Identification of a Reliable Biomarker Profile for the Diagnosis of Gaucher Disease Type 1 Patients Using a Mass Spectrometry-Based Metabolomic Approach

 , ,
, ,
Abstract
:
1. Introduction
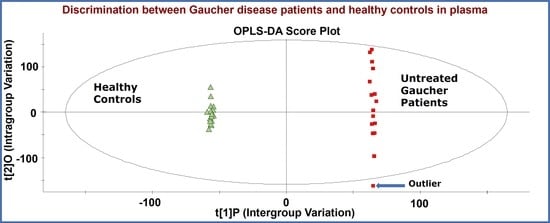
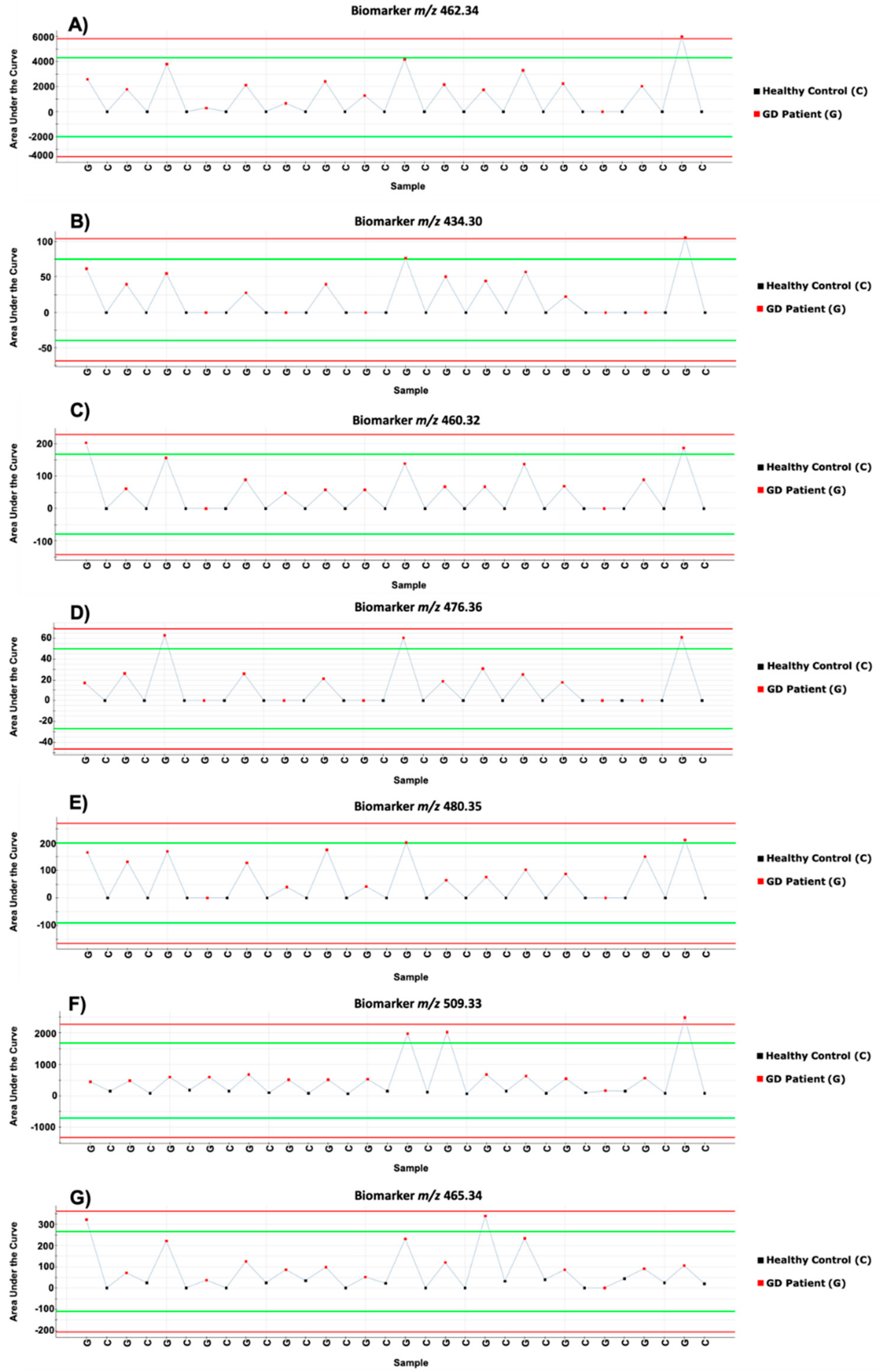
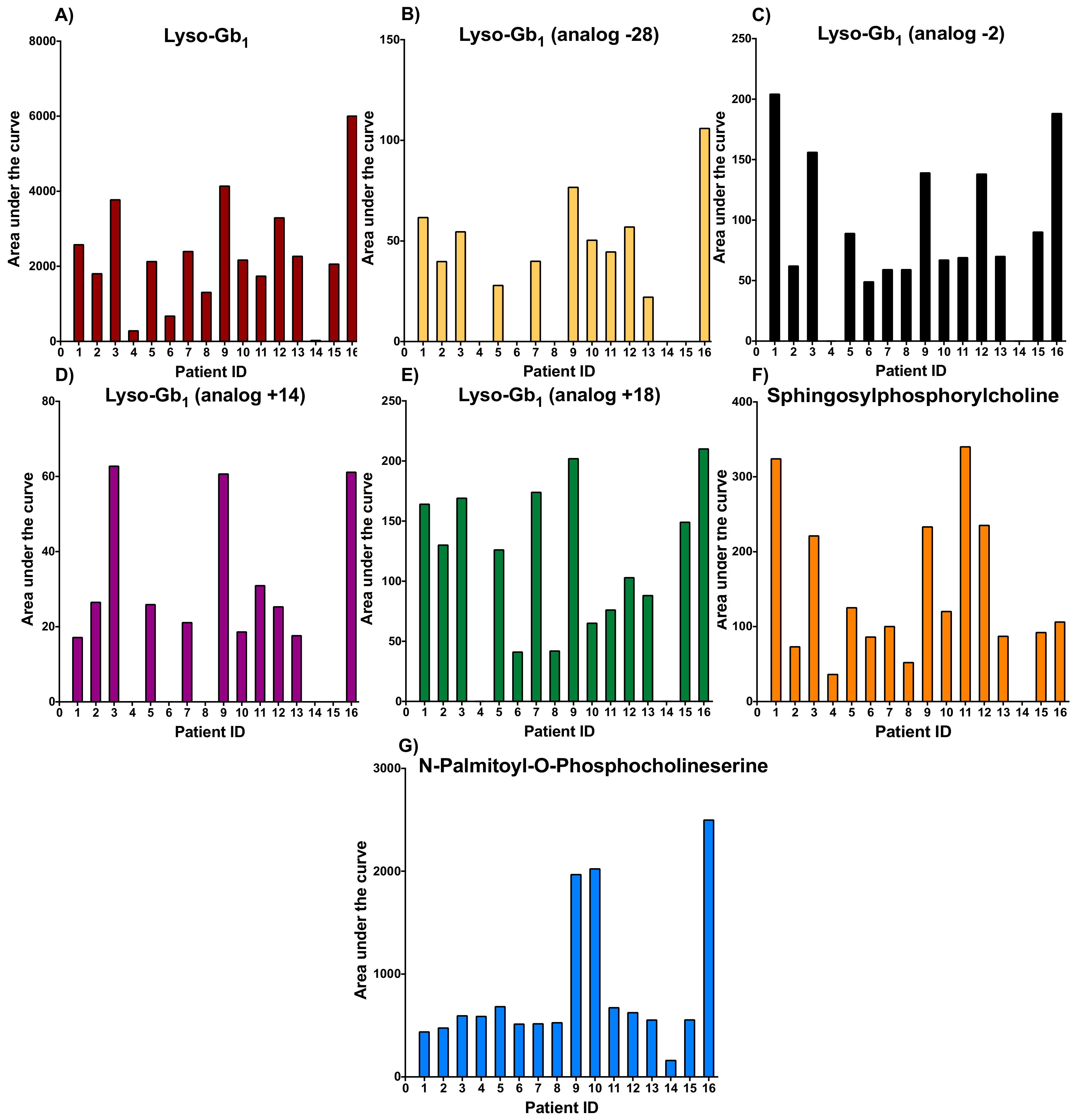
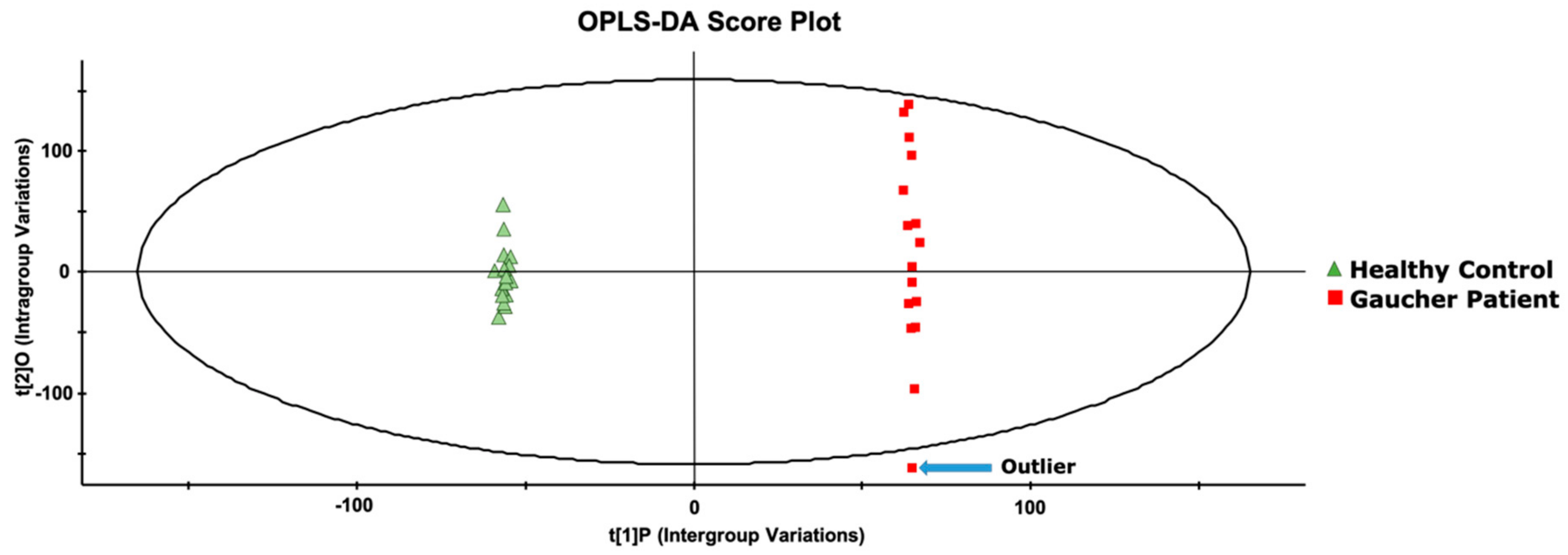
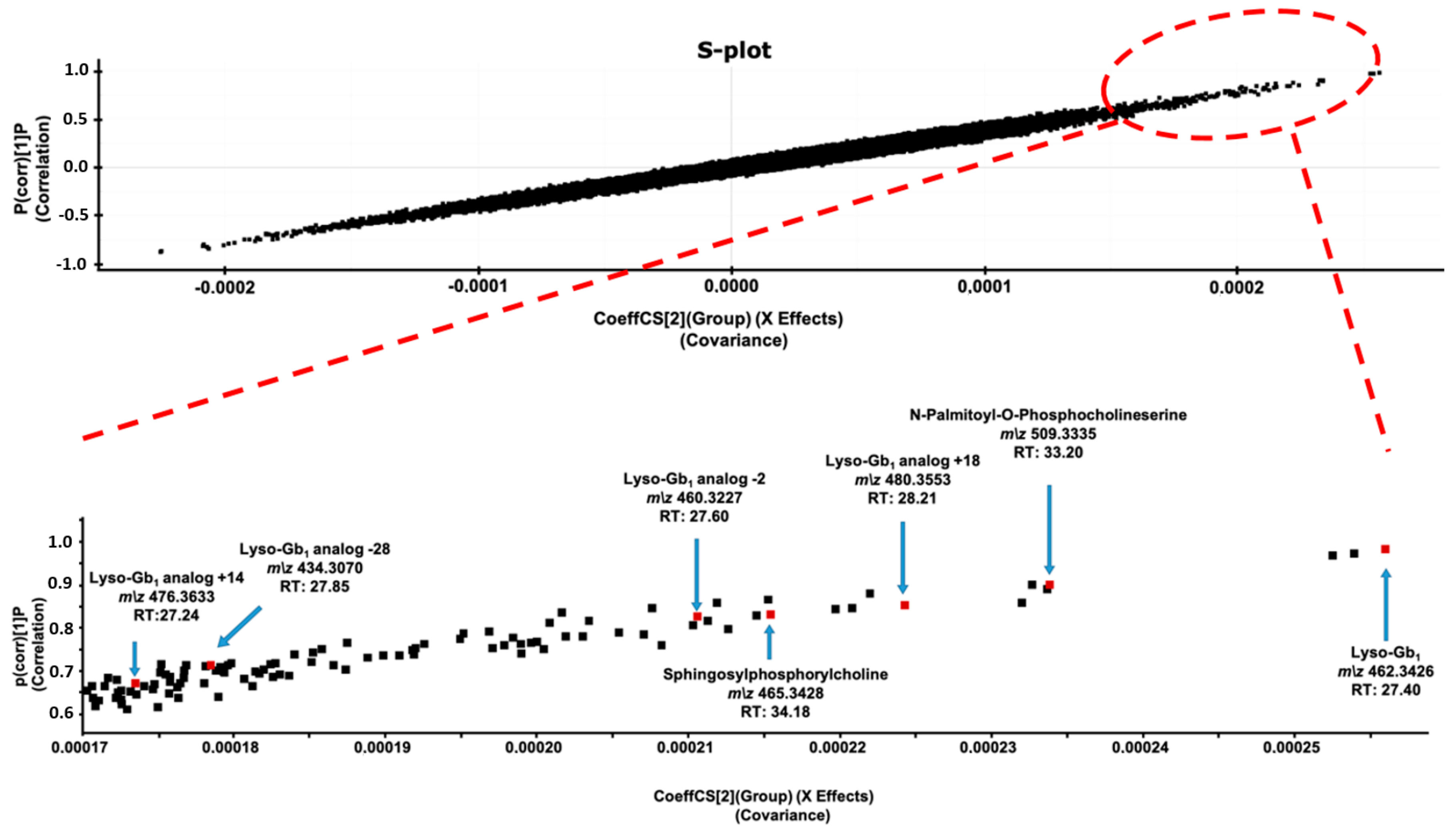
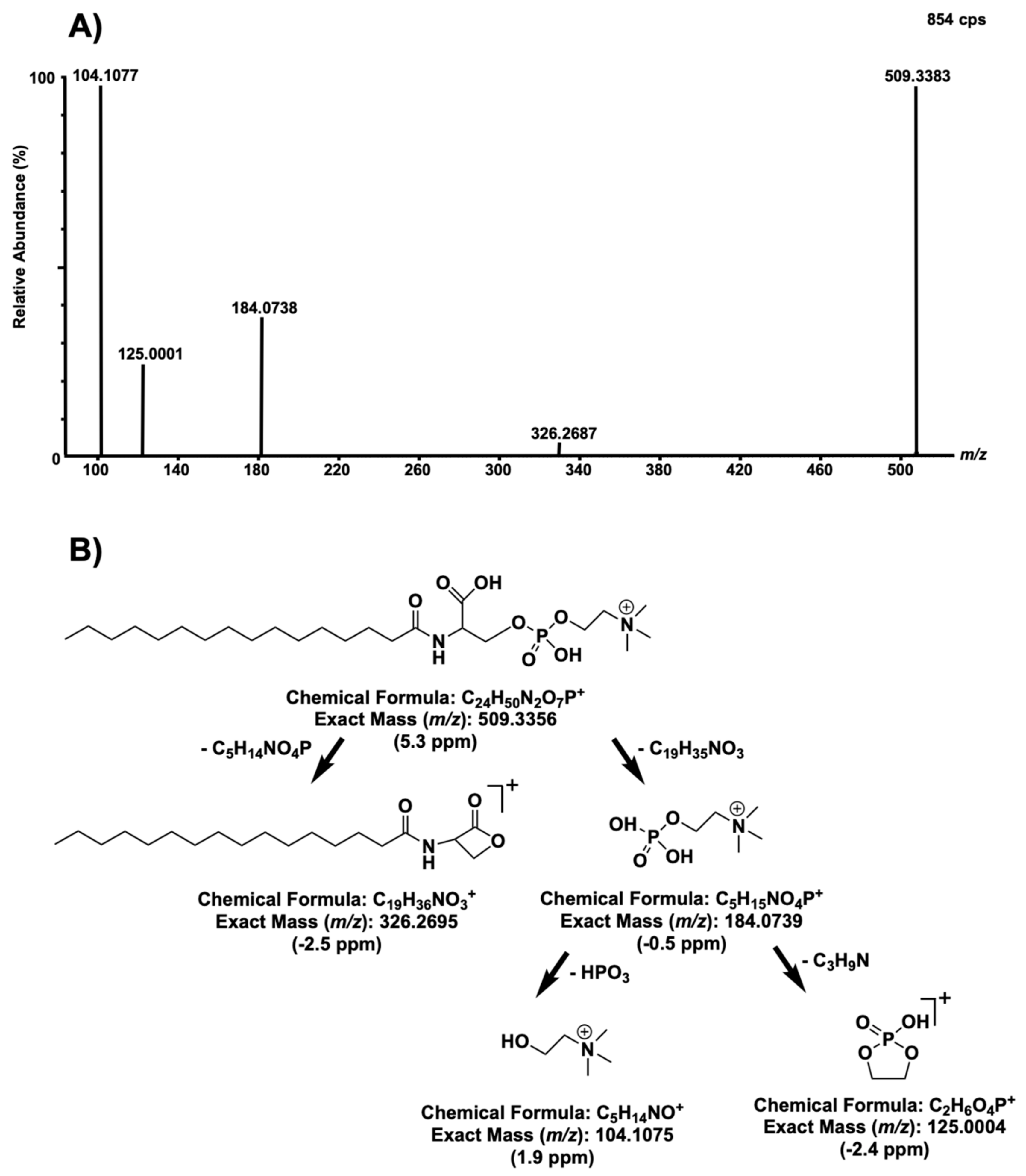
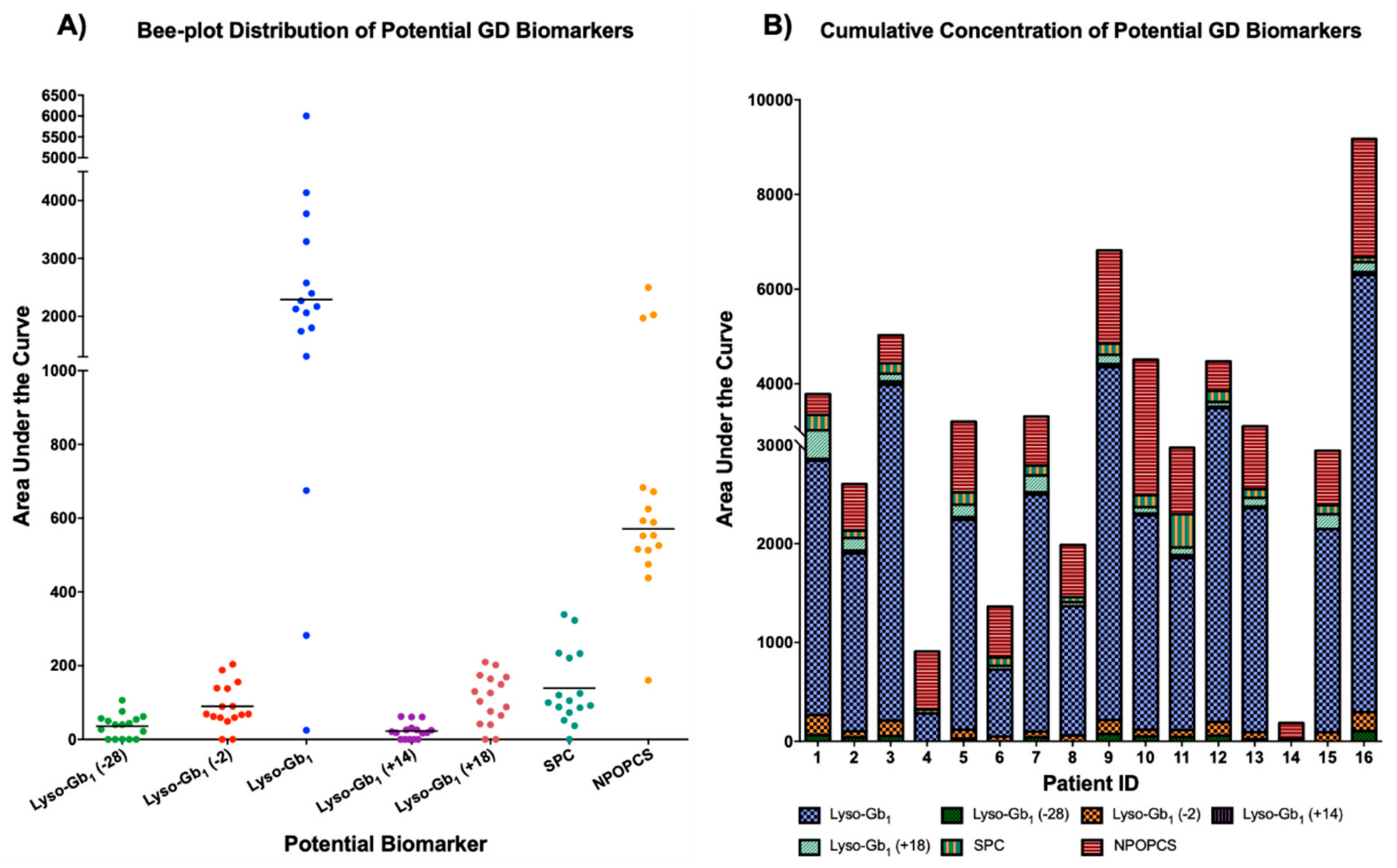
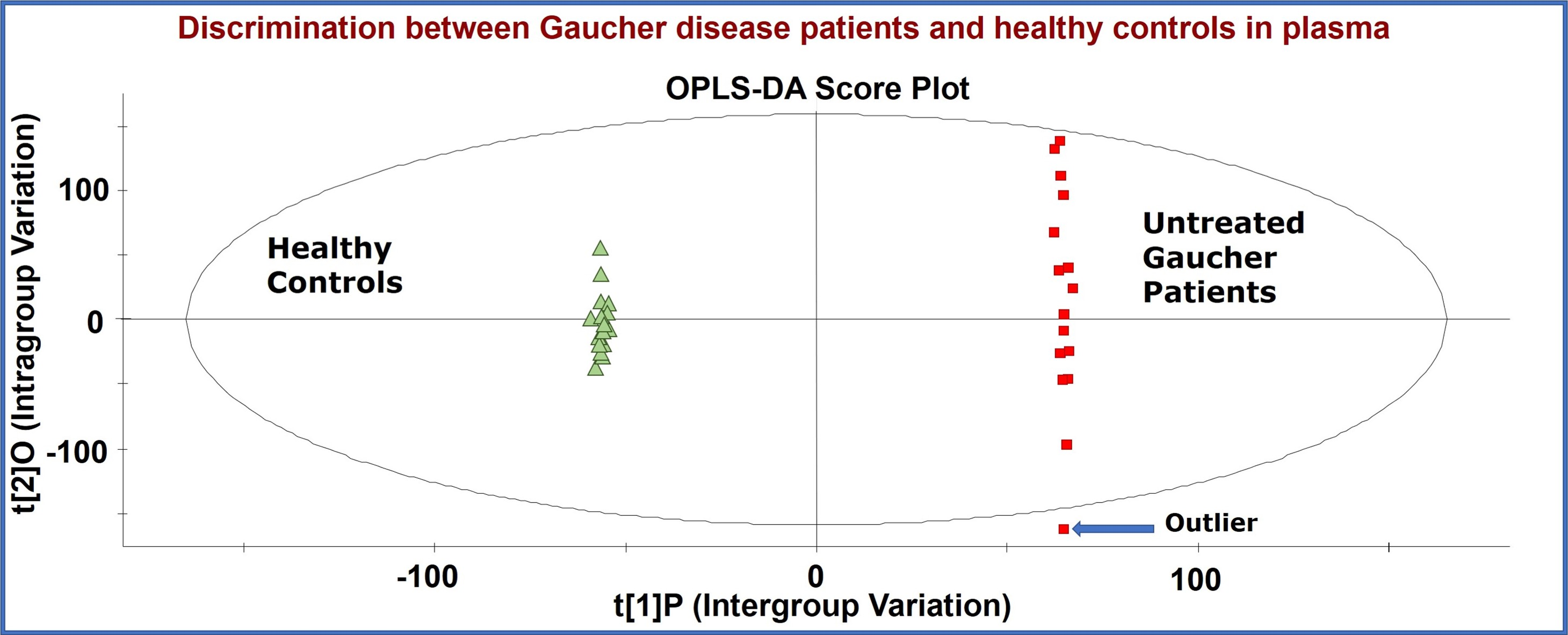
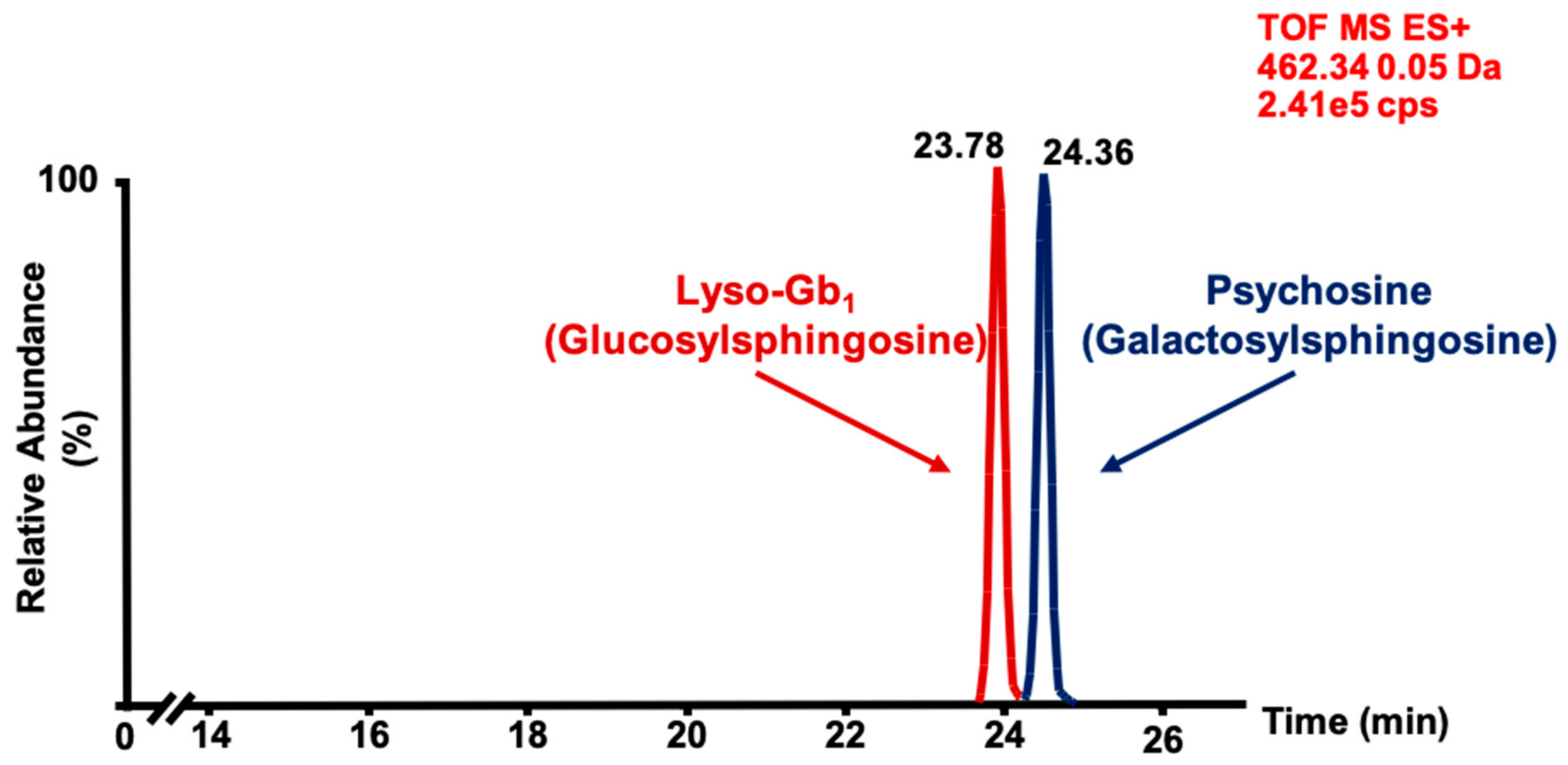
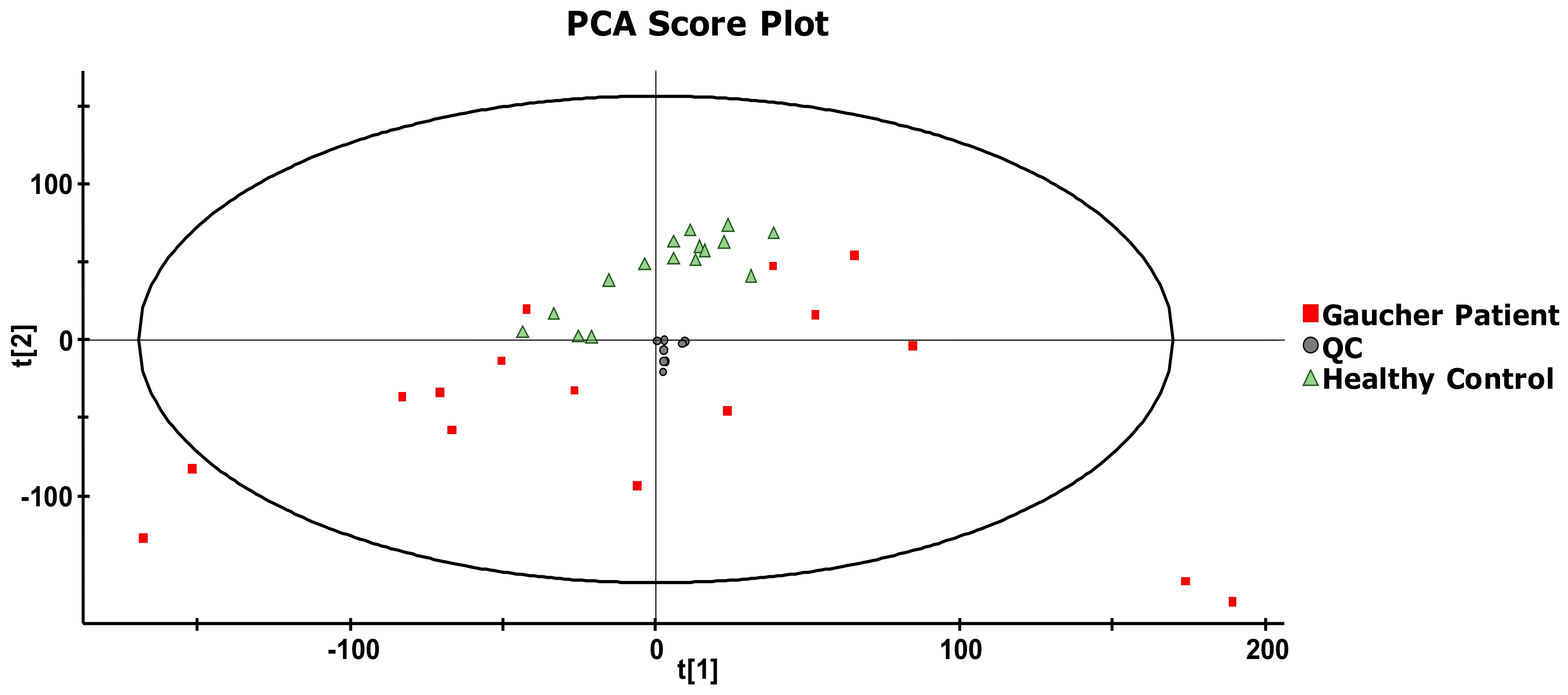
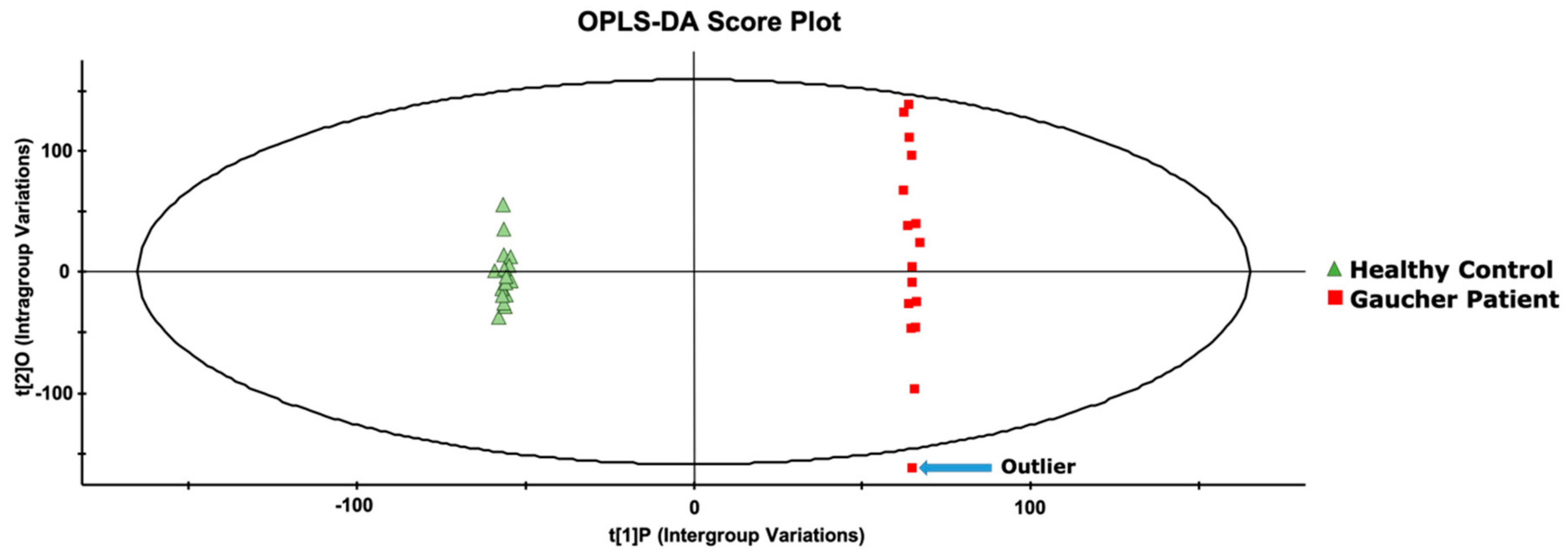
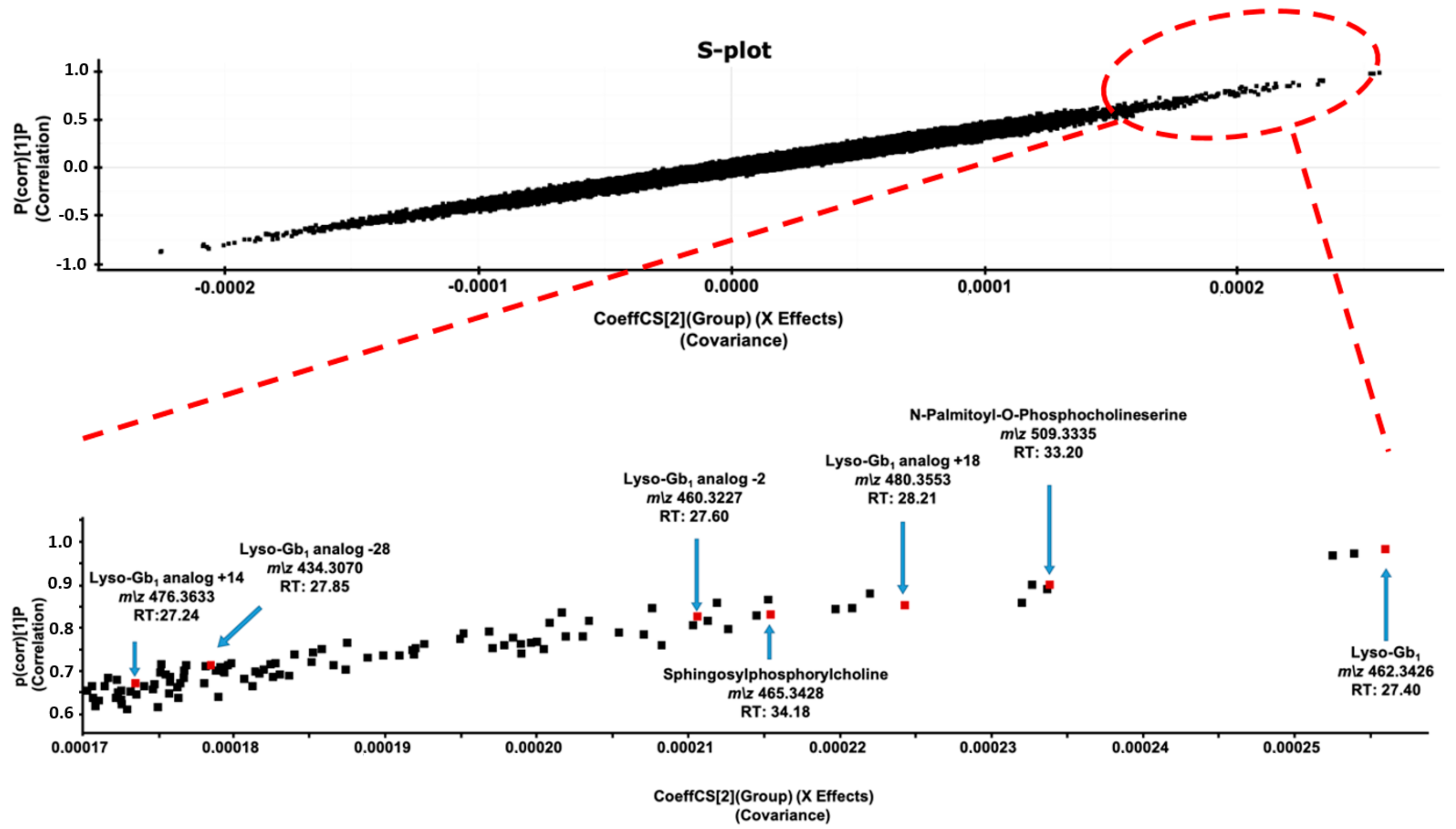
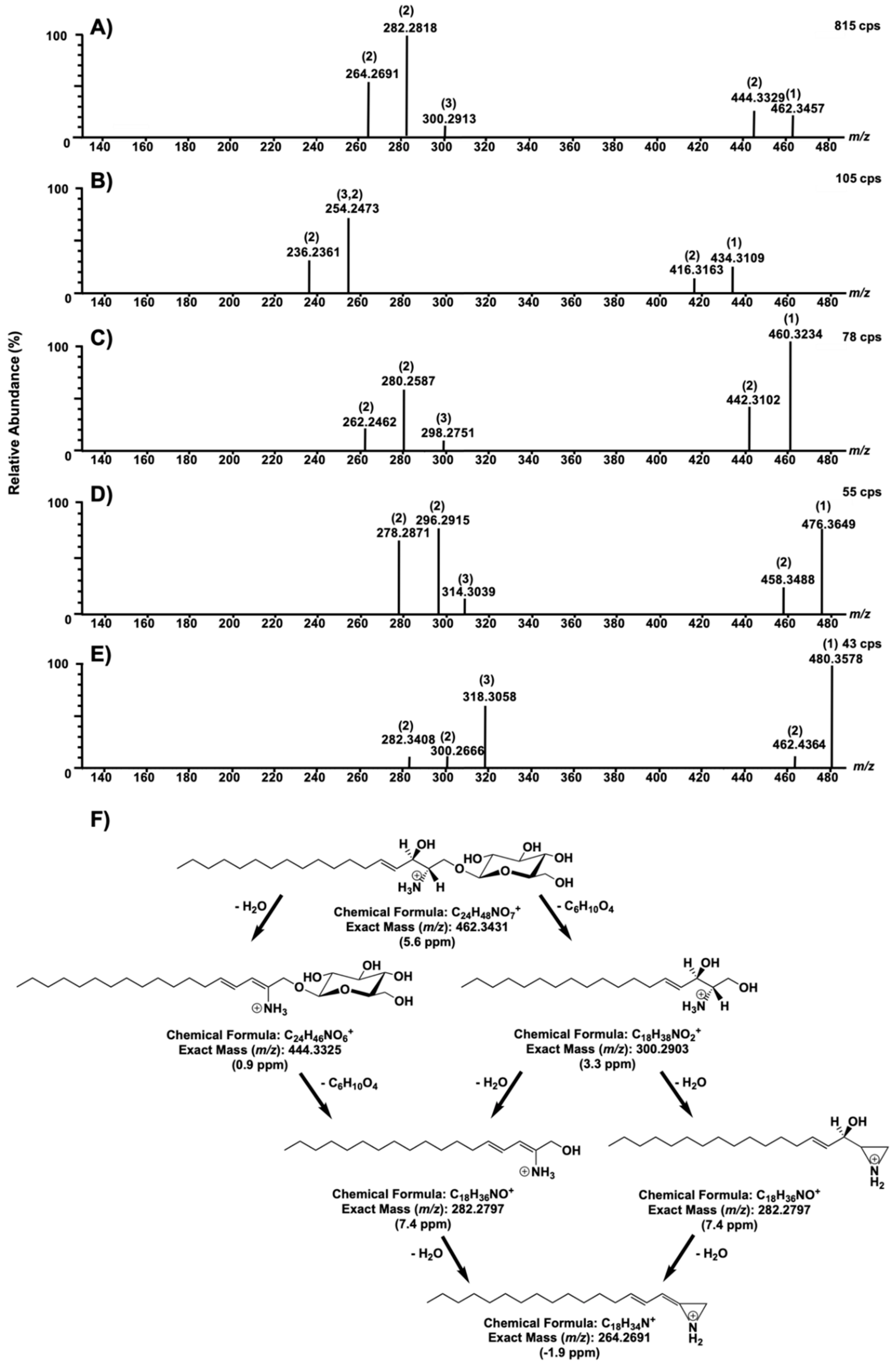
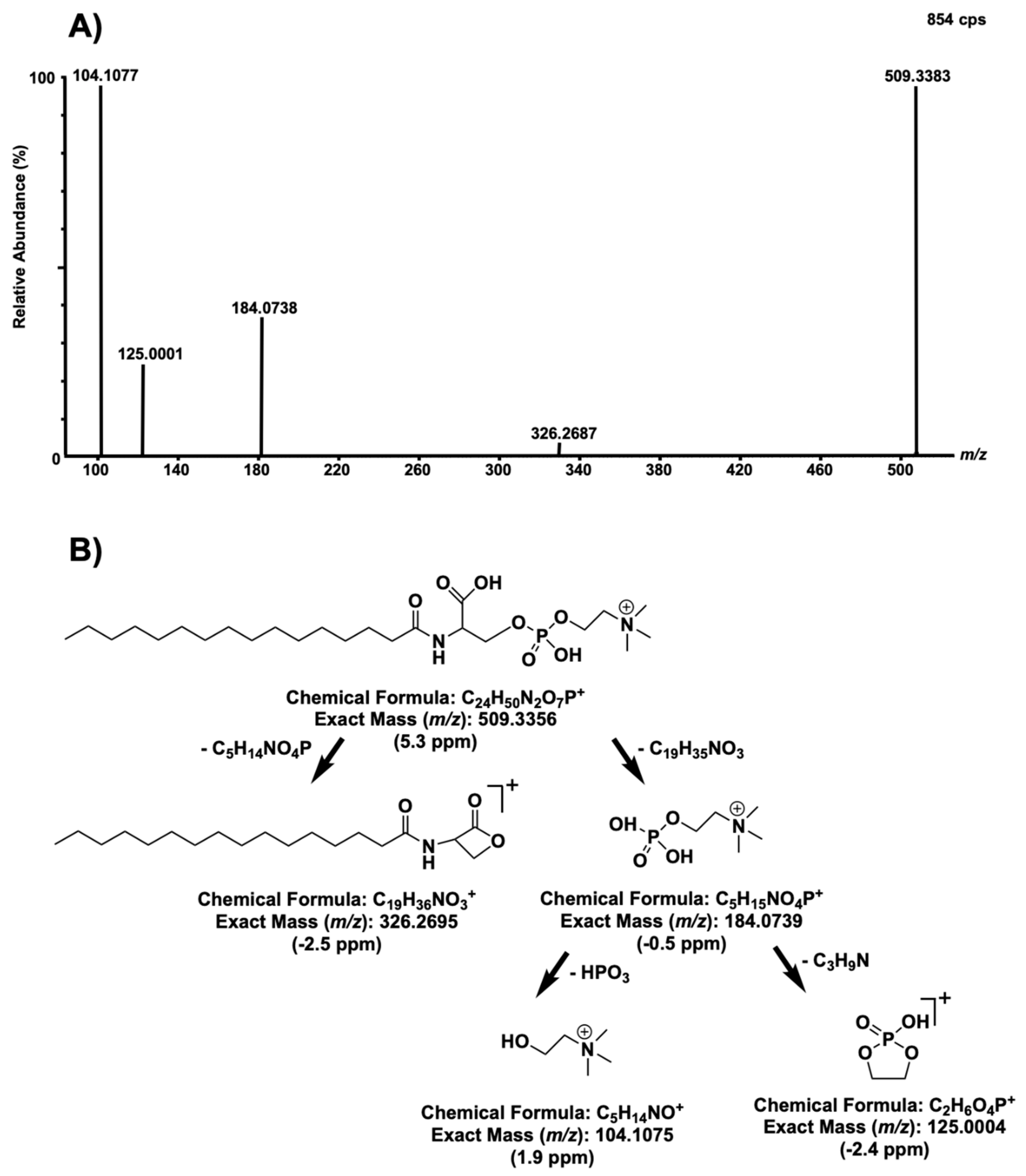
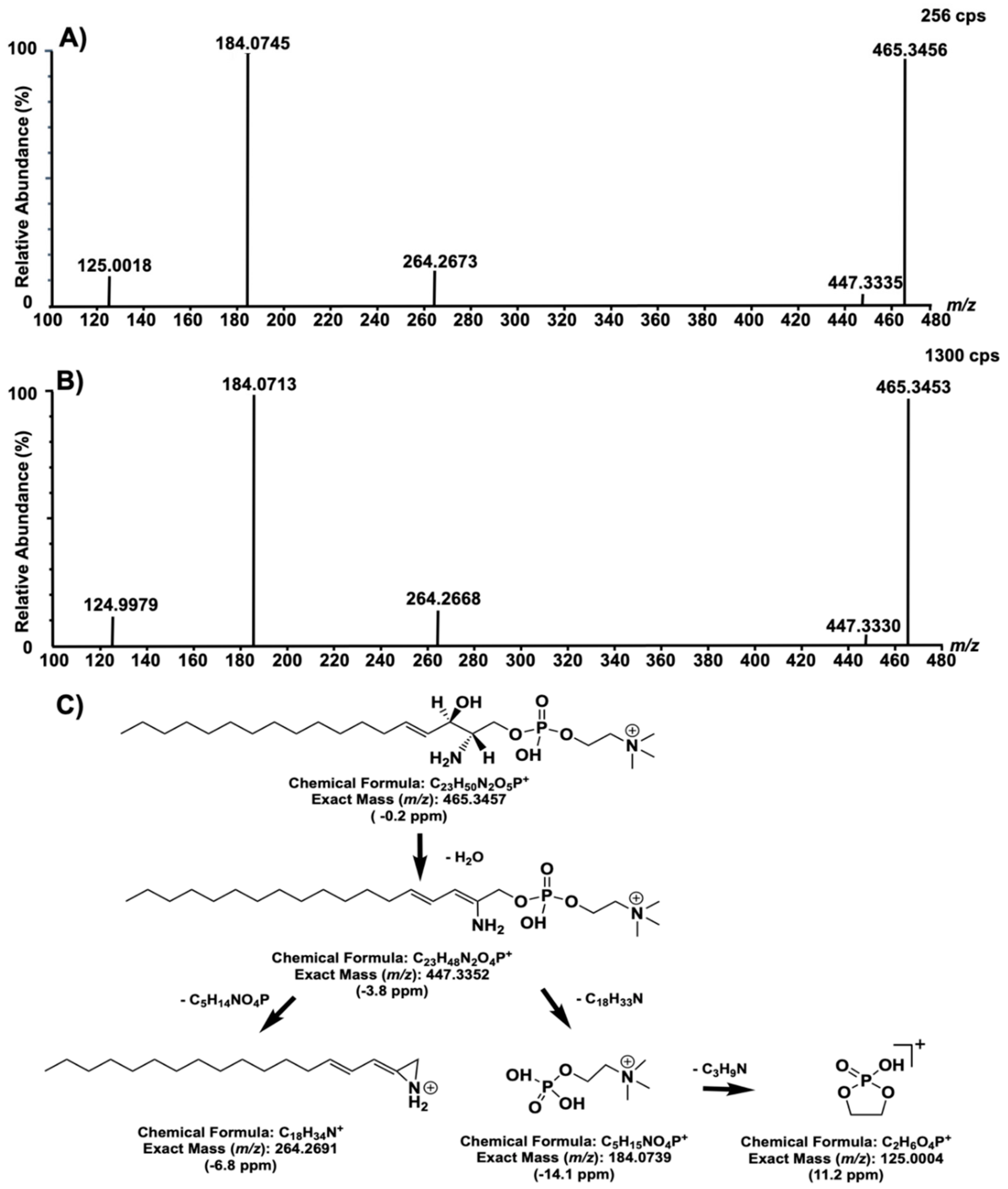
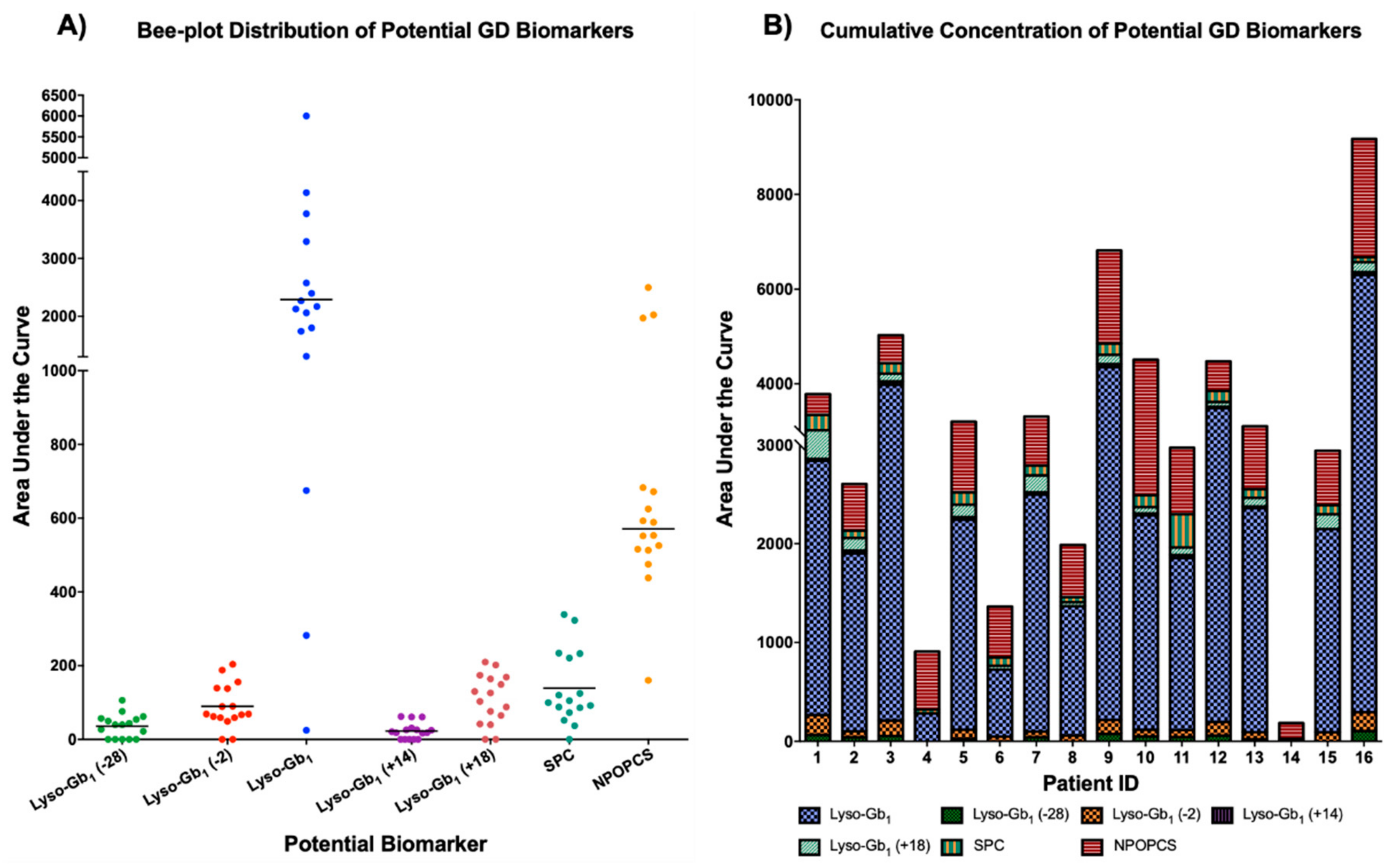
2. Results and Discussion
3. Materials and Methods
3.1. Ethics Approval
3.2. Patients and Controls
Clinical Features and Biochemical Parameters
3.3. Whole Blood and Plasma Specimen Collection and Processing
3.4. Reagents
3.5. Sample Preparation
3.6. Instrumentation and Parameters
4. Conclusions
5. Study Limitations
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Smith, L.; Mullin, S.; Schapira, A. Insights into the structural biology of Gaucher disease. Exp. Neurol. 2017, 298 Pt B, 180–190. [Google Scholar] [CrossRef] [Green Version]
- Aerts, J.M.; Kuo, C.L.; Lelieveld, L.T.; Boer, D.E.; Lienden, M.J.V.D.; Overkleeft, H.S.; Artola, M. Glycosphingolipids and lysosomal storage disorders as illustrated by Gaucher disease. Curr. Opin. Chem. Biol. 2019, 53, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Campos, M.; Alfonso, P.; Irun, P.; Armstrong, J.; Calvo, C.; Dalmau, J.; Domingo, M.R.; Barbera, J.L.; Cano, H.; Fernandez-Galán, M.A.; et al. Diagnosis features of pediatric Gaucher disease patients in the era of enzymatic therapy, a national-base study from the Spanish Registry of Gaucher disease. Orphanet. J. Rare Dis. 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Zeid, N.; Stauffer, C.; Yang, A.; Naik, H.; Fierro, L.; Ganesh, J.; Balwani, M. The N370S/R496H genotype in type 1 Gaucher disease—Natural history and implications for pre-symptomatic diagnosis and counseling. Mol. Genet. Metab. Rep. 2020, 22, 100567. [Google Scholar] [CrossRef] [PubMed]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. Review of Gaucher disease pathophysiology, clinical presentation and treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef] [PubMed]
- Fraga, C.; Medeiros, S.; Serpa, S.; Silva, D. Case Series Synopsis: Gaucher disease type 1 patients treated with eliglustat over 6 years. Int. J. Rare Dis. Disord. 2020, 3. [Google Scholar] [CrossRef]
- Mistry, P.K.; Lopez, G.; Schiffmann, R.; Barton, N.W.; Weinreb, N.J.; Sidransky, E. Gaucher disease: Progress and ongoing challenges. Mol. Genet. Metab. 2017, 120, 8–21. [Google Scholar] [CrossRef] [Green Version]
- Dandana, A.; Khelifa, S.B.; Chahed, H.; Miled, A.; Ferchichi, S. Gaucher disease: Clinical, biological and therapeutic aspects. Pathobiology 2015, 83, 13–23. [Google Scholar] [CrossRef]
- Lal, T.R.; Sidransky, E. The spectrum of neurological manifestations associated with Gaucher disease. Diseases 2017, 5, 10. [Google Scholar] [CrossRef]
- Schiffmann, R.; Sevigny, J.; Rolfs, A.; Davies, E.H.; Goker-Alpan, O.; Abdelwahab, M.; Vellodi, A.; Mengel, E.; Lukina, E.; Yoo, H.W.; et al. The definition of neuronopathic Gaucher disease. J. Inherit. Metab. Dis. 2020, 1–4. [Google Scholar] [CrossRef]
- Van Rossum, A.; Holsopple, M. Enzyme replacement or substrate reduction? A Review of Gaucher disease treatment options. Hosp. Pharm. 2016, 51, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Gary, S.E.; Ryan, E.; Steward, A.M.; Sidransky, E. Recent advances in the diagnosis and management of Gaucher disease. Expert Rev. Endocrinol. Metab. 2018, 13, 107–118. [Google Scholar] [CrossRef]
- Giraldo, P.; Frutos, L.L.D.; Cebolla, J.J. Biomarker combination is necessary for the assessment of Gaucher disease? Ann. Transl. Med. 2018, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raskovalova, T.; Deegan, P.B.; Yang, R.; Pavlova, E.; Stirnemann, J.; Labarère, J.; Zimran, A.; Mistry, P.; Berger, M. Plasma chitotriosidase activity versus CCL18 level for assessing type I Gaucher disease severity: Protocol for a systematic review with meta-analysis of individual participant data. Syst. Rev. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, K.E.; Khan, Z.; Giani, J.F.; Cao, D.Y.; Bernstein, E.A.; Shen, X.Z. Angiotensin-converting enzyme in innate and adaptive immunity. Nat. Rev. Nephrol. 2018, 14, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Van Dussen, L.; Lips, P.; Everts, V.E.; Bravenboer, N.; Jansen, I.D.; Groener, J.E.; Maas, M.; Blokland, J.A.; Aerts, J.M.; Hollak, C.E. Markers of bone turnover in Gaucher disease: Modeling the evolution of bone disease. J. Clin. Endocrinol. Metab. 2011, 96, 2194–2205. [Google Scholar] [CrossRef] [Green Version]
- Murugesan, V.; Chuang, W.L.; Liu, J.; Lischuk, A.; Kacena, K.; Lin, H.; Pastores, G.M.; Yang, R.; Keutzer, J.; Zhang, K.; et al. Glucosylsphingosine is a key biomarker of Gaucher disease. Am. J. Hematol. 2016, 91, 1082–1089. [Google Scholar] [CrossRef] [Green Version]
- Ferraz, M.J.; Marques, A.R.; Appelman, M.D.; Verhoek, M.; Strijland, A.; Mirzaian, M.; Scheij, S.; Ouairy, C.M.; Lahav, D.; Wisse, P.; et al. Lysosomal glycosphingolipid catabolism by acid ceramidase: Formation of glycosphingoid bases during deficiency of glycosidases. FEBS Lett. 2016, 590, 716–725. [Google Scholar] [CrossRef] [Green Version]
- Rolfs, A.; Giese, A.K.; Grittner, U.; Mascher, D.; Elstein, D.; Zimran, A.; Böttcher, T.; Lukas, J.; Hübner, R.; Gölnitz, U.; et al. Glucosylsphingosine is a highly sensitive and specific biomarker for primary diagnostic and follow-up monitoring in Gaucher disease in a non-Jewish, Caucasian cohort of Gaucher disease patients. PLoS ONE 2013, 8, e79732. [Google Scholar] [CrossRef] [PubMed]
- Dekker, N.; van Dussen, L.; Hollak, C.E.; Overkleeft, H.; Scheij, S.; Ghauharali, K.; van Breemen, M.J.; Ferraz, M.J.; Groener, J.E.; Maas, M.; et al. Elevated plasma glucosylsphingosine in Gaucher disease: Relation to phenotype, storage cell markers, and therapeutic response. Blood 2011, 118, e118–e127. [Google Scholar] [CrossRef] [Green Version]
- Arkadir, D.; Dinur, T.; Revel-Vilk, S.; Becker Cohen, M.; Cozma, C.; Hovakimyan, M.; Eichler, S.; Rolfs, A.; Zimran, A. Glucosylsphingosine is a reliable response biomarker in Gaucher disease. Am. J. Hematol. 2018, 93, E140–E142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mucci, J.M.; Rozenfeld, P. Pathogenesis of bone alterations in Gaucher disease: The role of immune System. J. Immunol. Res. 2015, 2015, 192761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irún, P.; Cebolla, J.J.; López de Frutos, L.; De Castro-Orós, I.; Roca-Espiau, M.; Giraldo, P. LC-MS/MS analysis of plasma glucosylsphingosine as a biomarker for diagnosis and follow-up monitoring in Gaucher disease in the Spanish population. Clin. Chem. Lab. Med. 2020, 58, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Cozma, C.; Cullufi, P.; Kramp, G.; Hovakimyan, M.; Velmishi, V.; Gjikopulli, A.; Tomori, S.; Fischer, S.; Oppermann, S.; Grittner, U.; et al. Treatment efficiency in Gaucher patients can reliably be monitored by quantification of Lyso-Gb1 concentrations in dried blood spots. Int. J. Mol. Sci. 2020, 21, 4577. [Google Scholar] [CrossRef]
- Dinur, T.; Zimran, A.; Becker-Cohen, M.; Arkadir, D.; Cozma, C.; Hovakimyan, M.; Oppermann, S.; Demuth, L.; Rolfs, A.; Revel-Vilk, S. Long term follow-up of 103 untreated adult patients with type 1 Gaucher disease. J. Clin. Med. 2019, 8, 1662. [Google Scholar] [CrossRef] [Green Version]
- Lavoie, P.; Boutin, M.; Auray-Blais, C. Multiplex analysis of novel urinary lyso-Gb3-related biomarkers for Fabry disease by tandem mass spectrometry. Anal Chem. 2013, 85, 1743–1752. [Google Scholar] [CrossRef]
- Boutin, M.; Auray-Blais, C. Multiplex tandem mass spectrometry analysis of novel plasma lyso-Gb3-related analogues in Fabry disease. Anal Chem. 2014, 86, 3476–3483. [Google Scholar] [CrossRef] [PubMed]
- Boutin, M.; Auray-Blais, C. Metabolomic Discovery of novel urinary galabiosylceramide analogs as Fabry disease biomarkers. J. Am. Soc. Mass. Spectrom. 2015, 26, 499–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auray-Blais, C.; Boutin, M.; Gagnon, R.; Dupont, F.O.; Lavoie, P.; Clarke, J.T. Urinary globotriaosylsphingosine-related biomarkers for Fabry disease targeted by metabolomics. Anal Chem. 2012, 84, 2745–2753. [Google Scholar] [CrossRef] [PubMed]
- Mirzaian, M.; Wisse, P.; Ferraz, M.J.; Gold, H.; Donker-Koopman, W.E.; Verhoek, M.; Overkleeft, H.S.; Boot, R.G.; Kramer, G.; Dekker, N.; et al. Mass spectrometric quantification of glucosylsphingosine in plasma and urine of type 1 Gaucher patients using an isotope standard. Blood Cells Mol. Dis. 2015, 54, 307–314. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, C.; Dev, K.K. Galactosylsphingosine (psychosine)-induced demyelination is attenuated by sphingosine 1-phosphate signalling. J. Cell Sci. 2015, 128, 3878–3887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, Z.; Turgeon, C.T.; Biski, C.; Khaledi, H.; Shoemaker, N.B.; Dearmond, P.D.; Smith, S.; Orsini, J.; Matern, D.; Gelb, M.H. Achieving congruence among reference laboratories for absolute abundance measurement of analytes for rare diseases: Psychosine for diagnosis and prognosis of Krabbe disease. Int. J. Neonatal. Screen 2020, 6, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutin, M.; Sun, Y.; Shacka, J.J.; Auray-Blais, C. Tandem mass spectrometry multiplex analysis of glucosylceramide and galactosylceramide isoforms in brain tissues at different stages of Parkinson disease. Anal Chem. 2016, 88, 1856–1863. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, R.; Mondjinou, Y.; Qian, M.; Song, H.; Kumar, A.B.; Hong, X.; Hsu, F.F.; Dietzen, D.J.; Yanjanin, N.M.; Porter, F.D.; et al. N-acyl-O-phosphocholineserines: Structures of a novel class of lipids that are biomarkers for Niemann-Pick C1 disease. J. Lipid Res. 2019, 60, 1410–1424. [Google Scholar] [CrossRef]
- Sidhu, R.; Kell, P.; Dietzen, D.J.; Farhat, N.Y.; Do, A.; Porter, F.D.; Berry-Kravis, E.; Vite, C.H.; Reunert, J.; Marquardt, T.; et al. Application of N-palmitoyl-O-phosphocholineserine for diagnosis and assessment of response to treatment in Niemann-Pick type C disease. Mol. Genet. Metab. 2020, 129, 292–302. [Google Scholar] [CrossRef]
- Thomas, G.D.; Snetkov, V.A.; Patel, R.; Leach, R.M.; Aaronson, P.I.; Ward, J.P. Sphingosylphosphorylcholine-induced vasoconstriction of pulmonary artery: Activation of non-store-operated Ca2+ entry. Cardiovasc. Res. 2005, 68, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Ługowska, A.; Hetmańczyk-Sawicka, K.; Iwanicka-Nowicka, R.; Fogtman, A.; Cieśla, J.; Purzycka-Olewiecka, J.K.; Sitarska, D.; Płoski, R.; Filocamo, M.; Lualdi, S.; et al. Gene expression profile in patients with Gaucher disease indicates activation of inflammatory processes. Sci. Rep. 2019, 9, 6060. [Google Scholar] [CrossRef] [Green Version]
- Deodato, F.; Boenzi, S.; Taurisano, R.; Semeraro, M.; Sacchetti, E.; Carrozzo, R.; Dionisi-Vici, C. The impact of biomarkers analysis in the diagnosis of Niemann-Pick C disease and acid sphingomyelinase deficiency. Clin. Chim. Acta 2018, 486, 387–394. [Google Scholar] [CrossRef]
- Zhang, K.; Chuang, W.-L.; Pacheco, J.; Cooper, S.; Mcgovern, M.; Cox, G.; Keutzer, J. Lyso-sphingomyelin is elevated in dried blood spots of Niemann-Pick disease type B patients. Mol. Genet. Metab. 2014, 111, 209–211. [Google Scholar] [CrossRef]
- Saville, J.T.; McDermott, B.K.; Chin, S.J.; Fletcher, J.M.; Fuller, M. Expanding the clinical utility of glucosylsphingosine for Gaucher disease. J. Inherit. Metab. Dis. 2020, 43, 558–563. [Google Scholar] [CrossRef]
- Hurvitz, L.; Dinur, T.; Becker-Cohen, M.; Cozma, C.; Hovakimyan, M.; Oppermann, S.; Demuth, L.; Rolfs, A.; Abramov, A.; Zimran, A.; et al. Glucosylsphingosine (lyso-Gb1) as a Biomarker for Monitoring Treated and Untreated Children with Gaucher Disease. Int. J. Mol. Sci. 2019, 20, 3033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giese, A.K.; Mascher, H.; Grittner, U.; Eichler, S.; Kramp, G.; Lukas, J.; te Vruchte, D.; Al Eisa, N.; Cortina-Borja, M.; Porter, F.D.; et al. A novel, highly sensitive and specific biomarker for Niemann-Pick type C1 disease. Orphanet. J. Rare Dis. 2015, 10, 78. [Google Scholar] [CrossRef] [Green Version]
- Auray-Blais, C.; Lavoie, P.; Boutin, M.; Ntwari, A.; Hsu, T.; Huang, C.; Niu, D. Biomarkers associated with clinical manifestations in Fabry disease patients with a late-onset cardiac variant mutation. Clin. Chim. Acta 2017, 466, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, R.A.; Hoefsloot, H.C.; Westerhuis, J.A.; Smilde, A.K.; van der Werf, M.J. Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genom. 2006, 7, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
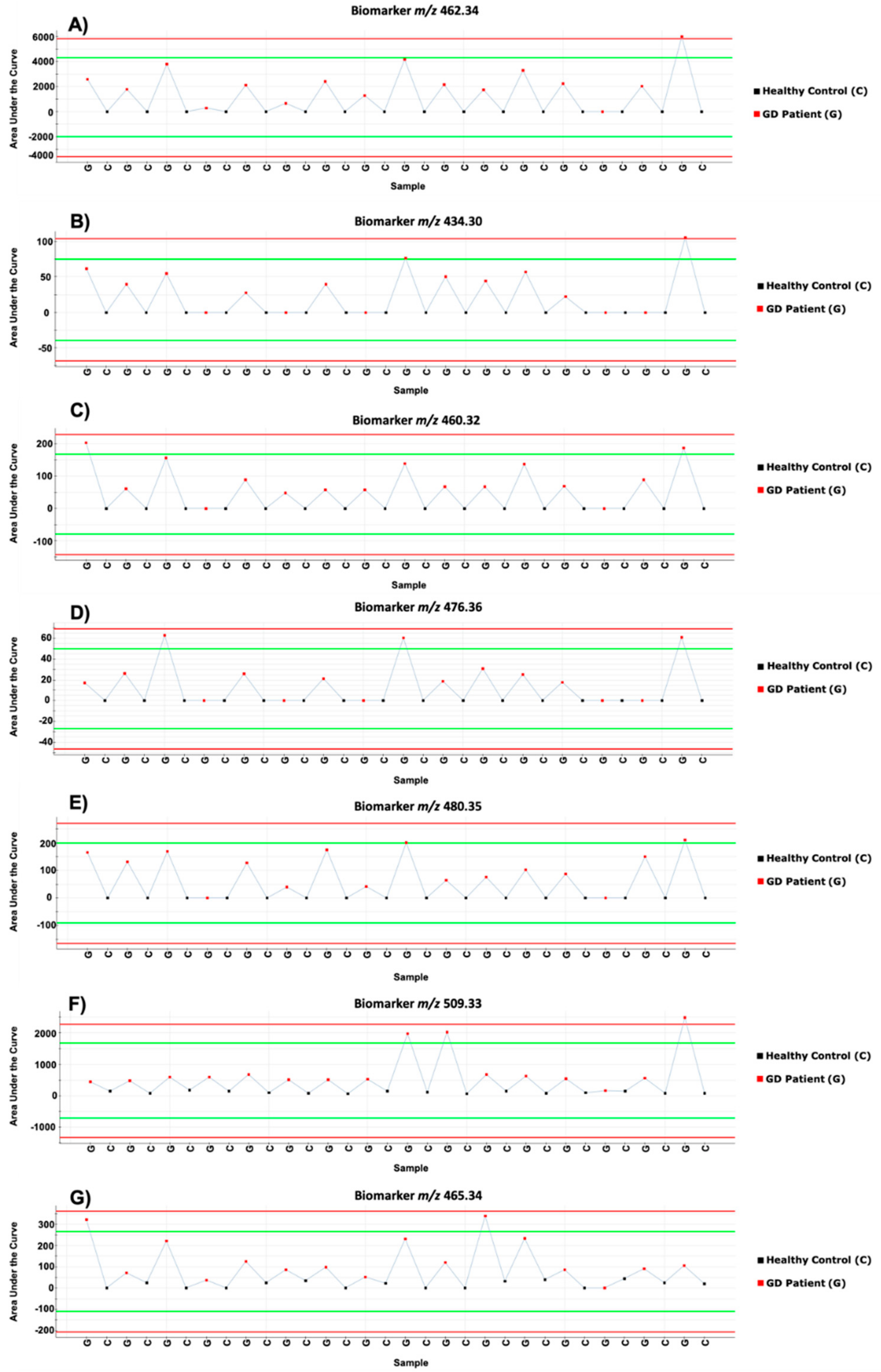{kind=link}
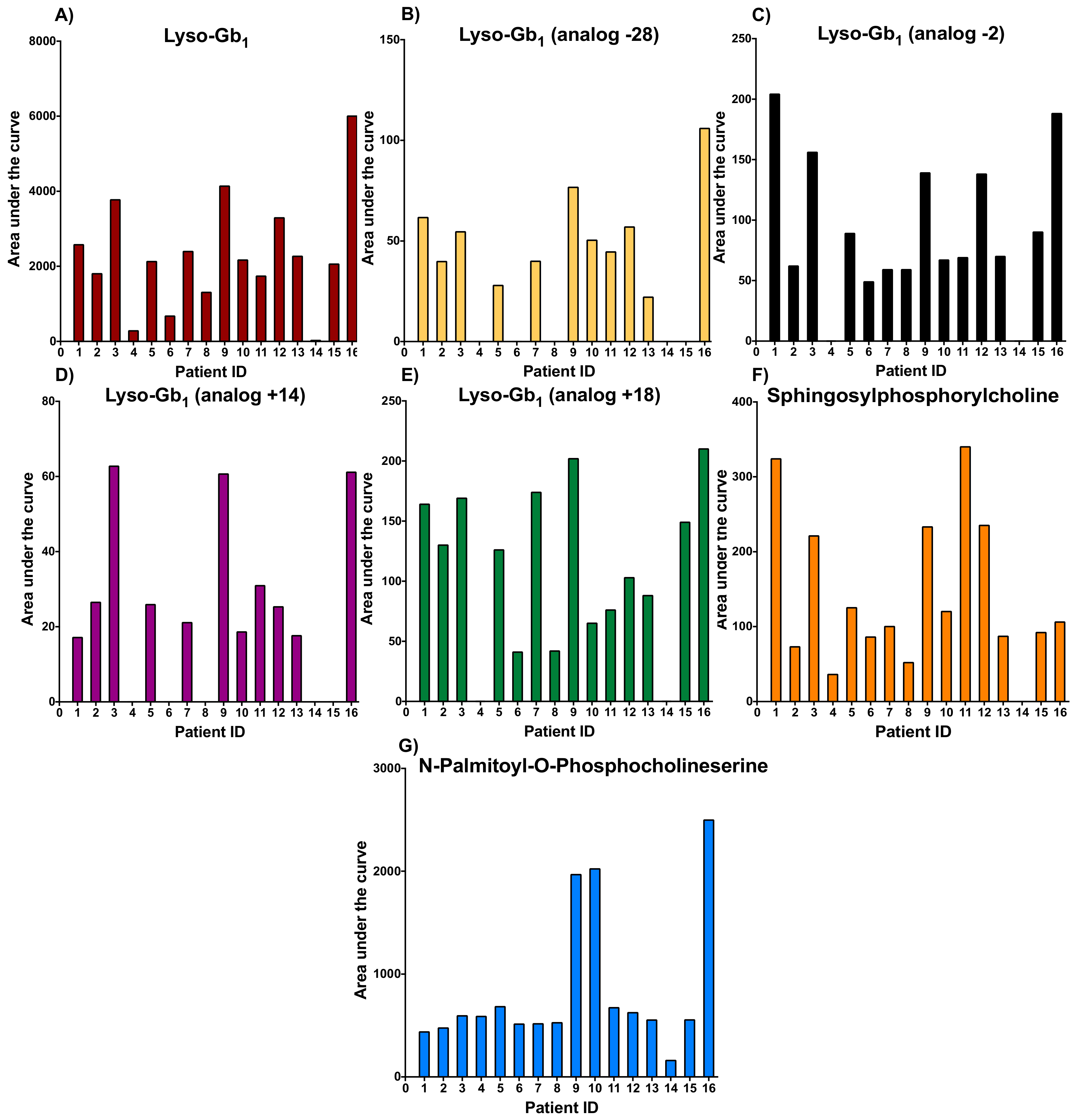{kind=link}
| Patient ID | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age | 62 | 49 | 43 | 62 | 22 | 47 | 49 | 70 | 63 | 71 | 62 | 43 | 70 | 53 | 24 | 44 |
| Gender | F | F | F | F | F | M | F | F | M | M | F | F | F | F | M | F |
| Age at diagnosis | 28 | 40 | 36 | 16 | N/E | 12 | 40 | 42 | 19 | 21 | 3 | 36 | N/E | 44 | 13 | 27 |
| Mutations | N370S N370S | N370S N370S | N370S L444P | N370S N370S | N370S W184 | N370S L444P | N370S N370S | N370S L444P | N370S R120W | N370S W378G | L444P L444P | N370S N370S | N370S L444P | N370S R496H | N370S L444P | L444P 84GG |
| Treatment | No | No | No | No | No | No | No | No | No | No | No | No | No | No | No | No |
| Bone disease | ||||||||||||||||
| Bone pain | - | Mild | Moderate | Mild | - | Moderate | - | - | - | Mild | Moderate | Mild | Moderate | - | - | Mild |
| Bone crisis | - | - | - | - | - | Moderate | - | - | Moderate | - | - | - | - | - | - | - |
| Erlenmeyer flask deformity | x | - | - | - | - | - | - | - | - | x | x | - | - | x | ||
| Bone marrow infiltration | x | x | x | x | - | x | - | - | - | x | - | x | x | - | x | x |
| Osteopenia | - | - | - | - | - | x | - | - | x | x | x | - | x | x | - | - |
| Infarction | - | - | - | - | - | x | - | - | x | - | - | - | x | - | - | x |
| Avascular necrosis | - | - | - | - | - | x | - | - | - | - | - | - | - | - | - | - |
| New fractures | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| Lytic lesions | x | - | - | - | - | - | - | - | x | - | - | - | - | - | - | - |
| Decreased bone mineral density | - | - | - | - | - | x | - | - | - | x | - | - | - | x | - | - |
| Bone marrow burden | DGS 5 | LL 5 S 6–7 | N/E | LL 4 S 3 | N/E | LL 4 S 3 | N/E | LL 7 S 5 | DGS 7 | N/E | N/E | LL 4 S 5–6 | DGS 7 | LL 2 S 3 | N/E | LL 7 S 7 |
| Hematological and visceral manifestations | ||||||||||||||||
| Anemia | - | - | x | - | - | - | - | - | x | - | x | x | - | - | - | - |
| Hemoglobin (g/L) | 125 | 125 | 118 | 140 | 127 | 149 | 122 | 132 | 103 | 140 | 110 | 113 | 123 | 117 | 124 | 127 |
| Thrombocytopenia and coagulopathy | - | x | x | x | - | - | x | - | x | - | - | x | x | - | x | x |
| Platelet counts (/L) | 156 | 87 | 65 | 95 | 123 | 197 | 117 | 144 | 63 | 182 | 185 | 82 | 130 | 254 | 103 | 71 |
| Splenomegaly | Mild | Mild | Mild | - | - | - | x | N/A | Moderate | N/A | N/A | - | N/A | - | - | N/A |
| Hepatomegaly | - | Mild | Mild | Mild | - | Mild | - | Mild | Moderate | N/E | Mild | Moderate | Mild | Moderate | Severe | - |
| Splenectomized | - | - | - | - | - | - | - | x | - | x | x | - | x | - | - | x |
| Gammopathies and malignancy | - | - | - | - | - | - | - | - | - | x | - | - | - | - | - | - |
| Pulmonary disease | - | - | ||||||||||||||
| Elevated arterial pressure | - | - | - | - | - | - | - | - | - | x | x | - | - | - | - | - |
| Severe pulmonary hypertension | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| Hepatopulmonary syndrome | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| Respiratory failure | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| Other | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| Parameters | Description |
|---|---|
| Column | Halo HILIC 2.7 |
| ID x Length | 4.6 × 150 mm |
| Particle size | 2.7 μm |
| Column temperature | 30 °C |
| Weak Wash solvent | 94.5:2.5:2.5:0.5 ACN:MeOH:H2O:FA + 5 mM Amm. Form. |
| Strong Wash solvent | ACN |
| Injection volume | 10 μL |
| Injection mode | Partial Loop |
| Autosampler temperature | 10 °C |
| Mobile phase A | 94.5:2.5:2.5:0.5 ACN:MeOH:H2O:FA + 5 mM Amm. Form. |
| Mobile phase B | 10:89.5:0.5 ACN:H2O:FA + 5 mM Amm. Form. |
| Flow rate | 0.5 mL/min |
| Gradient (% mobile phase B) | 0 to 5min: 0% |
| 5.0 to 20.0 min: 0–10% (Linear gradient) | |
| 20.0 to 25.0 min: 10% | |
| 25.0 to 35.0 min: 10–60% (Linear gradient) | |
| 35.0 to 40.0 min: 60 | |
| 40.0 to 45.0 min: 0% |
| Parameters | Description |
|---|---|
| Scan Mode | MS-TOF |
| Ionization Mode | Electrospray Ionization |
| Polarity | Positive |
| Analyzer Mode | V |
| Dynamic Range | Extended |
| Capillary Voltage | 1.4 kV |
| Sampling Cone Voltage | 10 V |
| Extraction Cone Voltage | 5.0 V |
| Source Temperature | 150 °C |
| Desolvation Temperature | 450 °C |
| Cone Gas Flow | 30 L/h |
| Desolvation Gas Flow | 700 L/h |
| Trap Collision Energy | 4.0 V |
| Transfer Collision Energy | 2.0 V |
| Data Format | Centroid |
| Mass Range | 50–1000 Da |
| Scan Time | 0.1 s |
| Lock mass | |
| Compound | Terfenadine (500 nM) |
| Exact Mass | 472.3215 Da |
| Solvent | 94.5:5:0.5 H2O:ACN:FA |
| Scan Time | 0.5 s |
| Interval | 5.0 s |
| Sampling Cone Voltage | 5 V |
| Trap Collision Energy | 10 |
| Mass Window | ±0.2 Da |
| Scan Average | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menkovic, I.; Boutin, M.; Alayoubi, A.; Mercier, F.E.; Rivard, G.-É.; Auray-Blais, C. Identification of a Reliable Biomarker Profile for the Diagnosis of Gaucher Disease Type 1 Patients Using a Mass Spectrometry-Based Metabolomic Approach. Int. J. Mol. Sci. 2020, 21, 7869. https://doi.org/10.3390/ijms21217869
Menkovic I, Boutin M, Alayoubi A, Mercier FE, Rivard G-É, Auray-Blais C. Identification of a Reliable Biomarker Profile for the Diagnosis of Gaucher Disease Type 1 Patients Using a Mass Spectrometry-Based Metabolomic Approach. International Journal of Molecular Sciences. 2020; 21(21):7869. https://doi.org/10.3390/ijms21217869
Chicago/Turabian StyleMenkovic, Iskren, Michel Boutin, Abdulfatah Alayoubi, François E. Mercier, Georges-Étienne Rivard, and Christiane Auray-Blais. 2020. "Identification of a Reliable Biomarker Profile for the Diagnosis of Gaucher Disease Type 1 Patients Using a Mass Spectrometry-Based Metabolomic Approach" International Journal of Molecular Sciences 21, no. 21: 7869. https://doi.org/10.3390/ijms21217869