice daily for 1 week, then 5 mg twice daily 2.5 mg twice daily for 2–5 weeks | FXa inhibitor | oral anticoagulant | [155,178,179,180] |
| Edoxaban DU-176 | 15, 30, and 60 mg daily | FXa inhibitor | oral anticoagulant | [178,179,181,182] |
| Rivaroxaban BAY59-7939 | 20 mg daily 15 mg BD for 3 weeks, then 20 mg 10 mg daily for 2–5 weeks | FXa inhibitor | oral anticoagulant | [155,178,179,180] |
| Low molecular weight heparin (LMWH) Anti-Xa | Dosing of LMWH depends on the drug used: Dalteparin: 200 U/kg OD or 100 U/kg BID Enoxaparin: 1.5 mg/kg OD or 1 mg/kg BID Tinzaparin: 175 U/kg OD Nadroparin: 171 U/kg OD | Indirect FXa inhibitor | parenteral formulation | [155,183] |
| Fondaparinux | - Under 50 kg: 5 mg once a day - 50 to 100 kg: 7.5 mg once a day - Over 100 kg: 10 mg once a day | Indirect FXa inhibitor | parenteral formulation | [155,169,184] |
| Idraparinux SR34006 | 2.5 mg once weekly | Indirect FXa inhibitor | parenteral formulation | [169] |
| Darexaban YM-150 | 30 mg or 60 mg daily | FXa inhibitor | oral anticoagulant | [155,185] |
| Betrixaban | 15 mg twice daily or 40 mg twice daily | FXa inhibitor | oral anticoagulant | [155] |
| Nokxaban GC-2107 GCC-4401C | 20 mg and 40 mg administered in a fasted state is comparable to rivaroxaban 10 mg and 20 mg in a fed state 2.5–80 mg single dose | FXa inhibitor | oral anticoagulant | [155,186,187,188] |
| Hirudin | 1.75 mg/kg/h | Thrombin inhibitor | parenteral formulation | [169] |
| Bivalirudin | 0.75 mg/kg intravenous bolus dose followed immediately 1.75 mg/kg/h intravenous infusion for the duration of the procedure | Thrombin inhibitor | parenteral formulation | [169] |
| Argatroban | 2 µg/kg/min administered as a constant infusion | Thrombin inhibitor | parenteral formulation | [169] |
| Unfractionated heparin UFH | 80 units/kg | Indirect FXa inhibitor, thrombin inhibitor | parenteral formulation | [169,189] |
| Dabigatran BIBR 1048 BIBR 953 | 150 mg twice daily (after 5 days of parenteral anticoagulation) 110 mg loading dose then 220 mg daily for 9–34 days | FIIa, thrombin inhibitor | oral anticoagulant | [155,169,178,179,190] |
| Warfarin | 5–10 mg daily | FIIa, FVIIa, FIXa, FXa inhibitor | oral anticoagulant | [155,178] |
| Antiplatelet Drugs | ||||
|---|---|---|---|---|
| Drug | Formulation | Mechanisms of Action | Route of Administration | References |
| Abciximab | Bolus 0.25 mg/kg injection then 0.125 µg/kg/min infusion | Glycoprotein IIb/IIIa inhibitor | parenteral formulation | [202,209] |
| Eptifibatide | 180 µg/kg before the start of percutaneous coronary intervention followed by a continuous infusion of 2 µg/kg/min with another 180 µg/kg IV bolus given 10 min | Glycoprotein IIb/IIIa inhibitor | parenteral formulation | [202,210] |
| Tirofiban | 0.15 µg/kg/min for 24–36 h | Glycoprotein IIb/IIIa inhibitor | parenteral formulation | [202,211] |
| Clopidogrel | 300 mg–600 mg loading dose followed by 75 mg daily, in conjunction with aspirin, ideally for up to 12 months | P2Y12 receptor antagonist | oral anticoagulant | [202,212] |
| Prasugrel LY-640315 | 60 mg loading dose 10 mg once-daily maintenance dose | P2Y12 receptor antagonist | oral anticoagulant | [202,213] |
| Cangrelor | Bolus 30 µg/kg injection then 4 µg/kg/min infusion | P2Y12 receptor blockade | parenteral formulation | [202,214] |
| Acetylsalicylic Acid ASA | In Europe loading dose 300 mg, maintenance dose 75 mg | Irreversible acetylation and inhibition of COX enzyme | oral anticoagulant | [202,213] |
| Ticagrelor | 180 mg loading dose 90 mg twice-daily maintenance dose | P2Y12 receptor blockade | oral anticoagulant | [202,213] |
| Cilostazol | 100 mg twice daily | PDE inhibitor | oral anticoagulant | [215] |
| Dipyridamole | ASA 25 mg and dipyridamole 200 mg 225 mg with 1000 mg ASA | PDE inhibitor | oral anticoagulant | [216] |
| Ticlopidine | 250 mg twice daily for 4 weeks 300 mg daily | ADP receptor inhibitor | oral anticoagulant | [217] |
| Selatogrel ACT-246475 | 8 mg or 16 mg single dose | P2Y12 receptor antagonist | oral anticoagulant | [218,219] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lichota, A.; Szewczyk, E.M.; Gwozdzinski, K. Factors Affecting the Formation and Treatment of Thrombosis by Natural and Synthetic Compounds. Int. J. Mol. Sci. 2020, 21, 7975. https://doi.org/10.3390/ijms21217975
Lichota A, Szewczyk EM, Gwozdzinski K. Factors Affecting the Formation and Treatment of Thrombosis by Natural and Synthetic Compounds. International Journal of Molecular Sciences. 2020; 21(21):7975. https://doi.org/10.3390/ijms21217975
Chicago/Turabian StyleLichota, Anna, Eligia M. Szewczyk, and Krzysztof Gwozdzinski. 2020. "Factors Affecting the Formation and Treatment of Thrombosis by Natural and Synthetic Compounds" International Journal of Molecular Sciences 21, no. 21: 7975. https://doi.org/10.3390/ijms21217975
APA StyleLichota, A., Szewczyk, E. M., & Gwozdzinski, K. (2020). Factors Affecting the Formation and Treatment of Thrombosis by Natural and Synthetic Compounds. International Journal of Molecular Sciences, 21(21), 7975. https://doi.org/10.3390/ijms21217975