Targeting Autophagy in Breast Cancer

 , , , ,
, , , , 

Abstract
:1. Introduction
2. Main Mechanisms Inducing Autophagy
3. Role of Autophagy in Cancer Diseases
Targeting Autophagy as a Cancer Treatment
4. Targeting Autophagy in Breast Cancer
4.1. ER+, PgR+, HER2-Subtype
4.2. HER2-Positive Subtype
4.3. TNBC Subtype
4.4. Clinical Trials
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 3-MA | 3-methyladenine |
| AI | Aromatase Inhibitor |
| ALDH | Aldehyde Dehydrogenase |
| AMPK | 5’ AMP-activated protein kinase |
| ATG | Autophagy related genes |
| ACD | Autophagic cell death |
| BC | Breast Cancer |
| CQ | Chloroquine |
| CSCs | Cancer Stem Cells |
| DNA | Deoxyribonucleic Acid |
| ECM | Extracellular matrix |
| EGFR | Epidermal Growth Factor Receptor |
| EMT | Epithelial Mesenchymal Transition |
| ER | Estrogen Receptor |
| EXE | Exemestane |
| HCQ | Hidroxychloroquine |
| HER2 | Human Epidermal Growth Factor Receptor 2 |
| HIF-1 | Hipoxia-inducible factor 1 |
| YAP | Yes-associated protein |
| LC3 | Light chain 3 |
| MAPK | Mitogen-activated protein kinase |
| MSL | Mesenchymal Stem-Like |
| mTOR | Mammalian Target Of Rapamycin |
| mTORC1 | mTOR complex 1 |
| NFKB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| PAS | Phagophore assembly site |
| PDL1 | Programmed Cell Death Ligand-1 |
| PDX | Patient-Derived-Xenograft |
| PE | Phosphatidylethanolamine |
| PI3K | Phosphatidylinositol 3-kinase |
| PIK3CA | Phosphatidylinositol-4, 5-bisphosphate 3-kinase, catalytic subunit, alpha |
| PIP3 | Phosphatidylinositol (3,4,5)-trisphosphate |
| PTEN | Phosphatase and Tensin Homolog |
| PgR | Progesteron Receptor |
| RB | Retinoblastoma protein |
| ROS | Reactive Oxygen Species |
| T-DM1 | Trastuzumab emtansine |
| TFEB | Transcription factor EB |
| TNBC | Triple-Negative Breast Cancer |
| ULK1 | Unc-51 like autophagy activating kinase |
References
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Pedro, J.M.B.-S.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular Definitions of Autophagy and Related Processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the Integrated Stress Response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and Medical Implications of Mammalian Autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Jeong, H.; Yu, S.-W. Autophagy as a Decisive Process for Cell Death. Exp. Mol. Med. 2020, 52, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Khandia, R.; Dadar, M.; Munjal, A.; Dhama, K.; Karthik, K.; Tiwari, R.; Yatoo, M.I.; Iqbal, H.M.N.; Singh, K.P.; Joshi, S.K.; et al. A Comprehensive Review of Autophagy and Its Various Roles in Infectious, Non-Infectious, and Lifestyle Diseases: Current Knowledge and Prospects for Disease Prevention, Novel Drug Design, and Therapy. Cells 2019, 8, 674. [Google Scholar] [CrossRef] [Green Version]
- Bonam, S.R.; Wang, F.; Muller, S. Autophagy: A New Concept in Autoimmunity Regulation and a Novel Therapeutic Option. J. Autoimmun. 2018, 94, 16–32. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Klionsky, D.J. An Overview of the Molecular Mechanism of Autophagy. Curr. Top. Microbiol. Immunol. 2009, 335, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Mercer, T.J.; Gubas, A.; Tooze, S.A. A Molecular Perspective of Mammalian Autophagosome Biogenesis. J. Biol. Chem. 2018, 293, 5386–5395. [Google Scholar] [CrossRef] [Green Version]
- Cecconi, F.; Levine, B. The Role of Autophagy in Mammalian Development: Cell Makeover Rather than Cell Death. Dev. Cell 2008, 15, 344–357. [Google Scholar] [CrossRef] [Green Version]
- Bonam, S.R.; Bayry, J.; Tschan, M.P.; Muller, S. Progress and Challenges in The Use of MAP1LC3 as a Legitimate Marker for Measuring Dynamic Autophagy In Vivo. Cells 2020, 9, 1321. [Google Scholar] [CrossRef]
- Corona Velazquez, A.F.; Jackson, W.T. So Many Roads: The Multifaceted Regulation of Autophagy Induction. Mol. Cell. Biol. 2018, 38, e00303-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A Lysosome-to-Nucleus Signalling Mechanism Senses and Regulates the Lysosome via MTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ao, X.; Zou, L.; Wu, Y. Regulation of Autophagy by the Rab GTPase Network. Cell Death Differ. 2014, 21, 348–358. [Google Scholar] [CrossRef] [Green Version]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-MTOR-Ulk1/2 in the Regulation of Autophagy: Cross Talk, Shortcuts, and Feedbacks. Mol. Cell. Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chen, Y. AMPK and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 85–108. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Klisch, T.J.; et al. TFEB Controls Cellular Lipid Metabolism through a Starvation-Induced Autoregulatory Loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo Recognition and Trafficking in Selective Autophagy. Nat. Cell Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef]
- Germain, K.; Kim, P.K. Pexophagy: A Model for Selective Autophagy. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.; Wilkinson, S. ER Homeostasis and Autophagy. Essays Biochem. 2017, 61, 625–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denton, D.; Kumar, S. Ribophagy: New Receptor Discovered. Cell Res. 2018, 28, 699–700. [Google Scholar] [CrossRef]
- Kounakis, K.; Chaniotakis, M.; Markaki, M.; Tavernarakis, N. Emerging Roles of Lipophagy in Health and Disease. Front. Cell Dev. Biol. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Levine, B. Eating Oneself and Uninvited Guests: Autophagy-Related Pathways in Cellular Defense. Cell 2005, 120, 159–162. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.-X.; Ni, H.-M.; Gao, W.; Yoshimori, T.; Stolz, D.B.; Ron, D.; Yin, X.-M. Linking of Autophagy to Ubiquitin-Proteasome System Is Important for the Regulation of Endoplasmic Reticulum Stress and Cell Viability. Am. J. Pathol. 2007, 171, 513–524. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, G.; Wada, K.; Okuno, M.; Kurosawa, M.; Nukina, N. Serine 403 Phosphorylation of P62/SQSTM1 Regulates Selective Autophagic Clearance of Ubiquitinated Proteins. Mol. Cell 2011, 44, 279–289. [Google Scholar] [CrossRef]
- Bernardini, J.P.; Lazarou, M.; Dewson, G. Parkin and Mitophagy in Cancer. Oncogene 2017, 36, 1315–1327. [Google Scholar] [CrossRef]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The Role of Autophagy in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2011, 10, 1533. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Cescon, M.; Bonaldo, P. Autophagy-Mediated Regulation of Macrophages and Its Applications for Cancer. Autophagy 2014, 10, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Barnard, R.A.; Regan, D.P.; Hansen, R.J.; Maycotte, P.; Thorburn, A.; Gustafson, D.L. Autophagy Inhibition Delays Early but Not Late-Stage Metastatic Disease. J. Pharmacol. Exp. Ther. 2016, 358, 282–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Klionsky, D.J. Mitochondria Removal by Autophagy. Autophagy 2011, 7, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.-Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy Suppresses Tumorigenesis through Elimination of P62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of Autophagy and Inhibition of Tumorigenesis by Beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of Autophagy in Cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef]
- Das, C.K.; Mandal, M.; Kögel, D. Pro-Survival Autophagy and Cancer Cell Resistance to Therapy. Cancer Metastasis Rev. 2018, 37, 749–766. [Google Scholar] [CrossRef]
- Thomas, M.; Davis, T.; Loos, B.; Sishi, B.; Huisamen, B.; Strijdom, H.; Engelbrecht, A.-M. Autophagy Is Essential for the Maintenance of Amino Acids and ATP Levels during Acute Amino Acid Starvation in MDAMB231 Cells. Cell Biochem. Funct. 2018, 36, 65–79. [Google Scholar] [CrossRef]
- Martin, S.; Dudek-Peric, A.M.; Garg, A.D.; Roose, H.; Demirsoy, S.; Van Eygen, S.; Mertens, F.; Vangheluwe, P.; Vankelecom, H.; Agostinis, P. An Autophagy-Driven Pathway of ATP Secretion Supports the Aggressive Phenotype of BRAFV600E Inhibitor-Resistant Metastatic Melanoma Cells. Autophagy 2017, 13, 1512–1527. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; He, S.; Ma, B. Autophagy and Autophagy-Related Proteins in Cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef] [PubMed]
- Ahn, C.H.; Jeong, E.G.; Lee, J.W.; Kim, M.S.; Kim, S.H.; Kim, S.S.; Yoo, N.J.; Lee, S.H. Expression of Beclin-1, an Autophagy-Related Protein, in Gastric and Colorectal Cancers. APMIS Acta Pathol. Microbiol. Immunol. Scand. 2007, 115, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Da, L.; Mao, Y.; Li, Y.; Li, D.; Xu, Z.; Li, F.; Wang, Y.; Tiollais, P.; Li, T.; et al. Hepatitis B Virus X Protein Sensitizes Cells to Starvation-Induced Autophagy via up-Regulation of Beclin 1 Expression. Hepatol. Baltim. MD 2009, 49, 60–71. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, J.-H.; Jin, L.; Lin, S.-M.; Yang, Y.; Sui, Y.-X.; Shi, H. Over-Expression of the Beclin1 Gene Upregulates Chemosensitivity to Anti-Cancer Drugs by Enhancing Therapy-Induced Apoptosis in Cervix Squamous Carcinoma CaSki Cells. Cancer Lett. 2010, 294, 204–210. [Google Scholar] [CrossRef]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.-L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of Tumorigenesis by Heterozygous Disruption of the Beclin 1 Autophagy Gene. J. Clin. Invest. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Hu, L.; Zheng, H.; Mao, C.; Hu, W.; Xiong, K.; Wang, F.; Liu, C. Application and Interpretation of Current Autophagy Inhibitors and Activators. Acta Pharmacol. Sin. 2013, 34, 625–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.Y.; Chen, H.-Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.S.; Karantza, V.; et al. Activated Ras Requires Autophagy to Maintain Oxidative Metabolism and Tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Xiong, H.; Liu, D.; Hill, C.; Ertay, A.; Li, J.; Zou, Y.; Miller, P.; White, E.; Downward, J.; et al. Autophagy Inhibition Specifically Promotes Epithelial-Mesenchymal Transition and Invasion in RAS-Mutated Cancer Cells. Autophagy 2019, 15, 886–899. [Google Scholar] [CrossRef] [PubMed]
- Seton-Rogers, S. Eliminating Protective Autophagy in KRAS-Mutant Cancers. Nat. Rev. Cancer 2019, 19, 247. [Google Scholar] [CrossRef]
- Lazova, R.; Camp, R.L.; Klump, V.; Siddiqui, S.F.; Amaravadi, R.K.; Pawelek, J.M. Punctate LC3B Expression Is a Common Feature of Solid Tumors and Associated with Proliferation, Metastasis, and Poor Outcome. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 370–379. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Yang, M.; Zhao, J.; Wang, J.; Zhang, Y.; Zhang, Q. High Expression of LC3B Is Associated with Progression and Poor Outcome in Triple-Negative Breast Cancer. Med. Oncol. Northwood Lond. Engl. 2013, 30, 475. [Google Scholar] [CrossRef] [PubMed]
- Lazova, R.; Klump, V.; Pawelek, J. Autophagy in Cutaneous Malignant Melanoma. J. Cutan. Pathol. 2010, 37, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.-F.; Shi, Y.-H.; Ding, Z.-B.; Ke, A.-W.; Gu, C.-Y.; Hui, B.; Zhou, J.; Qiu, S.-J.; Dai, Z.; Fan, J. Autophagy Inhibition Suppresses Pulmonary Metastasis of HCC in Mice via Impairing Anoikis Resistance and Colonization of HCC Cells. Autophagy 2013, 9, 2056–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galavotti, S.; Bartesaghi, S.; Faccenda, D.; Shaked-Rabi, M.; Sanzone, S.; McEvoy, A.; Dinsdale, D.; Condorelli, F.; Brandner, S.; Campanella, M.; et al. The Autophagy-Associated Factors DRAM1 and P62 Regulate Cell Migration and Invasion in Glioblastoma Stem Cells. Oncogene 2013, 32, 699–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of Autophagy during Extracellular Matrix Detachment Promotes Cell Survival. Mol. Biol. Cell 2008, 19, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Chaffer, C.L.; Weinberg, R.A. A Perspective on Cancer Cell Metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef]
- Valastyan, S.; Weinberg, R.A. Tumor Metastasis: Molecular Insights and Evolving Paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [Green Version]
- Ravanan, P.; Srikumar, I.F.; Talwar, P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017, 188, 53–67. [Google Scholar] [CrossRef]
- Kroemer, G.; Levine, B. Autophagic Cell Death: The Story of a Misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-Consumption: The Interplay of Autophagy and Apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Shoji-Kawata, S.; Sumpter, R.M.; Wei, Y.; Ginet, V.; Zhang, L.; Posner, B.; Tran, K.A.; Green, D.R.; Xavier, R.J.; et al. Autosis Is a Na+,K+-ATPase-Regulated Form of Cell Death Triggered by Autophagy-Inducing Peptides, Starvation, and Hypoxia-Ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, 20364–20371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.; Ginet, V.; Sun, Y.; Koike, M.; Zhou, K.; Li, T.; Li, H.; Li, Q.; Wang, X.; Uchiyama, Y.; et al. Neuroprotection by Selective Neuronal Deletion of Atg7 in Neonatal Brain Injury. Autophagy 2016, 12, 410–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gump, J.M.; Thorburn, A. Autophagy and apoptosis: What is the connection? Trends Cell Biol. 2011, 21, 387–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napoletano, F.; Baron, O.; Vandenabeele, P.; Mollereau, B.; Fanto, M. Intersections between Regulated Cell Death and Autophagy. Trends Cell Biol. 2019, 29, 323–338. [Google Scholar] [CrossRef] [Green Version]
- Bialik, S.; Dasari, S.K.; Kimchi, A. Autophagy-dependent cell death - where, how and why a cell eats itself to death. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luciani, M.-F.; Giusti, C.; Harms, B.; Oshima, Y.; Kikuchi, H.; Kubohara, Y.; Golstein, P. Atg1 Allows Second-Signaled Autophagic Cell Death in Dictyostelium. Autophagy 2011, 7, 501–508. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Yang, J.; Liao, W.; Liu, X.; Zhang, H.; Wang, S.; Wang, D.; Feng, J.; Yu, L.; Zhu, W.-G. Cytosolic FoxO1 Is Essential for the Induction of Autophagy and Tumour Suppressor Activity. Nat. Cell Biol. 2010, 12, 665–675. [Google Scholar] [CrossRef]
- Law, B.Y.K.; Michelangeli, F.; Qu, Y.Q.; Xu, S.-W.; Han, Y.; Mok, S.W.F.; de Seabra Rodrigues Dias, I.R.; Javed, M.-H.; Chan, W.-K.; Xue, W.-W.; et al. Neferine Induces Autophagy-Dependent Cell Death in Apoptosis-Resistant Cancers via Ryanodine Receptor and Ca2+-Dependent Mechanism. Sci. Rep. 2019, 9, 20034. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, N.; Liu, W.; Nakamura, K.; Yoshida, K.; Ikeda, Y.; Tanaka, H.; Mizuno, M.; Toyokuni, S.; Hori, M.; Kikkawa, F.; et al. Plasma-Activated Medium Promotes Autophagic Cell Death along with Alteration of the MTOR Pathway. Sci. Rep. 2020, 10, 1614. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, S.; Kanaseki, T.; Mizushima, N.; Mizuta, T.; Arakawa-Kobayashi, S.; Thompson, C.B.; Tsujimoto, Y. Role of Bcl-2 Family Proteins in a Non-Apoptotic Programmed Cell Death Dependent on Autophagy Genes. Nat. Cell Biol. 2004, 6, 1221–1228. [Google Scholar] [CrossRef]
- Law, B.Y.K.; Chan, W.K.; Xu, S.W.; Wang, J.R.; Bai, L.P.; Liu, L.; Wong, V.K.W. Natural Small-Molecule Enhancers of Autophagy Induce Autophagic Cell Death in Apoptosis-Defective Cells. Sci. Rep. 2014, 4, 5510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauckman, K.A.; Haller, E.; Flores, I.; Nanjundan, M. Iron Modulates Cell Survival in a Ras- and MAPK-Dependent Manner in Ovarian Cells. Cell Death Dis. 2013, 4, e592. [Google Scholar] [CrossRef] [Green Version]
- Kocaturk, N.M.; Akkoc, Y.; Kig, C.; Bayraktar, O.; Gozuacik, D.; Kutlu, O. Autophagy as a Molecular Target for Cancer Treatment. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2019, 134, 116–137. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Shi, Y.; Tao, M.; Liu, N.; Zhuang, S.; Yuan, W. Pharmacological Inhibition of Autophagy by 3-MA Attenuates Hyperuricemic Nephropathy. Clin. Sci. Lond. Engl. 2018, 132, 2299–2322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Bel, M.; Abela, A.R.; Ng, J.D.; Guerrero, C.A. Enantioselective Chemical Syntheses of the Furanosteroids (−)-Viridin and (−)-Viridiol. J. Am. Chem. Soc. 2017, 139, 6819–6822. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Jiang, C.; Xia, W.; Ju, H.; Jin, S.; Liu, S.; Zhang, L.; Ren, G.; Ma, H.; Ruan, M.; et al. Blocking Autophagy Flux Promotes Interferon-Alpha-Mediated Apoptosis in Head and Neck Squamous Cell Carcinoma. Cancer Lett. 2019, 451, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Florey, O.; Gammoh, N.; Kim, S.E.; Jiang, X.; Overholtzer, M. V-ATPase and Osmotic Imbalances Activate Endolysosomal LC3 Lipidation. Autophagy 2015, 11, 88–99. [Google Scholar] [CrossRef] [Green Version]
- Mauvezin, C.; Neufeld, T.P. Bafilomycin A1 Disrupts Autophagic Flux by Inhibiting Both V-ATPase-Dependent Acidification and Ca-P60A/SERCA-Dependent Autophagosome-Lysosome Fusion. Autophagy 2015, 11, 1437–1438. [Google Scholar] [CrossRef] [Green Version]
- Mauthe, M.; Langereis, M.; Jung, J.; Zhou, X.; Jones, A.; Omta, W.; Tooze, S.A.; Stork, B.; Paludan, S.R.; Ahola, T.; et al. An SiRNA Screen for ATG Protein Depletion Reveals the Extent of the Unconventional Functions of the Autophagy Proteome in Virus Replication. J. Cell Biol. 2016, 214, 619–635. [Google Scholar] [CrossRef]
- Verbaanderd, C.; Maes, H.; Schaaf, M.B.; Sukhatme, V.P.; Pantziarka, P.; Sukhatme, V.; Agostinis, P.; Bouche, G. Repurposing Drugs in Oncology (ReDO)—Chloroquine and Hydroxychloroquine as Anti-cancer Agents. Available online: http://ecancer.org/en/journal/article/781-repurposing-drugs-in-oncology-redo-chloroquine-and-hydroxychloroquine-as-anti-cancer-agents (accessed on 25 September 2020). [CrossRef] [Green Version]
- Zhang, Y.; Wang, Q.; Ma, A.; Li, Y.; Li, R.; Wang, Y. Functional Expression of TLR9 in Esophageal Cancer. Oncol. Rep. 2014, 31, 2298–2304. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Yip, M.L.R.; Shen, X.; Li, H.; Hsin, L.-Y.C.; Labarge, S.; Heinrich, E.L.; Lee, W.; Lu, J.; Vaidehi, N. Identification of Anti-Malarial Compounds as Novel Antagonists to Chemokine Receptor CXCR4 in Pancreatic Cancer Cells. PLoS ONE 2012, 7, e31004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, Z.; Li, Q.; Wu, P.; Ye, Y.; Tseng, H.-Y.; Zhang, L.; Zhang, X.D. Autophagy-Mediated HMGB1 Release Antagonizes Apoptosis of Gastric Cancer Cells Induced by Vincristine via Transcriptional Regulation of Mcl-1. Autophagy 2012, 8, 109–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wu, G.S. Role of Autophagy in Cisplatin Resistance in Ovarian Cancer Cells. J. Biol. Chem. 2014, 289, 17163–17173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, L.; Jaramillo, M.C.; Zhang, Z.; Zheng, Y.; Yao, M.; Zhang, D.D.; Yi, X. Induction of Autophagy Contributes to Cisplatin Resistance in Human Ovarian Cancer Cells. Mol. Med. Rep. 2015, 11, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, T.; Oda, K.; Wada-Hiraike, O.; Sone, K.; Inaba, K.; Ikeda, Y.; Miyasaka, A.; Kashiyama, T.; Tanikawa, M.; Arimoto, T.; et al. The Anti-Malarial Chloroquine Suppresses Proliferation and Overcomes Cisplatin Resistance of Endometrial Cancer Cells via Autophagy Inhibition. Gynecol. Oncol. 2015, 137, 538–545. [Google Scholar] [CrossRef]
- Wu, T.; Wang, M.-C.; Jing, L.; Liu, Z.-Y.; Guo, H.; Liu, Y.; Bai, Y.-Y.; Cheng, Y.-Z.; Nan, K.-J.; Liang, X. Autophagy Facilitates Lung Adenocarcinoma Resistance to Cisplatin Treatment by Activation of AMPK/MTOR Signaling Pathway. Drug Des. Devel. Ther. 2015, 9, 6421–6431. [Google Scholar] [CrossRef] [Green Version]
- Grimaldi, A.; Santini, D.; Zappavigna, S.; Lombardi, A.; Misso, G.; Boccellino, M.; Desiderio, V.; Vitiello, P.P.; Di Lorenzo, G.; Zoccoli, A.; et al. Antagonistic Effects of Chloroquine on Autophagy Occurrence Potentiate the Anticancer Effects of Everolimus on Renal Cancer Cells. Cancer Biol. Ther. 2015, 16, 567–579. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhu, Y.-R.; Wang, S.; Zhao, S. Autophagy Inhibition Sensitizes WYE-354-Induced Anti-Colon Cancer Activity in Vitro and in Vivo. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 11743–11752. [Google Scholar] [CrossRef]
- Elmansi, A.M.; El-Karef, A.A.; El-Shishtawy, M.M.; Eissa, L.A. Hepatoprotective Effect of Curcumin on Hepatocellular Carcinoma Through Autophagic and Apoptic Pathways. Ann. Hepatol. 2017, 16, 607–618. [Google Scholar] [CrossRef]
- Misirkic, M.; Janjetovic, K.; Vucicevic, L.; Tovilovic, G.; Ristic, B.; Vilimanovich, U.; Harhaji-Trajkovic, L.; Sumarac-Dumanovic, M.; Micic, D.; Bumbasirevic, V.; et al. Inhibition of AMPK-Dependent Autophagy Enhances in Vitro Antiglioma Effect of Simvastatin. Pharmacol. Res. 2012, 65, 111–119. [Google Scholar] [CrossRef]
- Lamoureux, F.; Thomas, C.; Crafter, C.; Kumano, M.; Zhang, F.; Davies, B.R.; Gleave, M.E.; Zoubeidi, A. Blocked Autophagy Using Lysosomotropic Agents Sensitizes Resistant Prostate Tumor Cells to the Novel Akt Inhibitor AZD5363. Clin. Cancer Res. 2013, 19, 833. [Google Scholar] [CrossRef] [Green Version]
- PRIME PubMed|Antitumor Activity of Chloroquine in Combination with Cisplatin in Human Gastric Cancer Xenografts. Available online: https://www.unboundmedicine.com/medline/citation/25987058/Antitumor_activity_of_chloroquine_in_combination_with_Cisplatin_in_human_gastric_cancer_xenografts_ (accessed on 28 September 2020).
- Abdel-Aziz, A.K.; Shouman, S.; El-Demerdash, E.; Elgendy, M.; Abdel-Naim, A.B. Chloroquine Synergizes Sunitinib Cytotoxicity via Modulating Autophagic, Apoptotic and Angiogenic Machineries. Chem. Biol. Interact. 2014, 217, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Honma, Y.; Harada, M. Sorafenib Enhances Proteasome Inhibitor-Mediated Cytotoxicity via Inhibition of Unfolded Protein Response and Keratin Phosphorylation. Exp. Cell Res. 2013, 319, 2166–2178. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.-C.; Lin, L.-D.; Wu, M.-T.; Chan, C.-P.; Chang, H.-H.; Lee, M.-S.; Sun, T.-Y.; Jeng, P.-Y.; Yeung, S.-Y.; Lin, H.-J.; et al. Effects of Camphorquinone on Cytotoxicity, Cell Cycle Regulation and Prostaglandin E2 Production of Dental Pulp Cells: Role of ROS, ATM/Chk2, MEK/ERK and Hemeoxygenase-1. PLoS ONE 2015, 10, e0143663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michael, R.O.; Williams, G.M. Choloroquine Inhibition of Repair of DNA Damage Induced in Mammalian Cells by Methyl Methanesulfonate. Mutat. Res. Mol. Mech. Mutagen. 1974, 25, 391–396. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020, CA. Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Redig, A.J.; McAllister, S.S. Breast Cancer as a Systemic Disease: A View of Metastasis. J. Intern. Med. 2013, 274, 113–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prat, A.; Perou, C.M. Deconstructing the Molecular Portraits of Breast Cancer. Mol. Oncol. 2011, 5, 5–23. [Google Scholar] [CrossRef]
- Cardoso, F.; Costa, A.; Senkus, E.; Aapro, M.; André, F.; Barrios, C.H.; Bergh, J.; Bhattacharyya, G.; Biganzoli, L.; Cardoso, M.J.; et al. 3rd ESO-ESMO International Consensus Guidelines for Advanced Breast Cancer (ABC 3). Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017, 28, 16–33. [Google Scholar] [CrossRef]
- Wang, J.; Xu, B. Targeted Therapeutic Options and Future Perspectives for HER2-Positive Breast Cancer. Signal Transduct. Target. Ther. 2019, 4, 34. [Google Scholar] [CrossRef] [Green Version]
- Cocco, S.; Piezzo, M.; Calabrese, A.; Cianniello, D.; Caputo, R.; Lauro, V.D.; Fusco, G.; di Gioia, G.; Licenziato, M.; de Laurentiis, M. Biomarkers in Triple-Negative Breast Cancer: State-of-the-Art and Future Perspectives. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. IMpassion130 Trial Investigators. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Piezzo, M.; Chiodini, P.; Riemma, M.; Cocco, S.; Caputo, R.; Cianniello, D.; Di Gioia, G.; Di Lauro, V.; Rella, F.D.; Fusco, G.; et al. Progression-Free Survival and Overall Survival of CDK 4/6 Inhibitors Plus Endocrine Therapy in Metastatic Breast Cancer: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Heimes, A.-S.; Schmidt, M. Atezolizumab for the Treatment of Triple-Negative Breast Cancer. Expert Opin. Investig. Drugs 2019, 28, 1–5. [Google Scholar] [CrossRef]
- Pistelli, M.; Mora, A.D.; Ballatore, Z.; Berardi, R. Aromatase Inhibitors in Premenopausal Women with Breast Cancer: The State of the Art and Future Prospects. Curr. Oncol. Tor. Ont. 2018, 25, e168–e175. [Google Scholar] [CrossRef] [Green Version]
- Arpino, G.; De Angelis, C.; Giuliano, M.; Giordano, A.; Falato, C.; De Laurentiis, M.; De Placido, S. Molecular Mechanism and Clinical Implications of Endocrine Therapy Resistance in Breast Cancer. Oncology 2009, 77, 23–37. [Google Scholar] [CrossRef]
- Duan, L.; Motchoulski, N.; Danzer, B.; Davidovich, I.; Shariat-Madar, Z.; Levenson, V.V. Prolylcarboxypeptidase Regulates Proliferation, Autophagy, and Resistance to 4-Hydroxytamoxifen-Induced Cytotoxicity in Estrogen Receptor-Positive Breast Cancer Cells. J. Biol. Chem. 2011, 286, 2864–2876. [Google Scholar] [CrossRef] [Green Version]
- Samaddar, J.S.; Gaddy, V.T.; Duplantier, J.; Thandavan, S.P.; Shah, M.; Smith, M.J.; Browning, D.; Rawson, J.; Smith, S.B.; Barrett, J.T.; et al. A Role for Macroautophagy in Protection against 4-Hydroxytamoxifen–Induced Cell Death and the Development of Antiestrogen Resistance. Mol. Cancer Ther. 2008, 7, 2977. [Google Scholar] [CrossRef] [Green Version]
- Cook, K.L.; Wärri, A.; Soto-Pantoja, D.R.; Clarke, P.A.; Cruz, M.I.; Zwart, A.; Clarke, R. Hydroxychloroquine Inhibits Autophagy to Potentiate Antiestrogen Responsiveness in ER+ Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 3222–3232. [Google Scholar] [CrossRef] [Green Version]
- Amaral, C.; Augusto, T.V.; Tavares-da-Silva, E.; Roleira, F.M.F.; Correia-da-Silva, G.; Teixeira, N. Hormone-Dependent Breast Cancer: Targeting Autophagy and PI3K Overcomes Exemestane-Acquired Resistance. J. Steroid Biochem. Mol. Biol. 2018, 183, 51–61. [Google Scholar] [CrossRef]
- Fruman, D.A.; Rommel, C. PI3K and Cancer: Lessons, Challenges and Opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loehberg, C.R.; Strissel, P.L.; Dittrich, R.; Strick, R.; Dittmer, J.; Dittmer, A.; Fabry, B.; Kalender, W.A.; Koch, T.; Wachter, D.L.; et al. Akt and P53 Are Potential Mediators of Reduced Mammary Tumor Growth by Cloroquine and the MTOR Inhibitor RAD001. Biochem. Pharmacol. 2012, 83, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, J.; Qiu, Y.; Jin, M.; Chen, X.; Fan, G.; Wang, R.; Kong, D. ZSTK474, a Specific Class I Phosphatidylinositol 3-Kinase Inhibitor, Induces G1 Arrest and Autophagy in Human Breast Cancer MCF-7 Cells. Oncotarget 2016, 7, 19897–19909. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Xia, J.; Zou, J.; Wang, Q.; Ma, Q.; Sun, R.; Liao, H.; Xu, L.; Wang, D.; Guo, X. The PI3K/MTOR Dual Inhibitor NVP-BEZ235 Stimulates Mutant P53 Degradation to Exert Anti-Tumor Effects on Triple-Negative Breast Cancer Cells. FEBS Open Bio 2020, 10, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Di, W.; Yang, Q.; Lu, Z.; Cai, W.; Wu, J. Inhibition of Autophagy Increases Proliferation Inhibition and Apoptosis Induced by the PI3K/MTOR Inhibitor NVP-BEZ235 in Breast Cancer Cells. Clin. Lab. 2015, 61, 1043–1051. [Google Scholar] [CrossRef]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. SOLAR-1 Study Group. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Zhai, B.; Hu, F.; Jiang, X.; Xu, J.; Zhao, D.; Liu, B.; Pan, S.; Dong, X.; Tan, G.; Wei, Z.; et al. Inhibition of Akt Reverses the Acquired Resistance to Sorafenib by Switching Protective Autophagy to Autophagic Cell Death in Hepatocellular Carcinoma. Mol. Cancer Ther. 2014, 13, 1589–1598. [Google Scholar] [CrossRef] [Green Version]
- IGF1R Upregulation Confers Resistance to Isoform-specific Inhibitors of PI3K in PIK3CA-driven Ovarian Cancer—Abstract—Europe PMC. Available online: https://europepmc.org/article/pmc/pmc6148236 (accessed on 28 September 2020).
- Im, S.-A.; Lu, Y.-S.; Bardia, A.; Harbeck, N.; Colleoni, M.; Franke, F.; Chow, L.; Sohn, J.; Lee, K.-S.; Campos-Gomez, S.; et al. Overall Survival with Ribociclib plus Endocrine Therapy in Breast Cancer. N. Engl. J. Med. 2019, 381, 307–316. [Google Scholar] [CrossRef]
- Piezzo, M.; Cocco, S.; Caputo, R.; Cianniello, D.; Gioia, G.D.; Lauro, V.D.; Fusco, G.; Martinelli, C.; Nuzzo, F.; Pensabene, M.; et al. Targeting Cell Cycle in Breast Cancer: CDK4/6 Inhibitors. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, Q.; Peng, X.; Zhou, C.; Zhong, Y.; Chen, X.; Qiu, Y.; Jin, M.; Gong, M.; Kong, D. Stellettin B Induces G1 Arrest, Apoptosis and Autophagy in Human Non-Small Cell Lung Cancer A549 Cells via Blocking PI3K/Akt/MTOR Pathway. Sci. Rep. 2016, 6, 27071. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, F.-S.; Chen, Y.-L.; Hung, M.-H.; Chu, P.-Y.; Tsai, M.-H.; Chen, L.-J.; Hsiao, Y.-J.; Shih, C.-T.; Chang, M.-J.; Chao, T.-I.; et al. Palbociclib Induces Activation of AMPK and Inhibits Hepatocellular Carcinoma in a CDK4/6-Independent Manner. Mol. Oncol. 2017, 11, 1035–1049. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, C.A.; Vargas, L.; Martinez, V.; Bravo, S.; Brown, N.E. Palbociclib-Induced Autophagy and Senescence in Gastric Cancer Cells. Exp. Cell Res. 2017, 360, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Vijayaraghavan, S.; Karakas, C.; Doostan, I.; Chen, X.; Bui, T.; Yi, M.; Raghavendra, A.S.; Zhao, Y.; Bashour, S.I.; Ibrahim, N.K.; et al. CDK4/6 and Autophagy Inhibitors Synergistically Induce Senescence in Rb Positive Cytoplasmic Cyclin E Negative Cancers. Nat. Commun. 2017, 8, 15916. [Google Scholar] [CrossRef]
- Okada, Y.; Kato, S.; Sakamoto, Y.; Oishi, T.; Ishioka, C. Synthetic Lethal Interaction of CDK Inhibition and Autophagy Inhibition in Human Solid Cancer Cell Lines. Oncol. Rep. 2017, 38, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iriyama, N.; Hino, H.; Moriya, S.; Hiramoto, M.; Hatta, Y.; Takei, M.; Miyazawa, K. The Cyclin-Dependent Kinase 4/6 Inhibitor, Abemaciclib, Exerts Dose-Dependent Cytostatic and Cytocidal Effects and Induces Autophagy in Multiple Myeloma Cells. Leuk. Lymphoma 2018, 59, 1439–1450. [Google Scholar] [CrossRef]
- Small, J.; Washburn, E.; Millington, K.; Zhu, J.; Holder, S.L. The Addition of Abemaciclib to Sunitinib Induces Regression of Renal Cell Carcinoma Xenograft Tumors. Oncotarget 2017, 8, 95116–95134. [Google Scholar] [CrossRef]
- Hino, H.; Iriyama, N.; Kokuba, H.; Kazama, H.; Moriya, S.; Takano, N.; Hiramoto, M.; Aizawa, S.; Miyazawa, K. Abemaciclib Induces Atypical Cell Death in Cancer Cells Characterized by Formation of Cytoplasmic Vacuoles Derived from Lysosomes. Cancer Sci. 2020, 111, 2132–2145. [Google Scholar] [CrossRef]
- Yin, Q.; Jian, Y.; Xu, M.; Huang, X.; Wang, N.; Liu, Z.; Li, Q.; Li, J.; Zhou, H.; Xu, L.; et al. CDK4/6 Regulate Lysosome Biogenesis through TFEB/TFE3. J. Cell Biol. 2020, 219, e201911036. [Google Scholar] [CrossRef]
- Hsu, J.L.; Hung, M.-C. The Role of HER2, EGFR, and Other Receptor Tyrosine Kinases in Breast Cancer. Cancer Metastasis Rev. 2016, 35, 575–588. [Google Scholar] [CrossRef] [Green Version]
- Meric-Bernstam, F.; Johnson, A.M.; Dumbrava, E.E.I.; Raghav, K.; Balaji, K.; Bhatt, M.; Murthy, R.K.; Rodon, J.; Piha-Paul, S.A. Advances in HER2-Targeted Therapy: Novel Agents and Opportunities Beyond Breast and Gastric Cancer. Clin. Cancer Res. 2019, 25(7), 2033–2041. [Google Scholar]
- Negri, T.; Tarantino, E.; Orsenigo, M.; Reid, J.F.; Gariboldi, M.; Zambetti, M.; Pierotti, M.A.; Pilotti, S. Chromosome Band 17q21 in Breast Cancer: Significant Association between Beclin 1 Loss and HER2/NEU Amplification. Genes. Chromosomes Cancer 2010, 49, 901–909. [Google Scholar] [CrossRef]
- Tang, H.; Sebti, S.; Titone, R.; Zhou, Y.; Isidoro, C.; Ross, T.S.; Hibshoosh, H.; Xiao, G.; Packer, M.; Xie, Y.; et al. Decreased BECN1 MRNA Expression in Human Breast Cancer Is Associated with Estrogen Receptor-Negative Subtypes and Poor Prognosis. EBioMedicine 2015, 2, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Lozy, F.; Cai-McRae, X.; Teplova, I.; Price, S.; Reddy, A.; Bhanot, G.; Ganesan, S.; Vazquez, A.; Karantza, V. ERBB2 Overexpression Suppresses Stress-Induced Autophagy and Renders ERBB2-Induced Mammary Tumorigenesis Independent of Monoallelic Becn1 Loss. Autophagy 2014, 10, 662–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vega-Rubín-de-Celis, S.; Zou, Z.; Fernández, Á.F.; Ci, B.; Kim, M.; Xiao, G.; Xie, Y.; Levine, B. Increased Autophagy Blocks HER2-Mediated Breast Tumorigenesis. Proc. Natl. Acad. Sci. USA 2018, 115, 4176–4181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janser, F.A.; Adams, O.; Bütler, V.; Schläfli, A.M.; Dislich, B.; Seiler, C.A.; Kröll, D.; Langer, R.; Tschan, M.P. Her2-Targeted Therapy Induces Autophagy in Esophageal Adenocarcinoma Cells. Int. J. Mol. Sci. 2018, 19, 3069. [Google Scholar] [CrossRef] [Green Version]
- Autophagy-related Gene 12 (ATG12) is a Novel Determinant of Primary Resistance to HER2-targeted Therapies: Utility of Transcriptome Analysis of the Autophagy Interactome to Guide Breast Cancer Treatment—Abstract—Europe PMC. Available online: https://europepmc.org/article/pmc/3681498 (accessed on 28 September 2020).
- Chen, S.; Zhu, X.; Qiao, H.; Ye, M.; Lai, X.; Yu, S.; Ding, L.; Wen, A.; Zhang, J. Protective Autophagy Promotes the Resistance of HER2-Positive Breast Cancer Cells to Lapatinib. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 2321–2331. [Google Scholar] [CrossRef]
- Chen, S.; Li, X.; Feng, J.; Chang, Y.; Wang, Z.; Wen, A. Autophagy Facilitates the Lapatinib Resistance of HER2 Positive Breast Cancer Cells. Med. Hypotheses 2011, 77, 206–208. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A Nutrient and Energy Sensor That Maintains Energy Homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Cotzomi-Ortega, I.; Rosas-Cruz, A.; Ramírez-Ramírez, D.; Reyes-Leyva, J.; Rodriguez-Sosa, M.; Aguilar-Alonso, P.; Maycotte, P. Autophagy Inhibition Induces the Secretion of Macrophage Migration Inhibitory Factor (MIF) with Autocrine and Paracrine Effects on the Promotion of Malignancy in Breast Cancer. Biology 2020, 9, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Reilly, E.A.; Gubbins, L.; Sharma, S.; Tully, R.; Guang, M.H.Z.; Weiner-Gorzel, K.; McCaffrey, J.; Harrison, M.; Furlong, F.; Kell, M.; et al. The Fate of Chemoresistance in Triple Negative Breast Cancer (TNBC). BBA Clin. 2015, 3, 257–275. [Google Scholar] [CrossRef] [Green Version]
- Overgaard, J.; Yilmaz, M.; Guldberg, P.; Hansen, L.L.; Alsner, J. TP53 Mutation Is an Independent Prognostic Marker for Poor Outcome in Both Node-Negative and Node-Positive Breast Cancer. Acta Oncol. Stockh. Swed. 2000, 39, 327–333. [Google Scholar] [CrossRef]
- ATG9A Is Overexpressed in Triple Negative Breast Cancer and Its In Vitro Extinction Leads to the Inhibition of Pro-Cancer Phenotypes—Abstract—Europe PMC. Available online: https://europepmc.org/article/med/30563263 (accessed on 28 September 2020).
- Hamurcu, Z.; Delibaşı, N.; Geçene, S.; Şener, E.F.; Dönmez-Altuntaş, H.; Özkul, Y.; Canatan, H.; Ozpolat, B. Targeting LC3 and Beclin-1 Autophagy Genes Suppresses Proliferation, Survival, Migration and Invasion by Inhibition of Cyclin-D1 and UPAR/Integrin Β1/ Src Signaling in Triple Negative Breast Cancer Cells. J. Cancer Res. Clin. Oncol. 2018, 144, 415–430. [Google Scholar] [CrossRef] [PubMed]
- Maycotte, P.; Gearheart, C.M.; Barnard, R.; Aryal, S.; Mulcahy Levy, J.M.; Fosmire, S.P.; Hansen, R.J.; Morgan, M.J.; Porter, C.C.; Gustafson, D.L.; et al. STAT3-Mediated Autophagy Dependence Identifies Subtypes of Breast Cancer Where Autophagy Inhibition Can Be Efficacious. Cancer Res. 2014, 74, 2579. [Google Scholar] [CrossRef] [Green Version]
- Liang, D.H.; Choi, D.S.; Ensor, J.E.; Kaipparettu, B.A.; Bass, B.L.; Chang, J.C. The Autophagy Inhibitor Chloroquine Targets Cancer Stem Cells in Triple Negative Breast Cancer by Inducing Mitochondrial Damage and Impairing DNA Break Repair. Cancer Lett. 2016, 376, 249–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Bai, Y.; Patel, C.; Geng, F. Autophagy Promotes Triple Negative Breast Cancer Metastasis via YAP Nuclear Localization. Biochem. Biophys. Res. Commun. 2019, 520, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Maycotte, P.; Thorburn, A. Targeting Autophagy in Breast Cancer. World J. Clin. Oncol. 2014, 5, 224–240. [Google Scholar] [CrossRef]
- Viry, E.; Noman, M.Z.; Arakelian, T.; Lequeux, A.; Chouaib, S.; Berchem, G.; Moussay, E.; Paggetti, J.; Janji, B. Hijacker of the Antitumor Immune Response: Autophagy Is Showing Its Worst Facet. Front. Oncol. 2016, 6, 246. [Google Scholar] [CrossRef]
- Clark, C.A.; Gupta, H.B.; Sareddy, G.; Pandeswara, S.; Lao, S.; Yuan, B.; Drerup, J.M.; Padron, A.; Conejo-Garcia, J.; Murthy, K.; et al. Tumor-Intrinsic PD-L1 Signals Regulate Cell Growth, Pathogenesis, and Autophagy in Ovarian Cancer and Melanoma. Cancer Res. 2016, 76, 6964–6974. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wu, W.K.K.; Gao, J.; Li, Z.; Dong, B.; Lin, X.; Li, Y.; Li, Y.; Gong, J.; Qi, C.; et al. Autophagy Inhibition Enhances PD-L1 Expression in Gastric Cancer. J. Exp. Clin. Cancer Res. CR 2019, 38, 140. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-Y.; Choi, J.-H.; Nam, J.-S. Targeting Cancer Stem Cells in Triple-Negative Breast Cancer. Cancers 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective Identification of Tumorigenic Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; et al. ALDH1 Is a Marker of Normal and Malignant Human Mammary Stem Cells and a Predictor of Poor Clinical Outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Ma, H.; Zhang, J.; Zhu, L.; Wang, C.; Yang, Y. Unraveling the Roles of CD44/CD24 and ALDH1 as Cancer Stem Cell Markers in Tumorigenesis and Metastasis. Sci. Rep. 2017, 7, 13856. [Google Scholar] [CrossRef] [Green Version]
- Van Phuc, P.; Nhan, P.L.; Nhung, T.H.; Tam, N.T.; Hoang, N.M.; Tue, V.G.; Thuy, D.T.; Ngoc, P.K. Downregulation of CD44 Reduces Doxorubicin Resistance of CD44CD24 Breast Cancer Cells. OncoTargets Ther. 2011, 4, 71–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartucci, M.; Dattilo, R.; Moriconi, C.; Pagliuca, A.; Mottolese, M.; Federici, G.; Benedetto, A.D.; Todaro, M.; Stassi, G.; Sperati, F.; et al. TAZ Is Required for Metastatic Activity and Chemoresistance of breast cancer stem cells. Oncogene 2015, 34, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Palomeras, S.; Ruiz-Martínez, S.; Puig, T. Targeting Breast Cancer Stem Cells to Overcome Treatment Resistance. Molecules 2018, 23, 2193. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast Cancer Stem Cells Transition between Epithelial and Mesenchymal States Reflective of Their Normal Counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef]
- Liu, H.; Patel, M.R.; Prescher, J.A.; Patsialou, A.; Qian, D.; Lin, J.; Wen, S.; Chang, Y.F.; Bachmann, M.H.; Shimono, Y.; et al. Cancer Stem Cells from Human Breast Tumors Are Involved in Spontaneous Metastases in Orthotopic Mouse Models. Proc. Natl. Acad. Sci. USA 2010, 107, 18115–18120. [Google Scholar] [CrossRef] [Green Version]
- Ohi, Y.; Umekita, Y.; Yoshioka, T.; Souda, M.; Rai, Y.; Sagara, Y.; Sagara, Y.; Sagara, Y.; Tanimoto, A. Aldehyde Dehydrogenase 1 Expression Predicts Poor Prognosis in Triple-Negative Breast Cancer. Histopathology 2011, 59, 776–780. [Google Scholar] [CrossRef]
- Wu, Q.; Wang, J.; Liu, Y.; Gong, X. Epithelial Cell Adhesion Molecule and Epithelial-Mesenchymal Transition Are Associated with Vasculogenic Mimicry, Poor Prognosis, and Metastasis of Triple Negative Breast Cancer. Int. J. Clin. Exp. Pathol. 2019, 12, 1678–1689. [Google Scholar]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michiels, C. Physiological and Pathological Responses to Hypoxia. Am. J. Pathol. 2004, 164, 1875–1882. [Google Scholar] [CrossRef] [Green Version]
- Samanta, D.; Gilkes, D.M.; Chaturvedi, P.; Xiang, L.; Semenza, G.L. Hypoxia-Inducible Factors Are Required for Chemotherapy Resistance of Breast Cancer Stem Cells. Proc. Natl. Acad. Sci. USA 2014, 111, E5429–E5438. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Salnikov, A.V.; Bauer, N.; Aleksandrowicz, E.; Labsch, S.; Nwaeburu, C.; Mattern, J.; Gladkich, J.; Schemmer, P.; Werner, J.; et al. Triptolide Reverses Hypoxia-Induced Epithelial-Mesenchymal Transition and Stem-like Features in Pancreatic Cancer by NF-KappaB Downregulation. Int. J. Cancer 2014, 134, 2489–2503. [Google Scholar] [CrossRef] [Green Version]
- Conley, S.J.; Gheordunescu, E.; Kakarala, P.; Newman, B.; Korkaya, H.; Heath, A.N.; Clouthier, S.G.; Wicha, M.S. Antiangiogenic Agents Increase Breast Cancer Stem Cells via the Generation of Tumor Hypoxia. Proc. Natl. Acad. Sci. USA 2012, 109, 2784–2789. [Google Scholar] [CrossRef] [Green Version]
- Schwab, L.P.; Peacock, D.L.; Majumdar, D.; Ingels, J.F.; Jensen, L.C.; Smith, K.D.; Cushing, R.C.; Seagroves, T.N. Hypoxia-Inducible Factor 1alpha Promotes Primary Tumor Growth and Tumor-Initiating Cell Activity in Breast Cancer. Breast Cancer Res. 2012, 14, R6. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Oxygen Sensing, Hypoxia-Inducible Factors, and Disease Pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71. [Google Scholar] [CrossRef]
- Buffa, F.M.; Harris, A.L.; West, C.M.; Miller, C.J. Large Meta-Analysis of Multiple Cancers Reveals a Common, Compact and Highly Prognostic Hypoxia Metagene. Br. J. Cancer 2010, 102, 428–435. [Google Scholar] [CrossRef]
- Cao, Y.; Eble, J.M.; Moon, E.; Yuan, H.; Weitzel, D.H.; Landon, C.D.; Yu-Chih Nien, C.; Hanna, G.; Rich, J.N.; Provenzale, J.M.; et al. Tumor Cells Upregulate Normoxic HIF-1α in Response to Doxorubicin. Cancer Res. 2013, 73, 6230. [Google Scholar] [CrossRef] [Green Version]
- Daskalaki, I.; Gkikas, I.; Tavernarakis, N. Hypoxia and Selective Autophagy in Cancer Development and Therapy. Front. Cell Dev. Biol. 2018, 6, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazio, F.; Bordi, M.; Cianfanelli, V.; Locatelli, F.; Cecconi, F. Autophagy and Cancer Stem Cells: Molecular Mechanisms and Therapeutic Applications. Cell Death Differ. 2019, 26, 690–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaterjee, M.; van Golen, K.L. Breast Cancer Stem Cells Survive Periods of Farnesyl-Transferase Inhibitor-Induced Dormancy by Undergoing Autophagy. Bone Marrow Res. 2011, 2011, 362938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.; Song, E.; Codogno, P.; Mehrpour, M. The Roles of BECN1 and Autophagy in Cancer Are Context Dependent. Autophagy 2012, 8, 1853–1855. [Google Scholar] [CrossRef] [Green Version]
- Sk, Y.; J, W.; S, C.; Jl, G. Autophagy Differentially Regulates Distinct Breast Cancer Stem-like Cells in Murine Models via EGFR/Stat3 and Tgfβ/Smad Signaling. Available online: https://pubmed.ncbi.nlm.nih.gov/27197172/ (accessed on 28 September 2020). [CrossRef] [Green Version]
- Golden, E.B.; Cho, H.-Y.; Jahanian, A.; Hofman, F.M.; Louie, S.G.; Schönthal, A.H.; Chen, T.C. Chloroquine Enhances Temozolomide Cytotoxicity in Malignant Gliomas by Blocking Autophagy. Neurosurg. Focus 2014, 37, E12. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Xia, L.; Klaiber, U.; Sachsenmaier, M.; Hinz, U.; Bergmann, F.; Strobel, O.; Büchler, M.W.; Neoptolemos, J.P.; Fortunato, F.; et al. Effects of Neoadjuvant FOLFIRINOX and Gemcitabine-Based Chemotherapy on Cancer Cell Survival and Death in Patients with Pancreatic Ductal Adenocarcinoma. Oncotarget 2019, 10, 7276–7287. [Google Scholar] [CrossRef]
- Karasic, T.B.; O’Hara, M.H.; Loaiza-Bonilla, A.; Reiss, K.A.; Teitelbaum, U.R.; Borazanci, E.; De Jesus-Acosta, A.; Redlinger, C.; Burrell, J.A.; Laheru, D.A.; et al. Effect of Gemcitabine and Nab-Paclitaxel With or Without Hydroxychloroquine on Patients With Advanced Pancreatic Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 993–998. [Google Scholar] [CrossRef]
- Malhotra, J.; Jabbour, S.; Orlick, M.; Riedlinger, G.; Guo, Y.; White, E.; Aisner, J. Phase Ib/II Study of Hydroxychloroquine in Combination with Chemotherapy in Patients with Metastatic Non-Small Cell Lung Cancer (NSCLC). Cancer Treat. Res. Commun. 2019, 21, 100158. [Google Scholar] [CrossRef]
- A Phase I Trial of MK-2206 and Hydroxychloroquine in Patients with Advanced Solid Tumors—Abstract—Europe PMC. Available online: https://europepmc.org/article/med/31463691 (accessed on 28 September 2020).
- Xu, R.; Ji, Z.; Xu, C.; Zhu, J. The Clinical Value of Using Chloroquine or Hydroxychloroquine as Autophagy Inhibitors in the Treatment of Cancers: A Systematic Review and Meta-Analysis. Medicine 2018, 97, e12912. [Google Scholar] [CrossRef]
- Arnaout, A.; Robertson, S.J.; Pond, G.R.; Lee, H.; Jeong, A.; Ianni, L.; Kroeger, L.; Hilton, J.; Coupland, S.; Gottlieb, C.; et al. A Randomized, Double-Blind, Window of Opportunity Trial Evaluating the Effects of Chloroquine in Breast Cancer Patients. Breast Cancer Res. Treat. 2019, 178, 327–335. [Google Scholar] [CrossRef]
- Augustijns, P.; Geusens, P.; Verbeke, N. Chloroquine Levels in Blood during Chronic Treatment of Patients with Rheumatoid Arthritis. Eur. J. Clin. Pharmacol. 1992, 42, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Schrezenmeier, E.; Dörner, T. Mechanisms of Action of Hydroxychloroquine and Chloroquine: Implications for Rheumatology. Nat. Rev. Rheumatol. 2020, 16, 155–166. [Google Scholar] [CrossRef] [PubMed]
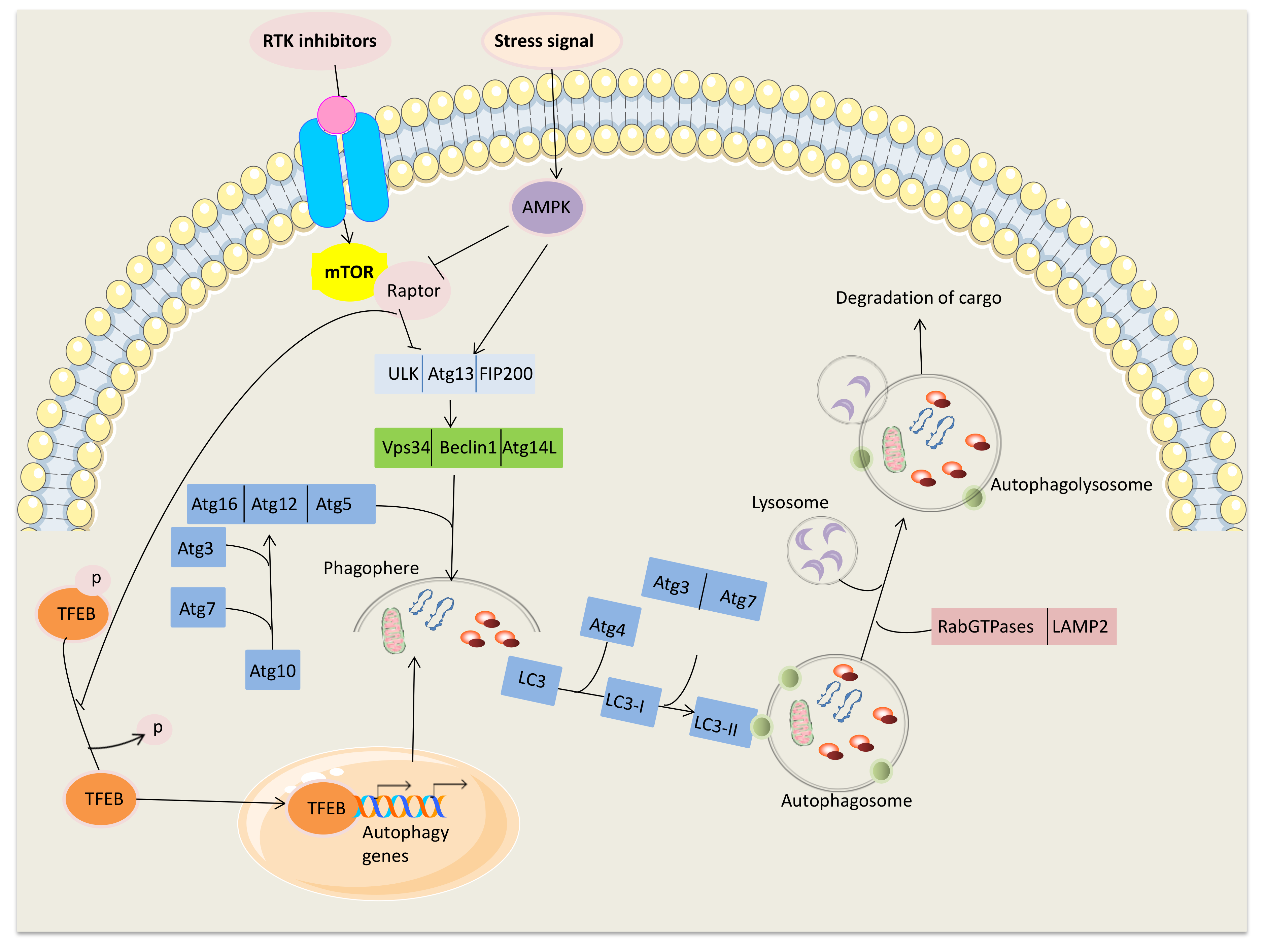

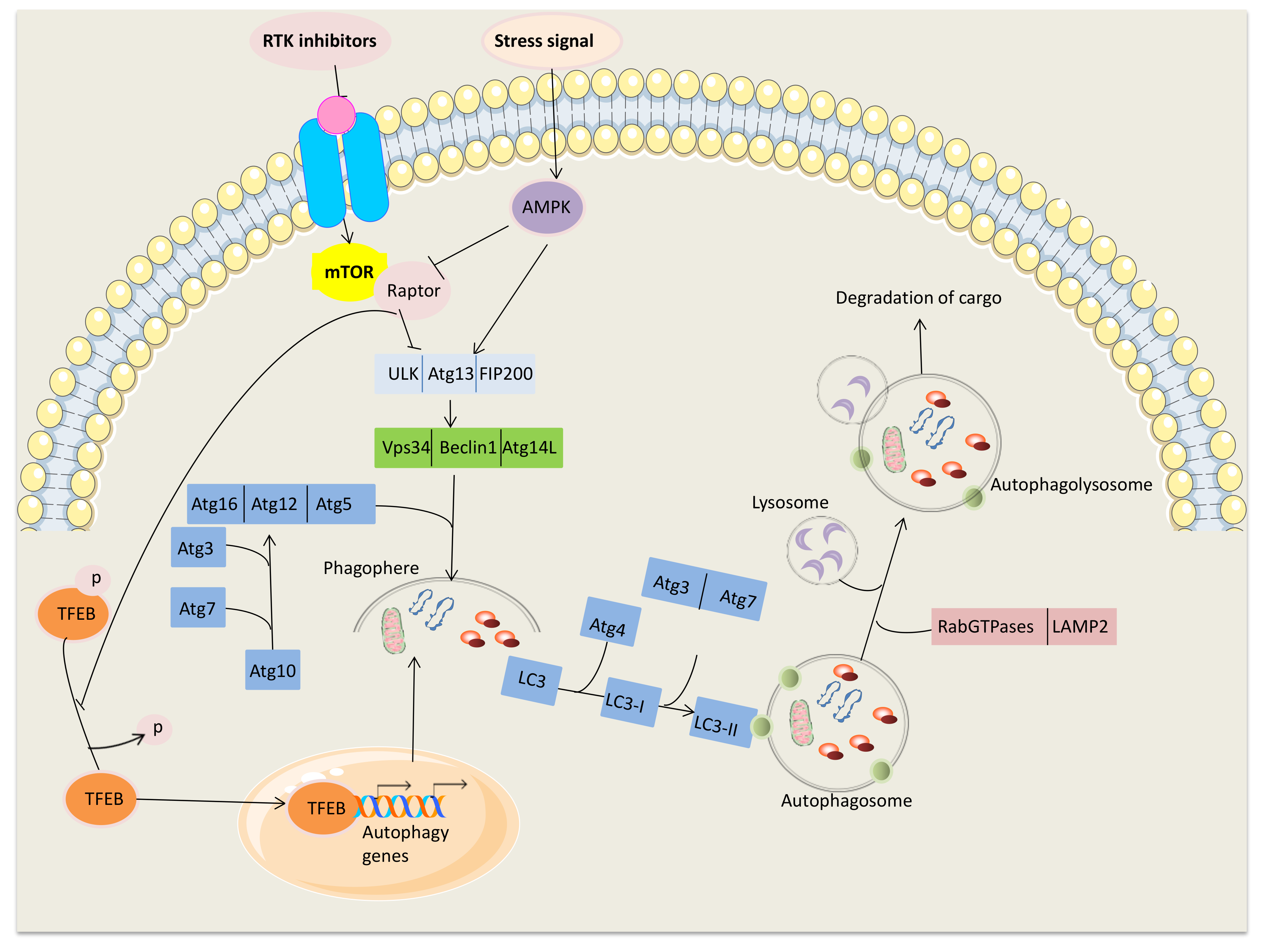{kind=link}
| Breast Cancer Stage/Subtype/Setting | Intervention | Study Design | Dosage | Clinicaltrial.gov |
|---|---|---|---|---|
| Invasive Breast Cancer/Neoadjuvant | Drug: Chloroquine Drug: Placebo | Phase 2 Randomized, Double-blind, placebo-controlled | chloroquine 500 mg daily as an oral capsule during the wait time to surgery. | * NCT02333890 |
| Advanced or Metastatic Breast Cancer | Chloroquine in Combination with taxane or taxane-like Chemo Agents | Phase II Non-Randomized, open label | chloroquine 250 mg daily as an oral capsule | NCT01446016 |
| Carcinoma, Intraductal, Noninfiltrating DCIS Ductal Carcinoma In Situ Neoadjuvant | Chloroquine | Phase 1 Phase 2 Non-Randomized open label | chloroquine standard dose (500mg/week) or chloroquine low dose (250mg/week) for 1 month prior to surgical removal of the tumor. | NCT01023477 |
| Metastatic Breast Cancer | Hydroxychloroquine + Ixabepilone | Phase 1 Phase 2 Non-Randomized, open label | Dose escalation from 200 mg po qd to 200 mg po bid. | # NCT00765765 |
| Estrogen Receptor-Positive Metastatic Breast Cancer | Hydroxychloroquine in combination with the current hormonal therapy | Phase Ib/II Study Non-Randomized, open label | Not available | NCT02414776 |
| Invasive breast cancer | Arm A: Abemaciclib Arm B: Abemaciclib + Hydroxychloroquine | Phase II Pilot Trial, Randomized, open lable | Hydroxychloroquine 600 mg BID | NCT04523857 |
| Recurrent Breast Cancer | Phase II Arm A: Hydroxychloroquine for 24 weeks Arm B: Hydroxychloroquine for 24 weeks + Gedatolisib and × 2 weeks administered weekly Arm C: Hydroxychloroquine for 24 weeks + Gedatolisib and × 6 weeks administered weekly Arm D: Hydroxychloroquine for 24 weeks + Gedatolisib and × 12 weeks administered weekly Phase Ib: Hydroxychloroquine for 24 weeks + Gedatolisib and × 6 weeks administered weekly | Phase 1 Phase 2 Randomized open label | Hydroxychloroquine 600 mg BID | NCT03400254 |
| Breast Cancer Stage IIB | Arm A: Hydroxychloroquine Arm B: Everolimus Arm C: Hydroxychloroquine + Everolimus Arm D: Observation | Phase 2 Pilot Randomized | Not available | NCT03032406 |
| HR+/Her 2- Advanced Breast Cancer | Arm A: Abemaciclib + Hydroxychloroquine 200 mg b.i.d. Arm B. Abemaciclib + Hydroxychloroquine 400 mg b.i.d. Arm C: Abemaciclib + Hydroxychloroquine 600 mg b.i.d. Arm D: Abemaciclib + Hydroxychloroquine + endocrine therapy. | Phase 1 Non-Randomized Sequential Assignment Open Label | Hydroxychloroquine 200 mg Hydroxychloroquine 400 mg Hydroxychloroquine 600 mg | NCT04316169 |
| Participants with advanced, metastatic (stage IV) breast cancer (phase I) Participants with early stage (stage I-III) breast cancer (phase II) | Hydroxychloroquine, Palbociclib, and Letrozole | Phase 1 Phase 2 Open label | Hydroxychloroquine 400 mg | NCT03774472 |
| Breast Cancer Neoadijuvant | Hydrochloroquine | Phase II Pilot Open Label | Hydrochloroquine 800 mg per os once, and then 400 mg per day | NCT01292408 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cocco, S.; Leone, A.; Piezzo, M.; Caputo, R.; Di Lauro, V.; Di Rella, F.; Fusco, G.; Capozzi, M.; Gioia, G.d.; Budillon, A.; et al. Targeting Autophagy in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 7836. https://doi.org/10.3390/ijms21217836
Cocco S, Leone A, Piezzo M, Caputo R, Di Lauro V, Di Rella F, Fusco G, Capozzi M, Gioia Gd, Budillon A, et al. Targeting Autophagy in Breast Cancer. International Journal of Molecular Sciences. 2020; 21(21):7836. https://doi.org/10.3390/ijms21217836
Chicago/Turabian StyleCocco, Stefania, Alessandra Leone, Michela Piezzo, Roberta Caputo, Vincenzo Di Lauro, Francesca Di Rella, Giuseppina Fusco, Monica Capozzi, Germira di Gioia, Alfredo Budillon, and et al. 2020. "Targeting Autophagy in Breast Cancer" International Journal of Molecular Sciences 21, no. 21: 7836. https://doi.org/10.3390/ijms21217836
APA StyleCocco, S., Leone, A., Piezzo, M., Caputo, R., Di Lauro, V., Di Rella, F., Fusco, G., Capozzi, M., Gioia, G. d., Budillon, A., & De Laurentiis, M. (2020). Targeting Autophagy in Breast Cancer. International Journal of Molecular Sciences, 21(21), 7836. https://doi.org/10.3390/ijms21217836