Neurobiology of Cancer: The Role of β-Adrenergic Receptor Signaling in Various Tumor Environments


Abstract
:
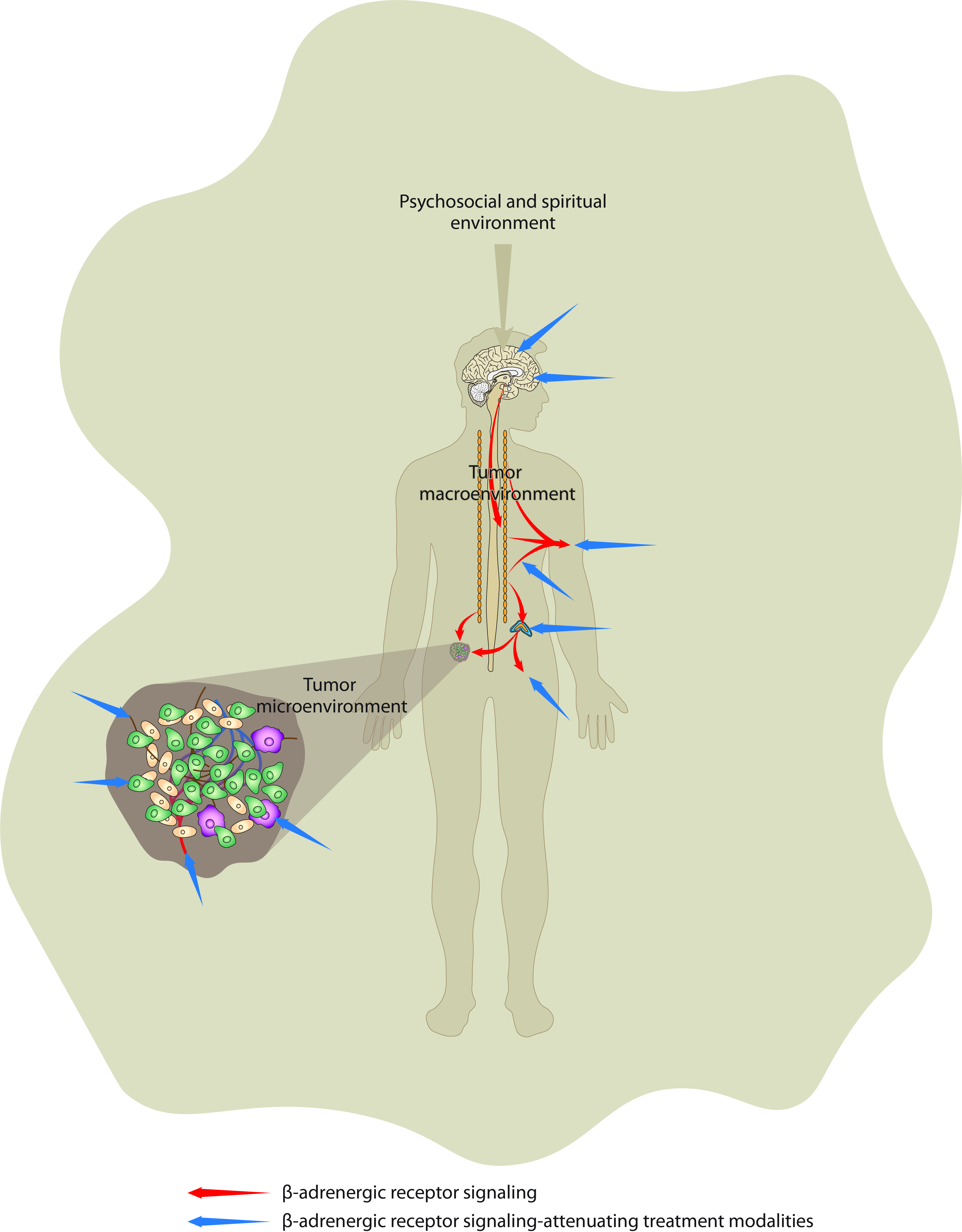{kind=link}
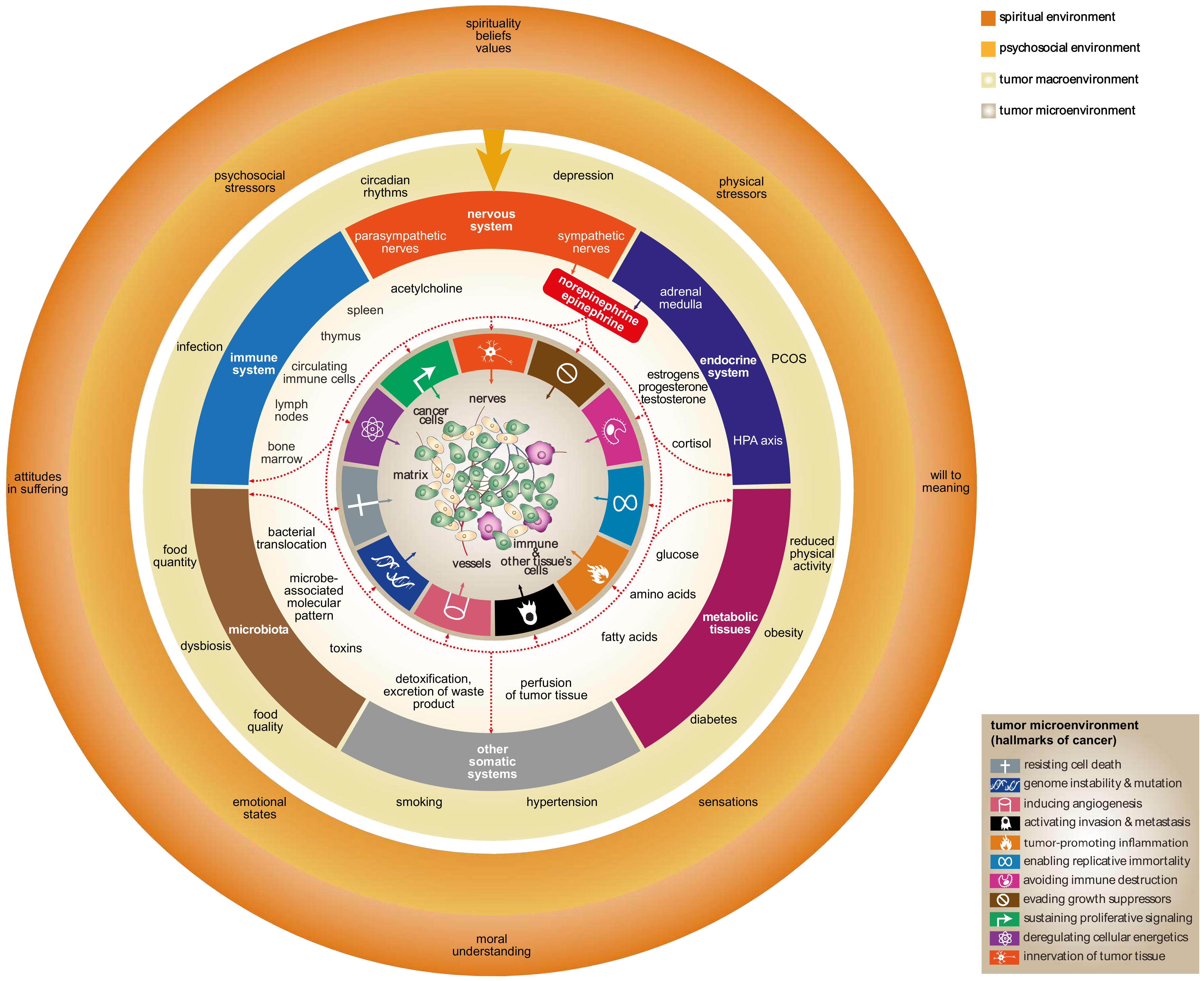
{kind=link}
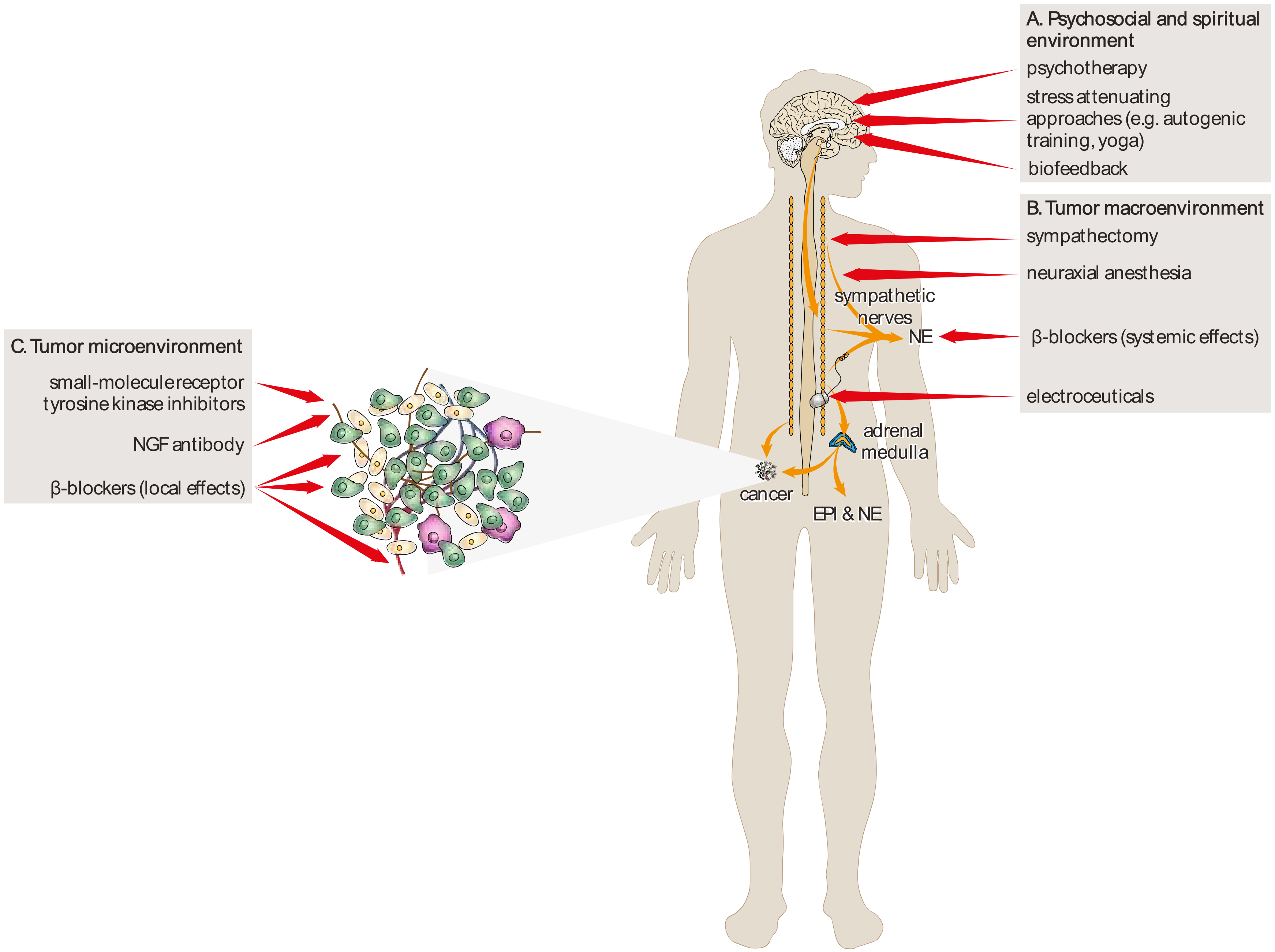
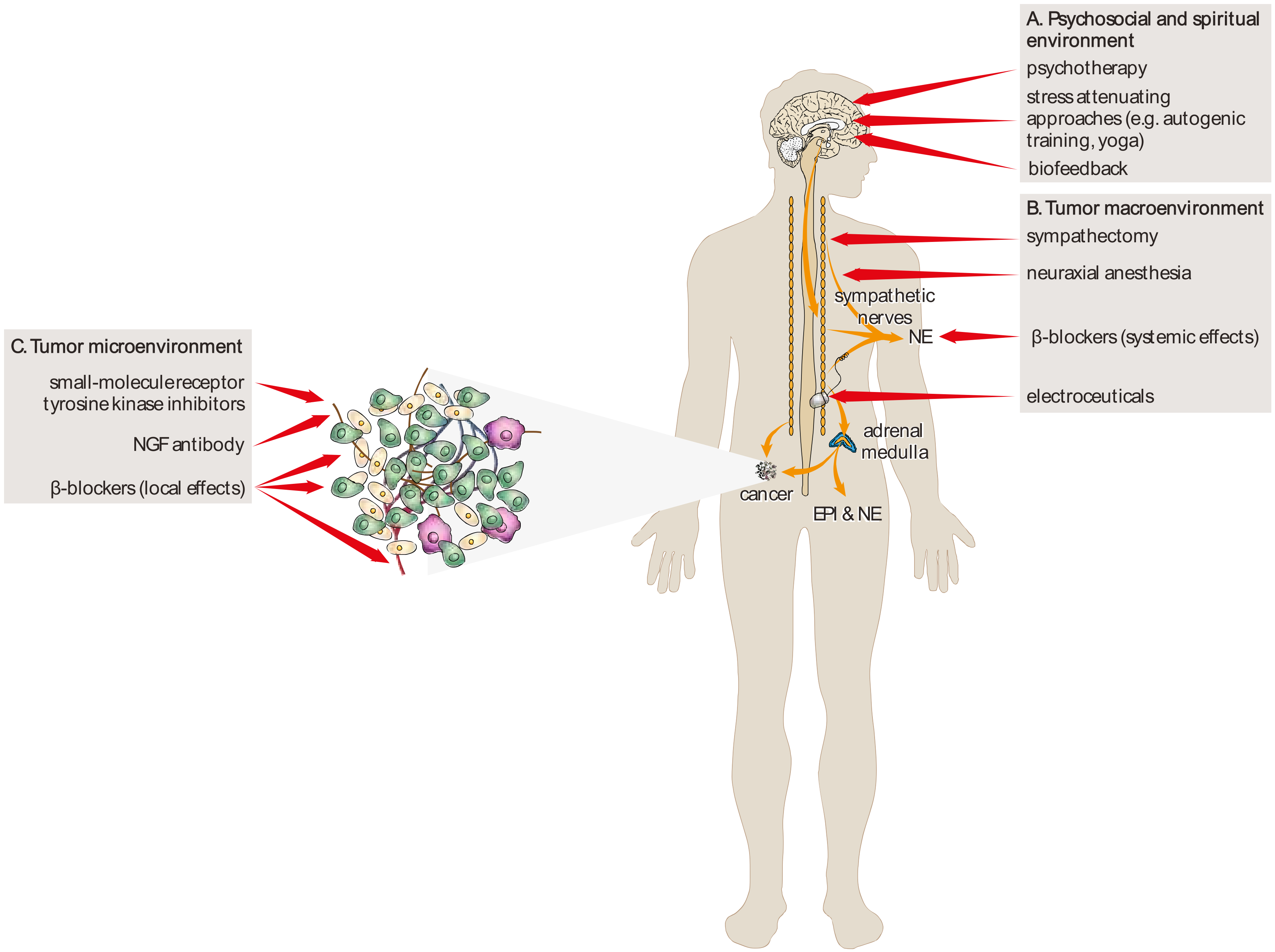{kind=link}
1. Introduction
2. Norepinephrine and Epinephrine, the Effector Molecules of Sympathoadrenal System
3. Norepinephrine and Epinephrine Effects on Cancer
3.1. Effects of β-Adrenergic Signaling at the Level of Tumor Microenvironment
3.1.1. β-Adrenergic Signaling Induces Genome Instability and Mutation, and Attenuates DNA Damage Repair Mechanisms
3.1.2. β-Adrenergic Signaling Potentiates Sustained Proliferative Signaling
3.1.3. β-Adrenergic Signaling Increases Resistance to Cell Death
3.1.4. β-Adrenergic Signaling Induces Cell Motility and Trafficking and Activates Invasion and Metastasis
3.1.5. β-Adrenergic Signaling Induces Vascular Remodeling and Stimulates Angiogenesis
3.1.6. β-Adrenergic Signaling Induces Avoidance to Immune Destruction
3.1.7. β-Adrenergic Signaling Induces Tumor-Promoting Inflammation
3.1.8. β-Adrenergic Signaling Affects Cancer Cell Energetics
3.2. Effects of β-Adrenergic Signaling at the Level of the Tumor Macroenvironment
3.2.1. Nervous System
The Sympathoadrenal System Affects Metastasis
3.2.2. Immune System
3.2.3. Endocrine System
3.2.4. Metabolism
3.2.5. Microbiota
3.2.6. Somatic Diseases Predisposing to Cancer that Are Characterized by Increased β-Adrenergic Signaling
3.3. Effects of β-Adrenergic Signaling at the Level of Psychosocial and Spiritual/Noetic Environments
4. Therapeutic Implications and Future Directions
4.1. Pharmacological Approaches
4.2. Non-Pharmacological Approaches
4.3. The Source of Ambiguity
4.4. Future Directions
5. Conclusions
Funding
Conflicts of Interest
References
- Lamboy-Caraballo, R.; Ortiz, C.; Acevedo-Santiago, A.; Matta, J.; Monteiro, A.N.A.; Armaiz-Pena, G.N. Norepinephrine-Induced DNA Damage in Ovarian Cancer Cells. Int. J. Mol. Sci. 2020, 21, 2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhang, Y.; He, Z.; Yin, K.; Li, B.; Zhang, L.; Xu, Z. Chronic stress promotes gastric cancer progression and metastasis: An essential role for ADRB2. Cell Death Dis. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, T.; Cui, X.; Li, W.; Lin, W.; Li, Y.; Chen, X.; Wu, T. Novel regulatory program for norepinephrine-induced epithelial–mesenchymal transition in gastric adenocarcinoma cell lines. Cancer Sci. 2014, 105, 847–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbieri, A.; Bimonte, S.; De Palma, G.; Luciano, A.; Rea, D.; Giudice, A.; Scognamiglio, G.; La Mantia, E.; Franco, R.; Perdonà, S.; et al. The stress hormone norepinephrine increases migration of prostate cancer cells in vitro and in vivo. Int. J. Oncol. 2015, 47, 527–534. [Google Scholar] [CrossRef] [Green Version]
- Bastos, D.B.; Sarafim-Silva, B.A.M.; Sundefeld, M.L.M.M.; Ribeiro, A.A.; Brandão, J.D.P.; Éder, B.R.; Miyahara, G.I.; Casarini, D.E.; Bernabé, D.G. Circulating catecholamines are associated with biobehavioral factors and anxiety symptoms in head and neck cancer patients. PLoS ONE 2018, 13, e0202515. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Wu, C.; Chen, W.; Qiu, L.; Li, S.; Wang, T.; Xie, H.; Li, Y.; Li, C.; Li, L. The stress hormone norepinephrine promotes tumor progression through beta2-adrenoreceptors in oral cancer. Arch. Oral Biol. 2020, 113, 104712. [Google Scholar] [CrossRef]
- Rains, S.L.; Amaya, C.N.; Bryan, B.A. Beta-adrenergic receptors are expressed across diverse cancers. Oncoscience 2017, 4, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Kurozumi, S.; Kaira, K.; Matsumoto, H.; Hirakata, T.; Yokobori, T.; Inoue, K.; Horiguchi, J.; Katayama, A.; Koshi, H.; Shimizu, A.; et al. Beta2-Adrenergic receptor expression is associated with biomarkers of tumor immunity and predicts poor prognosis in estrogen receptor-negative breast cancer. Breast Cancer Res. Treat. 2019, 177, 603–610. [Google Scholar] [CrossRef]
- Yazawa, T.; Kaira, K.; Shimizu, K.; Shimizu, A.; Mori, K.; Nagashima, T.; Ohtaki, Y.; Oyama, T.; Mogi, A.; Kuwano, H. Prognostic significance of beta2-adrenergic receptor expression in non-small cell lung cancer. Am. J. Transl. Res. 2016, 8, 5059–5070. [Google Scholar]
- Mravec, B.; Tibensky, M.; Horvathova, L. Stress and cancer. Part I: Mechanisms mediating the effect of stressors on cancer. J. Neuroimmunol. 2020, 346, 577311. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Kvetnansky, R.; Sabban, E.L.; Palkovits, M. Catecholaminergic Systems in Stress: Structural and Molecular Genetic Approaches. Physiol. Rev. 2009, 89, 535–606. [Google Scholar] [CrossRef] [PubMed]
- Tank, A.W.; Wong, D.L. Peripheral and Central Effects of Circulating Catecholamines. Compr. Physiol. 2014, 5, 1–15. [Google Scholar] [CrossRef]
- Marino, F.; Cosentino, M. Adrenergic modulation of immune cells: An update. Amino Acids 2011, 45, 55–71. [Google Scholar] [CrossRef]
- Jänig, W. Integrative Action of the Autonomic Nervous System; Cambridge University Press (CUP): Cambridge, UK, 2006; p. 610. [Google Scholar]
- Bleich, H.L.; Moore, M.J.; Cryer, P.E. Physiology and Pathophysiology of the Human Sympathoadrenal Neuroendocrine System. N. Engl. J. Med. 1980, 303, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Hulsurkar, M.; Li, Z.; Zhang, Y.; Li, X.; Zheng, D.; Li, W. Beta-adrenergic signaling promotes tumor angiogenesis and prostate cancer progression through HDAC2-mediated suppression of thrombospondin-1. Oncogene 2016, 36, 1525–1536. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Gill, N.K.; Nyberg, K.D.; Nguyen, A.V.; Hohlbauch, S.V.; Geisse, N.A.; Nowell, C.J.; Sloan, E.K.; Rowat, A.C. Cancer cells become less deformable and more invasive with activation of beta-adrenergic signaling. J. Cell Sci. 2016, 129, 4563–4575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.; Wei, Y.; Li, Z.-Y.; Cai, X.-Y.; Zhang, L.-L.; Dong, X.-R.; Zhang, S.; Zhang, R.-G.; Meng, R.; Zhu, F.; et al. Catecholamines contribute to the neovascularization of lung cancer via tumor-associated macrophages. Brain Behav. Immun. 2019, 81, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Li, Y.; Li, Z.Z.; Sun, J.; Li, J.W.; Wei, W.; Li, L.; Zhang, C.; Huang, C.; Yang, S.Y.; et al. Chronic restraint stress promotes hepatocellular carcinoma growth by mobilizing splenic myeloid cells through activating beta-adrenergic signaling. Brain Behav. Immun. 2019, 80, 825–838. [Google Scholar] [CrossRef]
- Horvathova, L.; Padova, A.; Tillinger, A.; Osacka, J.; Bizik, J.; Mravec, B. Sympathectomy reduces tumor weight and affects expression of tumor-related genes in melanoma tissue in the mouse. Stress 2016, 19, 528–534. [Google Scholar] [CrossRef]
- Horvathova, L.; Tillinger, A.; Padova, A.; Mravec, B. Sympathectomized tumor-bearing mice survive longer but develop bigger melanomas. Endocr. Regul. 2016, 50, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Raju, B.; Hultström, M.; Haug, S.R.; Ibrahim, S.O.; Heyeraas, K.J. Sympathectomy suppresses tumor growth and alters gene-expression profiles in rat tongue cancer. Eur. J. Oral Sci. 2009, 117, 351–361. [Google Scholar] [CrossRef]
- Magnon, C.; Hall, S.J.; Lin, J.; Xue, X.; Gerber, L.; Freedland, S.J.; Frenette, P.S. Autonomic Nerve Development Contributes to Prostate Cancer Progression. Science 2013, 341, 1236361. [Google Scholar] [CrossRef] [Green Version]
- Ben-Eliyahu, S.; Shakhar, G.; Page, G.G.; Stefanski, V.; Shakhar, K. Suppression of NK cell activity and of resistance to metastasis by stress: A role for adrenal catecholamines and beta-adrenoceptors. Neuroimmunomodulation 2000, 8, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Montoya, A.; Varela-Ramírez, A.; Dickerson, E.; Pasquier, Y.; Torabi, A.; Aguilera, R.; Nahleh, Z.; Bryan, B. The beta adrenergic receptor antagonist propranolol alters mitogenic and apoptotic signaling in late stage breast cancer. Biomed. J. 2019, 42, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Shaashua, L.; Shabat-Simon, M.; Haldar, R.; Matzner, P.; Zmora, O.; Shabtai, M.; Sharon, E.; Allweis, T.; Barshack, I.; Hayman, L.; et al. Perioperative COX-2 and beta-Adrenergic Blockade Improves Metastatic Biomarkers in Breast Cancer Patients in a Phase-II Randomized Trial. Clin. Cancer Res. 2017, 23, 4651–4661. [Google Scholar] [CrossRef] [Green Version]
- Daher, C.; Vimeux, L.; Stoeva, R.; Peranzoni, E.; Bismuth, G.; Wieduwild, E.; Lucas, B.; Donnadieu, E.; Bercovici, N.; Trautmann, A.; et al. Blockade of beta-Adrenergic Receptors Improves CD8(+) T-cell Priming and Cancer Vaccine Efficacy. Cancer Immunol. Res. 2019, 7, 1849–1863. [Google Scholar] [CrossRef] [PubMed]
- Udumyan, R.; Montgomery, S.; Duberg, A.-S.; Fang, F.; Valdimarsdottir, U.; Ekbom, A.; Smedby, K.E.; Fall, K. Beta-adrenergic receptor blockers and liver cancer mortality in a national cohort of hepatocellular carcinoma patients. Scand. J. Gastroenterol. 2020, 55, 597–605. [Google Scholar] [CrossRef]
- Cole, S.W.; Sood, A.K. Molecular Pathways: Beta-Adrenergic Signaling in Cancer: Figure 1. Clin. Cancer Res. 2011, 18, 1201–1206. [Google Scholar] [CrossRef] [Green Version]
- Krizanova, O.; Babula, P.; Pacak, K. Stress, catecholaminergic system and cancer. Stress 2016, 19, 419–428. [Google Scholar] [CrossRef]
- Qiao, G.; Chen, M.; Bucsek, M.J.; Repasky, E.A.; Hylander, B.L. Adrenergic Signaling: A Targetable Checkpoint Limiting Development of the Antitumor Immune Response. Front. Immunol. 2018, 9, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Cao, X. Beta-Adrenergic Signaling in Tumor Immunology and Immunotherapy. Crit. Rev. Immunol. 2019, 39, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Lutgendorf, S.K.; DeGeest, K.; Dahmoush, L.; Farley, D.; Penedo, F.; Bender, D.; Goodheart, M.; Buekers, T.E.; Mendez, L.; Krueger, G.; et al. Social isolation is associated with elevated tumor norepinephrine in ovarian carcinoma patients. Brain Behav. Immun. 2011, 25, 250–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szpunar, M.J.; Belcher, E.K.; Dawes, R.P.; Madden, K.S. Sympathetic innervation, norepinephrine content, and norepinephrine turnover in orthotopic and spontaneous models of breast cancer. Brain Behav. Immun. 2016, 53, 223–233. [Google Scholar] [CrossRef] [Green Version]
- Sone, Y.; Takatori, S.; Ochi, E.; Zamami, Y.; Matsuyama, A.; Fukuhara, S.; Goda, M.; Kitamura, Y.; Kawasaki, H. Nerve Growth Factor Facilitates the Innervation of Perivascular Nerves in Tumor-Derived Neovasculature in the Mouse Cornea. Pharmacology 2016, 99, 57–66. [Google Scholar] [CrossRef]
- March, B.; Faulkner, S.; Jobling, P.; Steigler, A.; Blatt, A.; Denham, J.; Hondermarck, H. Tumour innervation and neurosignalling in prostate cancer. Nat. Rev. Urol. 2020, 17, 119–130. [Google Scholar] [CrossRef]
- Zahalka, A.H.; Frenette, P.S. Nerves in cancer. Nat. Rev. Cancer 2020, 20, 143–157. [Google Scholar] [CrossRef]
- Nance, D.M.; Sanders, V.M. Autonomic innervation and regulation of the immune system (1987–2007). Brain Behav. Immun. 2007, 21, 736–745. [Google Scholar] [CrossRef] [Green Version]
- Stavropoulos, I.; Sarantopoulos, A.; Liverezas, A. Does sympathetic nervous system modulate tumor progression? A narrative review of the literature. J. Drug Assess. 2020, 9, 106–116. [Google Scholar] [CrossRef]
- Vermeer, P.D. Exosomal Induction of Tumor Innervation. Cancer Res. 2019, 79, 3529–3535. [Google Scholar] [CrossRef]
- Mauffrey, P.; Tchitchek, N.; Barroca, V.; Bemelmans, A.-P.; Firlej, V.; Allory, Y.; Roméo, P.-H.; Magnon, C. Progenitors from the central nervous system drive neurogenesis in cancer. Nat. Cell Biol. 2019, 569, 672–678. [Google Scholar] [CrossRef]
- Amit, M.; Takahashi, H.; Dragomir, M.P.; Lindemann, A.; Gleber-Netto, F.O.; Pickering, C.R.; Anfossi, S.; Osman, A.A.; Cai, Y.; Wang, R.; et al. Loss of p53 drives neuron reprogramming in head and neck cancer. Nat. Cell Biol. 2020, 578, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Broustas, C.G.; Lieberman, H.B. DNA Damage Response Genes and the Development of Cancer Metastasis. Radiat. Res. 2014, 181, 111–130. [Google Scholar] [CrossRef] [Green Version]
- Flint, M.S.; Baum, A.; Chambers, W.H.; Jenkins, F.J. Induction of DNA damage, alteration of DNA repair and transcriptional activation by stress hormones. Psychoneuroendocrinology 2007, 32, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.R.; Kovacs, J.J.; Whalen, E.J.; Rajagopal, S.; Strachan, R.T.; Grant, W.; Towers, A.J.; Williams, B.; Lam, C.M.; Xiao, K.; et al. A stress response pathway regulates DNA damage through beta2-adrenoreceptors and beta-arrestin-1. Nature 2011, 477, 349–353. [Google Scholar] [CrossRef] [Green Version]
- Coelho, M.; Soares-Silva, C.; Brandao, D.; Marino, F.; Cosentino, M.; Ribeiro, L. Beta-Adrenergic modulation of cancer cell proliferation: Available evidence and clinical perspectives. J. Cancer Res. Clin. Oncol. 2017, 143, 275–291. [Google Scholar] [CrossRef]
- Liao, X.; Che, X.; Zhao, W.; Zhang, D.; Bi, T.; Wang, G. The beta-adrenoceptor antagonist, propranolol, induces human gastric cancer cell apoptosis and cell cycle arrest via inhibiting nuclear factor kappaB signaling. Oncol. Rep. 2010, 24, 1669–1676. [Google Scholar]
- Lin, Q.; Wang, F.; Yang, R.; Zheng, X.; Gao, H.; Zhang, P. Effect of Chronic Restraint Stress on Human Colorectal Carcinoma Growth in Mice. PLoS ONE 2013, 8, e61435. [Google Scholar] [CrossRef] [Green Version]
- Bernabé, D.G.; Tamae, A.C.; Biasoli, É.R.; Oliveira, S.H. Stress hormones increase cell proliferation and regulates interleukin-6 secretion in human oral squamous cell carcinoma cells. Brain Behav. Immun. 2011, 25, 574–583. [Google Scholar] [CrossRef] [Green Version]
- Coelho, M.; Moz, M.; Correia, G.; Teixeira, A.; Medeiros, R.; Ribeiro, L. Antiproliferative effects of beta-blockers on human colorectal cancer cells. Oncol. Rep. 2015, 33, 2513–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; He, X.; Tan, J.; Zhou, X.; Zou, L. Beta-arrestin2 mediates beta-2 adrenergic receptor signaling inducing prostate cancer cell progression. Oncol. Rep. 2011, 26, 1471–1477. [Google Scholar] [PubMed] [Green Version]
- Surcel, M. Adrenergic Modulation of Melanoma Cells Proliferation. Farmacia 2018, 66, 820–825. [Google Scholar] [CrossRef]
- He, J.J.; Zhang, W.H.; Liu, S.L.; Chen, Y.F.; Liao, C.X.; Shen, Q.Q.; Hu, P. Activation of beta-adrenergic receptor promotes cellular proliferation in human glioblastoma. Oncol. Lett. 2017, 14, 3846–3852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dal Monte, M.; Fornaciari, I.; Nicchia, G.P.; Svelto, M.; Casini, G.; Bagnoli, P. Beta3-adrenergic receptor activity modulates melanoma cell proliferation and survival through nitric oxide signaling. Naunyn Schmiedebergs Arch. Pharmacol. 2014, 387, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Luthy, I.A.; Bruzzone, A.; Pinero, C.P.; Castillo, L.F.; Chiesa, I.J.; Vazquez, S.M.; Sarappa, M.G. Adrenoceptors: Non conventional target for breast cancer? Curr. Med. Chem. 2009, 16, 1850–1862. [Google Scholar] [CrossRef]
- Calvani, M.; Pelon, F.; Comito, G.; Taddei, M.L.; Moretti, S.; Innocenti, S.; Nassini, R.; Gerlini, G.; Borgognoni, L.; Bambi, F.; et al. Norepinephrine promotes tumor microenvironment reactivity through beta3-adrenoreceptors during melanoma progression. Oncotarget 2015, 6, 4615–4632. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Ma, Q.; Wang, Z.; Zhang, M.; Guo, K.; Wang, F.; Wu, E. Beta2-adrenoceptor blockage induces G1/S phase arrest and apoptosis in pancreatic cancer cells via Ras/Akt/NFkappaB pathway. Mol. Cancer 2011, 10, 146. [Google Scholar] [CrossRef] [Green Version]
- Chin, C.C.; Li, J.M.; Lee, K.F.; Huang, Y.C.; Wang, K.C.; Lai, H.C.; Cheng, C.C.; Kuo, Y.H.; Shi, C.S. Selective beta2-AR Blockage Suppresses Colorectal Cancer Growth Through Regulation of EGFR-Akt/ERK1/2 Signaling, G1-Phase Arrest, and Apoptosis. J. Cell. Physiol. 2016, 231, 459–472. [Google Scholar] [CrossRef]
- Wang, F.; Liu, H.; Wang, F.; Xu, R.; Wang, P.; Tang, F.; Zhang, X.; Zhu, Z.; Lv, H.; Han, T. Propranolol suppresses the proliferation and induces the apoptosis of liver cancer cells. Mol. Med. Rep. 2018, 17, 5213–5221. [Google Scholar] [CrossRef] [Green Version]
- Gruet, M.; Cotton, D.; Coveney, C.; Boocock, D.J.; Wagner, S.; Komorowski, L.; Rees, R.C.; Pockley, A.G.; Garner, A.C.; Wallis, J.D.; et al. Beta2-Adrenergic Signalling Promotes Cell Migration by Upregulating Expression of the Metastasis-Associated Molecule LYPD3. Biology (Basel) 2020, 9, 39. [Google Scholar]
- Bravo-Calderón, D.M.; Assao, A.; Garcia, N.G.; Coutinho-Camillo, C.M.; Roffé, M.; Germano, J.N.; Oliveira, D.T. Beta adrenergic receptor activation inhibits oral cancer migration and invasiveness. Arch. Oral Biol. 2020, 118, 104865. [Google Scholar] [CrossRef] [PubMed]
- Rivero, E.M. The ?2-Adrenergic Agonist Salbutamol Inhibits Migration, Invasion and Metastasis of the Human Breast Cancer MDA-MB- 231 Cell Line. Curr. Cancer Drug Targets 2017, 17, 1. [Google Scholar] [CrossRef] [PubMed]
- Le, C.P.; Nowell, C.J.; Kim-Fuchs, C.; Botteri, E.; Hiller, J.G.; Ismail, H.; Pimentel, M.A.; Chai, M.G.; Karnezis, T.; Rotmensz, N.; et al. Chronic stress in mice remodels lymph vasculature to promote tumour cell dissemination. Nat. Commun. 2016, 7, 10634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, A.; Le, C.P.; Walker, A.K.; Creed, S.J.; Pon, C.K.; Albold, S.; Carroll, D.; Halls, M.L.; Lane, J.R.; Riedel, B.; et al. Beta2-Adrenoceptors on tumor cells play a critical role in stress-enhanced metastasis in a mouse model of breast cancer. Brain Behav. Immun. 2016, 57, 106–115. [Google Scholar] [CrossRef] [Green Version]
- Kuol, N.; Stojanovska, L.; Apostolopoulos, V.; Nurgali, K. Role of the Nervous System in Tumor Angiogenesis. Cancer Microenviron. 2018, 11, 1–11. [Google Scholar] [CrossRef]
- De Palma, M.; Biziato, D.; Petrova, T.V. Microenvironmental regulation of tumour angiogenesis. Nat. Rev. Cancer 2017, 17, 457–474. [Google Scholar] [CrossRef]
- Li, W.; Zhao, Y. Beta-adrenergic signaling on neuroendocrine differentiation, angiogenesis, and metastasis in prostate cancer progression. Asian J. Androl. 2019, 21, 253–259. [Google Scholar] [CrossRef]
- Nuevo-Tapioles, C.; Santacatterina, F.; Stamatakis, K.; Nunez de Arenas, C.; Gomez de Cedron, M.; Formentini, L.; Cuezva, J.M. Coordinate beta-adrenergic inhibition of mitochondrial activity and angiogenesis arrest tumor growth. Nat. Commun. 2020, 11, 3606. [Google Scholar] [CrossRef]
- Dieterich, L.C.; Bikfalvi, A. The tumor organismal environment: Role in tumor development and cancer immunotherapy. Semin. Cancer Biol. 2019, 65, 197–206. [Google Scholar] [CrossRef]
- Stacker, S.A.; Williams, S.P.; Karnezis, T.; Shayan, R.; Fox, S.B.; Achen, M.G. Lymphangiogenesis and lymphatic vessel remodelling in cancer. Nat. Rev. Cancer 2014, 14, 159–172. [Google Scholar] [CrossRef]
- Farnsworth, R.H.; Achen, M.G.; Stacker, S.A. The evolving role of lymphatics in cancer metastasis. Curr. Opin. Immunol. 2018, 53, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, S.B.; Gsponer, D.; Montoya-Zegarra, J.A.; Schneider, M.; Scholkmann, F.; Tacconi, C.; Noerrelykke, S.F.; Proulx, S.T.; Detmar, M. A Distinct Role of the Autonomic Nervous System in Modulating the Function of Lymphatic Vessels under Physiological and Tumor-Draining Conditions. Cell Rep. 2019, 27, 3305–3314. [Google Scholar] [CrossRef] [PubMed]
- Kaira, K.; Kamiyoshihara, M.; Kawashima, O.; Endoh, H.; Imaizumi, K.; Sugano, M.; Tanaka, S.; Fujita, A.; Kogure, Y.; Shimizu, A.; et al. Prognostic Impact of beta2 Adrenergic Receptor Expression in Surgically Resected Pulmonary Pleomorphic Carcinoma. Anticancer Res. 2019, 39, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Swann, J.B.; Smyth, M.J. Immune surveillance of tumors. J. Clin. Investig. 2007, 117, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Zitvogel, L.; Tesniere, A.; Kroemer, G. Cancer despite immunosurveillance: Immunoselection and immunosubversion. Nat. Rev. Immunol. 2006, 6, 715–727. [Google Scholar] [CrossRef]
- Bernstorff, W.V.; Glickman, J.N.; Odze, R.D.; Farraye, F.A.; Joo, H.G.; Goedegebuure, P.S.; Eberlein, T.J. Fas (CD95/APO-1) and Fas ligand expression in normal pancreas and pancreatic tumors. Implications for immune privilege and immune escape. Cancer 2002, 94, 2552–2560. [Google Scholar] [CrossRef]
- Bennett, M.W.; O’Connell, J.; O’Sullivan, G.C.; Roche, D.; Brady, C.; Kelly, J.; Collins, J.K.; Shanahan, F. Expression of Fas ligand by human gastric adenocarcinomas: A potential mechanism of immune escape in stomach cancer. Gut 1999, 44, 156–162. [Google Scholar] [CrossRef]
- Qiao, G.; Bucsek, M.J.; Winder, N.M.; Chen, M.; Giridharan, T.; Olejniczak, S.H.; Hylander, B.L.; Repasky, E.A. Beta-Adrenergic signaling blocks murine CD8(+) T-cell metabolic reprogramming during activation: A mechanism for immunosuppression by adrenergic stress. Cancer Immunol. Immunother. 2019, 68, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Nissen, M.D.; Sloan, E.K.; Mattarollo, S.R. Beta-adrenergic signaling impairs anti-tumor CD8+ T cell responses to B cell lymphoma immunotherapy. Cancer Immunol. Res. 2017, 6, 98–109. [Google Scholar] [CrossRef] [Green Version]
- Nagaraja, A.S.; Dorniak, P.L.; Sadaoui, N.C.; Kang, Y.; Lin, T.; Armaiz-Pena, G.N.; Wu, S.Y.; Rupaimoole, R.; Allen, J.K.; Gharpure, K.M.; et al. Sustained adrenergic signaling leads to increased metastasis in ovarian cancer via increased PGE2 synthesis. Oncogene 2015, 35, 2390–2397. [Google Scholar] [CrossRef] [Green Version]
- Thaker, P.H.; Han, L.Y.; Kamat, A.A.; Arevalo, J.M.; Takahashi, R.; Lu, C.; Jennings, N.B.; Armaiz-Pena, G.A.; Bankson, J.; Ravoori, M.; et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat. Med. 2006, 12, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Haldar, R.; Ricon-Becker, I.; Radin, A.; Gutman, M.; Cole, S.W.; Zmora, O.; Ben-Eliyahu, S. Perioperative COX2 and beta-adrenergic blockade improves biomarkers of tumor metastasis, immunity, and inflammation in colorectal cancer: A randomized controlled trial. Cancer 2020, 126, 3991–4001. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Liu, D.; Yang, Z.; Guo, N. Central and peripheral nervous systems: Master controllers in cancer metastasis. Cancer Metastasis Rev. 2013, 32, 603–621. [Google Scholar] [CrossRef] [PubMed]
- Al-Zhoughbi, W.; Huang, J.; Paramasivan, G.S.; Till, H.; Pichler, M.; Guertl-Lackner, B.; Hoefler, G. Tumor Macroenvironment and Metabolism. Semin. Oncol. 2014, 41, 281–295. [Google Scholar] [CrossRef] [Green Version]
- Al-Zoughbi, W.; Hoefler, G. Tumor Macroenvironment: An Update. Pathobiology 2019, 87, 58–60. [Google Scholar] [CrossRef] [PubMed]
- De Assis, L.V.M.; Moraes, M.N.; Magalhães-Marques, K.K.; Kinker, G.S.; Cruz-Machado, S.D.S.; Castrucci, A.M.D.L. Non-Metastatic Cutaneous Melanoma Induces Chronodisruption in Central and Peripheral Circadian Clocks. Int. J. Mol. Sci. 2018, 19, 1065. [Google Scholar] [CrossRef] [Green Version]
- Masri, S.; Sassone-Corsi, P. The emerging link between cancer, metabolism, and circadian rhythms. Nat. Med. 2018, 24, 1795–1803. [Google Scholar] [CrossRef]
- Porporato, P. Understanding cachexia as a cancer metabolism syndrome. Oncogenesis 2016, 5, e200. [Google Scholar] [CrossRef] [Green Version]
- Faulkner, S.; Jobling, P.; March, B.; Jiang, C.C.; Hondermarck, H. Tumor Neurobiology and the War of Nerves in Cancer. Cancer Discov. 2019, 9, 702–710. [Google Scholar] [CrossRef] [Green Version]
- Mravec, B.; Gidron, Y.; Hulin, I. Neurobiology of cancer: Interactions between nervous, endocrine and immune systems as a base for monitoring and modulating the tumorigenesis by the brain. Semin. Cancer Biol. 2008, 18, 150–163. [Google Scholar] [CrossRef]
- Chen, H.; Liu, D.; Guo, L.; Cheng, X.; Guo, N.; Shi, M. Chronic psychological stress promotes lung metastatic colonization of circulating breast cancer cells by decorating a pre-metastatic niche through activating beta-adrenergic signaling. J. Pathol. 2018, 244, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Zukowska-Grojec, Z.; Karwatowska-Prokopczuk, E.; Rose, W.; Rone, J.; Movafagh, S.; Ji, H.; Yeh, Y.; Chen, W.T.; Kleinman, H.K.; Grouzmann, E.; et al. Neuropeptide Y: A novel angiogenic factor from the sympathetic nerves and endothelium. Circ. Res. 1998, 83, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Neeman, E.; Zmora, O.; Ben-Eliyahu, S. A new approach to reducing postsurgical cancer recurrence: Perioperative targeting of catecholamines and prostaglandins. Clin. Cancer Res. 2012, 18, 4895–4902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiller, J.G.; Cole, S.W.; Crone, E.M.; Byrne, D.J.; Shackleford, D.M.; Pang, J.B.; Henderson, M.A.; Nightingale, S.S.; Ho, K.M.; Myles, P.S.; et al. Preoperative beta-Blockade with Propranolol Reduces Biomarkers of Metastasis in Breast Cancer: A Phase II Randomized Trial. Clin. Cancer Res. 2020, 26, 1803–1811. [Google Scholar] [CrossRef] [PubMed]
- Ricon, I.; Hanalis-Miller, T.; Haldar, R.; Jacoby, R.; Ben-Eliyahu, S. Perioperative biobehavioral interventions to prevent cancer recurrence through combined inhibition of beta-adrenergic and cyclooxygenase 2 signaling. Cancer 2019, 125, 45–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laplane, L.; Duluc, D.; Larmonier, N.; Pradeu, T.; Bikfalvi, A. The Multiple Layers of the Tumor Environment. Trends Cancer 2018, 4, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Elenkov, I.J.; Wilder, R.L.; Chrousos, G.P.; Vizi, E.S. The sympathetic nerve--an integrative interface between two supersystems: The brain and the immune system. Pharmacol. Rev. 2000, 52, 595–638. [Google Scholar]
- Qin, J.-F.; Jin, F.-J.; Li, N.; Guan, H.-T.; Lan, L.; Ni, H.; Wang, Y. Adrenergic receptor β2 activation by stress promotes breast cancer progression through macrophages M2 polarization in tumor microenvironment. BMB Rep. 2015, 48, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Le, W.; Chen, Q.; Chen, J.; Zhu, Y.; Shi, D.; Chen, B.; Cui, Z. Suppression of the innate cancer-killing activity in human granulocytes by stress reaction as a possible mechanism for affecting cancer development. Stress 2019, 23, 87–96. [Google Scholar] [CrossRef]
- Kuol, N.; Stojanovska, L.; Apostolopoulos, V.; Nurgali, K. Crosstalk between cancer and the neuro-immune system. J. Neuroimmunol. 2018, 315, 15–23. [Google Scholar] [CrossRef]
- Wilson, J.D.; Foster, D.W.; Kronenberg, H.K.; Larsen, P.R. Williams Textbook of Endocrinology, 9th ed.; Saunders: Philadelphia, PA, USA, 1998; p. 1819. [Google Scholar]
- Nonogaki, K. New insights into sympathetic regulation of glucose and fat metabolism. Diabetology 2000, 43, 533–549. [Google Scholar] [CrossRef]
- Petruzzelli, M.; Wagner, E.F. Mechanisms of metabolic dysfunction in cancer-associated cachexia. Genes Dev. 2016, 30, 489–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, F.; Nilsson, S.K.; Nilsson, J.A. Beta-3 adrenergic antagonism can alleviate energy wasting associated with cancer-induced cachexia. In Proceedings of the Molecular and Cellular Biology, Washington, DC, USA, 1–5 April 2017; American Association for Cancer Research (AACR): Philadelphia, PA, USA, 2017; Volume 77, p. 5435, Abstract 5435. [Google Scholar]
- Salazar-Degracia, A.; Busquets, S.; Argilés, J.M.; Bargalló-Gispert, N.; López-Soriano, F.J.; Barreiro, E. Effects of the beta2 agonist formoterol on atrophy signaling, autophagy, and muscle phenotype in respiratory and limb muscles of rats with cancer-induced cachexia. Biochimie 2018, 149, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Baffy, G. Gut Microbiota and Cancer of the Host: Colliding Interests. Adv. Exp. Med. Biol. 2020, 1219, 93–107. [Google Scholar] [CrossRef]
- Vivarelli, S.; Salemi, R.; Candido, S.; Falzone, L.; Santagati, M.; Stefani, S.; Torino, F.; Banna, G.L.; Tonini, G.; Libra, M. Gut Microbiota and Cancer: From Pathogenesis to Therapy. Cancers 2019, 11, 38. [Google Scholar] [CrossRef] [Green Version]
- Herremans, K.M.; Riner, A.; Cameron, M.E.; Trevino, J.G. The Microbiota and Cancer Cachexia. Int. J. Mol. Sci. 2019, 20, 6267. [Google Scholar] [CrossRef] [Green Version]
- Williams, P. Quorum sensing, communication and cross-kingdom signalling in the bacterial world. Microbiology 2007, 153, 3923–3938. [Google Scholar] [CrossRef] [Green Version]
- Waldor, M.K.; Sperandio, V. Adrenergic Regulation of Bacterial Virulence. J. Infect. Dis. 2007, 195, 1248–1249. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, M.M.; Pina-Vaz, C.; Baltazar, F. Microbes and Cancer: Friends or Faux? Int. J. Mol. Sci. 2020, 21, 3115. [Google Scholar] [CrossRef]
- Seretis, A.; Cividini, S.; Markozannes, G.; Tseretopoulou, X.; Lopez, D.S.; Ntzani, E.E.; Tsilidis, K.K. Association between blood pressure and risk of cancer development: A systematic review and meta-analysis of observational studies. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- De Pergola, G.; Silvestris, F. Obesity as a Major Risk Factor for Cancer. J. Obes. 2013, 2013, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- US Department of Health and Human Services; Centers for Disease Control and Prevention; National Center for Chronic Disease Prevention and Health Promotion; Office on Smoking and Health. The Health Consequences of Smoking–50 Years of Progress: A Report of the Surgeon General; Centers for Disease Control and Prevention (US): Atlanta, GA, USA, 2014.
- Yin, W.; Falconer, H.; Yin, L.; Xu, L.; Ye, W. Association Between Polycystic Ovary Syndrome and Cancer Risk. JAMA Oncol. 2019, 5, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalil, G.Z.; Haynes, W.G. Sympathetic nervous system in obesity-related hypertension: Mechanisms and clinical implications. Hypertens. Res. 2011, 35, 4–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thackeray, J.T.; Beanlands, R.S.; DaSilva, J.N. Altered sympathetic nervous system signaling in the diabetic heart: Emerging targets for molecular imaging. Am. J. Nucl. Med. Mol. Imaging 2012, 2, 314–334. [Google Scholar] [PubMed]
- Grassi, G.; Seravalle, G.; Calhoun, D.A.; Bolla, G.B.; Cristina, G.; Marabini, M.; Del Bo, A.; Mancia, G. Mechanisms responsible for sympathetic activation by cigarette smoking in humans. Circulation 1994, 90, 248–253. [Google Scholar] [CrossRef] [Green Version]
- Frankl, V.E. Man’s search for meaning; Pocket Books Washington Square Press: New York, NY, USA, 1985; p. 1959. [Google Scholar]
- Sperry, L.; Shafranske, E.P. Spiritually Oriented Psychotherapy; American Psychological Association: Washington, DC, USA, 2004; p. 368. [Google Scholar] [CrossRef]
- Glaw, X.; Kable, A.; Hazelton, M.; Inder, K. Meaning in Life and Meaning of Life in Mental Health Care: An Integrative Literature Review. Issues Ment. Health Nurs. 2016, 38, 1–13. [Google Scholar] [CrossRef]
- Scrignaro, M.; Msc, E.B.; Brunelli, C.; Miccinesi, G.; Ripamonti, C.; Magrin, M.E.; Borreani, C. Seeking and experiencing meaning: Exploring the role of meaning in promoting mental adjustment and eudaimonic well-being in cancer patients. Palliat. Support. Care 2014, 13, 673–681. [Google Scholar] [CrossRef]
- Halama, P. Meaning in Life and Coping: Sense of Meaning as a Buffer Against Stress. In Meaning in Positive and Existential Psychology; Batthyany, A., Russo-Netzer, P., Eds.; Springer: New York, NY, USA, 2014; p. 484. [Google Scholar]
- Vehling, S.; Kissane, D. Existential distress in cancer: Alleviating suffering from fundamental loss and change. Psycho Oncol. 2018, 27, 2525–2530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vehling, S.; Philipp, R. Existential distress and meaning-focused interventions in cancer survivorship. Curr. Opin. Support. Palliat. Care 2018, 12, 46–51. [Google Scholar] [CrossRef]
- Putranto, R.; Mudjaddid, E.; Shatri, H.; Adli, M.; Martina, D. Development and challenges of palliative care in Indonesia: Role of psychosomatic medicine. Biopsychosoc. Med. 2017, 11, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasuo, H.; Kanbara, K.; Fukunaga, M. Effect of Heart Rate Variability Biofeedback Sessions With Resonant Frequency Breathing on Sleep: A Pilot Study Among Family Caregivers of Patients With Cancer. Front. Med. 2020, 7. [Google Scholar] [CrossRef]
- Manzini, C.S.S.; Damasceno, V.A.M.; Elias, A.C.A.; Orlandi, F.D.S. The brief psychotherapeutic intervention “relaxation, mental images and spirituality”: A systematic review. Sao Paulo Med. J. 2020, 138, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Tonhajzerova, I.; Mestanik, M. New Perspectives in the Model of Stress Response. Physiol. Res. 2017, 66, S173–S185. [Google Scholar] [CrossRef] [PubMed]
- Charmandari, E.; Tsigos, C.; Chrousos, G. Endocrinology of the Stress Response. Annu. Rev. Physiol. 2005, 67, 259–284. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, M.; Neeman, E.; Sharon, E.; Ben-Eliyahu, S. Exploiting the critical perioperative period to improve long-term cancer outcomes. Nat. Rev. Clin. Oncol. 2015, 12, 213–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplon, H.; Muralidharan, M.; Schneider, Z.; Reichert, J.M. Antibodies to watch in 2020. mAbs 2019, 12, 1703531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhangoo, M.S.; Sigal, D.S. TRK Inhibitors: Clinical Development of Larotrectinib. Curr. Oncol. Rep. 2019, 21, 14. [Google Scholar] [CrossRef]
- Asmus, S.E.; Tian, H.; Landis, S.C. Induction of Cholinergic Function in Cultured Sympathetic Neurons by Periosteal Cells: Cellular Mechanisms. Dev. Biol. 2001, 235, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Hiller, J.G.; Perry, N.J.; Poulogiannis, G.; Riedel, B.; Sloan, E.K. Perioperative events influence cancer recurrence risk after surgery. Nat. Rev. Clin. Oncol. 2017, 15, 205–218. [Google Scholar] [CrossRef]
- Eilami, O.; Moslemirad, M.; Naimi, E.; Babuei, A.; Rezaei, K. The Effect of Religious Psychotherapy Emphasizing the Importance of Prayers on Mental Health and Pain in Cancer Patients. J. Relig. Health 2018, 58, 444–451. [Google Scholar] [CrossRef]
- Dowlatabadi, M.M.; Ahmadi, S.M.; Sorbi, M.H.; Beiki, O.; Razavi, T.K.; Bidaki, R. The effectiveness of group positive psychotherapy on depression and happiness in breast cancer patients: A randomized controlled trial. Electron. Physician 2016, 8, 2175–2180. [Google Scholar] [CrossRef] [Green Version]
- Mirosevic, S.; Jo, B.; Kraemer, H.C.; Ershadi, M.; Neri, E.; Spiegel, D. “Not just another meta-analysis”: Sources of heterogeneity in psychosocial treatment effect on cancer survival. Cancer Med. 2019, 8, 363–373. [Google Scholar] [CrossRef]
- Burch, J.B.; Ginsberg, J.P.; McLain, A.C.; Franco, R.; Stokes, S.; Susko, K.; Hendry, W.; Crowley, E.; Christ, A.; Hanna, J.; et al. Symptom Management Among Cancer Survivors: Randomized Pilot Intervention Trial of Heart Rate Variability Biofeedback. Appl. Psychophysiol. Biofeedback 2020, 45, 99–108. [Google Scholar] [CrossRef]
- Famm, K.; Litt, B.; Tracey, K.J.; Boyden, E.S.; Slaoui, M. Drug discovery: A jump-start for electroceuticals. Nature 2013, 496, 159–161. [Google Scholar] [CrossRef] [Green Version]
- Mravec, B.; Tibensky, M.; Horvathova, L. Stress and cancer. Part II: Therapeutic implications for oncology. J. Neuroimmunol. 2020, 346, 577312. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mravec, B.; Horvathova, L.; Hunakova, L. Neurobiology of Cancer: The Role of β-Adrenergic Receptor Signaling in Various Tumor Environments. Int. J. Mol. Sci. 2020, 21, 7958. https://doi.org/10.3390/ijms21217958
Mravec B, Horvathova L, Hunakova L. Neurobiology of Cancer: The Role of β-Adrenergic Receptor Signaling in Various Tumor Environments. International Journal of Molecular Sciences. 2020; 21(21):7958. https://doi.org/10.3390/ijms21217958
Chicago/Turabian StyleMravec, Boris, Lubica Horvathova, and Luba Hunakova. 2020. "Neurobiology of Cancer: The Role of β-Adrenergic Receptor Signaling in Various Tumor Environments" International Journal of Molecular Sciences 21, no. 21: 7958. https://doi.org/10.3390/ijms21217958
APA StyleMravec, B., Horvathova, L., & Hunakova, L. (2020). Neurobiology of Cancer: The Role of β-Adrenergic Receptor Signaling in Various Tumor Environments. International Journal of Molecular Sciences, 21(21), 7958. https://doi.org/10.3390/ijms21217958