New Insights into Therapy-Induced Progression of Cancer

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
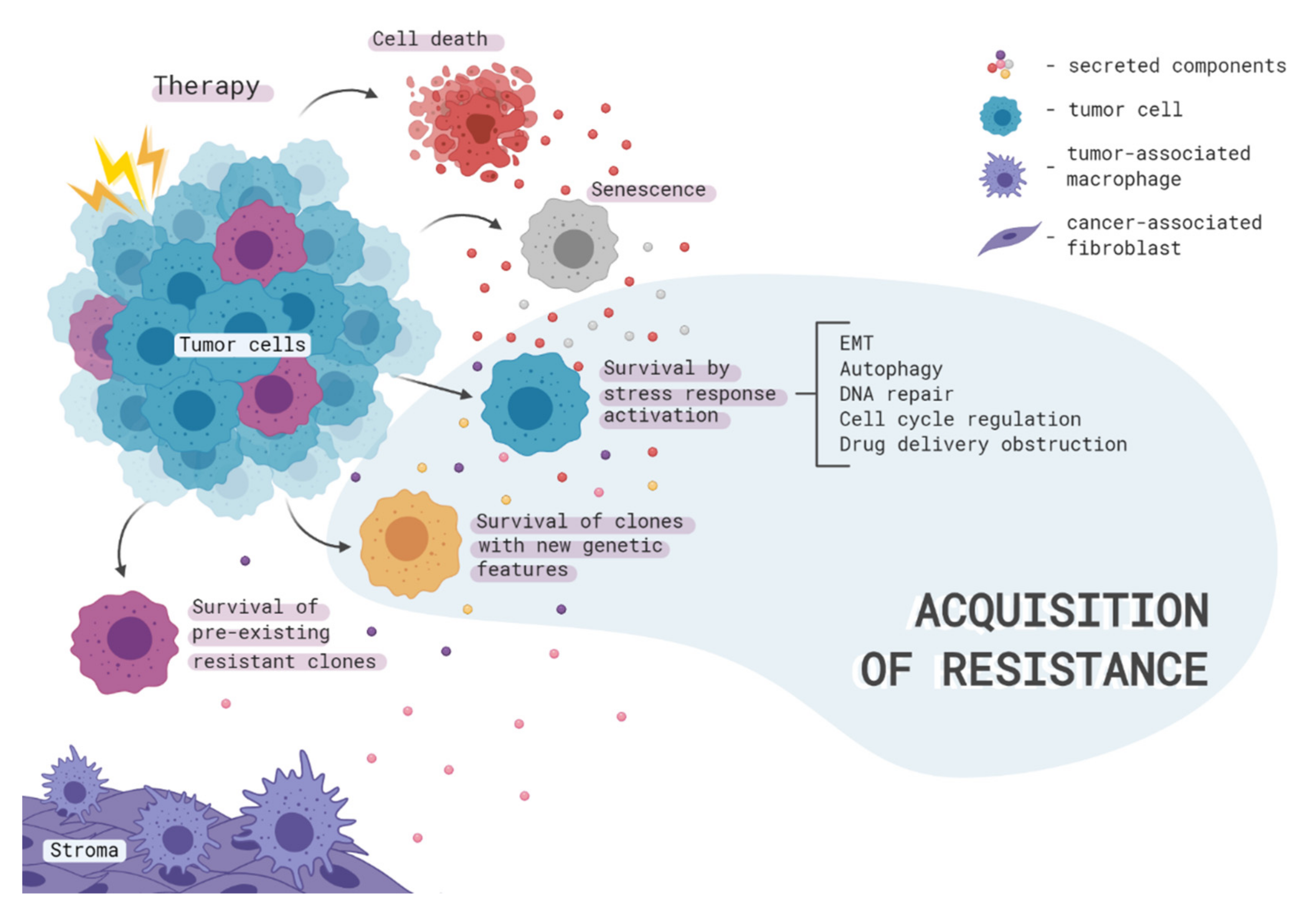
2. Therapy-Induced Clonal Selection
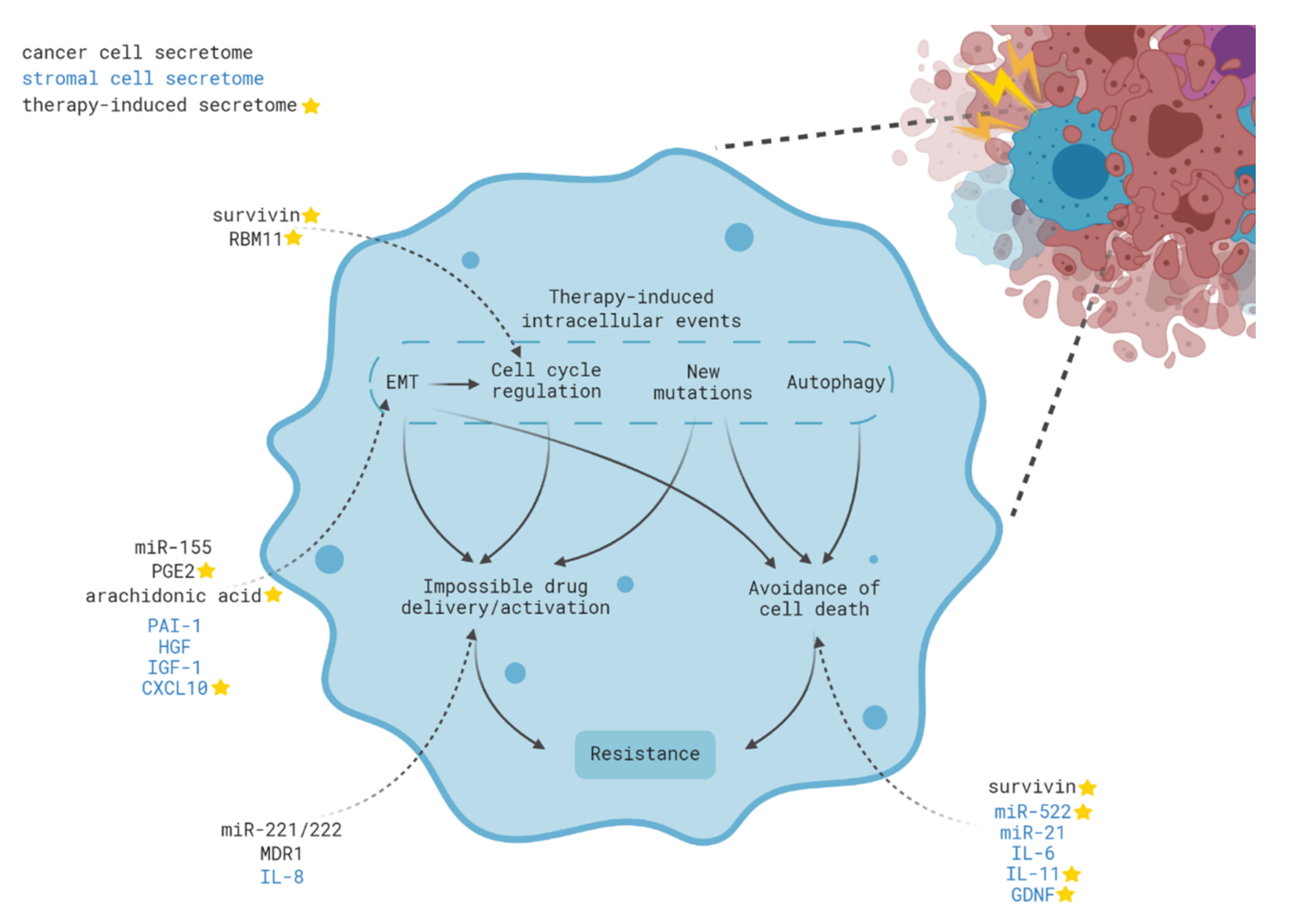
3. Intracellular Mechanisms of Acquired Therapy Resistance
3.1. Aspects of Epithelial-to-Mesenchymal Transition
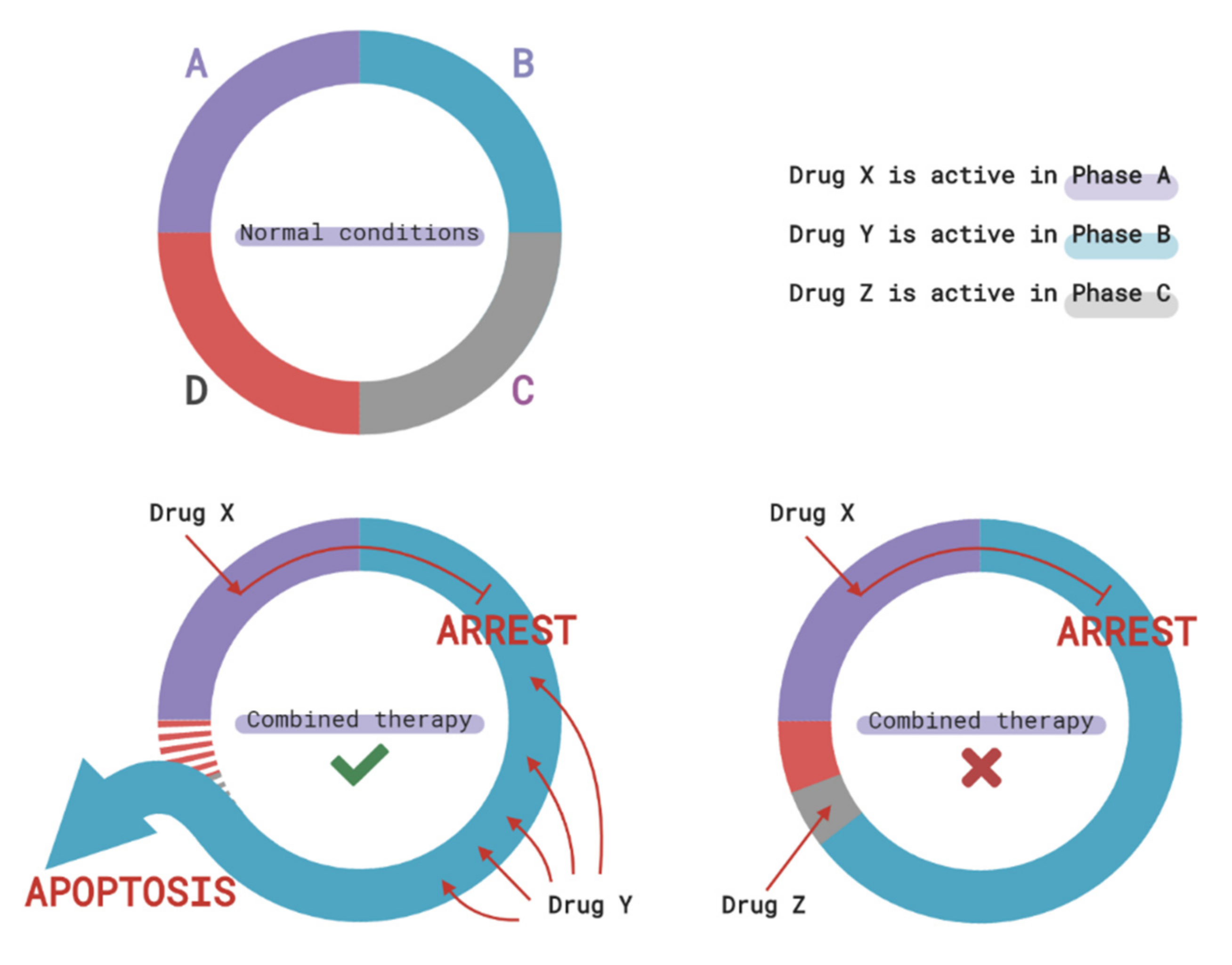
3.2. Cell Cycle-Mediated Chemoresistance of Cancer Cells
3.3. Autophagy as a Way to Avoid Therapy-Induced Cell Death
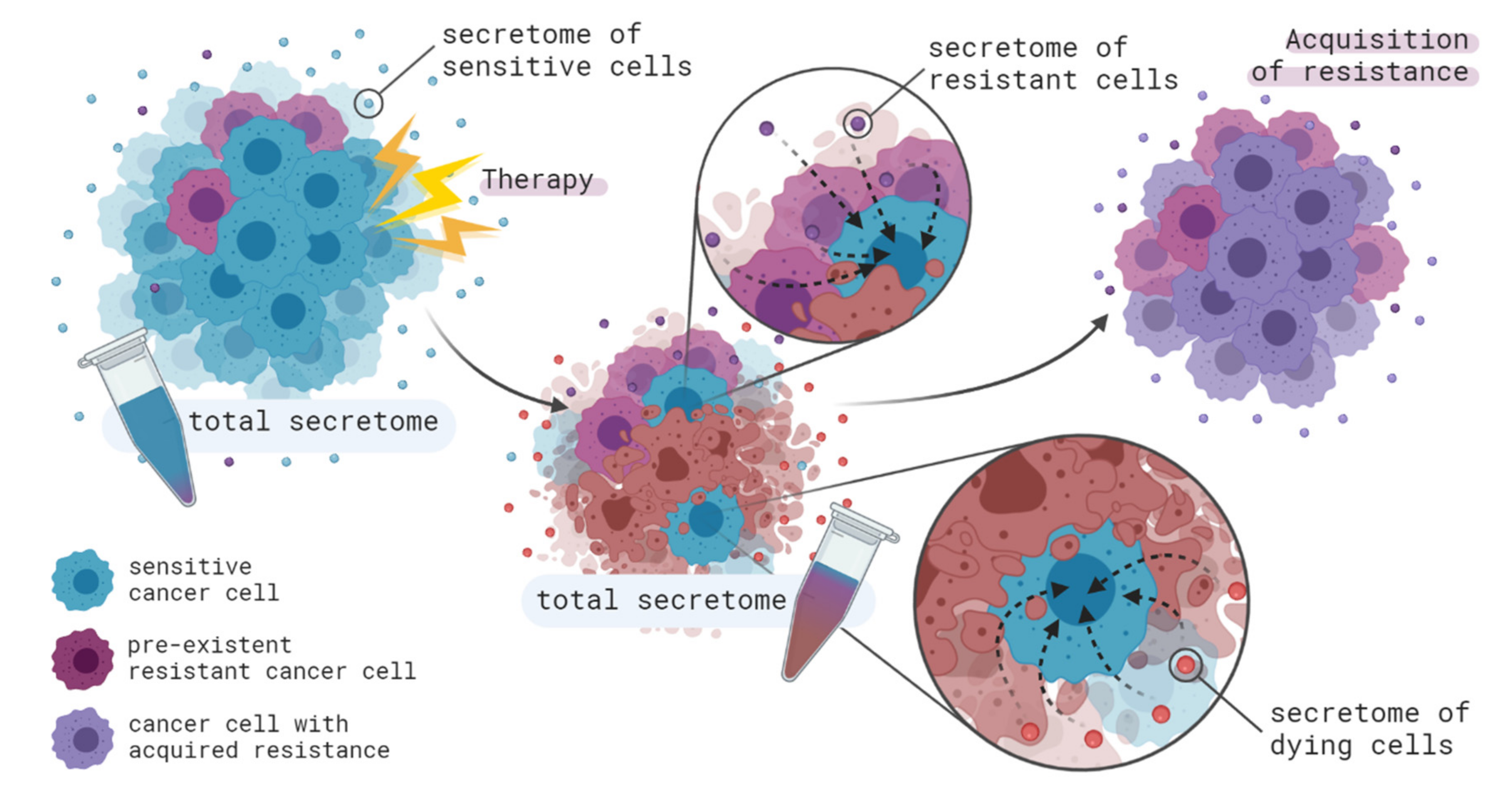
4. Contribution of Intercellular Communication to Acquired Therapy Resistance
4.1. From Cancer Cells to Cancer Cells
4.2. Communication between Cancer Cells and Surrounding Stromal Cells
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 14-3-3ζ | 14-3-3 protein zeta/delta |
| ADAM10 | Disintegrin and metalloproteinase domain-containing protein 1 |
| AKT | Protein kinase B |
| ALK | ALK tyrosine kinase receptor |
| ALOX15 | Polyunsaturated fatty acid lipoxygenase ALOX15 |
| AMPK | AMP-activated protein kinase |
| APAF1 | Apoptotic protease-activating factor 1 |
| ATR | Serine/threonine-protein kinase ATR |
| Axl | Tyrosine-protein kinase receptor UFO |
| B2M | Beta-2-microglobulin |
| Bcl | B-cell lymphoma protein |
| BRCA1/2 | Breast cancer type 1/2 susceptibility protein |
| CAFs | Cancer-associated fibroblasts |
| CCL20 | C-C motif chemokine 20 |
| CDKN1A | Cyclin-dependent kinase inhibitor 1 |
| CEBPB | CCAAT/enhancer-binding protein beta |
| CHK1 | Serine/threonine-protein kinase Chk1 |
| CXCL10 | C-X-C motif chemokine 10 |
| CXCR3 | C-X-C chemokine receptor type 3 |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial-to-mesenchymal transition |
| EpCAM | Epithelial cell adhesion molecule |
| EphA2 | Ephrin type-A receptor 2 |
| ERK1/2 | Mitogen-activated protein kinase 3/1 |
| ER-α | Estrogen receptor alpha |
| ESRP1 | Epithelial splicing regulatory protein 1 |
| FBXW7 | F-box/WD repeat-containing protein 7 |
| FOXC2 | Forkhead box protein C2 |
| FOXO1 | Forkhead box protein O1 |
| G3BP2 | Ras GTPase-activating protein-binding protein 2 |
| GDNF | Glial cell line-derived neurotrophic factor |
| GSTP1 | Glutathione S-transferase P |
| HER2 | Receptor tyrosine-protein kinase erbB-2 |
| HGF | Hepatocyte growth factor |
| HIF-1α | Hypoxia-inducible factor 1-alpha |
| HMGB1 | High mobility group protein B1 |
| HMMR | Hyaluronan mediated motility receptor |
| IGF-1 | Insulin-like growth factor I |
| IGFBP7 | Insulin-like growth factor-binding protein 7 |
| Ils | Interleukins |
| iPLA2 | Calcium independent phospholipase A2 |
| Jak | Tyrosine-protein kinase JAK |
| JAM-A | Junctional adhesion molecule A |
| JNK | c-Jun N-terminal kinase |
| lncRNAs | Long non-coding RNAs |
| MAPK | Mitogen-activated protein kinase |
| MDK | Midkine |
| MDM4 | Protein Mdm4 |
| MDR1 | ATP-dependent translocase ABCB1 |
| miR | microRNA |
| MOAP1 | Modulator of apoptosis 1 |
| MRP1 | Multidrug resistance-associated protein 1 |
| MSCs | Mesenchymal stem cells |
| mTOR | Mammalian target of rapamycin |
| MXR | Broad substrate specificity ATP-binding cassette transporter ABCG2 |
| MYCN | N-myc proto-oncogene protein |
| NEK2 | Serine/threonine-protein kinase Nek2 |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| p38-MAPK | p38 mitogen-activated protein kinase |
| p62 | Ubiquitin-binding protein p62 |
| PAI-1 | Plasminogen activator inhibitor 1 |
| PD1 | Programmed cell death-1 |
| PDGFRb | Platelet-derived growth factor receptor beta |
| PD1L1 | Programmed cell death 1 ligand 1 |
| PGE2 | Prostaglandin E2 |
| PI3K | Phosphoinositide 3-kinase |
| p-STAT3 | Phospho-Stat3, Signal transducer and activator of transcription 3 |
| RAB25 | Ras-related protein Rab-25 |
| RAS | Ras GTPase |
| RBM11 | Splicing regulator RBM11 |
| SASP | Senescence-associated secretory phenotype |
| SLUG | Zinc finger protein SNAI2 |
| SMAD2/3 | Mothers against decapentaplegic homolog 2/3 |
| SNAIL | Zinc finger protein SNAI1 |
| ST14 | Suppressor of tumorigenicity 14 protein |
| STAT | Signal transducer and activator of transcription |
| TAMs | Tumor-associated macrophages |
| TGFβ1 | Transforming growth factor beta-1 proprotein |
| TME | Tumor microenvironment |
| TP53 | Cellular tumor antigen p53 |
| TP53INP1 | Tumor protein p53-inducible nuclear protein 1 |
| TWIST | Twist-related protein |
| UCH-L1 | Ubiquitin carboxyl-terminal hydrolase isozyme L1 |
| UTR | Untranslated region |
| VEGF | Vascular endothelial growth factor A |
| WEE1 | Wee1-like protein kinase |
| WNT | Proto-oncogene Wnt |
| ZEB1 | Zinc finger E-box-binding homeobox 1 |
References
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Dillekås, H.; Rogers, M.S.; Straume, O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019, 8, 5574–5576. [Google Scholar] [CrossRef] [PubMed]
- Welch, D.R.; Hurst, D.R. Defining the hallmarks of metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal. Transduct. Target Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Miller, K.D.; Nogueira, L.; Mariotto, A.B.; Rowland, J.H.; Yabroff, K.R.; Alfano, C.M.; Jemal, A.; Kramer, J.L.; Siegel, R.L. Cancer treatment and survivorship statistics, 2019. CA Cancer J. Clin. 2019, 69, 363–385. [Google Scholar] [CrossRef] [PubMed]
- Nabors, L.B.; Portnow, J.; Ammirati, M.; Baehring, J.; Brem, H.; Butowski, N.; Fenstermaker, R.A.; Forsyth, P.; Hattangadi-Gluth, J.; Holdhoff, M.; et al. NCCN guidelines insights: Central nervous system cancers, version 1.2017. J. Natl. Compr. Cancer Netw. 2017, 15, 1331–1345. [Google Scholar] [CrossRef]
- Weller, M.; Felsberg, J.; Hartmann, C.; Berger, H.; Steinbach, J.P.; Schramm, J.; Westphal, M.; Schackert, G.; Simon, M.; Tonn, J.C.; et al. Molecular predictors of progression-free and overall survival in patients with newly diagnosed glioblastoma: A prospective translational study of the German Glioma Network. J. Clin. Oncol. 2009, 27, 5743–5750. [Google Scholar] [CrossRef]
- Corrado, G.; Salutari, V.; Palluzzi, E.; Distefano, M.G.; Scambia, G.; Ferrandina, G. Optimizing treatment in recurrent epithelial ovarian cancer. Expert Rev. Anticancer Ther. 2017, 17, 1147–1158. [Google Scholar] [CrossRef]
- Sjoquist, K.M.; Lord, S.J.; Friedlander, M.L.; Simes, J.R.; Marschner, I.C.; Lee, C.K. Progression-free survival as a surrogate endpoint for overall survival in modern ovarian cancer trials: A meta-analysis. Ther. Adv. Med. Oncol. 2018, 10, 1758835918788500. [Google Scholar] [CrossRef]
- Goss, P.E.; Ingle, J.N.; Pritchard, K.I.; Robert, N.J.; Muss, H.; Gralow, J.; Gelmon, K.; Whelan, T.; Strasser-Weippl, K.; Rubin, S.; et al. Extending aromatase-inhibitor adjuvant therapy to 10 years. N. Engl. J. Med. 2016, 375, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Brookman-May, S.D.; May, M.; Shariat, S.F.; Novara, G.; Zigeuner, R.; Cindolo, L.; De Cobelli, O.; De Nunzio, C.; Pahernik, S.; Wirth, M.P.; et al. Time to recurrence is a significant predictor of cancer-specific survival after recurrence in patients with recurrent renal cell carcinoma--results from a comprehensive multi-centre database (CORONA/SATURN-Project). BJU Int. 2013, 112, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef] [PubMed]
- Madden, E.C.; Gorman, A.M.; Logue, S.E.; Samali, A. Tumour cell secretome in chemoresistance and tumour recurrence. Trends Cancer Res. 2020, 6, 489–505. [Google Scholar] [CrossRef] [PubMed]
- Morrissy, A.S.; Garzia, L.; Shih, D.J.H.; Zuyderduyn, S.; Huang, X.; Skowron, P.; Remke, M.; Cavalli, F.M.G.; Ramaswamy, V.; Lindsay, P.E.; et al. Divergent clonal selection dominates medulloblastoma at recurrence. Nature 2016, 529, 351–357. [Google Scholar] [CrossRef]
- Shlush, L.I.; Mitchell, A.; Heisler, L.; Abelson, S.; Ng, S.W.K.; Trotman-Grant, A.; Medeiros, J.J.F.; Rao-Bhatia, A.; Jaciw-Zurakowsky, I.; Marke, R.; et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 2017, 547, 104–108. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Salichos, L.; Meyerson, W.; Warrell, J.; Gerstein, M. Estimating growth patterns and driver effects in tumor evolution from individual samples. Nat. Commun. 2020, 11, 732. [Google Scholar] [CrossRef]
- Ben-David, U.; Beroukhim, R.; Golub, T.R. Genomic evolution of cancer models: Perils and opportunities. Nat. Rev. Cancer 2019, 19, 97–109. [Google Scholar] [CrossRef]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing. Cell 2018, 173, 879–893.e13. [Google Scholar] [CrossRef]
- Maynard, A.; McCoach, C.E.; Rotow, J.K.; Harris, L.; Haderk, F.; Kerr, D.L.; Yu, E.A.; Schenk, E.L.; Tan, W.; Zee, A.; et al. Therapy-induced evolution of human lung cancer revealed by single-cell RNA sequencing. Cell 2020, 182, 1232–1251.e22. [Google Scholar] [CrossRef] [PubMed]
- Izar, B.; Tirosh, I.; Stover, E.H.; Wakiro, I.; Cuoco, M.S.; Alter, I.; Rodman, C.; Leeson, R.; Su, M.-J.; Shah, P.; et al. A single-cell landscape of high-grade serous ovarian cancer. Nat. Med. 2020, 26, 1271–1279. [Google Scholar] [CrossRef]
- Park, S.R.; Namkoong, S.; Friesen, L.; Cho, C.-S.; Zhang, Z.Z.; Chen, Y.-C.; Yoon, E.; Kim, C.H.; Kwak, H.; Kang, H.M.; et al. Single-cell transcriptome analysis of colon cancer cell response to 5-fluorouracil-induced DNA damage. Cell Rep. 2020, 32, 108077. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Chung, W.; Lee, H.-O.; Jeong, D.E.; Jo, A.; Lim, J.E.; Hong, J.H.; Nam, D.-H.; Jeong, B.C.; Park, S.H.; et al. Single-cell RNA sequencing reveals the tumor microenvironment and facilitates strategic choices to circumvent treatment failure in a chemorefractory bladder cancer patient. Genome Med. 2020, 12, 47. [Google Scholar] [CrossRef]
- Roh, V.; Abramowski, P.; Hiou-Feige, A.; Cornils, K.; Rivals, J.-P.; Zougman, A.; Aranyossy, T.; Thielecke, L.; Truan, Z.; Mermod, M.; et al. Cellular barcoding identifies clonal substitution as a hallmark of local recurrence in a surgical model of head and neck squamous cell carcinoma. Cell Rep. 2018, 25, 2208–2222.e7. [Google Scholar] [CrossRef]
- Bhang, H.-E.C.; Ruddy, D.A.; Radhakrishna, K.V.; Caushi, J.X.; Zhao, R.; Hims, M.M.; Singh, A.P.; Kao, I.; Rakiec, D.; Shaw, P.; et al. Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat. Med. 2015, 21, 440–448. [Google Scholar] [CrossRef]
- Venkatesan, S.; Swanton, C.; Taylor, B.S.; Costello, J.F. Treatment-induced mutagenesis and selective pressures sculpt cancer evolution. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Szikriszt, B.; Póti, Á.; Pipek, O.; Krzystanek, M.; Kanu, N.; Molnár, J.; Ribli, D.; Szeltner, Z.; Tusnády, G.E.; Csabai, I.; et al. A comprehensive survey of the mutagenic impact of common cancer cytotoxics. Genome Biol. 2016, 17, 99. [Google Scholar] [CrossRef] [PubMed]
- Patch, A.-M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Zheng, S.; Amini, S.S.; Virk, S.M.; Mikkelsen, T.; Brat, D.J.; Grimsby, J.; Sougnez, C.; Muller, F.; Hu, J.; et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015, 25, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Kurtova, A.V.; Xiao, J.; Mo, Q.; Pazhanisamy, S.; Krasnow, R.; Lerner, S.P.; Chen, F.; Roh, T.T.; Lay, E.; Ho, P.L.; et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 2015, 517, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Abbosh, P.; Keliher, D.; Reardon, B.; Miao, D.; Mouw, K.; Weiner-Taylor, A.; Wankowicz, S.; Han, G.; Teo, M.Y.; et al. Mutational patterns in chemotherapy resistant muscle-invasive bladder cancer. Nat. Commun. 2017, 8, 2193. [Google Scholar] [CrossRef] [PubMed]
- Stiehl, T.; Baran, N.; Ho, A.D.; Marciniak-Czochra, A. Clonal selection and therapy resistance in acute leukaemias: Mathematical modelling explains different proliferation patterns at diagnosis and relapse. J. R. Soc. Interface 2014, 11, 20140079. [Google Scholar] [CrossRef] [PubMed]
- Vosberg, S.; Greif, P.A. Clonal evolution of acute myeloid leukemia from diagnosis to relapse. Genes Chromosomes Cancer 2019, 58, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Suvà, M.L.; Tirosh, I. The glioma stem cell model in the era of single-cell genomics. Cancer Cell 2020, 37, 630–636. [Google Scholar] [CrossRef]
- Vetrie, D.; Helgason, G.V.; Copland, M. The leukaemia stem cell: Similarities, differences and clinical prospects in CML and AML. Nat. Rev. Cancer 2020, 20, 158–173. [Google Scholar] [CrossRef]
- Giovannelli, P.; Di Donato, M.; Galasso, G.; Di Zazzo, E.; Medici, N.; Bilancio, A.; Migliaccio, A.; Castoria, G. Breast cancer stem cells: The role of sex steroid receptors. World J. Stem Cells 2019, 11, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Durinikova, E.; Kozovska, Z.; Poturnajova, M.; Plava, J.; Cierna, Z.; Babelova, A.; Bohovic, R.; Schmidtova, S.; Tomas, M.; Kucerova, L.; et al. ALDH1A3 upregulation and spontaneous metastasis formation is associated with acquired chemoresistance in colorectal cancer cells. BMC Cancer 2018, 18, 848. [Google Scholar] [CrossRef]
- Wang, J.; Sakariassen, P.Ø.; Tsinkalovsky, O.; Immervoll, H.; Bøe, S.O.; Svendsen, A.; Prestegarden, L.; Røsland, G.; Thorsen, F.; Stuhr, L.; et al. CD133 negative glioma cells form tumors in nude rats and give rise to CD133 positive cells. Int. J. Cancer 2008, 122, 761–768. [Google Scholar] [CrossRef]
- Brescia, P.; Ortensi, B.; Fornasari, L.; Levi, D.; Broggi, G.; Pelicci, G. CD133 is essential for glioblastoma stem cell maintenance. Stem Cells 2013, 31, 857–869. [Google Scholar] [CrossRef]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef] [PubMed]
- Capp, J.-P. Cancer stem cells: From historical roots to a new perspective. J. Oncol. 2019, 2019, 5189232. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, B.; Tobias, A.L.; Han, Y.; Lee, G.; Guo, D.; Dey, M.; Lesniak, M.S.; Ahmed, A.U. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014, 21, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Kester, L.; van Oudenaarden, A. Single-cell transcriptomics meets lineage tracing. Cell Stem Cell 2018, 23, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Karimi Roshan, M.; Soltani, A.; Soleimani, A.; Kahkhaie, R.K.; Afshari, A.R.; Soukhtanloo, M. Role of AKT and mTOR signaling pathways in the induction of epithelial-mesenchymal transition (EMT) process. Biochimie 2019, 165, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Jin, W. Role of JAK/STAT3 signaling in the regulation of metastasis, the transition of cancer stem cells, and chemoresistance of cancer by epithelial-mesenchymal transition. Cells 2020, 9, 217. [Google Scholar] [CrossRef]
- Hu, W.; Wang, Z.; Zhang, S.; Lu, X.; Wu, J.; Yu, K.; Ji, A.; Lu, W.; Wang, Z.; Wu, J.; et al. IQGAP1 promotes pancreatic cancer progression and epithelial-mesenchymal transition (EMT) through Wnt/β-catenin signaling. Sci. Rep. 2019, 9, 7539. [Google Scholar] [CrossRef]
- Marusyk, A.; Janiszewska, M.; Polyak, K. Intratumor heterogeneity: The rosetta stone of therapy resistance. Cancer Cell 2020, 37, 471–484. [Google Scholar] [CrossRef]
- Zahreddine, H.; Borden, K.L.B. Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 2013, 4, 28. [Google Scholar] [CrossRef]
- Yoshida, A.; Bu, Y.; Qie, S.; Wrangle, J.; Camp, E.R.; Hazard, E.S.; Hardiman, G.; de Leeuw, R.; Knudsen, K.E.; Diehl, J.A. SLC36A1-mTORC1 signaling drives acquired resistance to CDK4/6 inhibitors. Sci. Adv. 2019, 5, eaax6352. [Google Scholar] [CrossRef]
- Muley, H.; Fadó, R.; Rodríguez-Rodríguez, R.; Casals, N. Drug uptake-based chemoresistance in breast cancer treatment. Biochem. Pharmacol. 2020, 177, 113959. [Google Scholar] [CrossRef]
- Baxter, D.E.; Kim, B.; Hanby, A.M.; Verghese, E.T.; Sims, A.H.; Hughes, T.A. Neoadjuvant endocrine therapy in breast cancer upregulates the cytotoxic drug pump ABCG2/BCRP, and may lead to resistance to subsequent chemotherapy. Clin. Breast Cancer 2018, 18, 481–488. [Google Scholar] [CrossRef]
- Yu, T.; Cheng, H.; Ding, Z.; Wang, Z.; Zhou, L.; Zhao, P.; Tan, S.; Xu, X.; Huang, X.; Liu, M.; et al. GPER mediates decreased chemosensitivity via regulation of ABCG2 expression and localization in tamoxifen-resistant breast cancer cells. Mol. Cell. Endocrinol. 2020, 506, 110762. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Leech, A.O.; Vellanki, S.H.; Rutherford, E.J.; Keogh, A.; Jahns, H.; Hudson, L.; O’Donovan, N.; Sabri, S.; Abdulkarim, B.; Sheehan, K.M.; et al. Cleavage of the extracellular domain of junctional adhesion molecule-A is associated with resistance to anti-HER2 therapies in breast cancer settings. Breast Cancer Res. 2018, 20, 140. [Google Scholar] [CrossRef]
- Brennan, K.; McSherry, E.A.; Hudson, L.; Kay, E.W.; Hill, A.D.K.; Young, L.S.; Hopkins, A.M. Junctional adhesion molecule-A is co-expressed with HER2 in breast tumors and acts as a novel regulator of HER2 protein degradation and signaling. Oncogene 2013, 32, 2799–2804. [Google Scholar] [CrossRef]
- Gerstung, M.; Jolly, C.; Leshchiner, I.; Dentro, S.C.; Gonzalez, S.; Rosebrock, D.; Mitchell, T.J.; Rubanova, Y.; Anur, P.; Yu, K.; et al. The evolutionary history of 2,658 cancers. Nature 2020, 578, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.T.; Vyse, S.; Huang, P.H. Rare epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer. Semin. Cancer Biol. 2020, 61, 167–179. [Google Scholar] [CrossRef]
- Sampera, A.; Sánchez-Martín, F.J.; Arpí, O.; Visa, L.; Iglesias, M.; Menéndez, S.; Gaye, É.; Dalmases, A.; Clavé, S.; Gelabert-Baldrich, M.; et al. HER-family ligands promote acquired resistance to trastuzumab in gastric cancer. Mol. Cancer Ther. 2019, 18, 2135–2145. [Google Scholar] [CrossRef]
- Bray, S.M.; Lee, J.; Kim, S.T.; Hur, J.Y.; Ebert, P.J.; Calley, J.N.; Wulur, I.H.; Gopalappa, T.; Wong, S.S.; Qian, H.-R.; et al. Genomic characterization of intrinsic and acquired resistance to cetuximab in colorectal cancer patients. Sci. Rep. 2019, 9, 15365. [Google Scholar] [CrossRef]
- Del Re, M.; Rofi, E.; Restante, G.; Crucitta, S.; Arrigoni, E.; Fogli, S.; Di Maio, M.; Petrini, I.; Danesi, R. Implications of KRAS mutations in acquired resistance to treatment in NSCLC. Oncotarget 2018, 9, 6630–6643. [Google Scholar] [CrossRef] [PubMed]
- Gornstein, E.L.; Sandefur, S.; Chung, J.H.; Gay, L.M.; Holmes, O.; Erlich, R.L.; Soman, S.; Martin, L.K.; Rose, A.V.; Stephens, P.J.; et al. BRCA2 reversion mutation associated with acquired resistance to olaparib in estrogen receptor-positive breast cancer detected by genomic profiling of tissue and liquid biopsy. Clin. Breast Cancer 2018, 18, 184–188. [Google Scholar] [CrossRef]
- Faraoni, I.; Graziani, G. Role of BRCA mutations in cancer treatment with poly(ADP-ribose) polymerase (PARP) inhibitors. Cancers 2018, 10, 487. [Google Scholar] [CrossRef]
- Tassone, P.; Tagliaferri, P.; Perricelli, A.; Blotta, S.; Quaresima, B.; Martelli, M.L.; Goel, A.; Barbieri, V.; Costanzo, F.; Boland, C.R.; et al. BRCA1 expression modulates chemosensitivity of BRCA1-defective HCC1937 human breast cancer cells. Br. J. Cancer 2003, 88, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Kwa, M.; Edwards, S.; Downey, A.; Reich, E.; Wallach, R.; Curtin, J.; Muggia, F. Ovarian cancer in BRCA mutation carriers: Improved outcome after intraperitoneal (IP) cisplatin. Ann. Surg. Oncol. 2014, 21, 1468–1473. [Google Scholar] [CrossRef]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Pishvaian, M.J.; Biankin, A.V.; Bailey, P.; Chang, D.K.; Laheru, D.; Wolfgang, C.L.; Brody, J.R. BRCA2 secondary mutation-mediated resistance to platinum and PARP inhibitor-based therapy in pancreatic cancer. Br. J. Cancer 2017, 116, 1021–1026. [Google Scholar] [CrossRef]
- Lorenzon, I.; Pellarin, I.; Pellizzari, I.; D’Andrea, S.; Belletti, B.; Sonego, M.; Baldassarre, G.; Schiappacassi, M. Identification and characterization of a new platinum-induced TP53 mutation in MDAH ovarian cancer cells. Cells 2019, 9, 36. [Google Scholar] [CrossRef]
- Williams, D.S.; Mouradov, D.; Browne, C.; Palmieri, M.; Elliott, M.J.; Nightingale, R.; Fang, C.G.; Li, R.; Mariadason, J.M.; Faragher, I.; et al. Overexpression of TP53 protein is associated with the lack of adjuvant chemotherapy benefit in patients with stage III colorectal cancer. Mod. Pathol. 2020, 33, 483–495. [Google Scholar] [CrossRef]
- Brown, R.; Curry, E.; Magnani, L.; Wilhelm-Benartzi, C.S.; Borley, J. Poised epigenetic states and acquired drug resistance in cancer. Nat. Rev. Cancer 2014, 14, 747–753. [Google Scholar] [CrossRef]
- Romero-Garcia, S.; Prado-Garcia, H.; Carlos-Reyes, A. Role of DNA methylation in the resistance to therapy in solid tumors. Front. Oncol. 2020, 10, 1152. [Google Scholar] [CrossRef]
- Ponnusamy, L.; Mahalingaiah, P.K.S.; Chang, Y.-W.; Singh, K.P. Role of cellular reprogramming and epigenetic dysregulation in acquired chemoresistance in breast cancer. CDR 2019. [Google Scholar] [CrossRef]
- Heijink, A.M.; Everts, M.; Honeywell, M.E.; Richards, R.; Kok, Y.P.; de Vries, E.G.E.; Lee, M.J.; van Vugt, M.A.T.M. Modeling of cisplatin-induced signaling dynamics in triple-negative breast cancer cells reveals mediators of sensitivity. Cell Rep. 2019, 28, 2345–2357.e5. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Pradella, D.; Naro, C.; Sette, C.; Ghigna, C. EMT and stemness: Flexible processes tuned by alternative splicing in development and cancer progression. Mol. Cancer 2017, 16, 8. [Google Scholar] [CrossRef]
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Choi, W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009, 69, 5820–5828. [Google Scholar] [CrossRef]
- He, Y.; Xie, H.; Yu, P.; Jiang, S.; Wei, L. FOXC2 promotes epithelial-mesenchymal transition and cisplatin resistance of non-small cell lung cancer cells. Cancer Chemother. Pharmacol. 2018, 82, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Jahangiri, A.; Chen, W.; Nguyen, A.T.; Yagnik, G.; Pereira, M.P.; Jain, S.; Garcia, J.H.; Shah, S.S.; Wadhwa, H.; et al. Clonal ZEB1-driven mesenchymal transition promotes targetable oncologic antiangiogenic therapy resistance. Cancer Res. 2020, 80, 1498–1511. [Google Scholar] [CrossRef]
- Marín-Aguilera, M.; Codony-Servat, J.; Reig, Ò.; Lozano, J.J.; Fernández, P.L.; Pereira, M.V.; Jiménez, N.; Donovan, M.; Puig, P.; Mengual, L.; et al. Epithelial-to-mesenchymal transition mediates docetaxel resistance and high risk of relapse in prostate cancer. Mol. Cancer Ther. 2014, 13, 1270–1284. [Google Scholar] [CrossRef]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef]
- Weng, C.-H.; Chen, L.-Y.; Lin, Y.-C.; Shih, J.-Y.; Lin, Y.-C.; Tseng, R.-Y.; Chiu, A.-C.; Yeh, Y.-H.; Liu, C.; Lin, Y.-T.; et al. Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for acquired resistance to EGFR TKI. Oncogene 2019, 38, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, L.; Li, A.; Han, X. The roles of ZEB1 in tumorigenic progression and epigenetic modifications. Biomed. Pharmacother. 2019, 110, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Sun, Y.; Ma, L. ZEB1: At the crossroads of epithelial-mesenchymal transition, metastasis and therapy resistance. Cell Cycle 2015, 14, 481–487. [Google Scholar] [CrossRef]
- Zhang, P.; Wei, Y.; Wang, L.; Debeb, B.G.; Yuan, Y.; Zhang, J.; Yuan, J.; Wang, M.; Chen, D.; Sun, Y.; et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat. Cell Biol. 2014, 16, 864–875. [Google Scholar] [CrossRef]
- Cortez, M.A.; Valdecanas, D.; Zhang, X.; Zhan, Y.; Bhardwaj, V.; Calin, G.A.; Komaki, R.; Giri, D.K.; Quini, C.C.; Wolfe, T.; et al. Therapeutic delivery of miR-200c enhances radiosensitivity in lung cancer. Mol. Ther. 2014, 22, 1494–1503. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.-S.; Sun, Y.-Z.; Wang, S.-M.; Ruan, J.-S. Epithelial-mesenchymal transition: Potential regulator of ABC transporters in tumor progression. J. Cancer 2017, 8, 2319–2327. [Google Scholar] [CrossRef]
- Wu, Y.; Jin, D.; Wang, X.; Du, J.; Di, W.; An, J.; Shao, C.; Guo, J. UBE2C induces cisplatin resistance via ZEB1/2-dependent upregulation of ABCG2 and ERCC1 in NSCLC cells. J. Oncol. 2019, 2019, 8607859. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Z.; Zhang, Q.; Zhang, Q.; Sun, P.; Xiang, R.; Ren, G.; Yang, S. ZEB1 confers chemotherapeutic resistance to breast cancer by activating ATM. Cell Death Dis. 2018, 9, 57. [Google Scholar] [CrossRef]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef]
- Gubelmann, C.; Schwalie, P.C.; Raghav, S.K.; Röder, E.; Delessa, T.; Kiehlmann, E.; Waszak, S.M.; Corsinotti, A.; Udin, G.; Holcombe, W.; et al. Identification of the transcription factor ZEB1 as a central component of the adipogenic gene regulatory network. Elife 2014, 3, e03346. [Google Scholar] [CrossRef]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT subtype influences epithelial plasticity and mode of cell migration. Dev. Cell 2018, 45, 681–695.e4. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Loret, N.; Denys, H.; Tummers, P.; Berx, G. The role of epithelial-to-mesenchymal plasticity in ovarian cancer progression and therapy resistance. Cancers 2019, 11, 838. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Lee, D.-K.; Feng, Z.; Xu, Y.; Bu, W.; Li, Y.; Liao, L.; Xu, J. Breast tumor cell-specific knockout of Twist1 inhibits cancer cell plasticity, dissemination, and lung metastasis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 11494–11499. [Google Scholar] [CrossRef]
- Tran, H.D.; Luitel, K.; Kim, M.; Zhang, K.; Longmore, G.D.; Tran, D.D. Transient SNAIL1 expression is necessary for metastatic competence in breast cancer. Cancer Res. 2014, 74, 6330–6340. [Google Scholar] [CrossRef]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef] [PubMed]
- Díaz, V.M.; de Herreros, A.G. F-box proteins: Keeping the epithelial-to-mesenchymal transition (EMT) in check. Semin. Cancer Biol. 2016, 36, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di Donato, M.; Bilancio, A.; Perillo, B.; Sinisi, A.A.; Migliaccio, A.; Castoria, G. Estrogen receptors in epithelial-mesenchymal transition of prostate cancer. Cancers 2019, 11, 1418. [Google Scholar] [CrossRef]
- Hausser, J.; Szekely, P.; Bar, N.; Zimmer, A.; Sheftel, H.; Caldas, C.; Alon, U. Tumor diversity and the trade-off between universal cancer tasks. Nat. Commun. 2019, 10, 5423. [Google Scholar] [CrossRef]
- Celià-Terrassa, T.; Meca-Cortés, O.; Mateo, F.; Martínez de Paz, A.; Rubio, N.; Arnal-Estapé, A.; Ell, B.J.; Bermudo, R.; Díaz, A.; Guerra-Rebollo, M.; et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J. Clin. Investig. 2012, 122, 1849–1868. [Google Scholar] [CrossRef]
- Tsuji, T.; Ibaragi, S.; Shima, K.; Hu, M.G.; Katsurano, M.; Sasaki, A.; Hu, G.-F. Epithelial-mesenchymal transition induced by growth suppressor p12CDK2-AP1 promotes tumor cell local invasion but suppresses distant colony growth. Cancer Res. 2008, 68, 10377–10386. [Google Scholar] [CrossRef]
- Hoek, K.S.; Eichhoff, O.M.; Schlegel, N.C.; Döbbeling, U.; Kobert, N.; Schaerer, L.; Hemmi, S.; Dummer, R. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008, 68, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, R.M.; Yano, S. Tumor-specific S/G2-phase cell cycle arrest of cancer cells by methionine restriction. Methods Mol. Biol. 2019, 1866, 49–60. [Google Scholar] [CrossRef]
- Zhou, J.; Giannakakou, P. Targeting microtubules for cancer chemotherapy. Curr. Med. Chem. Anticancer Agents 2005, 5, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, K.A.; Hill, D.S.; Daignault, S.M.; Lui, G.Y.L.; Sharp, D.M.; Gabrielli, B.; Weninger, W.; Haass, N.K. Cell cycle phase-specific drug resistance as an escape mechanism of melanoma cells. J. Investig. Dermatol. 2016, 136, 1479–1489. [Google Scholar] [CrossRef] [PubMed]
- Mao, Q.-Q.; Bai, Y.; Lin, Y.-W.; Zheng, X.-Y.; Qin, J.; Yang, K.; Xie, L.-P. Resveratrol confers resistance against taxol via induction of cell cycle arrest in human cancer cell lines. Mol. Nutr. Food Res. 2010, 54, 1574–1584. [Google Scholar] [CrossRef]
- Wang, X.; Pan, L.; Mao, N.; Sun, L.; Qin, X.; Yin, J. Cell-cycle synchronization reverses Taxol resistance of human ovarian cancer cell lines. Cancer Cell Int. 2013, 13, 77. [Google Scholar] [CrossRef] [PubMed]
- Yoshiba, S.; Ito, D.; Nagumo, T.; Shirota, T.; Hatori, M.; Shintani, S. Hypoxia induces resistance to 5-fluorouracil in oral cancer cells via G(1) phase cell cycle arrest. Oral Oncol. 2009, 45, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Shao, F.; Martin, S.; Xu, X.; Deng, C.-X. WEE1 inhibition targets cell cycle checkpoints for triple negative breast cancers to overcome cisplatin resistance. Sci. Rep. 2017, 7, 43517. [Google Scholar] [CrossRef] [PubMed]
- Sarin, N.; Engel, F.; Kalayda, G.V.; Mannewitz, M.; Cinatl, J., Jr.; Rothweiler, F.; Michaelis, M.; Saafan, H.; Ritter, C.A.; Jaehde, U.; et al. Cisplatin resistance in non-small cell lung cancer cells is associated with an abrogation of cisplatin-induced G2/M cell cycle arrest. PLoS ONE 2017, 12, e0181081. [Google Scholar] [CrossRef] [PubMed]
- Horibe, S.; Matsuda, A.; Tanahashi, T.; Inoue, J.; Kawauchi, S.; Mizuno, S.; Ueno, M.; Takahashi, K.; Maeda, Y.; Maegouchi, T.; et al. Cisplatin resistance in human lung cancer cells is linked with dysregulation of cell cycle associated proteins. Life Sci. 2015, 124, 31–40. [Google Scholar] [CrossRef]
- Tormo, E.; Ballester, S.; Adam-Artigues, A.; Burgués, O.; Alonso, E.; Bermejo, B.; Menéndez, S.; Zazo, S.; Madoz-Gúrpide, J.; Rovira, A.; et al. The miRNA-449 family mediates doxorubicin resistance in triple-negative breast cancer by regulating cell cycle factors. Sci. Rep. 2019, 9, 5316. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef]
- Kim, T.W.; Lee, S.Y.; Kim, M.; Cheon, C.; Ko, S.-G. Kaempferol induces autophagic cell death via IRE1-JNK-CHOP pathway and inhibition of G9a in gastric cancer cells. Cell Death Dis. 2018, 9, 875. [Google Scholar] [CrossRef]
- Sun, D.; Zhu, L.; Zhao, Y.; Jiang, Y.; Chen, L.; Yu, Y.; Ouyang, L. Fluoxetine induces autophagic cell death via eEF2K-AMPK-mTOR-ULK complex axis in triple negative breast cancer. Cell Prolif. 2018, 51, e12402. [Google Scholar] [CrossRef]
- Lee, Y.J.; Won, A.J.; Lee, J.; Jung, J.H.; Yoon, S.; Lee, B.M.; Kim, H.S. Molecular mechanism of SAHA on regulation of autophagic cell death in tamoxifen-resistant MCF-7 breast cancer cells. Int. J. Med. Sci. 2012, 9, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Kögel, D. Cell death by autophagy: Emerging molecular mechanisms and implications for cancer therapy. Oncogene 2015, 34, 5105–5113. [Google Scholar] [CrossRef]
- Sui, X.; Kong, N.; Ye, L.; Han, W.; Zhou, J.; Zhang, Q.; He, C.; Pan, H. p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 2014, 344, 174–179. [Google Scholar] [CrossRef]
- Li, X.; Zhou, Y.; Li, Y.; Yang, L.; Ma, Y.; Peng, X.; Yang, S.; Liu, J.; Li, H. Autophagy: A novel mechanism of chemoresistance in cancers. Biomed. Pharmacother. 2019, 119, 109415. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Gu, C.; Zhong, D.; Shi, L.; Kong, Y.; Zhou, Z.; Liu, S. Induction of autophagy counteracts the anticancer effect of cisplatin in human esophageal cancer cells with acquired drug resistance. Cancer Lett. 2014, 355, 34–45. [Google Scholar] [CrossRef]
- O’Donovan, T.R.; O’Sullivan, G.C.; McKenna, S.L. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy 2011, 7, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Panda, P.K.; Mukhopadhyay, S.; Das, D.N.; Sinha, N.; Naik, P.P.; Bhutia, S.K. Mechanism of autophagic regulation in carcinogenesis and cancer therapeutics. Semin. Cell Dev. Biol. 2015, 39, 43–55. [Google Scholar] [CrossRef]
- Choi, C.-H.; Jung, Y.-K.; Oh, S.-H. Autophagy induction by capsaicin in malignant human breast cells is modulated by p38 and extracellular signal-regulated mitogen-activated protein kinases and retards cell death by suppressing endoplasmic reticulum stress-mediated apoptosis. Mol. Pharmacol. 2010, 78, 114–125. [Google Scholar] [CrossRef]
- Sun, W.-L.; Chen, J.; Wang, Y.-P.; Zheng, H. Autophagy protects breast cancer cells from epirubicin-induced apoptosis and facilitates epirubicin-resistance development. Autophagy 2011, 7, 1035–1044. [Google Scholar] [CrossRef]
- Xu, L.; Liu, J.-H.; Zhang, J.; Zhang, N.; Wang, Z.-H. Blockade of autophagy aggravates endoplasmic reticulum stress and improves Paclitaxel cytotoxicity in human cervical cancer cells. Cancer Res. Treat. 2015, 47, 313–321. [Google Scholar] [CrossRef]
- Yoon, J.-H.; Ahn, S.-G.; Lee, B.-H.; Jung, S.-H.; Oh, S.-H. Role of autophagy in chemoresistance: Regulation of the ATM-mediated DNA-damage signaling pathway through activation of DNA-PKcs and PARP-1. Biochem. Pharmacol. 2012, 83, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Han, M.W.; Lee, J.C.; Choi, J.-Y.; Kim, G.C.; Chang, H.W.; Nam, H.Y.; Kim, S.W.; Kim, S.Y. Autophagy inhibition can overcome radioresistance in breast cancer cells through suppression of TAK1 activation. Anticancer Res. 2014, 34, 1449–1455. [Google Scholar] [PubMed]
- Min, H.; Xu, M.; Chen, Z.-R.; Zhou, J.-D.; Huang, M.; Zheng, K.; Zou, X.-P. Bortezomib induces protective autophagy through AMP-activated protein kinase activation in cultured pancreatic and colorectal cancer cells. Cancer Chemother. Pharmacol. 2014, 74, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zheng, H.; Ruan, J.; Fang, W.; Li, A.; Tian, G.; Niu, X.; Luo, S.; Zhao, P. Autophagy inhibition induces enhanced proapoptotic effects of ZD6474 in glioblastoma. Br. J. Cancer 2013, 109, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, G.S. Role of autophagy in cisplatin resistance in ovarian cancer cells. J. Biol. Chem. 2014, 289, 17163–17173. [Google Scholar] [CrossRef]
- Chen, X.; Tan, M.; Xie, Z.; Feng, B.; Zhao, Z.; Yang, K.; Hu, C.; Liao, N.; Wang, T.; Chen, D.; et al. Inhibiting ROS-STAT3-dependent autophagy enhanced capsaicin-induced apoptosis in human hepatocellular carcinoma cells. Free Radic. Res. 2016, 50, 744–755. [Google Scholar] [CrossRef]
- Masui, A.; Hamada, M.; Kameyama, H.; Wakabayashi, K.; Takasu, A.; Imai, T.; Iwai, S.; Yura, Y. Autophagy as a survival mechanism for squamous cell carcinoma cells in endonuclease g-mediated apoptosis. PLoS ONE 2016, 11, e0162786. [Google Scholar] [CrossRef]
- Song, P.; Ye, L.; Fan, J.; Li, Y.; Zeng, X.; Wang, Z.; Wang, S.; Zhang, G.; Yang, P.; Cao, Z.; et al. Asparaginase induces apoptosis and cytoprotective autophagy in chronic myeloid leukemia cells. Oncotarget 2015, 6, 3861–3873. [Google Scholar] [CrossRef]
- Chen, Y.S.; Song, H.X.; Lu, Y.; Li, X.; Chen, T.; Zhang, Y.; Xue, J.X.; Liu, H.; Kan, B.; Yang, G.; et al. Autophagy inhibition contributes to radiation sensitization of esophageal squamous carcinoma cells. Dis. Esophagus 2011, 24, 437–443. [Google Scholar] [CrossRef]
- Lin, J.-F.; Tsai, T.-F.; Liao, P.-C.; Lin, Y.-H.; Lin, Y.-C.; Chen, H.-E.; Chou, K.-Y.; Hwang, T.I.-S. Benzyl isothiocyanate induces protective autophagy in human prostate cancer cells via inhibition of mTOR signaling. Carcinogenesis 2013, 34, 406–414. [Google Scholar] [CrossRef]
- Lin, C.-J.; Lee, C.-C.; Shih, Y.-L.; Lin, T.-Y.; Wang, S.-H.; Lin, Y.-F.; Shih, C.-M. Resveratrol enhances the therapeutic effect of temozolomide against malignant glioma in vitro and in vivo by inhibiting autophagy. Free Radic. Biol. Med. 2012, 52, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Kong, N.; Wang, X.; Fang, Y.; Hu, X.; Xu, Y.; Chen, W.; Wang, K.; Li, D.; Jin, W.; et al. JNK confers 5-fluorouracil resistance in p53-deficient and mutant p53-expressing colon cancer cells by inducing survival autophagy. Sci. Rep. 2014, 4, 4694. [Google Scholar] [CrossRef]
- Lang, F.; Qin, Z.; Li, F.; Zhang, H.; Fang, Z.; Hao, E. Apoptotic cell death induced by resveratrol is partially mediated by the autophagy pathway in human ovarian cancer cells. PLoS ONE 2015, 10, e0129196. [Google Scholar] [CrossRef] [PubMed]
- Kaewpiboon, C.; Surapinit, S.; Malilas, W.; Moon, J.; Phuwapraisirisan, P.; Tip-Pyang, S.; Johnston, R.N.; Koh, S.S.; Assavalapsakul, W.; Chung, Y.-H. Feroniellin A-induced autophagy causes apoptosis in multidrug-resistant human A549 lung cancer cells. Int. J. Oncol. 2014, 44, 1233–1242. [Google Scholar] [CrossRef]
- Han, W.; Pan, H.; Chen, Y.; Sun, J.; Wang, Y.; Li, J.; Ge, W.; Feng, L.; Lin, X.; Wang, X.; et al. EGFR tyrosine kinase inhibitors activate autophagy as a cytoprotective response in human lung cancer cells. PLoS ONE 2011, 6, e18691. [Google Scholar] [CrossRef]
- Notte, A.; Ninane, N.; Arnould, T.; Michiels, C. Hypoxia counteracts taxol-induced apoptosis in MDA-MB-231 breast cancer cells: Role of autophagy and JNK activation. Cell Death Dis. 2013, 4, e638. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Jung, J.-Y.; Choi, S.; Lee, H.; Morales, L.D.; Koh, J.-T.; Kim, S.H.; Choi, Y.-D.; Choi, C.; Slaga, T.J.; et al. GFRA1 promotes cisplatin-induced chemoresistance in osteosarcoma by inducing autophagy. Autophagy 2017, 13, 149–168. [Google Scholar] [CrossRef]
- Cheng, X.; Liu, H.; Jiang, C.-C.; Fang, L.; Chen, C.; Zhang, X.-D.; Jiang, Z.-W. Connecting endoplasmic reticulum stress to autophagy through IRE1/JNK/beclin-1 in breast cancer cells. Int. J. Mol. Med. 2014, 34, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Chen, D.; Huang, J.; Wang, R.; Feng, B.; Song, H.; Chen, L. HMGB1-mediated autophagy promotes docetaxel resistance in human lung adenocarcinoma. Mol. Cancer 2014, 13, 165. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.-L.; DeLay, M.; Jahangiri, A.; Molinaro, A.M.; Rose, S.D.; Carbonell, W.S.; Aghi, M.K. Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma. Cancer Res. 2012, 72, 1773–1783. [Google Scholar] [CrossRef]
- Wu, H.-M.; Jiang, Z.-F.; Ding, P.-S.; Shao, L.-J.; Liu, R.-Y. Hypoxia-induced autophagy mediates cisplatin resistance in lung cancer cells. Sci. Rep. 2015, 5, 12291. [Google Scholar] [CrossRef]
- Xu, N.; Zhang, J.; Shen, C.; Luo, Y.; Xia, L.; Xue, F.; Xia, Q. Cisplatin-induced downregulation of miR-199a-5p increases drug resistance by activating autophagy in HCC cell. Biochem. Biophys. Res. Commun. 2012, 423, 826–831. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, W.; Yan, Z.; Zhao, W.; Mi, J.; Li, J.; Yan, H. Metformin induces autophagy and G0/G1 phase cell cycle arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways. J. Exp. Clin. Cancer Res. 2018, 37, 63. [Google Scholar] [CrossRef]
- Tan, M.; Wu, A.; Liao, N.; Liu, M.; Guo, Q.; Yi, J.; Wang, T.; Huang, Y.; Qiu, B.; Zhou, W. Inhibiting ROS-TFE3-dependent autophagy enhances the therapeutic response to metformin in breast cancer. Free Radic. Res. 2018, 52, 872–886. [Google Scholar] [CrossRef]
- Tai, W.-T.; Shiau, C.-W.; Chen, H.-L.; Liu, C.-Y.; Lin, C.-S.; Cheng, A.-L.; Chen, P.-J.; Chen, K.-F. Mcl-1-dependent activation of Beclin 1 mediates autophagic cell death induced by sorafenib and SC-59 in hepatocellular carcinoma cells. Cell Death Dis. 2013, 4, e485. [Google Scholar] [CrossRef]
- Shi, Y.-H.; Ding, Z.-B.; Zhou, J.; Hui, B.; Shi, G.-M.; Ke, A.-W.; Wang, X.-Y.; Dai, Z.; Peng, Y.-F.; Gu, C.-Y.; et al. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy 2011, 7, 1159–1172. [Google Scholar] [CrossRef] [PubMed]
- Shingu, T.; Fujiwara, K.; Bögler, O.; Akiyama, Y.; Moritake, K.; Shinojima, N.; Tamada, Y.; Yokoyama, T.; Kondo, S. Inhibition of autophagy at a late stage enhances imatinib-induced cytotoxicity in human malignant glioma cells. Int. J. Cancer 2009, 124, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Dominiak, A.; Chełstowska, B.; Olejarz, W.; Nowicka, G. Communication in the cancer microenvironment as a target for therapeutic interventions. Cancers 2020, 12, 1232. [Google Scholar] [CrossRef]
- Xiang, Y.; Liu, Y.; Yang, Y.; Hu, H.; Hu, P.; Ren, H.; Zhang, D. A secretomic study on human hepatocellular carcinoma multiple drug-resistant cell lines. Oncol. Rep. 2015, 34, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Bosse, K.; Haneder, S.; Arlt, C.; Ihling, C.H.; Seufferlein, T.; Sinz, A. Mass spectrometry-based secretome analysis of non-small cell lung cancer cell lines. Proteomics 2016, 16, 2801–2814. [Google Scholar] [CrossRef]
- Böttger, F.; Schaaij-Visser, T.B.; de Reus, I.; Piersma, S.R.; Pham, T.V.; Nagel, R.; Brakenhoff, R.H.; Thunnissen, E.; Smit, E.F.; Jimenez, C.R. Proteome analysis of non-small cell lung cancer cell line secretomes and patient sputum reveals biofluid biomarker candidates for cisplatin response prediction. J. Proteomics 2019, 196, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Bateman, N.W.; Jaworski, E.; Ao, W.; Wang, G.; Litzi, T.; Dubil, E.; Marcus, C.; Conrads, K.A.; Teng, P.-N.; Hood, B.L.; et al. Elevated AKAP12 in paclitaxel-resistant serous ovarian cancer cells is prognostic and predictive of poor survival in patients. J. Proteome Res. 2015, 14, 1900–1910. [Google Scholar] [CrossRef]
- Teng, P.-N.; Wang, G.; Hood, B.L.; Conrads, K.A.; Hamilton, C.A.; Maxwell, G.L.; Darcy, K.M.; Conrads, T.P. Identification of candidate circulating cisplatin-resistant biomarkers from epithelial ovarian carcinoma cell secretomes. Br. J. Cancer 2014, 110, 123–132. [Google Scholar] [CrossRef]
- Yao, L.; Zhang, Y.; Chen, K.; Hu, X.; Xu, L.X. Discovery of IL-18 as a novel secreted protein contributing to doxorubicin resistance by comparative secretome analysis of MCF-7 and MCF-7/Dox. PLoS ONE 2011, 6, e24684. [Google Scholar] [CrossRef]
- Wojtuszkiewicz, A.; Schuurhuis, G.J.; Kessler, F.L.; Piersma, S.R.; Knol, J.C.; Pham, T.V.; Jansen, G.; Musters, R.J.P.; van Meerloo, J.; Assaraf, Y.G.; et al. Exosomes secreted by apoptosis-resistant Acute Myeloid Leukemia (AML) blasts harbor regulatory network proteins potentially involved in antagonism of apoptosis. Mol. Cell. Proteomics 2016, 15, 1281–1298. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-X.; Liu, X.-M.; Lv, M.-M.; Chen, L.; Zhao, J.-H.; Zhong, S.-L.; Ji, M.-H.; Hu, Q.; Luo, Z.; Wu, J.-Z.; et al. Exosomes from drug-resistant breast cancer cells transmit chemoresistance by a horizontal transfer of microRNAs. PLoS ONE 2014, 9, e95240. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Lai, X.; Yu, S.; Chen, S.; Ma, Y.; Zhang, Y.; Li, H.; Zhu, X.; Yao, L.; Zhang, J. Exosomal miR-221/222 enhances tamoxifen resistance in recipient ER-positive breast cancer cells. Breast Cancer Res. Treat. 2014, 147, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.-M.; Zhu, X.-Y.; Chen, W.-X.; Zhong, S.-L.; Hu, Q.; Ma, T.-F.; Zhang, J.; Chen, L.; Tang, J.-H.; Zhao, J.-H. Exosomes mediate drug resistance transfer in MCF-7 breast cancer cells and a probable mechanism is delivery of P-glycoprotein. Tumour Biol. 2014, 35, 10773–10779. [Google Scholar] [CrossRef]
- Ning, K.; Wang, T.; Sun, X.; Zhang, P.; Chen, Y.; Jin, J.; Hua, D. UCH-L1-containing exosomes mediate chemotherapeutic resistance transfer in breast cancer. J. Surg. Oncol. 2017, 115, 932–940. [Google Scholar] [CrossRef]
- Yu, D.-D.; Wu, Y.; Zhang, X.-H.; Lv, M.-M.; Chen, W.-X.; Chen, X.; Yang, S.-J.; Shen, H.; Zhong, S.-L.; Tang, J.-H.; et al. Exosomes from adriamycin-resistant breast cancer cells transmit drug resistance partly by delivering miR-222. Tumour Biol. 2016, 37, 3227–3235. [Google Scholar] [CrossRef]
- Santos, J.C.; da Silva Lima, N.; Sarian, L.O.; Matheu, A.; Ribeiro, M.L.; Derchain, S.F.M. Exosome-mediated breast cancer chemoresistance via miR-155 transfer. Sci. Rep. 2018, 8, 829. [Google Scholar] [CrossRef] [PubMed]
- Crow, J.; Atay, S.; Banskota, S.; Artale, B.; Schmitt, S.; Godwin, A.K. Exosomes as mediators of platinum resistance in ovarian cancer. Oncotarget 2017, 8, 11917–11936. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.-F.; Zhu, Y.-F.; Zhao, Q.-N.; Yang, D.-T.; Dong, Y.-P.; Jiang, L.; Xing, W.-X.; Li, X.-Y.; Xing, H.; Shi, M.; et al. Microvesicles mediate transfer of P-glycoprotein to paclitaxel-sensitive A2780 human ovarian cancer cells, conferring paclitaxel-resistance. Eur. J. Pharmacol. 2014, 738, 83–90. [Google Scholar] [CrossRef]
- Corcoran, C.; Rani, S.; O’Brien, K.; O’Neill, A.; Prencipe, M.; Sheikh, R.; Webb, G.; McDermott, R.; Watson, W.; Crown, J.; et al. Docetaxel-resistance in prostate cancer: Evaluating associated phenotypic changes and potential for resistance transfer via exosomes. PLoS ONE 2012, 7, e50999. [Google Scholar] [CrossRef]
- Vella, L.J.; Behren, A.; Coleman, B.; Greening, D.W.; Hill, A.F.; Cebon, J. Intercellular resistance to BRAF inhibition can be mediated by extracellular vesicle-associated PDGFRβ. Neoplasia 2017, 19, 932–940. [Google Scholar] [CrossRef]
- Cesi, G.; Philippidou, D.; Kozar, I.; Kim, Y.J.; Bernardin, F.; Van Niel, G.; Wienecke-Baldacchino, A.; Felten, P.; Letellier, E.; Dengler, S.; et al. A new ALK isoform transported by extracellular vesicles confers drug resistance to melanoma cells. Mol. Cancer 2018, 17, 145. [Google Scholar] [CrossRef]
- Bebawy, M.; Combes, V.; Lee, E.; Jaiswal, R.; Gong, J.; Bonhoure, A.; Grau, G.E.R. Membrane microparticles mediate transfer of P-glycoprotein to drug sensitive cancer cells. Leukemia 2009, 23, 1643–1649. [Google Scholar] [CrossRef]
- Bouvy, C.; Wannez, A.; Laloy, J.; Chatelain, C.; Dogné, J.-M. Transfer of multidrug resistance among acute myeloid leukemia cells via extracellular vesicles and their microRNA cargo. Leuk. Res. 2017, 62, 70–76. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, R.-X.; Chan, K.-W.; Hu, J.; Zhang, J.; Wei, L.; Tan, H.; Yang, X.; Liu, H. Exosomal transfer of p-STAT3 promotes acquired 5-FU resistance in colorectal cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 320. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Huang, X.; Ma, X.; Zhu, X.; Zhang, Q. Research of the mechanism on miRNA193 in exosomes promotes cisplatin resistance in esophageal cancer cells. PLoS ONE 2020, 15, e0225290. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Ma, C.; Zhou, T.; Dong, X.; Luo, Q.; Geng, L.; Ding, L.; Zhang, Y.; Zhang, L.; Li, N.; et al. Exosomes derived from gemcitabine-resistant cells transfer malignant phenotypic traits via delivery of miRNA-222-3p. Mol. Cancer 2017, 16, 132. [Google Scholar] [CrossRef]
- Torreggiani, E.; Roncuzzi, L.; Perut, F.; Zini, N.; Baldini, N. Multimodal transfer of MDR by exosomes in human osteosarcoma. Int. J. Oncol. 2016, 49, 189–196. [Google Scholar] [CrossRef]
- Qu, L.; Ding, J.; Chen, C.; Wu, Z.-J.; Liu, B.; Gao, Y.; Chen, W.; Liu, F.; Sun, W.; Li, X.-F.; et al. Exosome-transmitted lncARSR promotes sunitinib resistance in renal cancer by acting as a competing endogenous RNA. Cancer Cell 2016, 29, 653–668. [Google Scholar] [CrossRef] [PubMed]
- Peak, T.C.; Panigrahi, G.K.; Praharaj, P.P.; Su, Y.; Shi, L.; Chyr, J.; Rivera-Chávez, J.; Flores-Bocanegra, L.; Singh, R.; Vander Griend, D.J.; et al. Syntaxin 6-mediated exosome secretion regulates enzalutamide resistance in prostate cancer. Mol. Carcinog. 2020, 59, 62–72. [Google Scholar] [CrossRef]
- Fan, J.; Wei, Q.; Koay, E.J.; Liu, Y.; Ning, B.; Bernard, P.W.; Zhang, N.; Han, H.; Katz, M.H.; Zhao, Z.; et al. Chemoresistance transmission via exosome-mediated EphA2 transfer in pancreatic cancer. Theranostics 2018, 8, 5986–5994. [Google Scholar] [CrossRef]
- Burt, R.; Dey, A.; Aref, S.; Aguiar, M.; Akarca, A.; Bailey, K.; Day, W.; Hooper, S.; Kirkwood, A.; Kirschner, K.; et al. Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood 2019, 134, 1415–1429. [Google Scholar] [CrossRef]
- Sahinbegovic, H.; Jelinek, T.; Hrdinka, M.; Bago, J.R.; Turi, M.; Sevcikova, T.; Kurtovic-Kozaric, A.; Hajek, R.; Simicek, M. Intercellular mitochondrial transfer in the tumor microenvironment. Cancers 2020, 12, 1787. [Google Scholar] [CrossRef]
- Desir, S.; Dickson, E.L.; Vogel, R.I.; Thayanithy, V.; Wong, P.; Teoh, D.; Geller, M.A.; Steer, C.J.; Subramanian, S.; Lou, E. Tunneling nanotube formation is stimulated by hypoxia in ovarian cancer cells. Oncotarget 2016, 7, 43150–43161. [Google Scholar] [CrossRef]
- Lu, J.; Zheng, X.; Li, F.; Yu, Y.; Chen, Z.; Liu, Z.; Wang, Z.; Xu, H.; Yang, W. Tunneling nanotubes promote intercellular mitochondria transfer followed by increased invasiveness in bladder cancer cells. Oncotarget 2017, 8, 15539–15552. [Google Scholar] [CrossRef] [PubMed]
- Hekmatshoar, Y.; Nakhle, J.; Galloni, M.; Vignais, M.-L. The role of metabolism and tunneling nanotube-mediated intercellular mitochondria exchange in cancer drug resistance. Biochem. J. 2018, 475, 2305–2328. [Google Scholar] [CrossRef] [PubMed]
- Obenauf, A.C.; Zou, Y.; Ji, A.L.; Vanharanta, S.; Shu, W.; Shi, H.; Kong, X.; Bosenberg, M.C.; Wiesner, T.; Rosen, N.; et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 2015, 520, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Pavlyukov, M.S.; Yu, H.; Bastola, S.; Minata, M.; Shender, V.O.; Lee, Y.; Zhang, S.; Wang, J.; Komarova, S.; Wang, J.; et al. Apoptotic cell-derived extracellular vesicles promote malignancy of glioblastoma via intercellular transfer of splicing factors. Cancer Cell 2018, 34, 119–135.e10. [Google Scholar] [CrossRef]
- Huang, Q.; Li, F.; Liu, X.; Li, W.; Shi, W.; Liu, F.-F.; O’Sullivan, B.; He, Z.; Peng, Y.; Tan, A.-C.; et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med. 2011, 17, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.-J.; Chen, Y.-Y.; Dai, J.-J.; Gu, D.-N.; Mei, Z.; Liu, F.-R.; Huang, Q.; Tian, L. Dying tumor cell-derived exosomal miR-194-5p potentiates survival and repopulation of tumor repopulating cells upon radiotherapy in pancreatic cancer. Mol. Cancer 2020, 19, 68. [Google Scholar] [CrossRef] [PubMed]
- Samuel, P.; Mulcahy, L.A.; Furlong, F.; McCarthy, H.O.; Brooks, S.A.; Fabbri, M.; Pink, R.C.; Carter, D.R.F. Cisplatin induces the release of extracellular vesicles from ovarian cancer cells that can induce invasiveness and drug resistance in bystander cells. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Yu, S.; Li, S.; Wu, J.; Ma, R.; Cao, H.; Zhu, Y.; Feng, J. Exosomes: Decreased sensitivity of lung cancer A549 cells to cisplatin. PLoS ONE 2014, 9, e89534. [Google Scholar] [CrossRef]
- Takahashi, K.; Yan, I.K.; Wood, J.; Haga, H.; Patel, T. Involvement of extracellular vesicle long noncoding RNA (linc-VLDLR) in tumor cell responses to chemotherapy. Mol. Cancer Res. 2014, 12, 1377–1387. [Google Scholar] [CrossRef]
- Patel, G.K.; Khan, M.A.; Bhardwaj, A.; Srivastava, S.K.; Zubair, H.; Patton, M.C.; Singh, S.; Khushman, M.D.; Singh, A.P. Exosomes confer chemoresistance to pancreatic cancer cells by promoting ROS detoxification and miR-155-mediated suppression of key gemcitabine-metabolising enzyme, DCK. Br. J. Cancer 2017, 116, 609–619. [Google Scholar] [CrossRef]
- Mikamori, M.; Yamada, D.; Eguchi, H.; Hasegawa, S.; Kishimoto, T.; Tomimaru, Y.; Asaoka, T.; Noda, T.; Wada, H.; Kawamoto, K.; et al. MicroRNA-155 controls exosome synthesis and promotes gemcitabine resistance in pancreatic ductal adenocarcinoma. Sci. Rep. 2017, 7, 42339. [Google Scholar] [CrossRef]
- Kreger, B.T.; Johansen, E.R.; Cerione, R.A.; Antonyak, M.A. The enrichment of survivin in exosomes from breast cancer cells treated with paclitaxel promotes cell survival and chemoresistance. Cancers 2016, 8, 111. [Google Scholar] [CrossRef]
- Aldonza, M.B.D.; Hong, J.-Y.; Lee, S.K. Paclitaxel-resistant cancer cell-derived secretomes elicit ABCB1-associated docetaxel cross-resistance and escape from apoptosis through FOXO3a-driven glycolytic regulation. Exp. Mol. Med. 2017, 49, e286. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Jutzy, J.M.S.; Aspe, J.R.; McGregor, D.W.; Neidigh, J.W.; Wall, N.R. Survivin is released from cancer cells via exosomes. Apoptosis 2011, 16, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Qiao, D.; Chen, L.; Xu, M.; Chen, S.; Huang, L.; Wang, F.; Chen, Z.; Cai, J.; Fu, L. Chemotherapeutic drugs stimulate the release and recycling of extracellular vesicles to assist cancer cells in developing an urgent chemoresistance. Mol. Cancer 2019, 18, 182. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Chiang, S.-F.; Chen, W.T.-L.; Ke, T.-W.; Chen, T.-W.; You, Y.-S.; Lin, C.-Y.; Chao, K.S.C.; Huang, C.-Y. HMGB1 promotes ERK-mediated mitochondrial Drp1 phosphorylation for chemoresistance through RAGE in colorectal cancer. Cell Death Dis. 2018, 9, 1004. [Google Scholar] [CrossRef]
- He, S.; Cheng, J.; Sun, L.; Wang, Y.; Wang, C.; Liu, X.; Zhang, Z.; Zhao, M.; Luo, Y.; Tian, L.; et al. HMGB1 released by irradiated tumor cells promotes living tumor cell proliferation via paracrine effect. Cell Death Dis. 2018, 9, 648. [Google Scholar] [CrossRef]
- Tanzer, M.C.; Frauenstein, A.; Stafford, C.A.; Phulphagar, K.; Mann, M.; Meissner, F. Quantitative and dynamic catalogs of proteins released during apoptotic and necroptotic cell death. Cell Rep. 2020, 30, 1260–1270.e5. [Google Scholar] [CrossRef] [PubMed]
- Mastri, M.; Tracz, A.; Lee, C.R.; Dolan, M.; Attwood, K.; Christensen, J.G.; Liu, S.; Ebos, J.M.L. A transient pseudosenescent secretome promotes tumor growth after antiangiogenic therapy withdrawal. Cell Rep. 2018, 25, 3706–3720.e8. [Google Scholar] [CrossRef]
- Ohanna, M.; Cheli, Y.; Bonet, C.; Bonazzi, V.F.; Allegra, M.; Giuliano, S.; Bille, K.; Bahadoran, P.; Giacchero, D.; Lacour, J.P.; et al. Secretome from senescent melanoma engages the STAT3 pathway to favor reprogramming of naive melanoma towards a tumor-initiating cell phenotype. Oncotarget 2013, 4, 2212–2224. [Google Scholar] [CrossRef]
- Ohanna, M.; Giuliano, S.; Bonet, C.; Imbert, V.; Hofman, V.; Zangari, J.; Bille, K.; Robert, C.; Bressac-de Paillerets, B.; Hofman, P.; et al. Senescent cells develop a PARP-1 and nuclear factor-{kappa}B-associated secretome (PNAS). Genes Dev. 2011, 25, 1245–1261. [Google Scholar] [CrossRef]
- Sun, X.; Shi, B.; Zheng, H.; Min, L.; Yang, J.; Li, X.; Liao, X.; Huang, W.; Zhang, M.; Xu, S.; et al. Senescence-associated secretory factors induced by cisplatin in melanoma cells promote non-senescent melanoma cell growth through activation of the ERK1/2-RSK1 pathway. Cell Death Dis. 2018, 9, 260. [Google Scholar] [CrossRef]
- Canino, C.; Mori, F.; Cambria, A.; Diamantini, A.; Germoni, S.; Alessandrini, G.; Borsellino, G.; Galati, R.; Battistini, L.; Blandino, R.; et al. SASP mediates chemoresistance and tumor-initiating-activity of mesothelioma cells. Oncogene 2012, 31, 3148–3163. [Google Scholar] [CrossRef]
- Barker, H.E.; Paget, J.T.E.; Khan, A.A.; Harrington, K.J. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef] [PubMed]
- Saleh, R.; Elkord, E. Acquired resistance to cancer immunotherapy: Role of tumor-mediated immunosuppression. Semin. Cancer Biol. 2020, 65, 13–27. [Google Scholar] [CrossRef]
- Wu, J.; Liang, C.; Chen, M.; Su, W. Association between tumor-stroma ratio and prognosis in solid tumor patients: A systematic review and meta-analysis. Oncotarget 2016, 7, 68954–68965. [Google Scholar] [CrossRef]
- Kramer, C.J.H.; Vangangelt, K.M.H.; van Pelt, G.W.; Dekker, T.J.A.; Tollenaar, R.A.E.M.; Mesker, W.E. The prognostic value of tumour-stroma ratio in primary breast cancer with special attention to triple-negative tumours: A review. Breast Cancer Res. Treat. 2019, 173, 55–64. [Google Scholar] [CrossRef]
- Jiang, M.-J.; Gu, D.-N.; Dai, J.-J.; Huang, Q.; Tian, L. Dark side of cytotoxic therapy: Chemoradiation-induced cell death and tumor repopulation. Trends Cancer Res. 2020, 6, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yang, L.; Yu, W.; Wu, Q.; Lian, J.; Li, F.; Liu, S.; Li, A.; He, Z.; Liu, J.; et al. Colorectal cancer cell-derived CCL20 recruits regulatory T cells to promote chemoresistance via FOXO1/CEBPB/NF-κB signaling. J. Immunother. Cancer 2019, 7, 215. [Google Scholar] [CrossRef]
- Rodriguez, Y.I.; Campos, L.E.; Castro, M.G.; Aladhami, A.; Oskeritzian, C.A.; Alvarez, S.E. Sphingosine-1 phosphate: A new modulator of immune plasticity in the tumor microenvironment. Front. Oncol. 2016, 6, 218. [Google Scholar] [CrossRef]
- Mizuno, R.; Kawada, K.; Sakai, Y. Prostaglandin E2/EP signaling in the tumor microenvironment of colorectal cancer. Int. J. Mol. Sci. 2019, 20, 6254. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Valcz, G.; Buzás, E.I.; Sebestyén, A.; Krenács, T.; Szállási, Z.; Igaz, P.; Molnár, B. Extracellular vesicle-based communication may contribute to the co-evolution of cancer stem cells and cancer-associated fibroblasts in anti-cancer therapy. Cancers 2020, 12, 2324. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Zhou, W.; Rong, Y.; Kuang, T.; Xu, X.; Wu, W.; Wang, D.; Lou, W. Exosomal miRNA-106b from cancer-associated fibroblast promotes gemcitabine resistance in pancreatic cancer. Exp. Cell Res. 2019, 383, 111543. [Google Scholar] [CrossRef]
- Yi, Y.; Zeng, S.; Wang, Z.; Wu, M.; Ma, Y.; Ye, X.; Zhang, B.; Liu, H. Cancer-associated fibroblasts promote epithelial-mesenchymal transition and EGFR-TKI resistance of non-small cell lung cancers via HGF/IGF-1/ANXA2 signaling. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Yan, C.; Mu, L.; Huang, K.; Li, X.; Tao, D.; Wu, Y.; Qin, J. Fibroblast-derived exosomes contribute to chemoresistance through priming cancer stem cells in colorectal cancer. PLoS ONE 2015, 10, e0125625. [Google Scholar] [CrossRef]
- Che, Y.; Wang, J.; Li, Y.; Lu, Z.; Huang, J.; Sun, S.; Mao, S.; Lei, Y.; Zang, R.; Sun, N.; et al. Cisplatin-activated PAI-1 secretion in the cancer-associated fibroblasts with paracrine effects promoting esophageal squamous cell carcinoma progression and causing chemoresistance. Cell Death Dis. 2018, 9, 759. [Google Scholar] [CrossRef]
- Zhang, H.; Xie, C.; Yue, J.; Jiang, Z.; Zhou, R.; Xie, R.; Wang, Y.; Wu, S. Cancer-associated fibroblasts mediated chemoresistance by a FOXO1/TGFβ1 signaling loop in esophageal squamous cell carcinoma. Mol. Carcinog. 2017, 56, 1150–1163. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Deng, T.; Liu, R.; Ning, T.; Yang, H.; Liu, D.; Zhang, Q.; Lin, D.; Ge, S.; Bai, M.; et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol. Cancer 2020, 19, 43. [Google Scholar] [CrossRef]
- Ham, I.-H.; Lee, D.; Hur, H. Role of cancer-associated fibroblast in gastric cancer progression and resistance to treatments. J. Oncol. 2019, 2019, 6270784. [Google Scholar] [CrossRef]
- Tao, L.; Huang, G.; Wang, R.; Pan, Y.; He, Z.; Chu, X.; Song, H.; Chen, L. Cancer-associated fibroblasts treated with cisplatin facilitates chemoresistance of lung adenocarcinoma through IL-11/IL-11R/STAT3 signaling pathway. Sci. Rep. 2016, 6, 38408. [Google Scholar] [CrossRef]
- Au Yeung, C.L.; Co, N.-N.; Tsuruga, T.; Yeung, T.-L.; Kwan, S.-Y.; Leung, C.S.; Li, Y.; Lu, E.S.; Kwan, K.; Wong, K.-K.; et al. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat. Commun. 2016, 7, 11150. [Google Scholar] [CrossRef]
- Hu, J.L.; Wang, W.; Lan, X.L.; Zeng, Z.C.; Liang, Y.S.; Yan, Y.R.; Song, F.Y.; Wang, F.F.; Zhu, X.H.; Liao, W.J.; et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol. Cancer 2019, 18, 91. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.-B.; Yan, C.; Mu, L.; Mi, Y.-L.; Zhao, H.; Hu, H.; Li, X.-L.; Tao, D.-D.; Wu, Y.-Q.; Gong, J.-P.; et al. Exosomal Wnt-induced dedifferentiation of colorectal cancer cells contributes to chemotherapy resistance. Oncogene 2019, 38, 1951–1965. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.; Shen, J.; Xie, G.; Wu, J.; He, M.; Gao, L.; Zhang, Y.; Yao, X.; Shen, L. Cancer-associated fibroblasts-derived IL-8 mediates resistance to cisplatin in human gastric cancer. Cancer Lett. 2019, 454, 37–43. [Google Scholar] [CrossRef]
- Shintani, Y.; Fujiwara, A.; Kimura, T.; Kawamura, T.; Funaki, S.; Minami, M.; Okumura, M. IL-6 secreted from cancer-associated fibroblasts mediates chemoresistance in NSCLC by increasing epithelial-mesenchymal transition signaling. J. Thorac. Oncol. 2016, 11, 1482–1492. [Google Scholar] [CrossRef]
- Qiao, Y.; Zhang, C.; Li, A.; Wang, D.; Luo, Z.; Ping, Y.; Zhou, B.; Liu, S.; Li, H.; Yue, D.; et al. IL6 derived from cancer-associated fibroblasts promotes chemoresistance via CXCR7 in esophageal squamous cell carcinoma. Oncogene 2018, 37, 873–883. [Google Scholar] [CrossRef]
- Huber, R.M.; Lucas, J.M.; Gomez-Sarosi, L.A.; Coleman, I.; Zhao, S.; Coleman, R.; Nelson, P.S. DNA damage induces GDNF secretion in the tumor microenvironment with paracrine effects promoting prostate cancer treatment resistance. Oncotarget 2015, 6, 2134–2147. [Google Scholar] [CrossRef]
- Hughes, R.; Qian, B.-Z.; Rowan, C.; Muthana, M.; Keklikoglou, I.; Olson, O.C.; Tazzyman, S.; Danson, S.; Addison, C.; Clemons, M.; et al. Perivascular M2 macrophages stimulate tumor relapse after chemotherapy. Cancer Res. 2015, 75, 3479–3491. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Yao, S.; Hu, Y.; Feng, Y.; Li, M.; Bian, Z.; Zhang, J.; Qin, Y.; Qi, X.; Zhou, L.; et al. The immune-microenvironment confers chemoresistance of colorectal cancer through macrophage-derived IL6. Clin. Cancer Res. 2017, 23, 7375–7387. [Google Scholar] [CrossRef] [PubMed]
- D’Errico, G.; Alonso-Nocelo, M.; Vallespinos, M.; Hermann, P.C.; Alcalá, S.; García, C.P.; Martin-Hijano, L.; Valle, S.; Earl, J.; Cassiano, C.; et al. Tumor-associated macrophage-secreted 14-3-3ζ signals via AXL to promote pancreatic cancer chemoresistance. Oncogene 2019, 38, 5469–5485. [Google Scholar] [CrossRef] [PubMed]
- Larionova, I.; Cherdyntseva, N.; Liu, T.; Patysheva, M.; Rakina, M.; Kzhyshkowska, J. Interaction of tumor-associated macrophages and cancer chemotherapy. Oncoimmunology 2019, 8, 1596004. [Google Scholar] [CrossRef]
- Timaner, M.; Letko-Khait, N.; Kotsofruk, R.; Benguigui, M.; Beyar-Katz, O.; Rachman-Tzemah, C.; Raviv, Z.; Bronshtein, T.; Machluf, M.; Shaked, Y. Therapy-educated mesenchymal stem cells enrich for tumor-initiating cells. Cancer Res. 2018, 78, 1253–1265. [Google Scholar] [CrossRef]
- He, W.; Liang, B.; Wang, C.; Li, S.; Zhao, Y.; Huang, Q.; Liu, Z.; Yao, Z.; Wu, Q.; Liao, W.; et al. MSC-regulated lncRNA MACC1-AS1 promotes stemness and chemoresistance through fatty acid oxidation in gastric cancer. Oncogene 2019, 38, 4637–4654. [Google Scholar] [CrossRef] [PubMed]
- Falzone, L.; Salomone, S.; Libra, M. Evolution of cancer pharmacological treatments at the turn of the third millennium. Front. Pharmacol. 2018, 9, 1300. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhang, L.; Yu, J.; Zhang, Y.; Pang, X.; Ma, C.; Shen, M.; Ruan, S.; Wasan, H.S.; Qiu, S. Clinical efficacy and safety of anti-PD-1/PD-L1 inhibitors for the treatment of advanced or metastatic cancer: A systematic review and meta-analysis. Sci. Rep. 2020, 10, 2083. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-Y.; Yan, Y.-Y.; Li, J.-J.; Adhikari, R.; Fu, L.-W. PD-1/PD-L1 based combinational cancer therapy: Icing on the cake. Front. Pharmacol. 2020, 11, 722. [Google Scholar] [CrossRef]
- Bertagnolli, M.M.; Eagle, C.J.; Zauber, A.G.; Redston, M.; Breazna, A.; Kim, K.; Tang, J.; Rosenstein, R.B.; Umar, A.; Bagheri, D.; et al. Five-year efficacy and safety analysis of the Adenoma Prevention with Celecoxib Trial. Cancer Prev. Res. 2009, 2, 310–321. [Google Scholar] [CrossRef]
- Li, F.; Aljahdali, I.; Ling, X. Cancer therapeutics using survivin BIRC5 as a target: What can we do after over two decades of study? J. Exp. Clin. Cancer Res. 2019, 38, 368. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shnaider, P.V.; Ivanova, O.M.; Malyants, I.K.; Anufrieva, K.S.; Semenov, I.A.; Pavlyukov, M.S.; Lagarkova, M.A.; Govorun, V.M.; Shender, V.O. New Insights into Therapy-Induced Progression of Cancer. Int. J. Mol. Sci. 2020, 21, 7872. https://doi.org/10.3390/ijms21217872
Shnaider PV, Ivanova OM, Malyants IK, Anufrieva KS, Semenov IA, Pavlyukov MS, Lagarkova MA, Govorun VM, Shender VO. New Insights into Therapy-Induced Progression of Cancer. International Journal of Molecular Sciences. 2020; 21(21):7872. https://doi.org/10.3390/ijms21217872
Chicago/Turabian StyleShnaider, Polina V., Olga M. Ivanova, Irina K. Malyants, Ksenia S. Anufrieva, Ilya A. Semenov, Marat S. Pavlyukov, Maria A. Lagarkova, Vadim M. Govorun, and Victoria O. Shender. 2020. "New Insights into Therapy-Induced Progression of Cancer" International Journal of Molecular Sciences 21, no. 21: 7872. https://doi.org/10.3390/ijms21217872
APA StyleShnaider, P. V., Ivanova, O. M., Malyants, I. K., Anufrieva, K. S., Semenov, I. A., Pavlyukov, M. S., Lagarkova, M. A., Govorun, V. M., & Shender, V. O. (2020). New Insights into Therapy-Induced Progression of Cancer. International Journal of Molecular Sciences, 21(21), 7872. https://doi.org/10.3390/ijms21217872